

Abstract
Cleavage and polyadenylation specificity factor 6 (CPSF6) is a cellular protein involved in mRNA processing. Emerging evidence suggests that CPSF6 also plays key roles in HIV-1 infection, specifically during nuclear import and integration targeting. However, the cellular and molecular mechanisms that regulate CPSF6 expression are largely unknown. In this study, we report a post-transcriptional mechanism that regulates CPSF6 via the cellular microRNA miR-125b. An in silico analysis revealed that the 3′UTR of CPSF6 contains a miR-125b–binding site that is conserved across several mammalian species. Because miRNAs repress protein expression, we tested the effects of miR-125b expression on CPSF6 levels in miR-125b knockdown and over-expression experiments, revealing that miR-125b and CPSF6 levels are inversely correlated. To determine whether miR-125b post-transcriptionally regulates CPSF6, we introduced the 3′UTR of CPSF6 mRNA into a luciferase reporter and found that miR-125b negatively regulates CPSF6 3′UTR-driven luciferase activity. Accordingly, mutations in the miR-125b seed sequence abrogated the regulatory effect of the miRNA on the CPSF6 3′UTR. Finally, pulldown experiments demonstrated that miR-125b physically interacts with CPSF6 3′UTR. Interestingly, HIV-1 infection down-regulated miR-125b expression concurrent with up-regulation of CPSF6. Notably, miR-125b down-regulation in infected cells was not due to reduced pri-miRNA or pre-miRNA levels. However, miR-125b down-regulation depended on HIV-1 reverse transcription but not viral DNA integration. These findings establish a post-transcriptional mechanism that controls CPSF6 expression and highlight a novel function of miR-125b during HIV-host interaction.
Keywords: human immunodeficiency virus (HIV), microRNA (miRNA), post-transcriptional regulation, epigenetics, RNA binding protein, host-pathogen interaction, 3′UTR, cleavage and polyadenylation specificity factor 6 (CPSF6), HIV, miR-125b, miRNA biogenesis
Introduction
Cleavage and polyadenylation specificity factor 6 (CPSF6) functions as part of the cleavage factor I mammalian (CFIm) complex (1, 2). CFIm is a RNA-binding protein complex and component of a multiprotein cleavage and adenylation complex that regulates mRNA processing and polyadenylation (3). CFIm is a heterotetrameric complex consisting of a dimer of CPSF5 and a dimer of either CPSF6 or CPSF7 (1, 4, 5). CFIm is also a component of paraspeckles, ribonucleoprotein complexes that are involved in regulating gene expression through nuclear retention of adenosine to inosine (A-to-I)–edited RNA (6, 7). A recent study suggested that through its association with paraspeckles, CPSF6 is involved in the regulation of breast cancer cell viability and tumorigenic capacity (8). In recent years, CPSF6 has also garnered a great deal of attention for its emerging role during the early steps of HIV 1 (HIV-1) infection (9–15).
HIV-1 has infected over 72 million people and has claimed more than 37 million lives worldwide (16). Untreated HIV-1 infection causes progressive CD4+ T-cell loss and a wide range of immunological abnormalities leading to acquired immune deficiency syndrome (AIDS) (17). Productive HIV-1 infection depends on integration of the reverse transcribed viral DNA into the host genome. The virally-encoded integrase enzyme carries out integration of the viral DNA in the context of a nucleoprotein complex called the preintegration complex (PIC)3 (18). The PIC, containing the viral DNA and other viral and cellular proteins, is actively transported into the nucleus of the target cell through the nuclear pore complex (19). A variety of cellular proteins including cyclophilin A (CypA), nucleoporin (NUP) proteins NUP358 and NUP153, the β-karyopherin transportin-3 (TNPO3), and CPSF6 have been implicated in PIC nuclear import (10, 11, 20–23). In the nucleus, HIV-1 DNA preferentially integrates into gene-dense regions of the host genome (24). Accumulating evidence suggests that many of the cellular factors involved in PIC nuclear import also facilitate targeting of the viral DNA integration into gene dense chromosomal regions (25–28). Although the exact mechanism of HIV-1 integration targeting is unclear, recent studies have clarified that CPSF6 is a key player in targeting HIV-1 DNA into the gene dense region of the host chromosomes (29, 30).
Despite the established role of CPSF6 in mRNA processing and HIV-1 infection, the cellular and molecular mechanism(s) by which CPSF6 expression is regulated remains largely unknown. In this study, we describe an miRNA-mediated post-transcriptional mechanism of CPSF6 regulation. Our bioinformatics analysis revealed that the CPSF6 3′UTR contains a binding site for the cellular miRNA miR-125b. This binding site is strikingly conserved across a number of mammalian species implying a regulatory role of this miRNA. Therefore, we examined miR-125b–mediated regulation of CPSF6 by carrying out knockdown and over-expression studies. Results from these genetic experiments revealed that miR-125b levels negatively associate with CPSF6 protein expression. To further pinpoint post-transcriptional regulation of CPSF6 by miR-125b, luciferase reporter constructs containing the 3′UTR sequences of CPSF6 mRNA were generated. Luciferase assays revealed that CPSF6 3′UTR activity is negatively regulated by miR-125b expression. Subsequently, physical interaction between the CPSF6 3′UTR and miR-125b was demonstrated by pulldown studies. Interestingly, upon HIV-1 infection, miR-125b levels were reduced concurrent with increased CPSF6 expression. The down-regulation of miR-125b in infected cells was not due to reduced levels of miR-125b precursors such as pre-miRNA or pri-miRNA. Interestingly, our results revealed that miR-125b down-regulation depended on reverse transcription of the viral genome but not on viral DNA integration. Finally, over-expression and knockdown of miR-125b in infected cells showed that the miR predominantly affected nuclear entry of HIV-1. Collectively, these studies describe: (a) post-transcriptional regulation of CPSF6, (b) identify CFSP6 as a direct target of miR-125b, (c) establish effects of miR-125b on early steps of HIV-1 infection, and (d) reveal a potential mechanism by which HIV-1 regulates miR-125b expression during early steps of infection.
Results
CPSF6 3′UTR contains a highly conserved binding site of miR-125b
CPSF6 is part of the CFIm complex and is well-established for its role in polyadenylation of cellular mRNAs (3). Emerging evidence also show that CPSF6 plays important roles in HIV-1 infection at the steps of PIC nuclear import (10, 11) and integration targeting (29, 30). However, the cellular and molecular mechanisms of regulating CPSF6 expression remain largely unknown. In this study, we examined the mechanisms involving post-transcriptional regulation of CPSF6 by initially subjecting its 3′UTR region to in silico analysis. CPSF6 mRNA is ∼6.58 kilobases (kb) with 10 exons and the 3′UTR consists of 4847 nucleotides (Fig. 1A). We used two different algorithms, RNAhybrid 2.1.2 (31, 32) and biFold:RNA structures (33), which employed intramolecular base-pairing of RNA:RNA interactions (RNAHybrid) and unimolecular and bimolecular interactions (biFold:RNA structures) to enhance in silico prediction accuracy. Interestingly, both platforms predicted a putative miR-125b–binding site within the CPSF6 3′UTR (Fig. 1, B and C). These studies also showed the formation of a hairpin-loop structure between the two RNA molecules that is essential for miRNA-mediated post-transcriptional regulation (Fig. 1, B and C). The 7-mer target site of miR-125b was located at nucleotide positions 2204–2211 of the CPSF6 mRNA within exon 10, corresponding to positions 471–477 of the 3′UTR (Fig. 1A). Importantly, the predicted miR-125b target sequence was highly conserved in the CPSF6 3′UTR of several mammalian species (Fig. 1D), another hallmark of miRNA-mediated post-transcriptional regulation (34).
Figure 1.

miR-125b negatively regulates CPSF6 expression. A, schematic representation of CPSF6 mRNA (∼6.6 kb) containing the miR-125b seed sequence in the 3′UTR (∼4.8 kb). Results from in silico analysis of CPSF6 3′UTR sequences for the presence of miR-125–binding sites and the formation of hairpin structures between the miR and the 3′UTR by using (B) RNAHybrid 2.1.2 and (C) RNAstructure-biFold prediction web servers. Mfe, minimum free energy of the predicted secondary structures. D, sequence alignment of 3′UTR of CPSF6 mRNA from different mammalian species. E and F, qPCR analyses of miR-125b levels in HEK-293 T cells treated with anti-miRs (E) or miR-mimics (F) of miR-125b or scrambled controls (F). G, representative immunoblot showing CPSF6 protein expression in miR-125b–knockdown and over-expressing cells. The graph above the immunoblot shows densitometry analysis of immunoblots from three independent experiments. H, endogenous levels of miR-125b in SupT1 and CEM cells as measured by qPCR. Results from qPCR analysis of knockdown of miR-125b levels in SupT1 cells (I) and over-expression of miR-125b via miR-mimics in CEM (J) and THP1 (K) cells. Representative immunoblots showing CPSF6 and GAPDH (loading control) protein expression in miR-125b–knockdown SupT1 cells (L), miR-125b over-expressing CEM cells (M), and miR-125b over-expressing THP1 cells (N). The graphs above the blots show densitometry analysis of immunoblots from three independent experiments. Error bars represent S.E. * represents p < 0.05 for the comparison of anti-miR and miR-mimics versus scrambled controls.
CPSF6 expression inversely correlates with miR-125b levels
To examine the biological relevance of the in silico prediction data described in Fig. 1, A–D, we tested whether miR-125b regulates CPSF6 protein expression in knockdown and over-expression studies. We transfected locked nucleic acid (LNA)-based miR-125b mimics and anti-miRs into HEK-293T cells and isolated total RNA after 24 h. qPCR analysis of reverse transcribed RNA showed a significant reduction in miR-125b levels in cells transfected with anti-miRs (Fig. 1E) and a marked increase in miR-125b levels in cells transfected with miR mimics (Fig. 1F). Measurement of CPSF6 protein levels in cellular lysates by Western blotting showed that cells with lower levels of miR-125b contained higher CPSF6 protein levels compared with control cells (Fig. 1G). Accordingly, higher miR-125b levels led to a reduced level of CPSF6 protein (Fig. 1G), supporting an inverse association between miR-125b expression and CPSF6 protein levels.
We also examined whether miR-125b expression negatively associates with CPSF6 protein levels in HIV-1 susceptible T and monocytic cell lines. SupT1 and CEM cell lines were specifically chosen as T cell models because SupT1 cells expressed miR-125b endogenously, whereas CEM cells contained very low or undetectable levels of miR-125b (Fig. 1H). Therefore, these two cell lines served as appropriate models for knockdown and over-expression studies, respectively. Knockdown of miR-125b was achieved by electroporation of anti-miRs into SupT1 cells (Fig. 1I) and over-expression of miR-125b was conducted in CEM cells using miR-125b mimics (Fig. 1J). Measurement of CPSF6 levels in SupT1 cells revealed that lower miR-125b levels were associated with higher CPSF6 protein (Fig. 1L). Conversely, increasing miR-125b levels resulted in a significant reduction in CPSF6 protein levels in CEM cells (Fig. 1M), consistent with the data obtained using HEK-293T cells (Fig. 1G). Similar inverse relationships between miR-125b and CPSF6 expression levels was also observed in THP1 monocytic cells (Fig. 1, K and N). Collectively, these results provide evidence that miR-125b levels inversely correlate with CPSF6 protein expression and support the in silico prediction of post-transcriptional regulation of CPSF6 by miR-125b.
CPSF6 3′UTR activity is regulated by miR-125b
miRNAs negatively regulate gene expression by usually binding to the 3′UTR of the target mRNA (35, 36). Therefore, we probed whether miR-125b targets the 3′UTR of CPSF6 mRNA. To test this, we introduced the CPSF6 3′UTR into a luciferase reporter construct. Because the 3′UTR of CPSF6 is about 4.9 kb (Fig. 2A), we generated three truncated constructs that retained the miR-125b–binding site: 1) 3′UTR-small containing 1–1011 bp from the 5′-end (3′UTR-S); 2) 3′UTR-medium containing 1–3114 bp (3′UTR-M); and 3) 3′UTR-large containing 1–4243 bp (3′UTR-L) (Fig. 2A). These 3′UTR reporter constructs or a control plasmid lacking the 3′UTR (ΔUTR) were transfected individually into HEK-293T cells. At 24 h post-transfection, cellular lysates were prepared for luciferase activity measurements. The reporter constructs that harbored the miR-125b–binding site showed less luciferase activity compared with the control construct that lacked the binding site (Fig. 2B). Because HEK-293T cells express miR-125b (Fig. 1, A and B), the reduced luciferase activity observed with the CPSF6 3′UTR constructs is likely due to the effects of the endogenous miRs on the CPSF6 3′UTR of the reporter constructs.
Figure 2.

miR-125b negatively regulates CPSF6 3′UTR activity. A, schematic representation of the three different sized DNA fragments of CPSF6 3′UTR that were cloned downstream of the luciferase stop codon to generate the corresponding reporter vectors: 3′UTR-small (3′UTR-S), 3′UTR-medium (3′UTR-M), and 3′UTR-large (3′UTR-L). The location of the miR-125b seed sequence is presented as an inverted black triangle. HEK-293T cells were transfected with either of these 3′UTR luciferase reporter vectors or the control pΔUTR (ΔUTR) vector in the presence or absence of miR-125b mimics or anti-miR-125b. 24 h post-transfection, luciferase activity was measured in the cellular lysates and plotted relative to the controls. B, luciferase activity of the CPSF6 3′UTRs and ΔUTR vectors. Luciferase activity of (C) ΔUTR, (D) 3′UTR-S, (E) 3′UTR-M, and (F) 3′UTR-L vector in the presence of anti-miRs or miR-mimics or scrambled controls. G–K, effects of altered miR-125b levels on CPSF6 3′UTR activity in CKO cells. Immunoblots showing CPSF6 expression in WT HEK-293T (CWT) (G, top panel) and CKO cells (G, middle panel), and GAPDH (loading control) expression in CKO cells (G, bottom panel). Luciferase activity in CKO cells transfected with (H) ΔUTR, (I) 3′UTR-S, (J) 3′UTR-M, and (K) 3′UTR-L vector in the presence of anti-miRs or miR-mimics or scrambled controls in CKO cells. Error bars represent S.E., whereas, *, p < 0.05 represents the comparison of anti-miR or miR mimic samples versus scrambled controls.
Each deletion construct supported a similar level of reduced luciferase activity (Fig. 2B), which is consistent with our in silico identification of the miR-125b–binding site within the upstream 1011-bp region of the 3′UTR (Figs. 1 and 2A). We next co-transfected the CPSF6 3′UTR constructs individually with miR-mimics or anti-miRs. miR-125b expression in co-transfected cells was confirmed by qPCR (data not shown). Alterations in miR-125b levels by knockdown or over-expression minimally affected the luciferase activity of the control reporter that lacked the 3′UTR (ΔUTR) (Fig. 2C). However, in the presence of anti-miRs, a significant increase in luciferase activity was observed in the lysate of cells individually transfected with the three different CPSF6 3′UTR constructs (Fig. 2, D–F). Conversely, increasing miR-125b levels resulted in a decrease in the luciferase activity of the three 3′UTR-containing constructs (Fig. 2, D–F). The modest decrease in luciferase activity in miR-125b over-expressed cells could be due to competition between the endogenous CPSF6 3′UTR and the 3′UTR of the reporter constructs for binding to the miR-mimics.
To address potential confounding effects from the 3′UTR of the endogenous CPSF6 mRNA, we next carried out co-transfection studies in CPSF6 knockout cells (29). Western blot analysis confirmed lack of CPSF6 protein expression in the knockout HEK-293T (CKO) cells compared with CWT cells that expressed CPSF6 (Fig. 2G). Knockdown and over-expression studies in these cells revealed that altering miR-125b expression levels in CKO cells minimally affected the luciferase activity of the CPSF6 ΔUTR control construct (Fig. 2H), consistent with the data in Fig. 2C. Accordingly, co-transfection of the three CPSF6 3′UTR constructs individually along with anti-miRs also showed significant increases in luciferase activity in CKO cells (Fig. 2, I–K) akin to the results with CWT cells (Fig. 2, D–F). Conversely, over-expression of miR-125b greatly reduced CPSF6 3′UTR driven luciferase activity in CKO cells compared with the scrambled controls (Fig. 2, I–K). Collectively, these results provide further evidence that the CPSF6 3′UTR is negatively regulated by miR-125b expression.
Mutations in the binding site abrogate miR-125b's effect on CPSF6 3′UTR
Our in silico studies identified that the miR-125b target site is located at nucleotides 2204–2211 of CPSF6 mRNA, corresponding to 471–477 positions within the 3′UTR (Fig. 1A). To determine the role of the genetic interaction between miR-125b and CPSF6 3′UTR in post-transcriptional regulation, we introduced specific mutations within the binding site. First, in silico mutational analyses revealed that introduction of two specific nucleotide substitutions CA > AT (Mut1) and CA > GT (Mut2) in the CPSF6 3′UTR would collapse the secondary structure formed by the interaction between the two RNA molecules (Fig. 3, A–C). Structural models also revealed that these mutations would reduce the stability and binding affinity between miR-125b and the CPSF6 3′UTR as measured by the mean free energy (Fig. 3, A–C). Therefore, we introduced these two specific substitutions into the CPSF6 3′UTR of the 3′UTR-S luciferase reporter construct by site-directed mutagenesis. Then, we studied the effects of miR-125b on the mutant 3′UTRs using the luciferase reporter assay. Transfection of the parental 3′UTR-S or mutant derivative (Mut1 and Mut2) constructs showed that the luciferase activity of the mutants was comparable with that of the CPSF6 ΔUTR control construct (Fig. 3, D and E). Importantly, co-transfection of the CPSF6 3′UTR mutant constructs either with the anti-miRs or miR-mimics showed minimal effect on luciferase activity (Fig. 3, G–I) even though the anti-miRs and miR-mimics, as expected, regulated parental 3′UTR-S activity (Fig. 3F). These results indicate that specific genetic interaction(s) between miR-125b and the 3′UTR sequences drive post-transcriptional regulation of CPSF6 expression.
Figure 3.

Mutations in the miR binding sequence abrogate the regulatory effects of miR-125b on the CPSF6 3′UTR. A–C, in silico analysis of the effects of two different mutations, Mut1 (CA > AT) and Mut2 (CA > GT), introduced at the miR-125b–binding site in CPSF6 3′UTR on their binding stability and affinity (as measured by mfe) and the secondary hairpin loop structure integrity. Shown are the effects of Mut1 (B) and Mut2 (C) compared with WT CPSF6 3′UTR (A). D and E, effects of endogenous miR-125b on the activity of the CPSF6 3′UTR mutants, Mut1 and Mut2. Shown is the comparison between the luciferase activity in the lysates of HEK-293T cells transfected with Mut1 (D) or Mut2 (E) and the luciferase activity of ΔUTR and 3′UTR-S. F–I, effects of miR-125b overexpression or down-regulation on the activity of the CPSF6 3′UTR mutants, Mut1 and Mut2. Luciferase activity in lysates prepared from HEK-293T cells transfected with (F) 3′UTR-S, (G) ΔUTR, (H) Mut1, or (I) Mut2 reporter constructs in the presence or absence of anti-miRs or miR-mimics or scrambled controls. Data presented are mean values of three independent experiments with error bars representing S.E. *, represents p < 0.05 for the comparison of miR-mimics or anti-miR versus scrambled controls.
miR-125b physically interacts with CPSF6 mRNA through complementary base pairing
miRNAs negatively regulate gene expression by directly binding to the 3′UTR of their target mRNA (35, 36). Our data in Figs. 1–3 provided compelling evidence that miR-125b post-transcriptionally regulates CPSF6 through a direct interaction involving complementary base pairing. To further probe whether miR-125b directly binds to the CPSF6 3′UTR, we employed a pulldown assay using biotinylated miR-mimics (37). The 3′UTR-S and 3′UTR-Mut1 constructs were co-transfected with either biotinylated miR-mimics or scrambled controls. The biotinylated miR-125b–CPSF6 mRNA complex was pulled down with streptavidin-coated magnetic beads. Enrichment of CPSF6 mRNA in the pulldown samples was quantified by qPCR using CPSF6 3′UTR-specific primers. PARP-1 was used as a positive control because it is a direct target of miR-125b (37, 38). In addition, β-actin was included as a negative control because the 3′UTR of its mRNA does not contain the binding site of miR-125b. The data in Fig. 4 show that in the biotinylated miR-125b pulldown complex, CPSF6 mRNA was significantly enriched compared with the scrambled control. As expected, PARP-1 mRNA was also highly enriched in the pulldown complex, whereas there was minimal enrichment of β-actin mRNA (Fig. 6). Finally, analysis of the CPSF6 3′UTR-Mut1 pulldown revealed that the specific mutation in the miR-125b–binding site abrogates enrichment of CPSF6 mRNA in the pulldown assay (Fig. 4). Collectively, these data establish that miR-125b physically interacts with the CPSF6 3′UTR via complementary base pairing to post-transcriptionally regulate CPSF6 expression.
Figure 4.
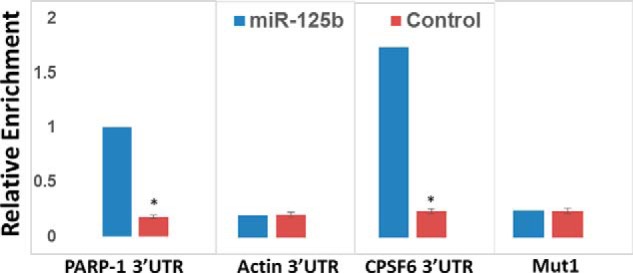
miR-125b physically interacts with CPSF6 mRNA. HEK-293T cells were transfected with biotinylated miR-125b mimics or scrambled controls and the miR-125b:mRNA complexes were pull down with streptavidin-coated magnetic beads. Enrichments of mRNAs of interest were quantified by qPCR with primers complementary to their respective 3′UTRs. Data shown are enrichment of PARP-1 (positive control), actin (negative control), and CPSF6 3′UTR levels. Blue bars represent pull downs with miR-mimics, whereas red bars represent pull downs with scrambled controls. Data presented are mean values of three independent experiments with error bars representing S.E. *, represents p < 0.05 for the comparison of miR-mimics versus scrambled controls.
Figure 6.

A–C, HIV-1 infection does not affect miR-125b precursors. HEK-293T cells were inoculated with pseudotyped HIV-1 GFP reporter virus and levels of (A) pri-, (B) pre-, and (C) mature miR-125b was measured in infected and uninfected cells by qPCR. D and E, down-regulation of miR-125b depends on HIV-1 reverse transcription. D, HEK-293T cells were inoculated with pseudotyped HIV-1 GFP virus in the presence of EFV or RAL and productive infection was assessed by GFP expression. E, RNA from these cells were analyzed for measuring miR-125b levels by qPCR. F-H, effects of miR-125b expression on early steps of HIV-1 infection. HEK-293T cells were transfected with either anti-miRs or mimics of miR-125b and after 24 h, inoculated with pseudotyped HIV-1 GFP virus. RNA from these cells was isolated and subjected to qPCR to measure copies of (F) reverse transcription, (G) 2-LTR circles, and (H) integration. Data presented are from three independent experiments. Error bars represent S.E. *, represents p < 0.05 for the comparison of uninfected versus infected cells.
HIV-1 infection activates miR-125b/CPSF6 pathway
miR-125b is a key cellular miRNA that plays important roles in various cellular functions (39–42). miR-125b has also been reported to inhibit HIV-1 replication by reducing viral protein translation (41, 43), which occurs after HIV-1 integration. Conversely, CPSF6 has been identified as an important cellular factor that facilitates HIV-1 nuclear import and integration targeting (10, 11, 29, 30). Given that miR-125b negatively regulates CPSF6 expression (Figs. 1–3), we tested if HIV-1 infection impacts the miR-125b–CPSF6 pathway. HEK-293T cells (Fig. 5, A and B), SupT1 cells (Fig. 5, C and D), and THP1 cells (Fig. 5, E and F) were infected with vesicular stomatitis virus G glycoprotein (VSV-G) pseudotyped HIV-1 particles that contained the GFP reporter gene. Productive infection of HEK-293T cells was confirmed by expression of HIV-1 capsid protein (p24) in the infected cells by Western blot analysis (Fig. 5A). The presence of higher molecular weight protein bands in the infected cell lane represents expected unprocessed HIV-1 Gag protein (p55) and other processing intermediates (44). In SupT1 (Fig. 5C) and THP1 cells (Fig. 5E), infection was probed by measuring GFP expression via flow cytometry. Then, total RNA from infected and uninfected control cells was subjected to qPCR to measure miR-125b levels. Interestingly, miR-125b expression was significantly reduced in HIV-1–infected HEK-293T (Fig. 5B), SupT1 cells (Fig. 5, D and F), and THP-1 cells (Fig. 5F) compared with the respective uninfected cells. Furthermore, Western blot analysis of the cellular lysates revealed that CPSF6 expression was up-regulated in HIV-1–infected cells compared with the uninfected cells (Fig. 5, G–J). These results indicate that HIV-1 infection down-regulates miR-125b concurrent with the induction of CPSF6 expression.
Figure 5.

HIV-1 infection down-regulates miR-125b expression. A and B, HEK-293T cells were inoculated with pseudotyped HIV-1 GFP reporter virus. A, infection was assessed by detecting HIV-1 p24 protein in the cellular lysate by Western blot analysis. Purified capsid (CA) protein served as a positive control. B, RNA isolated from these cells was subjected to qPCR to measure miR-125b levels. C and D, SupT1 cells, and E and F, THP1 monocytic cells were inoculated with pseudotyped HIV-1 GFP reporter virus. Productive infection in SupT1 (C) and THP1 (E) cells was assessed via GFP expression. miR-125b expression in uninfected and infected SupT1 (D) and THP1 (F) cells was measured by qPCR. Representative immunoblots showing CPSF6 protein expression in uninfected and infected HEK-293T (G) cells and SupT1 (I) cells. Graphs showing the densitometry analysis of CPSF6 expression in HEK-293T (H) and SupT1 (J) cells from three independent experiments. Error bars represent S.E. *, represents p < 0.05 for the comparison of uninfected versus infected cells.
HIV-1 infection does not affect the levels of pre- and pri-miR-125b levels
miRNAs are derived from primary transcripts termed “pri-miRNAs” that are subsequently processed to precursor miRNA (pre-miRNA) prior to the biogenesis of mature miRNAs (1). Therefore, to understand a possible mechanism of miR-125b down-regulation by HIV-1 infection, we measured the levels of miR-125b precursors in infected cells. HEK-293T cells were infected with VSV-G–pseudotyped HIV-1 particles. RNA isolated from the infected and uninfected controls cells were subjected to qPCR using primer sets designed to amplify specific regions of pri-miR-125b, pre-miR-125b, and mature miR-125b. Results from these analyses revealed that the levels of both pri-miR-125b (Fig. 6A) and pre-miR-125b (Fig. 6B) were not significantly reduced in the infected cells compared with the expression levels in uninfected controls. As expected, expression of mature miR-125b was significantly reduced in the infected cells relative to the controls (Fig. 6C). These observations strongly suggest that down-regulation of miR-125b in HIV-1–infected cells is not due to reduced biogenesis of the miR from its precursors.
Down-regulation of miR-125b depends on HIV-1 reverse transcription but not on viral DNA integration
HIV-1 replication depends on key early steps of infection including reverse transcription, nuclear entry, and viral DNA integration (45). To understand whether down-regulation of miR-125b depends on any of these early steps, we measured miR-125b levels in infected cells in the presence of inhibitors that block HIV-1 reverse transcription (EFV) or integration (RAL). HEK-293T cells were inoculated with VSV-G–pseudotyped HIV-1 particles and cultured in the presence of either EFV (100 μm) or RAL (100 μm). At these concentrations the inhibitor's HIV-1 infection was dramatically reduced (Fig. 6D). To measure miR-125b levels, RNA from infected and uninfected control cells was subjected to qPCR. As expected, HIV-1 infection resulted in the down-regulation of miR-125b compared with the uninfected cells (Fig. 6E). Interestingly, miR-125b expression was not significantly down-regulated in infected cells treated with EFV (Fig. 6E). Notably, the miR-125b level in the EFV-treated infected cells was comparable with its expression in uninfected control cells (Fig. 6E). Conversely, in infected cells treated with RAL, miR-125b expression remained significantly down-regulated to a level that was comparable with the infected cells cultured without the inhibitors (Fig. 6E). Collectively, these results indicate that reverse transcription of the viral genome is required for the down-regulation of miR-125b in HIV-1–infected cells, however, this effect is not dependent on the integration of the viral DNA into the host genome.
miR-125b expression affects nuclear entry step of HIV-1 infection
Our results in Fig. 5 demonstrated that miR-125b expression was down-regulated in HIV-1–infected cells. Therefore, to better understand the interaction between the miR and virus, we probed the effects of miR-125b expression on the early steps of HIV-1 infection including reverse transcription, nuclear entry, and integration. We transfected miR-mimics and anti-miRs into HEK-293T cells and after 24 h, these cells were inoculated with VSV-G–pseudotyped HIV-1 particles. DNA isolated from these cells were subjected to qPCR analysis to measure effects of miR-125b expression on accumulation of late reverse transcription products, formation of 2-LTR circles as a surrogate for viral nuclear entry, and integration. Results from these analyses revealed that altering miR-125b expression, either through over-expression or knockdown, minimally affected accumulation of reverse transcription products (Fig. 6F). Interestingly, measurement of 2-LTR circles (Fig. 6G) showed that reducing miR-125b expression resulted in an increase in HIV-1 nuclear entry. However, over-expression of miR-125b minimally changed the number of 2-LTR circles (Fig. 6G). Finally, qPCR analysis revealed that over-expression or knockdown of miR-125b did not significantly influence the level of integrated HIV-1 DNA significantly. Collectively, these studies suggest that miR-125b down-regulation enhances HIV-1 nuclear entry presumably due to increased CPSF6 levels.
Discussion
miRNA's are small (∼17–24 nucleotide) noncoding RNAs that post-transcriptionally regulate gene expression (46, 47). miRNAs negatively regulate gene expression by binding to the 3′UTR of the target mRNA that causes translational repression (34, 48, 49) or mRNA degradation (50, 51). Notably, a single miRNA can interact with multiple targets due to redundancy in base pairing between the two RNAs (48, 52, 53). Therefore, miRNAs regulate many cellular pathways and play widespread roles in cellular homeostasis including the regulation of host:pathogen interaction (35, 36, 48).
miR-125 is a family of highly conserved miRNAs found in species from nematodes to humans (54). In humans, miR-125 consists of three homologs, hsa-miR-125a, hsa-miR-125b-1, and hsa-miR-125b-2, that share the same seed sequence (55). miR-125b is transcribed from two loci located on chromosome 11q23 (hsa-miR-125b-1) and 21q21 (hsa-miR-125b-2) (55, 56). A body of evidence shows key functions of miR-125b in cell survival, differentiation, and multiple malignancies (39). Additionally, miR-125b regulates genes involved in innate immunity, inflammation, and hematopoietic differentiation (39). Interestingly, miR-125b also belongs to a group of cellular miRNAs that suppress HIV-1 replication (41, 42). The anti-HIV activity of these miRNAs was first reported in resting CD4+ T cells through their ability to suppress viral protein translation (41). This study reported that compared with activated CD4+ T cells, resting CD4+ T cells showed higher levels of anti-HIV miRNAs including miR-125b, miR-28, miR-150, miR-223, and miR-382. Accordingly, in resting CD4+ T cells that were infected with HIV-1, knockdown of these miRNAs enhanced viral protein production (41). Subsequent studies in T cell lines also demonstrated a negative association between miR-125b levels and HIV-1 protein translation, further supporting the antiviral activity of miR-125b (43). Nevertheless, the mechanism by which miR-125b and other cellular miRNAs confer anti-HIV activity is not clearly understood.
There are two key mechanisms by which miRNAs can confer antiviral activity (57–59). Through a direct mechanism, miRNAs can inhibit viral protein translation by targeting HIV-1 mRNAs. This is evident from studies showing that miR-125b and other anti-HIV miRNAs (miR-28, miR-150, miR-223, and miR-382) inhibit HIV-1 protein translation by directly targeting the 3′ end of HIV-1 RNAs (41). Additionally, miRNAs can indirectly inhibit HIV-1 by regulating expression of cellular factors that positively or negatively affect viral infection (58, 59). In this study, we have identified CPSF6 as a direct target of miR-125b. Because CPSF6 is a cellular factor that promotes HIV-1 infection, our data support an indirect mechanism for miR-125b–mediated anti-HIV activity. First, our in silico analysis predicted a highly conserved binding site of miR-125b within the CPSF6 3′UTR (Fig. 1D). Significant interspecies conservation of a miRNA-binding site in the 3′UTR of a target gene is an indicator of physiological functionality (34). Accordingly, secondary structure predictions supported that miR-125b could mediate post-transcriptional regulation of CPSF6, because a thermodynamically stable stem-loop hairpin structure was observed between miR-125b and the CPSF6 3′UTR (Fig. 1, B and C). To validate the in silico predictions and eliminate false-positive target identification, knockdown and over-expression studies of miR-125b were carried out. These studies provided experimental evidence that CPSF6 expression negatively correlates with miR-125b levels. This inverse relationship between miR-125b and CPSF6 was observed in cells that are either normally nonpermissive (HEK-293T) or permissive (SupT1, CEM, and THP1) to HIV-1 infection (Fig. 1).
Post-transcriptional regulation of gene expression by miRNAs depends on the interaction between the 3′UTR of the target mRNA and the miRNA (46, 47). Therefore, to probe the interaction between miR-125b and the 3′UTR of CPSF6, we created three different luciferase constructs containing varied lengths of the CPSF6 3′UTR that retained the miR-125b–binding site (Fig. 2A). Studies of the reporter constructs in the presence of miR-mimics or anti-miRs demonstrated a negative correlation between miR-125b expression and CPSF6 3′UTR-driven luciferase activity (Fig. 2). Accordingly, the regulatory effects of miR-125b on the CPSF6 3′UTR was abrogated when specific mutations were introduced in the miR-125b–binding site (Fig. 3). These results provided strong evidence that a post-transcriptional mechanism of regulation of CPSF6 is driven by specific genetic interaction between miR-125b and the CPSF6 3′UTR. Finally, a physical interaction between miR-125b and CPSF6 mRNA was verified by pulldown studies (Fig. 4). Biotinylated miR-125b mimics effectively pulled down the WT CPSF6 3′UTR but failed to enrich the mutant UTR (Fig. 4). These studies underscored that miR-125b physically interacts with the CPSF6 3′UTR for post-transcriptional regulation.
Identification of a post-transcriptional mechanism of CPSF6 regulation by miR-125b is highly significant because CPSF6 plays key roles in an array of cellular function. CPSF6 is well-established to regulate alternative cleavage and polyadenylation of cellular mRNA (1, 4, 5, 60, 61). Recently, an important role for CPSF6 is also described in cancer biology through its association with paraspeckles (8). Most noteworthy is the accumulating evidence showing key roles of CPSF6 during HIV-1 infection. Specifically, CPSF6 is emerging as a key host factor that promotes nuclear entry and integration targeting of HIV-1 (10, 11, 29, 30). Therefore, we attempted to understand the mechanism(s) by which CPSF6 expression is regulated during HIV-1 infection. Our results revealed that HIV-1 infection resulted in the down-regulation of miR-125b concurrent with increased CPSF6 protein levels (Fig. 5), suggesting that CPSF6 expression in HIV-1–infected cells is most likely regulated at post-transcriptional level by miR-125b.
To identify the mechanism of miR-125b down-regulation in HIV-1–infected cells, we focused on the miRNA biogenesis pathway. miRNA biogenesis depends on transcription of the pri-miRNAs from miRNA-encoding genes (34, 46–48). The pri-miRNAs contain an RNA hairpin that is cleaved from the pri-miRNA in the nucleus by Drosha, a double strand-specific RNase (34, 46–48). The resulting precursor “pre-miRNA” is then transported to the cytoplasm and cleaved by Dicer to generate single-stranded mature miRNA (34, 46–48). Interestingly, there is evidence that viruses such as Herpes simplex virus 1 (HSV-1) target miRNA biogenesis to block the synthesis of mature miRNAs from pre-miRNA (62). Interestingly, neither pri-miR-125b nor pre-miR-125b were reduced by HIV-1 infection even though miR-125b levels were significantly down-regulated in these cells (Fig. 6, A–C). These observations indicated that down-regulation of miR-125b in HIV-1–infected cells was not due to reduced transcript levels of the miR but involve an alternative mechanism. HIV-1 could potentially increase the rate of miR-125b turnover, a hypothesis consistent with observations that other viruses regulate the turnover of cellular miRNAs. For instance, human cytomegalovirus (HCMV) has been shown to bind and degrade the cellular mature miRNAs to promote virus production (63). Furthermore, Herpesvirus saimiri (HVS) has been reported to cause degradation of the cellular mature miR-27 (64).
Our studies with reverse transcriptase and integrase inhibitors provided further insights into the mechanism of miR-125b down-regulation in HIV-1–infected cells (Fig. 6, E and F). These data showed that HIV infection results in a 2-fold reduction in miR-125b levels. Similarly, HIV infection in the presence of RAL, an inhibitor of HIV integration, also resulted in roughly a 2-fold reduction in miR-125b levels (Fig. 6E). However, there was no alteration in miR-125b levels when HIV infection was carried out in the presence of EFV, an inhibitor of reverse transcription (Fig. 6E). These findings suggest that reverse transcription of HIV RNA, but not viral DNA integration, is necessary for HIV-induced down-regulation of miR-125b. Because miR-125b negatively regulates CPSF6 protein levels, these observations imply that an optimal level of CPSF6 protein is most likely necessary after reverse transcription of the viral genome. These results are consistent with the predominant role of CPSF6 after the reverse transcription step of HIV-1 infection (10, 11). Even though these results do not identify the factors involved in miR-125b down-regulation, we envision that reverse transcription of HIV-1 is most likely coupled with faster turnover of miR-125b levels because biogenesis of the miR was not altered during infection (Fig. 6, A–C).
A functional link between HIV capsid integrity and optimal reverse transcription is well-established. Also, the requirement of the functional interaction between capsid and cellular factor Cyclophilin A for optimal reverse transcription is reported by several groups (12, 23, 25, 65). Although a C-terminal truncation variant of CPSF6 has been shown to potently restrict HIV-1 infection at the reverse transcription step (14), a role of endogenous CPSF6 in reverse transcription has not yet been reported. Therefore, CPSF6 seems unlikely to play a role in the HIV reverse transcription-dependent down-regulation of miR-125b. It should be noted that an HIV-encoded protein or RNA element has not been shown previously to interfere with cellular miRNA turnover. Therefore, future studies are needed to identify the viral factors involved in the down-regulation of miR-125b. Finally, infection studies in miR-125b over-expressing or knockdown cells showed that reducing miR-125b expression increases nuclear entry of the virus without affecting reverse transcription (Fig. 6, F and G). However, enhanced nuclear entry in these cells did not result in increased viral DNA integration. It is possible that the up-regulated CPSF6 levels (due to the suppression of the endogenous miR-125b by the anti-miRs) leads to increased nuclear entry of PICs, but excess CPSF6 in these cells has no impact on the efficiency of the integration step. In this scenario there could be an accumulation of unintegrated viral DNA, thus higher levels 2-LTR circles. This hypothesis is consistent with the emerging role of CPSF6 in integration targeting to specific regions of chromatin but lack of a regulatory role of this cellular factor in HIV-1 integration efficiency.
In summary, our studies provide important insights into host miRNA and HIV-1 interaction. Specifically, we have: (a) identified CPSF6 as a direct target of the cellular miRNA “miR-125b,” (b) uncovered a post-transcriptional mechanism of regulation of CPSF6, (c) revealed activation of miR-125b–mediated regulation of CPSF6 to promote HIV-1 nuclear entry, and (d) suggest a mechanism by which HIV-1 regulates miR-125b expression during infection.
Experimental procedures
Reagents
Anti-CPSF6 polyclonal antibody and anti-GAPDH mAb were purchased from Proteintech (Rosemont, IL). Hsa–miR-125b-5p mimic and inhibitor were obtained from GE Dharmacon (MA). Control siRNA was purchased from Santa Cruz Biotechnology (USA). Anti-β-actin antibody was obtained from Sigma. Anti-HIV-1 p24 mAb (183-H12–5C), raltegravir (RAL), and efavirenz (EFV) were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, National Institutes of Health.
Cell culture
Human T-lymphoblast cell lines (SupT1 and CEM), monocytic cell line (THP1), and the human embryonic kidney epithelial cell line (HEK-293T) were purchased from American Type Culture Collection (ATCC, Manassas, VA). The TZM-bl reporter cell line was obtained from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc., through the NIH AIDS Reagent Program (USA). Although SupT1, CEM, and THP1 cells were maintained in Roswell Park Memorial Institute medium (RPMI) medium, the HEK-293T and the TZM-bl cells were maintained in Dulbecco's modified Eagle's medium. Both RPMI and Dulbecco's modified Eagle's medium were supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco, USA), 2 mm glutamine, and 1% antibiotics (penicillin-streptomycin) (Gibco, USA), and the cells were maintained at 37 °C in a humidified 5% CO2 atmosphere. The generation and culturing of HEK-293T CPSF6-knockout (CKO) and CRISPR-control variant (CWT) cell lines have been described previously (29).
In silico analysis
To predict hsa–miR-125b–binding sites, the miR-125b sequence, and the 3′UTR sequence of the CPSF6 gene (GenBank accession number: NM_007007.3) were queried on two independent web server platforms: RNAhybrid 2.1.2 (31, 32) and RNA Structures-BiFold (33). RNAhybrid predicts secondary structures between two RNA molecules through minimum free energy calculations, whereas the RNA Structures-BiFold algorithm considers intramolecular base pairings involved in secondary structure formation. The putative miR-125b–binding site was further analyzed for sequence conservation across mammalian species using Clustal Omega multiple sequence alignment program (66). RNAhybrid 2.1.2 and RNA Structures-BiFold were also used to probe the effects of nucleotide mutations on the secondary structure of miR-125b and the CPSF6 3′UTR. Mutations within the CPSF6 3′UTR included base substitutions and deletions in the predicted seed sequences.
Knockdown and over-expression of miR-125b
HEK-293T cells (5 × 105 cells) cultured in 6-well-culture plates overnight were transfected with 150 pmol of miR-mimics or anti-miRs or control siRNA using Polyfect transfection reagent (Qiagen, USA). Post-transfection, cells were cultured for 36 h. Thereafter, cells were gently washed with phosphate-buffered saline (PBS), scraped, aliquoted for RNA and protein extraction, and pelleted by centrifugation at 500 × g for 5 min. SupT1, CEM, and THP1 cells (2 × 105 cells) grown in 6-well-culture plates were transfected with 150 pmol of anti-miRNAs or mimics or control siRNA using Neon Transfection System (Thermo-Fisher, USA). SupT1 cells were electroporated using the following conditions: voltage, 1150 V; width, 20 ms; pulses, 2. CEM cells were electroporated under the following conditions: voltage, 1230 V; width, 40 ms; pulses, 1. THP1 cells were electroporated under the following conditions: voltage, 1400 V; width, 20 ms; pulses, 2. SupT1, CEM, and THP1 cells were cultured for 24–36 h, pelleted by centrifugation at 500 × g for 10 min, and subsequently aliquoted for RNA and protein extraction.
RNA isolation and qPCR
For measuring miRNA expression, total RNA was isolated from cells using Quick-RNA Plus kit (Zymo Research, USA) as per the manufacturer's instructions. cDNA was synthesized from the isolated RNA using the miRCURY LNA miRNA RT kit (Exiqon, USA). SYBR Green-based qPCR was used to quantify miR-125b-5p or 5s rRNA (5srRNA) in a reaction mixture containing 50 ng of the cDNA as template and 300 nm LNA-based primers specific for miR-125b and 5srRNA (Exiqon, USA). qPCR assay was performed using the C1000 Touch CFX96 system (Bio-Rad) and executed as per the manufacturer's instructions (Exiqon, USA). The expression levels (Ct values) of miR-125b-5p were normalized to expression levels of 5s rRNA as ΔCt values. For multiple samples involving miR-125b mimic and anti-miR along with control scramble siRNA, the relative expression levels of miR-125b were expressed as 2 − ΔCt values by comparing the ΔCt values of the control siRNA to the miR-125b mimic or anti-miR samples.
Western blotting
To detect protein levels by Western blotting, cell lysates were prepared using 1× RIPA buffer (Sigma) and total protein concentrations were quantified by BCA protein assay (Pierce). Equal amounts of total protein from cell lysates were electrophoresed on 4–12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes using a semidry blotter (Bio-Rad). Membranes were blocked with 5% (w/v) nonfat milk in TBS with Tween 20 (pH 8.0, Chem Cruz). Blots were then probed with the primary antibody in blocking buffer (with dilution of anti-CPSF6 at 1:4,000 and for anti-GAPDH at 1:25,000 (v/v)), and subsequently by a secondary antibody conjugated to horseradish peroxidase (anti-rabbit, 1:6,000; anti-mouse, 1:40,000). All blots were washed in TBS with Tween 20 and developed using the enhanced chemiluminescence (ECL) procedure (Bio-Rad). Densitometry analysis was performed through LI-COR Image Studio Digits version 5.2 software (LI-COR). Data were normalized to levels of GAPDH or β-actin.
Construction of luciferase reporter plasmids
Three luciferase reporter constructs encoding increasing lengths of the CPSF6 3′UTR, all containing the single candidate miR-125b–binding site, were generated by employing a cloning strategy based on PCR and the presence of specific internal restriction enzyme recognition sites within the CPSF6 3′UTR sequence. Total RNA, isolated from SupT1 cells using RNeasy mini kit (Qiagen), was used as template in a cDNA synthesis reaction containing oligo(dT) and random primers (iScript Select cDNA Synthesis Kit, Bio-Rad), and the resulting first-strand cDNA was used as template in PCR to amplify CPSF6 3′UTR sequences. All PCRs were performed using the high-fidelity Phusion DNA polymerase (New England Biolabs) and custom-made primers. Agarose gel-resolved DNAs were extracted using the QIAquick gel-purification kit (Qiagen), plasmid DNA and PCR amplicons were digested with restriction enzymes from New England Biolabs, DNAs were ligated using T4 DNA ligase (New England Biolabs), competent DH5α bacterial cells were used for bacterial transformation, plasmid DNAs were isolated using the Zyppy plasmid mini prep kit (Zymo Research, USA), and the recombinant plasmids were verified by restriction enzyme digestion and Sanger DNA sequencing. To construct the luciferase reporter plasmid containing a 1011-bp fragment spanning nucleotides 1–1011 of the CPSF6 3′UTR, the corresponding sequence was amplified using forward primer harboring EcoRI site (5′-TACCAGAATTCAAGCTGAAGGAAGAGGATCACC-3′) and reverse primer harboring consecutive PstI and MluI sites (5′-TATATACGCGTTGCTGCAGGGCTGTATTTACAC-3′). The gel-purified PCR amplicon was digested with EcoRI and MluI, and then ligated to EcoRI/MluI-cut pMirTarget plasmid (Origene Technologies, USA) to obtain pMirTarget-CPSF6.3′UTR-S (p3′UTR-S). To construct the luciferase reporter plasmid containing a 3114-bp fragment spanning nucleotides 1–3114 of the CPSF6 3′UTR, a 2111-bp fragment spanning 1004–3114 nucleotides was amplified using forward primer harboring PstI site (5′-TTATCTGCAGCAGGTAAATGTGAGTAAAGAGAGC-3′) and reverse primer harboring consecutive HindIII and MluI sites (5′-TTATACGCGTAAGCTTAAAACACAAAATTGGAGC-3′). The gel-purified PCR amplicon was digested with PstI and MluI and then ligated to PstI/MluI-cut pMirTarget-CPSF6.3′UTR-S plasmid, thereby positioning the DNA insert immediately downstream of the CPSF6.3′UTR-S, to obtain pMirTarget-CPSF6.3′UTR-M (p3′UTR-M). To construct the luciferase reporter plasmid containing a 4243-bp gene fragment spanning nucleotides 1–4243 of the CPSF6 3′UTR, a multifragment DNA ligation strategy was employed. First, the region encompassing 3109–4243 nucleotides of the CPSF6.3′UTR was amplified using forward primer harboring the HindIII site (5′-TACCAAGCTTCAGCTTAAGAGGAAGTTTATGTTC-3′) and reverse primer harboring consecutive BamHI and MluI sites (5′-TTATACGCGTGGATCCTTTCCCATTAAATACC-3′), and the gel-purified PCR amplicon was digested with HindIII and MluI. Second, the PstI/MluI-cut CPSF6.3′UTR-1004–3114 amplicon, which was generated during the construction of p3′UTR-M, was cut at the engineered HindIII site present upstream of the MluI site in the amplicon. Third, the p3′UTR-S plasmid was digested with PstI and MluI enzymes and gel-purified. Finally, the PstI/MluI-cut p3′UTR-S plasmid, the PstI/HindIII-cut CPSF6.3′UTR-1004–3114 amplicon, and the HindIII/MluI-cut CPSF6.3′UTR-3109–4243 amplicon were all ligated to yield pMirTarget-CPSF6.3′UTR-L (p3′UTR-L). To introduce site-specific mutations in the seed sequence in the CPSF6 3′UTR, we used the Q5 Site-directed Mutagenesis Kit (New England Biolabs). The CA > AT (pMut1) and the CA > GT (pMut2) mutations were introduced using forward primers containing the desired mutations (CA > AT, 5′-GTTTACACCTATGGGAAAGTCTTG-3′; CA > GT, 5′-GTTTACACCTGTGGGAAAGTCTTG-3′), and a reverse primer (5′-ATCAAATAACTTGAACAGCTTTAC-3′).
Luciferase assay
All transfections were performed using the polyethyleneimine transfection reagent (67). To determine whether CPSF6 3′UTR activity is regulated by miR-125b, HEK-293T, CKO, and CWT cells (1 × 105 cells/well) were cultured separately in 24-well-plates overnight and transfected with pMirTarget control vector (termed “pΔUTR”) or either of the CPSF63′UTR reporter constructs, p3′UTR-S, p3′UTR-M, or p3′UTR-L, and cultured for 24 h. To determine the effects of mutated seed sequence on CPSF6 3′UTR activity, HEK-293T cells (2.5 × 105 cells/well) cultured overnight in a 6-well-plate were transfected with either the pΔUTR or pUTR-S or CPSF6–3′UTR mutant plasmids, pMut1 or pMut2, independently or, in a miR-125b over-expression or knockdown background. Post-transfection plates were incubated for 48 h. Transfection efficiency was assessed by measuring the expression of the red fluorescent protein encoded in the pMirTarget vector. Transfected cells were lysed using 1× GLO lysis buffer (Life Technologies) and luciferase activity was measured using a plate reader (BioTek, USA). Samples were assayed in triplicate and the data are shown as luciferase activity normalized to red fluorescent protein expression.
Pulldown assays
Biotin-tagged LNA oligonucleotides of miR-125b (hsa-miR-125b-5p: 5′-UCCCUGAGACCCUAACUUGUGA/3′-Bio) and Scrambled control (5′-GAUGGCAUUCGAUCAGUUCUA/3′Bio) were purchased from Exiqon/Qiagen (USA). For pulldown experiments, HEK-293T cells (1 × 106 cells/well) cultured overnight were transfected with 100 pmol of oligonucleotide and the WT or mutant pCPSF6–3′UTRs and were incubated for 24 h. Cells were harvested by gentle scraping and lysates were prepared as per our published protocol (38). The freshly prepared lysates were incubated with the blocked streptavidin-coated magnetic beads on a bench top rotating mixer for 1 h at room temperature. Then the beads were washed using freshly prepared ice-cold pulldown wash buffer. Finally, the beads were resuspended in 100 μl of nuclease-free water. Half the volume of this reconstituted complex was processed for on-column DNase digestion (Ambion, USA) followed by column purification and enrichment using a Qiagen RNAeasy purification kit. Post-purification, the complexes were used as templates for cDNA synthesis using an iScript Reverse Transcriptase kit (Bio-Rad).
To detect the target mRNA from the pulldown mixture, we employed a qPCR assay. We designed primers to amplify the 3′-UTR of CPSF6 mRNA. We also included primer sets for detecting PARP-1 mRNA, a known target of miR-125b, as a positive control and Actin mRNA as a nonspecific negative control. In the cDNA synthesis reactions, 50 ng of column-purified RNA and 1 μl of crude bead eluate were used as templates in separate reactions to perform cDNA synthesis using oligo(dT) primers. The cDNA generated from these reactions served as templates for qPCR. All qPCR were performed in triplicate in a Bio-Rad 96-well clear-bottom plate in sterile conditions. 9 μl of the reaction mixture from the qPCR master mix was aliquoted into each well of the 96-well PCR plate. Then 1 μl of the cDNA was added to each well to achieve a final volume of 10 μl. Thereafter the PCR plate was sealed using heat-resistant PCR plate sealer and loaded into the CFX96 real-time PCR system (Bio-Rad) to perform thermal cycling. The thermal cycling conditions for qPCR analysis included an initial denaturation at 95 °C for 10 min followed by amplification and acquisition at 95 °C for 10 s, 56 °C for 30 s, and 72 °C for 30 s for 30 cycles, and the thermal profile for melt curve analysis was obtained by holding the sample at 65 °C for 31 s followed by a linear ramp in temperature from 65 to 95 °C with a ramp rate of 0.5 °C/s and acquisition at 0.5 °C intervals. The expression level of target genes was analyzed relative to their expression in scrambled controls. Data were plotted as fold-change, which was calculated by comparing the expression values of the target genes obtained in 3′-biotinylated miR-125b-5p pulldown with that of 3′-biotinylated scrambled control.
Virus stocks and infection assays
Virus stocks were generated using calcium phosphate-based transfection of HEK-293T cells (68, 69) with plasmids pNL4.3 GFP, which does not express the HIV-1 envelope but encodes the GFP in place of the nef gene (70), and pHCMV-G, which encodes VSV-G (71). Briefly, 2 × 106 cells were seeded per 10-cm dish and cultured overnight. Next day, cells in each culture dish were transfected with 20 μg of total plasmid DNA (9:1 ratio of pNL4.3 GFP to pHCMV-G). Twelve hours post-transfection, the cells were washed once with PBS, replenished with 6 ml of growth medium, and cultured further for 48 h. The virus-containing culture supernatants were harvested, cleared of cell debris by low-speed centrifugation, filtered through 0.45-μm filters, and treated with DNase I (Calbiochem; 20 μg/ml of supernatant) in the presence of 10 mm magnesium chloride for 1 h at 37 °C. The concentration of the virus stocks was quantified by the p24-specific ELISA using standard methods (68). Virus infectivity was determined using TZM-bl indicator cells as described (72).
For infection assay, cells (HEK-293T, SupT1, or THP1 cells) seeded in 96-well-plates (5 × 104 cells per well) were spinoculated with pseudotyped HIV-1 particles (equivalent to 15 ng of p24 per well) in the presence of 6 μg/ml of Polybrene (Sigma) for 2 h at 25 °C and then cultured for 48 h. Productive infection of HEK-293T cells was confirmed by Western blot analysis probing for HIV-1 p24 protein using anti-CA antibody. Infection of SupT1 and THP1 cells were confirmed by measuring GFP signal intensity using flow cytometry (Millipore GUAVA). For infection studies with HIV-1 inhibitors, cells were infected with VSV-G HIV-GFP virus and concurrently treated with either 1 μm RAL or 1 μm EFV or DMSO as a control. After 24–36 h, cells were harvested and total RNA was isolated followed by cDNA synthesis and evaluation of miR-125b levels using RT-PCR.
Quantification of reverse transcription, 2-LTR circles, and integration
Reverse transcription products, 2-LTR circles, and proviral integration in 100 ng of total DNA from infected and uninfected controls were measured by qPCR and analyzed using our published method (69). Briefly, reverse transcription products were quantified using SYBR Green-based qPCR, and 2-LTR circles were quantified using TaqMan probe-based qPCR. To quantify the copy number of chromosomally-integrated proviral DNA in HIV-infected cells, a nested PCR strategy wherein primers designed to amplify only the junctions of the chromosomal-integrated viral DNA but not any unintegrated viral DNA is used in the first round end point PCR, followed by the second round TaqMan probe-based qPCR with primers that amplify viral LTR-specific sequences present in the first round PCR amplicons (69). A standard curve was generated using 10-fold serial dilutions of known copy numbers of the HIV-1 molecular clone pNL43 (73) or p2LTR plasmid (74) during qPCR in tandem with measurement of sample quantification. Samples were evaluated against appropriate standard curves to generate values for copy number of late RT products and chromosomally integrated proviral DNA (pNL43 standard curve) or 2-LTR circle formation (p2LTR standard curve).
Quantification of primary (pri-) and pre-miR-125b levels
Primers were selected from the human miR-100–let-7a-2–miR-125b-1 cluster host gene sequence containing primary (pri-), precursor (pre-), and mature miR-125b-5p sequences. A region within 400 nucleotides of the mature sequence was chosen for designing primer sets for the pri-miR-125b sequence. These include: Pri-forward primer = 5′-CTGAGGTGATTGAGTATACCTCTGAGG-3′ and Pri-reverse primer = 5′-TCCAGGAGCTGCCACTCTCTG-3′. Pre-miR-125b primers were designed to encompass the entire 70-bp region of pre-miR-125b sequence: Pre-miR-125b forward primer: 5′-CCTCTCAGTCCCTGAGACCCTAAC-3′ and Pre-miR-125b reverse primer: 5′-GACTCGCAGCTCCCAAGAGC-3′.
To measure precursors of miR-125b, 293T cells (5 × 105 cells/ml) were cultured overnight in 6-well-plates and subsequently inoculated with VSV-G HIV-GFP virus as previously described. After 24–36 h, cells were harvested and RNA from these cells were isolated. 100 μg of total RNA was used to synthesize cDNA using ABMGood OneScript Plus cDNA synthesis kit (Vancouver, Canada) and random primers as per the manufacturer's instructions. 2 μl of cDNA were used in a RT-PCR using SYBR Green 2× Supermix (Bio-Rad). Samples were run in triplicates and normalized to 5S rRNA levels.
Statistical analysis
Data were expressed as mean ± S.E. obtained from three independent experiments. Significance of differences between control and treated samples was determined by Student's t test. Values of p < 0.05 were considered statistically significant.
Author contributions
E. C., S. D., M. B., A. P., J. H., and J. P. data curation; E. C., M. B., J. H., F. V., and C. D. formal analysis; E. C., S. D., and M. B. investigation; E. C., S. D., M. B., J. H., A. N. E., J. P., and C. D. methodology; E. C., S. D., and C. D. writing-original draft; S. D., M. B., F. V., J. P., and C. D. conceptualization; S. D., M. B., G. A. S., A. N. E., and C. D. resources; S. D., M. B., J. P., and C. D. supervision; S. D. validation; S. D., M. B., G. A. S., F. V., A. N. E., J. P., and C. D. writing-review and editing; C. D. funding acquisition; C. D. project administration.
This work was supported by National Institutes of Health Grants R01 AI136740, R56 AI122960, R24 DA036420, and U54 MD007593 (to C. D.), R01 AI052014 and P50 AI150481 (to A. N. E.), and T32 AI007245 (to G. A. S.), and the Research Centers in Minority Institutions (RCMI) U54MD007586-01 (to J. P.). This work is also in part supported by the Meharry Translational Research Center (MeTRC) Grant 5U54MD007593-10 and Tennessee CFAR Grant P30 AI110527 from the National Institutes of Health (to C. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PIC
- preintegration complex
- NUP
- nucleoporin
- miRNA
- microRNA
- LNA
- locked nucleic acid
- HCMV
- human cytomegalovirus
- RAL
- raltegravir
- EFV
- efavirenz
- CKO
- CPSF6-knockout
- CWT
- CRISPR-control variant
- VSV-G
- vesicular stomatitis virus G glycoprotein
- qPCR
- quantitative PCR
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Rüegsegger U., Beyer K., and Keller W. (1996) Purification and characterization of human cleavage factor Im involved in the 3′ end processing of messenger RNA precursors. J. Biol. Chem. 271, 6107–6113 10.1074/jbc.271.11.6107 [DOI] [PubMed] [Google Scholar]
- 2. Gruber A. R., Martin G., Keller W., and Zavolan M. (2012) Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol. 9, 1405–1412 10.4161/rna.22570 [DOI] [PubMed] [Google Scholar]
- 3. Hardy J. G., and Norbury C. J. (2016) Cleavage factor Im (CFIm) as a regulator of alternative polyadenylation. Biochem. Soc. Trans. 44, 1051–1057 10.1042/BST20160078 [DOI] [PubMed] [Google Scholar]
- 4. Brown K. M., and Gilmartin G. M. (2003) A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol. Cell 12, 1467–1476 10.1016/S1097-2765(03)00453-2 [DOI] [PubMed] [Google Scholar]
- 5. Millevoi S., and Vagner S. (2010) Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 38, 2757–2774 10.1093/nar/gkp1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dettwiler S., Aringhieri C., Cardinale S., Keller W., and Barabino S. M. (2004) Distinct sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA binding, protein-protein interactions, and subcellular localization. J. Biol. Chem. 279, 35788–35797 10.1074/jbc.M403927200 [DOI] [PubMed] [Google Scholar]
- 7. Naganuma T., Nakagawa S., Tanigawa A., Sasaki Y. F., Goshima N., and Hirose T. (2012) Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 31, 4020–4034 10.1038/emboj.2012.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Binothman N., Hachim I. Y., Lebrun J. J., and Ali S. (2017) CPSF6 is a clinically relevant breast cancer vulnerability target: role of CPSF6 in breast cancer. EBioMedicine 21, 65–78 10.1016/j.ebiom.2017.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Price A. J., Fletcher A. J., Schaller T., Elliott T., Lee K., KewalRamani V. N., Chin J. W., Towers G. J., and James L. C. (2012) CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 8, e1002896 10.1371/journal.ppat.1002896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chin C. R., Perreira J. M., Savidis G., Portmann J. M., Aker A. M., Feeley E. M., Smith M. C., and Brass A. L. (2015) Direct visualization of HIV-1 replication intermediates shows that capsid and CPSF6 modulate HIV-1 intra-nuclear invasion and integration. Cell Rep. 13, 1717–1731 10.1016/j.celrep.2015.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bejarano D. A., Peng K., Laketa V., Börner K., Jost K. L., Lucic B., Glass B., Lusic M., Müller B., and Kräusslich H. G. (2019) HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. Elife 8, e41800 10.7554/eLife.41800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ambrose Z., Lee K., Ndjomou J., Xu H., Oztop I., Matous J., Takemura T., Unutmaz D., Engelman A., Hughes S. H., and KewalRamani V. N. (2012) Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J. Virol. 86, 4708–4714 10.1128/JVI.05887-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhattacharya A., Alam S. L., Fricke T., Zadrozny K., Sedzicki J., Taylor A. B., Demeler B., Pornillos O., Ganser-Pornillos B. K., Diaz-Griffero F., Ivanov D. N., and Yeager M. (2014) Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. U.S.A. 111, 18625–18630 10.1073/pnas.1419945112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee K., Ambrose Z., Martin T. D., Oztop I., Mulky A., Julias J. G., Vandegraaff N., Baumann J. G., Wang R., Yuen W., Takemura T., Shelton K., Taniuchi I., Li Y., Sodroski J., Littman D. R., et al. (2010) Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7, 221–233 10.1016/j.chom.2010.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee K., Mulky A., Yuen W., Martin T. D., Meyerson N. R., Choi L., Yu H., Sawyer S. L., and Kewalramani V. N. (2012) HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J. Virol. 86, 3851–3860 10.1128/JVI.06607-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. UNAIDS (2018) Global HIV & AIDS statistics: 2018 fact sheet. UNAIDS [Google Scholar]
- 17. Swanstrom R., and Coffin J. (2012) HIV-1 pathogenesis: the virus. Cold Spring Harb. Perspect. Med. 2, a007443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Craigie R., and Bushman F. D. (2012) HIV DNA integration. Cold Spring Harb. Perspect. Med. 2, a006890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bukrinsky M. I., Sharova N., Dempsey M. P., Stanwick T. L., Bukrinskaya A. G., Haggerty S., and Stevenson M. (1992) Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. U.S.A. 89, 6580–6584 10.1073/pnas.89.14.6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Christ F., Thys W., De Rijck J., Gijsbers R., Albanese A., Arosio D., Emiliani S., Rain J. C., Benarous R., Cereseto A., and Debyser Z. (2008) Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 18, 1192–1202 10.1016/j.cub.2008.07.079 [DOI] [PubMed] [Google Scholar]
- 21. Zhang R., Mehla R., and Chauhan A. (2010) Perturbation of host nuclear membrane component RanBP2 impairs the nuclear import of human immunodeficiency virus-1 preintegration complex (DNA). PLoS ONE 5, e15620 10.1371/journal.pone.0015620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matreyek K. A., and Engelman A. (2011) The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 85, 7818–7827 10.1128/JVI.00325-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Iaco A., and Luban J. (2014) Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology 11, 11 10.1186/1742-4690-11-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schröder A. R., Shinn P., Chen H., Berry C., Ecker J. R., and Bushman F. (2002) HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110, 521–529 10.1016/S0092-8674(02)00864-4 [DOI] [PubMed] [Google Scholar]
- 25. Schaller T., Ocwieja K. E., Rasaiyaah J., Price A. J., Brady T. L., Roth S. L., Hué S., Fletcher A. J., Lee K., KewalRamani V. N., Noursadeghi M., Jenner R. G., James L. C., Bushman F. D., and Towers G. J. (2011) HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7, e1002439 10.1371/journal.ppat.1002439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ocwieja K. E., Brady T. L., Ronen K., Huegel A., Roth S. L., Schaller T., James L. C., Towers G. J., Young J. A., Chanda S. K., König R., Malani N., Berry C. C., and Bushman F. D. (2011) HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 7, e1001313 10.1371/journal.ppat.1001313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koh Y., Wu X., Ferris A. L., Matreyek K. A., Smith S. J., Lee K., KewalRamani V. N., Hughes S. H., and Engelman A. (2013) Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J. Virol. 87, 648–658 10.1128/JVI.01148-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Nunzio F., Fricke T., Miccio A., Valle-Casuso J. C., Perez P., Souque P., Rizzi E., Severgnini M., Mavilio F., Charneau P., and Diaz-Griffero F. (2013) Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 440, 8–18 10.1016/j.virol.2013.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sowd G. A., Serrao E., Wang H., Wang W., Fadel H. J., Poeschla E. M., and Engelman A. N. (2016) A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. U.S.A. 113, E1054–1063 10.1073/pnas.1524213113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Achuthan V., Perreira J. M., Sowd G. A., Puray-Chavez M., McDougall W. M., Paulucci-Holthauzen A., Wu X., Fadel H. J., Poeschla E. M., Multani A. S., Hughes S. H., Sarafianos S. G., Brass A. L., and Engelman A. N. (2018) Capsid-CPSF6 interaction licenses nuclear HIV-1 trafficking to sites of viral DNA integration. Cell Host Microbe 24, 392–404.e8 10.1016/j.chom.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rehmsmeier M., Steffen P., Hochsmann M., and Giegerich R. (2004) Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–1517 10.1261/rna.5248604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kruger J., and Rehmsmeier M. (2006) RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–W454 10.1093/nar/gkl243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reuter J. S., and Mathews D. H. (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129 10.1186/1471-2105-11-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bartel D. P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- 35. Pillai R. S., Bhattacharyya S. N., and Filipowicz W. (2007) Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 17, 118–126 10.1016/j.tcb.2006.12.007 [DOI] [PubMed] [Google Scholar]
- 36. Li S., Liu L., Zhuang X., Yu Y., Liu X., Cui X., Ji L., Pan Z., Cao X., Mo B., Zhang F., Raikhel N., Jiang L., and Chen X. (2013) MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 153, 562–574 10.1016/j.cell.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dash S., Balasubramaniam M., Rana T., Godino A., Peck E. G., Goodwin J. S., Villalta F., Calipari E. S., Nestler E. J., Dash C., and Pandhare J. (2017) Poly(ADP-ribose) polymerase-1 (PARP-1) induction by cocaine is post-transcriptionally regulated by miR-125b. eNeuro 4, e0089–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dash S., Balasubramaniam M., Dash C., and Pandhare J. (2018) Biotin-based pulldown assay to validate mRNA targets of cellular miRNAs. J. Vis. Exp. 2018, 136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shaham L., Binder V., Gefen N., Borkhardt A., and Izraeli S. (2012) MiR-125 in normal and malignant hematopoiesis. Leukemia 26, 2011–2018 10.1038/leu.2012.90 [DOI] [PubMed] [Google Scholar]
- 40. Rossi R. L., Rossetti G., Wenandy L., Curti S., Ripamonti A., Bonnal R. J., Birolo R. S., Moro M., Crosti M. C., Gruarin P., Maglie S., Marabita F., Mascheroni D., Parente V., Comelli M., et al. (2011) Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat. Immunol. 12, 796–803 10.1038/ni.2057 [DOI] [PubMed] [Google Scholar]
- 41. Huang J., Wang F., Argyris E., Chen K., Liang Z., Tian H., Huang W., Squires K., Verlinghieri G., and Zhang H. (2007) Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 13, 1241–1247 10.1038/nm1639 [DOI] [PubMed] [Google Scholar]
- 42. Wang X., Ye L., Hou W., Zhou Y., Wang Y. J., Metzger D. S., and Ho W. Z. (2009) Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 113, 671–674 10.1182/blood-2008-09-175000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mantri C. K., Pandhare Dash J., Mantri J. V., and Dash C. C. (2012) Cocaine enhances HIV-1 replication in CD4+ T cells by down-regulating miR-125b. PLoS ONE 7, e51387 10.1371/journal.pone.0051387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Engelman A., Englund G., Orenstein J. M., Martin M. A., and Craigie R. (1995) Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 69, 2729–2736 10.1128/JVI.69.5.2729-2736.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arhel N., and Kirchhoff F. (2010) Host proteins involved in HIV infection: new therapeutic targets. Biochim. Biophys. Acta 1802, 313–321 10.1016/j.bbadis.2009.12.003 [DOI] [PubMed] [Google Scholar]
- 46. Lee Y., Kim M., Han J., Yeom K. H., Lee S., Baek S. H., and Kim V. N. (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060 10.1038/sj.emboj.7600385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ha M., and Kim V. N. (2014) Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524 10.1038/nrm3838 [DOI] [PubMed] [Google Scholar]
- 48. Bartel D. P. (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cullen B. R. (2004) Transcription and processing of human microRNA precursors. Mol. Cell 16, 861–865 10.1016/j.molcel.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 50. Hutvágner G., and Zamore P. D. (2002) A microRNA in a multiple-turnover RNAi enzyme complex. Science 297, 2056–2060 10.1126/science.1073827 [DOI] [PubMed] [Google Scholar]
- 51. Zeng Y., Yi R., and Cullen B. R. (2005) Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 24, 138–148 10.1038/sj.emboj.7600491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hammond S. M., Bernstein E., Beach D., and Hannon G. J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296 10.1038/35005107 [DOI] [PubMed] [Google Scholar]
- 53. Umbach J. L., and Cullen B. R. (2009) The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 23, 1151–1164 10.1101/gad.1793309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun Y. M., Lin K. Y., and Chen Y. Q. (2013) Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 6, 6 10.1186/1756-8722-6-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rodriguez A., Griffiths-Jones S., Ashurst J. L., and Bradley A. (2004) Identification of mammalian microRNA host genes and transcription units. Genome Res. 14, 1902–1910 10.1101/gr.2722704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., and Tuschl T. (2002) Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739 10.1016/S0960-9822(02)00809-6 [DOI] [PubMed] [Google Scholar]
- 57. Balasubramaniam M., Pandhare J., and Dash C. (2018) Are microRNAs important players in HIV-1 infection?: an update. Viruses 10, E110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chiang K., and Rice A. P. (2011) Mini ways to stop a virus: microRNAs and HIV-1 replication. Future Virol. 6, 209–221 10.2217/fvl.10.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hariharan M., Scaria V., Pillai B., and Brahmachari S. K. (2005) Targets for human encoded microRNAs in HIV genes. Biochem. Biophys. Res. Commun. 337, 1214–1218 10.1016/j.bbrc.2005.09.183 [DOI] [PubMed] [Google Scholar]
- 60. Zhu Y., Wang X., Forouzmand E., Jeong J., Qiao F., Sowd G. A., Engelman A. N., Xie X., Hertel K. J., and Shi Y. (2018) Molecular mechanisms for CFIm-mediated regulation of mRNA alternative polyadenylation. Mol. Cell 69, 62–74.e64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jang S., Cook N. J., Pye V. E., Bedwell G. J., Dudek A. M., Singh P. K., Cherepanov P., and Engelman A. N. (2019) Differential role for phosphorylation in alternative polyadenylation function versus nuclear import of SR-like protein CPSF6. Nucleic Acids Res. 47, 4663–4683 10.1093/nar/gkz206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pan D., Li G., Morris-Love J., Qi S., Feng L., Mertens M. E., Jurak I., Knipe D. M., and Coen D. M. (2019) Herpes simplex virus 1 lytic infection blocks microRNA (miRNA) biogenesis at the stage of nuclear export of pre-miRNAs. mBio 10, e02856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee S., Song J., Kim S., Kim J., Hong Y., Kim Y., Kim D., Baek D., and Ahn K. (2013) Selective degradation of host MicroRNAs by an intergenic HCMV noncoding RNA accelerates virus production. Cell Host Microbe 13, 678–690 10.1016/j.chom.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 64. Cazalla D., Yario T., and Steitz J. (2010) Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328, 1563–1566 10.1126/science.1187197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luban J., Bossolt K. L., Franke E. K., Kalpana G. V., and Goff S. P. (1993) Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73, 1067–1078 10.1016/0092-8674(93)90637-6 [DOI] [PubMed] [Google Scholar]
- 66. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baker A., Saltik M., Lehrmann H., Killisch I., Mautner V., Lamm G., Christofori G., and Cotten M. (1997) Polyethylenimine (PEI) is a simple, inexpensive and effective reagent for condensing and linking plasmid DNA to adenovirus for gene delivery. Gene Ther. 4, 773–782 10.1038/sj.gt.3300471 [DOI] [PubMed] [Google Scholar]
- 68. Aiken C. (2009) Cell-free assays for HIV-1 uncoating. Methods Mol. Biol. 485, 41–53 10.1007/978-1-59745-170-3_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Balasubramaniam M., Zhou J., Addai A., Martinez P., Pandhare J., Aiken C., and Dash C. (2019) PF74 inhibits HIV-1 integration by altering the composition of the preintegration complex. J. Virol. 93, e01741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. He J., Chen Y., Farzan M., Choe H., Ohagen A., Gartner S., Busciglio J., Yang X., Hofmann W., Newman W., Mackay C. R., Sodroski J., and Gabuzda D. (1997) CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 385, 645–649 10.1038/385645a0 [DOI] [PubMed] [Google Scholar]
- 71. Burns J. C., Friedmann T., Driever W., Burrascano M., and Yee J. K. (1993) Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. U.S.A. 90, 8033–8037 10.1073/pnas.90.17.8033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Platt E. J., Wehrly K., Kuhmann S. E., Chesebro B., and Kabat D. (1998) Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72, 2855–2864 10.1128/JVI.72.4.2855-2864.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gallay P., Hope T., Chin D., and Trono D. (1997) HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. U.S.A. 94, 9825–9830 10.1073/pnas.94.18.9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Butler S. L., Hansen M. S., and Bushman F. D. (2001) A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7, 631–634 10.1038/87979 [DOI] [PubMed] [Google Scholar]