

Abstract
Monocytes are rapidly recruited to sites of diabetic complications and differentiate into macrophages. Previously, we showed that rat kidney mesangial cells dividing during hyperglycemic stress abnormally synthesize hyaluronan (HA) in intracellular compartments. This initiates a stress response, resulting in an extracellular HA matrix after division that recruits inflammatory cells. Cell–cell communication among macrophages that are recruited into the glomeruli and the damaged rat mesangial cells leads to diabetic nephropathy, fibrosis, and proteinurea, which are inhibited in heparin-treated diabetic rats. In this study, we found that murine bone marrow–derived macrophages (BMDMs) and a human leukemic cell line, U937 cells, dividing in hyperglycemia also accumulate intracellular HA and that heparin inhibits the HA accumulation. Both cell types expressed increased levels of proinflammatory markers: inducible nitric-oxide synthase and tumor necrosis factor-α, when cultured under hyperglycemic stress, which was inhibited by heparin. Furthermore, the abnormal intracellular HA was also observed in peripheral blood monocytes derived from three different hyperglycemic diabetic mouse models: streptozotocin-treated, high-fat fed, and Ins2Akita. Moreover, peripheral blood monocytes in humans with type 2 diabetes and poorly controlled blood glucose levels (hemoglobin A1c (HbA1c) levels of >7) also had intracellular HA, whereas those with HbA1c of <7, did not. Of note, heparin increased the anti-inflammatory markers arginase 1 and interleukin-10 in murine BMDMs. We conclude that heparin treatment of high glucose–exposed dividing BMDMs promotes an anti-inflammatory tissue-repair phenotype in these cells.
Keywords: macrophage, monocyte, hyaluronan, inflammation, heparin
Introduction
Previously we have shown that after the onset of diabetes in streptozotocin (STZ)–treated3 rats, their kidney glomerular mesangial cells (RMCs) that had divided in elevated blood glucose levels synthesized hyaluronan in intracellular compartments. This abnormal intracellular HA is extruded outside the cell after division in unique structures that recruit inflammatory cells forming a monocyte adhesive HA matrix that initiates a large stress response (1, 2). By week 6, kidney glomeruli have an extensive HA matrix around the RMCs with embedded recruited macrophages that are unable to remove the HA matrix effectively. The dialogue between the macrophages and the damaged RMCs initiates inflammatory and fibrotic responses that lead to diabetic nephropathy and proteinurea caused by tissue damage and loss of function. In contrast, RMCs in diabetic rats, injected intraperitoneally daily with a very low dose of heparin, do not synthesize HA in intracellular compartments nor undergo autophagy. Instead they synthesize an extensive monocyte-adhesive extracellular HA matrix after completing division that is present within 1 week. By week 6, kidney glomeruli in the uncontrolled diabetic rats treated with heparin have much less HA because of the influx of macrophages that effectively remove the HA matrix without initiating inflammatory and fibrotic responses. This allows the RMCs to maintain glomerular function while still synthesizing an extracellular monocyte-adhesive HA matrix in response to hyperglycemia (1).
It is known that monocytes are early infiltrating cells that arrive at sites of diabetic complications (3). Recruited circulating monocytes differentiate into macrophages. There are two main types of macrophages, the classically activated proinflammatory (Mpi) macrophages and the alternatively activated anti-inflammatory and tissue repair (Mtr) macrophages (4, 5). Mpi macrophages are characterized by their increased secretion of proinflammatory cytokines and production of nitric oxide (6) that lead to their enhanced microbicidal capacity. Mtr macrophages, on the other hand, have a high phagocytic capacity, can mitigate inflammatory responses, and can promote wound healing and tissue remodeling (7, 8). Based on that and our previous findings, we propose that monocytes and bone marrow–derived macrophages during uncontrolled high levels of glucose accumulate intracellular HA and are of a proinflammatory phenotype, whereas those treated with heparin are of a tissue repair phenotype, are anti-inflammatory, and do not accumulate intracellular HA. This provides a likely mechanism for the inability of macrophages to effectively remove the deposited HA matrix in kidneys from uncontrolled STZ diabetic rats compared with those treated with heparin.
Results
Heparin inhibits the expression of proinflammatory markers in dividing U937 cells under hyperglycemic stress
Mpi/Mtr macrophages are most commonly distinguished based on their catabolism of l-arginine and Th1/Th2 cytokine production (9). Mpi macrophages express increased levels of inducible nitric-oxide synthase (iNOS), which converts l-arginine to l-citrulline and nitric oxide (6). In contrast, Mtr macrophages express the cytosolic arginase 1 (ARG1), which catabolizes l-arginine to l-ornithine, a precursor of polyamines and proline (10, 11). Mpi macrophages express Th1 cytokines such as tumor necrosis factor α (TNFα), whereas M2 macrophages express Th2 cytokines such as interleukin-10 (IL-10) (9). To investigate the effect of heparin on monocytes under hyperglycemic stress, U937 cells, a human leukemic cell line commonly used to study monocyte biology, was investigated (12, 13). Fig. 1 (A and B) shows that hyperglycemia (5× normal glucose) induced the relative gene expression of proinflammatory markers iNOS and TNFα in dividing U937 cells, whereas a low concentration (2 μg/ml) of heparin inhibited the expression of those markers. No significant difference was observed for the relative gene expression of anti-inflammatory ARG1 and IL-10 in any of the treatments. Fig. 1C shows that heparin decreased the levels of a proinflammatory surface marker, CD80 (cluster of differentiation 80) in green (14) and instead increased the levels of an anti-inflammatory surface marker, CD163, in red in U937 cells dividing under hyperglycemic stress. These results indicate that hyperglycemia stimulates dividing U937 cells to become more of a proinflammatory phenotype, which the presence of heparin inhibited.
Figure 1.

Effect of heparin on proinflammatory and anti-inflammatory markers in dividing U937 cells under hyperglycemic stress. Heparin inhibits the relative gene expression of the proinflammatory markers iNOS (A) and TNFα (B); n = 6 (3). The fluorescent micrographs (C) show that heparin inhibits the proinflammatory surface marker, CD80 stained in green, and induces the anti-inflammatory surface marker, CD163, stained in red. Scale bars, 10 μm.
Heparin inhibits intracellular HA in U937 cells dividing under hyperglycemic stress
Our previous studies showed that rat mesangial cells dividing under hyperglycemic stress synthesize an abnormal HA matrix in intracellular compartments (1, 2). Fig. 2C shows that U937 cells dividing under hyperglycemic stress also have extensive HA (green) in intracellular compartments, which is not present in U937 cells dividing in normal glucose (Fig. 2A). This intracellular HA was also not observed in hyperglycemic dividing U937 cells treated with heparin (2 μg/ml) (Fig. 2D). CD44, a common HA receptor, formed clusters on the surface of U937 cells when they divided in hyperglycemia as shown in Fig. 2G (red). This phenomenon of CD44 clustering is known as “CD44 capping” (15, 16). However, the presence of heparin in the medium maintained a uniform distribution of CD44 on the cell surface of hyperglycemic stressed U937 cells (Fig. 2H) that is comparable with those cultured in normoglycemia with or without heparin (Fig. 2, E and F).
Figure 2.

Heparin inhibits intracellular hyaluronan in U937 cells dividing in hyperglycemia. These cells were permeabilized. The fluorescent micrographs show that heparin inhibits the induction of HA in hyperglycemic dividing U937 cells stained with a green fluorescent conjugate of biotinylated HA-binding protein (D compared with C). CD44 (red) remains uniformly distributed on the heparin-treated hyperglycemic U937 cells as on the normoglycemic and normoglycemic U937 cells treated with heparin (H, E, and F) but is punctate on the hyperglycemic dividing cells (G). Panels I–L represent the overlay images with DAPI staining of U937 cell nuclei. Scale bars, 10 μm.
U937 cells synthesize HA onto their cell surface in normoglycemia and into a more extensive matrix under hyperglycemic stress
We have previously shown that heparin prevents the synthesis of intracellular HA in dividing mesangial cells under hyperglycemic stress in vivo as well as in vitro. Instead a more extensive HA is extruded outside the cells as part of their extracellular matrix (1, 2, 17). Heparin has a similar effect on dividing U937 cells as shown in Fig. 3 (A–D). Cell surface–associated HA is increased under hyperglycemic stress (Fig. 3C, green), and a more extensive HA matrix is formed on those cells treated with heparin under hyperglycemic stress (Fig. 3D). Interestingly, U937 cells have small amounts of HA on their cell surface in normoglycemia (Fig. 3A). This HA is likely being synthesized by the HA synthases in the plasma membrane, which release the completed HA into the medium. This is supported by the results in Fig. 4. Heparin had no apparent increase in the amounts of HA on the U937 cell surface in normoglycemia (Fig. 3B).
Figure 3.

U937 cells have HA on their surfaces after division in normoglycemia or in hyperglycemia with or without heparin. These cells were not permeabilized. The fluorescent micrographs show the effect of hyperglycemia on the synthesis of HA in dividing U937 cells (A–D) stained with a green fluorescent conjugate of biotinylated HA-binding protein. Staining for CD44 is shown in red (E–H) and for nuclei stained with DAPI (blue, I–L). Scale bars for low-power images are 50 μm and for enlargements are 10 μm. The small dots of HA (green) on the normoglycemic U937 cells are close to the cell surface (A, enlarged box) as shown by comparison with the cell surface CD44 (I, red). In contrast, the HA on the hyperglycemic U937 cells with or without heparin have larger HA matrices (C and D), as shown by their extended distance from the CD44 (K and L).
Figure 4.
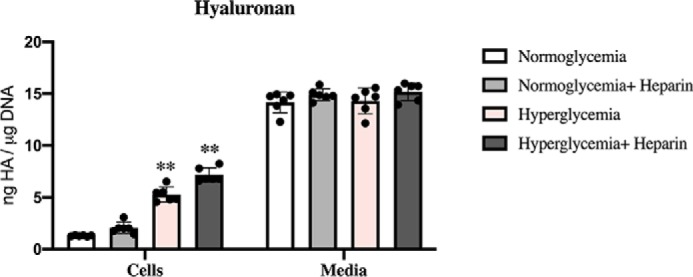
U937 cells synthesize HA that is mostly released into the culture media. The bar graphs show the amounts of HA determined by FACE in the cellular and medium compartments of U937 cells after 48 h of treatment under normoglycemic or hyperglycemic conditions with or without heparin. In all culture conditions, U937 cells synthesize equivalent amounts of HA that are released into the culture media (Media). The values of HA associated with U937 cells (Cells) cultured during hyperglycemia with and without heparin are statistically greater (p < 0.01) than those cultured during normoglycemia with or without heparin (n = 6). The amounts in the media are much more than the amounts associated with the cells.
Dividing U937 cells synthesize HA that is mostly released into the culture media
Measurements of HA in the media show that U937 cells in all four conditions secrete most of their synthesized HA into the medium, showing that HA synthesis continues during division in normal glucose with or without heparin (Fig. 4, Media), unlike mesangial cells (1). Further, the amounts of HA in the media were equivalent independent of glucose content and the presence of heparin (Fig. 4). As supported by Fig. 3, the cell matrix HA in the hyperglycemic cultures with or without heparin were significantly increased relative to the results in normoglycemia (Fig. 4, Cells).
Heparin inhibits the expression of proinflammatory markers in murine bone–derived macrophages under hyperglycemic stress
To investigate the effect of heparin on murine macrophages, BMDMs derived from their femur and tibia bones were cultured in high-glucose medium with or without heparin to compare with those cultured in normal glucose medium. Dividing hyperglycemic monocyte progenitor cells differentiated into macrophages that expressed higher levels of proinflammatory markers including iNOS (Fig. 5A) and TNFα (Fig. 5B) than in normoglycemia with or without heparin. Heparin inhibited the relative expression of these proinflammatory markers and instead promoted the relative gene expression of anti-inflammatory markers including ARG1 (Fig. 5A) and IL-10 (Fig. 5B). Furthermore, heparin decreased the protein levels of interferon regulatory factor-8 (IRF-8), a proinflammatory transcription factor (18), whereas it increased the levels of IRF-4, an anti-inflammatory transcription factor (19) as shown in Fig. 5C. Heparin had no significant effects on macrophage polarization in normoglycemia (Fig. 5).
Figure 5.

Effect of heparin on proinflammatory and anti-inflammatory markers in dividing murine bone marrow–derived macrophages under hyperglycemic stress. Heparin inhibits the relative gene expression of the proinflammatory markers iNOS (A) and TNFα (B) and induces the anti-inflammatory markers ARG1 (A) and IL-10 (B); n = 6. One representative Western blotting of three shows that heparin under high levels of glucose (25 mm) inhibits protein levels of proinflammatory transcription factor IRF-8 and induces anti-inflammatory transcription factor IRF-4 (C). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Heparin inhibits intracellular HA in dividing murine bone marrow–derived macrophages
Similar to U937 cells, there is intracellular HA in dividing BMDMs when cultured under hyperglycemic stress (Fig. 6C, green) compared with those cultured in normoglycemia (Fig. 6A). The addition of heparin prevented the intracellular HA in BMDMs cultured under hyperglycemic stress (Fig. 6D). Heparin had no significant effect on BMDMs cultured in normal glucose levels (Fig. 6B). F4/80, a murine macrophage marker, was stained in red (Fig. 6, E–H).
Figure 6.

Heparin inhibits intracellular hyaluronan in murine bone marrow–derived macrophages. These cells were permeabilized. The fluorescent micrographs show that heparin inhibits intracellular HA in hyperglycemic dividing murine BMDMs (D compared with C) as stained with a fluorescent conjugate (green) of biotinylated HA-binding protein. HA was also absent in normoglycemic and normoglycemic with heparin BMDMs (A and B). E–H show F4/80 staining for a macrophage marker (E–H, red). Overlay images and nuclei stained with DAPI are shown (I–L, blue). Scale bars, 50 μm.
Murine bone marrow–derived macrophages synthesize HA on their cell surface in normoglycemia and a more extensive extracellular matrix under hyperglycemic stress
Similar to U937 cells, BMDMs synthesize HA on their cell surface in normoglycemia (Fig. 7A, green). There is an increased synthesis of HA on BMDM cell surfaces when cultured under hyperglycemic stress (Fig. 7C) that is more extensive when treated with heparin (Fig. 7D). Heparin treatment did not significantly increase HA synthesis on BMDMs that were cultured in normoglycemia (Fig. 7B). F4/80, a murine macrophage marker, stained in red, showed the predominance of macrophages in the cultures (Fig. 7, E–H).
Figure 7.

Hyperglycemic dividing murine bone marrow–derived macrophages have hyaluronan on their cell surface in normoglycemia that is more extensive under hyperglycemic stress with or without heparin. These cells were nonpermeabilized. The fluorescent micrographs show that BMDMs have HA on their surface in normoglycemia that is more extensive and extended for BMDMs that divided in hyperglycemia without or with heparin (A compared with C and D). F4/80, a macrophage marker, is stained in red (E–H). Overlay images and nuclei stained with DAPI are shown (I–L, blue). Scale bars for low-power images are 50 μm and for enlargements are 10 μm.
Bone marrow–derived macrophages synthesize HA that is mostly released into the culture media
Similar to U937 cells, BMDMs synthesize and secrete most of their HA in all conditions (Fig. 8, Media), and they have cell surface HA when cultured in normal glucose levels that is significantly increased in the hyperglycemic cultures with or without heparin (Fig. 8, Cells).
Figure 8.
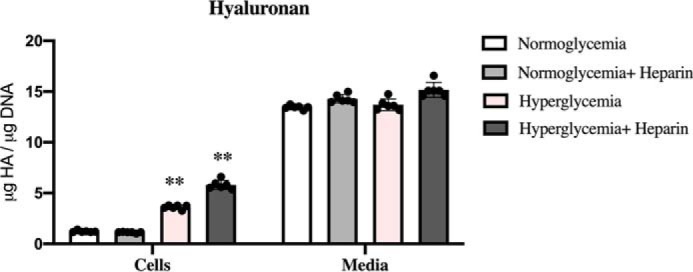
BMDMs synthesize HA that is mostly released into the culture media. The bar graphs show the amounts of HA determined by FACE in the cellular and medium compartments of BMDMs after 48 h of treatment in normoglycemia or hyperglycemia with or without heparin. In all culture conditions BMDMs synthesize the same amounts of HA that is released into the culture media (Media). The values of HA associated with BMDMs (Cells) cultured in hyperglycemia with or without heparin are statistically greater (p < 0.01) than those cultured in normoglycemia with or without heparin (n = 6).
Hyperglycemic stressed murine and human circulating monocytes contain an abnormal HA matrix in intracellular compartments
Ins2Akita mice (C57BL/6-Ins2Akita/J) are a monogenic model for phenotypes associated with type 1 diabetes that is useful for studying diabetic complications such as nephropathy, sympathetic autonomic neuropathy, and macrovascular disease (20–24). Circulating monocytes from a hyperglycemic Ins2Akita mouse stained for HA (green), but those from the control normoglycemia Ins2Akita mouse did not (Fig. 9C compared with Fig. 9A). Circulating monocytes from mice treated with streptozotocin, which is particularly toxic to the insulin-producing β cells of the pancreas in mammals and used to study type I diabetic issues (25), showed similar results as those from the hyperglycemic Ins2Akita mice (Fig. 9G compared with Fig. 9E). Circulating monocytes from high-fat diet–fed mice only stained for intracellular HA when blood glucose levels were high (Fig. 9K compared with Fig. 9I). Human circulating monocytes in blood smears from deidentified type 2 diabetic patients with HbA1c greater than 7, an indicator of uncontrolled high glucose levels in the blood, stained for HA in intracellular compartments in contrast with monocytes from deidentified type 2 diabetic patients with HbA1c lower than 7 (Fig. 9O compared with 9M, representative of four patients each). In summary, no intracellular HA was observed in any of the monocytes in the normoglycemic blood smears, whereas in the hyperglycemic ones, the vast majority did.
Figure 9.
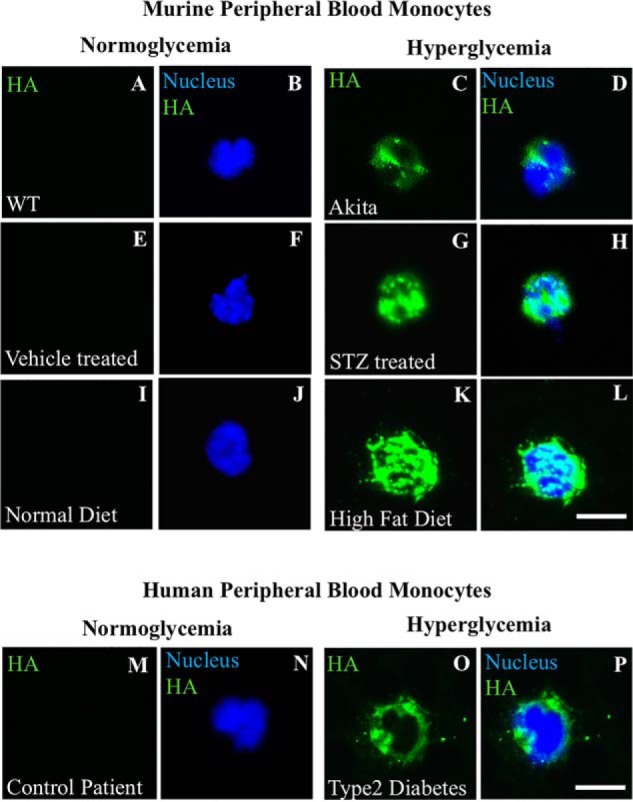
Hyperglycemic stressed murine circulating monocytes contain an abnormal HA matrix in intracellular compartments. The fluorescent micrographs show examples of circulating monocytes in blood smears from normoglycemic and hyperglycemic diabetic mice models that were stained for HA using a fluorescent conjugate of biotinylated HA-binding protein (green) and nuclei stained with DAPI (blue). Representative circulating monocytes in blood smears are shown for hyperglycemic mice from Ins2Akita (C and D), streptozotocin-treated (G and H), and high-fat diet–fed (K and L) mice compared with their controls (A and B, E and F, and I and J), n = 4. A representative circulating monocyte in blood smears are shown from deidentified type 2 diabetic patients of a HbA1c > 7, n = 4 (O and P), compared with (M and N) patients with a HbA1c <7; n = 4. Scale bars, 10 μm.
Discussion
Monocytes are rapidly recruited to the site of injury or inflammation where they are differentiated into macrophages. Classically activated proinflammatory (Mpi) macrophages are characterized by the increased secretion of inflammatory cytokines (Th1) and NO production required for an effective pathogen killing (5). Alternatively activated anti-inflammatory and tissue repair (Mtr) macrophages that have a high phagocytic capacity are characterized by the increased secretion of anti-inflammatory cytokines; produce ECM components, angiogenic, and chemotactic factors that mitigate inflammatory responses; promote wound healing; and facilitate the resolution of inflammation. Mpi and Mtr macrophages promote Th1 and Th2 responses respectively linking the innate to the adaptive immunity (5, 7, 9, 26–28). Our results show that hyperglycemic stressed monocytes such as leukemic U937 cells (Fig. 1) and BMDMs express higher levels of proinflammatory markers (Fig. 5). In contrast, heparin inhibits proinflammatory markers (iNOS and TNFα) and promotes anti-inflammatory markers (ARG1 and IL-10) in BMDMs, but not in U937 cells, dividing under hyperglycemic stress (Figs. 1 and 5). U937 cells are a leukemic monocytic cell line that is reprogrammed to sustain cell division and may no longer have the ability to promote an anti-inflammatory repair phenotype through the heparin-induced mechanism. IRF-4 has been shown to suppress M1 macrophage polarization (6, 29) and induce anti-inflammatory markers IL-10 (30) and ARG1 (14, 31). Fig. 5 shows that heparin-treated hyperglycemic BMDM cultures up-regulate anti-inflammatory IRF-4 and down-regulate proinflammatory IRF-8. In contrast, we have not been able to detect any levels of IRF-4 on Western blots of hyperglycemic U937 cells treated with heparin (data not shown). Unlike human monocytes, U937 cells are shown to not express IRF-4 (human protein atlas IRF-4). Therefore, the lack of expression of IRF-4 in U937 cells dividing in hyperglycemia could be a possible explanation for the inability of heparin to induce IL-10 and ARG1 expression.
Furthermore, our data show that intracellular HA is observed in hyperglycemic stressed U937 cells and BMDMs in vitro (Figs. 2 and 6), as well as in vivo in circulating monocytes in mouse diabetic models and in hyperglycemic human diabetic patients (Fig. 9) that results from the increased levels of cellular glucose. Glucose can be converted to the UDP sugars required for HA synthesis, which we propose is a mechanism by which cells such monocytes and macrophages deal with the increased levels of glucose in their environment. However, this intracellular HA accumulation, which is inhibited by heparin, in monocytes/macrophages is abnormal. It has been reported that peripheral blood monocytes in human subjects with poorly controlled diabetes, in a mechanism that is still unclear, are activated and functionally affected while showing some of the differentiation markers associated with macrophages (32, 33). We hypothesize that the abnormal intracellular accumulation of HA promotes the proinflammatory phenotype in monocytes/macrophages that compromises their function, whereas these effects are inhibited by heparin, which promotes the anti-inflammatory phenotype.
Our data show that U937 cells and BMDMs synthesize HA on their surface in normal glucose that is significantly increased in hyperglycemia, whereas heparin initiates a more extensive extracellular HA matrix than that in hyperglycemia alone. Interestingly, most of the HA, synthesized on the cell surface even in normoglycemia, is released into their environment (Figs. 4 and 8). This could be a process for circulating monocytes to deal with the increased levels of glucose in normal conditions such as after a meal or in diabetes. It is important for circulating monocytes not to retain the synthesized HA on their cell surface so as not to hinder their ability to roll and adhere on vascular endothelial cells, a process that is required for their extravasation into inflammatory regions (34).
It has been reported that adoptively transferred preactivated anti-inflammatory macrophages resulted in clinical abrogation of type 1 diabetes (35), which emphasizes on the importance of modulating the transition of Mpi and Mtr innate responses in resolving the inflammations caused by hyperglycemia (3). Our results show that heparin can promote Mtr macrophages, which explains their maintained ability to bind to and remove the HA matrix that is synthesized by the kidney glomerular mesangial cells of the uncontrolled diabetic rat (1, 2). Their ability to effectively remove the HA matrix helps resolve the inflammation without leading to fibrosis and renal failure.
Given the profound influence of macrophage polarization on the repair outcomes at the site of inflammation, it is important to focus on modulating Mpi and Mtr innate responses to achieve better health. Our results provide evidence that low concentrations of heparin can promote an Mtr while preventing an Mpi polarization, which can lead to a resolution in inflammation and provide a potential therapeutic agent that needs to be further investigated.
Experimental procedures
Mice
All mice were maintained in the Biological Resource Unit of the Cleveland Clinic Lerner Research Institute in a temperature-controlled facility with an automatic 12 h light-dark cycle and were given free access to food and water. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic (approval nos. 2015-1459, 2017-1894, and 2018-1944) in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. WT mice on C57BL/6J background were purchased from Jackson ImmunoResearch Laboratories and used for isolating murine femur bone marrow–derived macrophages (36).
Murine femur/tibia bone marrow–derived macrophages
Bone marrow cells were harvested from the tibia and femur bones and cultured in Dulbecco's modified Eagle's medium (1 g/liter glucose, 20% fetal bovine serum, 26140-079, Gibco) containing murine granulocyte macrophage–colony-stimulating factor (315-03; PeproTech) as previously described (36). After 24 h, the cells were treated for the duration of 7 days. The concentrations of glucose in the cultures were maintained for 7 days at 5 mm for the normal glucose medium (5) and at 25 mm for the high glucose medium. The concentration of heparin (375095, EMD Millipore, molecular mass of 13,500–15,000 kDa) was 2 μg/ml. At the end of day 7, media were collected, and adherent cells were harvested for various analytical assays.
Culturing U937 cells
U937 cells (ATCC® CRL-1593.2TM, a human leukemic cell line) were cultured in RPMI 1640 medium (1 g/liter glucose, 5% fetal bovine serum), serum starved (0.5% fetal bovine serum) for 24 h and then treated in the different media before the cells were harvested and separated from their media for various assays.
Circulating blood monocytes
Blood was collected from euthanized Ins2Akita mice (20–24, 37) versus their littermate controls, high-fat diet–fed mice, and STZ-treated mice versus wildtype via a cardiac puncture. Blood samples were obtained from deidentified type 2 diabetic patients with HbA1c greater versus lower than 7, by an approved protocol (institutional review board approval no. 06–050) at Cleveland Clinic with minimal risk, waived consent, and in accordance with the ethical guidelines of the 1975 Declaration of Helsinki. Blood was collected in tubes with K2 EDTA as an anticoagulant. Blood smears on glass slides from all groups were air-dried, then methanol-fixed, and stored at −20 °C before staining.
Fluorophore-assisted carbohydrate electrophoresis (FACE)
HA contents in the media and cellular compartments of murine macrophages and U937 cells were measured by FACE, as previously described (38). The DNA contents from identically treated cells were measured at the end of the duration of treatment and used for normalization purposes. Band intensities on the gel were measured using ImageJ software.
Western blotting analysis
The samples were electrophoresed on 4–5% Mini-PROTEAN TGX gels (Bio-Rad) and blotted using Bio-Rad polyvinylidene difluoride membranes and the Trans-Blot turbo system. All blots were blocked for 1 h with 5% milk and probed overnight at 4 °C with antibodies against IRF-4 (SC-28696) and IRF-8 (SC-6058) (1:1000; Santa Cruz Biotechnology). The blots were incubated with secondary antibody for 1 h at room temperature. All blots were developed using the ECL Prime Western blotting detection reagent (RPN2232; Amersham Biosciences).
Immunohistochemistry
U937 cells from different treatments were transferred to a glass slide via cytopsins. Cytospins as well as blood smears on glass slides were air-dried completely, then fixed with 100% methanol at −20 °C for 10 min, and air-dried for 1 h. In some experiments, BMDMs and U937 cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature and then permeabilized by treating them with 0.4% Triton X-100 for 10 min at room temperature. The slides were rehydrated in 1× PBS for 30 min and then blocked with 1% BSA in PBS. These slides were incubated with either HA biotinylated binding protein (5 μg/ml; 385911; EMD Chemicals), anti-human CD44 (C7923, 1:250; Sigma-Aldrich), anti-human CD80 (SC-1634), anti-human CD163 (SC-33559) (1:100; Santa Cruz Biotechnology), or anti-murine F4/80 (MAB5580, 1:100; R&D Systems) in blocking solution for 1 h, washed three times in 1× PBS, and then incubated with streptavidin conjugated to Alexa Fluorp® 488 (S1122, 1:500; Invitrogen), Alexa Fluor® 594 donkey anti-mouse secondary (A11058, 1:250; Invitrogen), CyTM3 AffiniPure goat anti-rat IgG (H+L) (112-165-167), or CyTM3 AffiniPure donkey anti-rabbit IgG (H+L) (711-165-152) (1:250; Jackson ImmunoResearch Laboratories) for 1 h. Vectashield mounting medium with DAPI (H-1200; Vector Laboratories) was applied, followed by coverslip application before visualizing using fluorescent microscopy.
PCR analyses
RNA was isolated using the RNAeasy kit (74104; Qiagen), and cDNA was prepared using Superscript first-strand synthesis system (11904-018; Invitrogen). The primers used are listed in Table 1. The PCR conditions were 1 cycle at 94 °C for 3 min, 40 cycles of 95 °C for 30 s, 55 °C for 45 s, 72 °C for 1 min, followed by 1 cycle of 72 °C for 1 min. These conditions were used for all primers.
Table 1.
Primers used

Statistical analysis
The data are presented as the means ± S.E.; n is indicated in figure legends in representative experiments.
Author contributions
A. A. and A. W. conceptualization; A. A. and J. L. data curation; A. A. formal analysis; A. A. validation; A. A. investigation; A. A. and M. Y. methodology; A. A. writing-original draft; A. A. and V. H. project administration; A. A. and V. H. writing-review and editing; Y. W., S. S., X. L., and E. M. resources; V. H. supervision; V. H. funding acquisition; A. W. provided Fig. 1C; Y. W. and S. S. performed the high-fat diet and streptozotocin experiments and provided the blood smears from those experiments; E. M. reviewing and editing the manuscript.
Acknowledgments
We thank Dr. Bela Anand-Apte and Mariya Ali (Cole Eye Institute, Cleveland Clinic) for providing us with the Ins2Akita mice. We thank Dr. Jennifer Ko (Dermatopathology and Central Biorepository, Cleveland Clinic) for providing us with blood samples from deidentified type 2 diabetic patients with HbA1c greater versus lower than 7.
This work was supported by Grants P01HL107147 and 1K12HL141952 from the NHLBI, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- STZ
- streptozotocin
- HA
- hyaluronan
- CD
- cluster of differentiation
- FACE
- fluorophore-assisted carbohydrate electrophoresis
- RMC
- rat mesangial cell
- TNFα
- tumor necrosis factor α
- iNOS
- inducible nitric-oxide synthase
- IL
- interleukin
- IRF
- interferon regulatory factor
- ARG1
- arginase 1
- BMDM
- murine bone marrow–derived macrophage
- HbA1c
- hemoglobin A1c
- Mpi
- proinflammatory macrophage(s)
- Mtr
- tissue repair macrophage(s)
- DAPI
- 4′,6′-diamino-2-phenylindole.
References
- 1. Wang A., Ren J., Wang C. P., and Hascall V. C. (2014) Heparin prevents intracellular hyaluronan synthesis and autophagy responses in hyperglycemic dividing mesangial cells and activates synthesis of an extensive extracellular monocyte-adhesive hyaluronan matrix after completing cell division. J. Biol. Chem. 289, 9418–9429 10.1074/jbc.M113.541441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang A., and Hascall V. C. (2004) Hyaluronan structures synthesized by rat mesangial cells in response to hyperglycemia induce monocyte adhesion. J. Biol. Chem. 279, 10279–10285 10.1074/jbc.M312045200 [DOI] [PubMed] [Google Scholar]
- 3. Forbes J. M., and Cooper M. E. (2013) Mechanisms of diabetic complications. Physiol. Rev. 93, 137–188 10.1152/physrev.00045.2011 [DOI] [PubMed] [Google Scholar]
- 4. Sica A., Erreni M., Allavena P., and Porta C. (2015) Macrophage polarization in pathology. Cell Mol. Life Sci. 72, 4111–4126 10.1007/s00018-015-1995-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murray P. J., Allen J. E., Biswas S. K., Fisher E. A., Gilroy D. W., Goerdt S., Gordon S., Hamilton J. A., Ivashkiv L. B., Lawrence T., Locati M., Mantovani A., Martinez F. O., Mege J. L., Mosser D. M., et al. (2014) Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Honma K., Udono H., Kohno T., Yamamoto K., Ogawa A., Takemori T., Kumatori A., Suzuki S., Matsuyama T., and Yui K. (2005) Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc. Natl. Acad. Sci. U.S.A. 102, 16001–16006 10.1073/pnas.0504226102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mills C. D. (2012) M1 and M2 Macrophages: oracles of health and disease. Crit. Rev. Immunol. 32, 463–488 10.1615/CritRevImmunol.v32.i6.10 [DOI] [PubMed] [Google Scholar]
- 8. Italiani P., and Boraschi D. (2014) From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front. Immunol. 5, 514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mills C. D., Kincaid K., Alt J. M., Heilman M. J., and Hill A. M. (2000) M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 164, 6166–6173 10.4049/jimmunol.164.12.6166 [DOI] [PubMed] [Google Scholar]
- 10. Mills C. D. (2001) Macrophage arginine metabolism to ornithine/urea or nitric oxide/citrulline: a life or death issue. Crit. Rev. Immunol. 21, 399–425 [PubMed] [Google Scholar]
- 11. Stempin C. C., Dulgerian L. R., Garrido V. V., and Cerban F. M. (2010) Arginase in parasitic infections: macrophage activation, immunosuppression, and intracellular signals. J. Biomed. Biotechnol. 2010, 683485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sundström C., and Nilsson K. (1976) Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565–577 10.1002/ijc.2910170504 [DOI] [PubMed] [Google Scholar]
- 13. Liu L., Zubik L., Collins F. W., Marko M., and Meydani M. (2004) The antiatherogenic potential of oat phenolic compounds. Atherosclerosis 175, 39–49 10.1016/j.atherosclerosis.2004.01.044 [DOI] [PubMed] [Google Scholar]
- 14. Jaguin M., Houlbert N., Fardel O., and Lecureur V. (2013) Polarization profiles of human M-CSF–generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol. 281, 51–61 10.1016/j.cellimm.2013.01.010 [DOI] [PubMed] [Google Scholar]
- 15. Wang A., de la Motte C., Lauer M., and Hascall V. (2011) Hyaluronan matrices in pathobiological processes. FEBS J. 278, 1412–1418 10.1111/j.1742-4658.2011.08069.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de la Motte C. A., Hascall V. C., Drazba J., Bandyopadhyay S. K., and Strong S. A. (2003) Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: inter-alpha-trypsin inhibitor is crucial to structure and function. Am. J. Pathol. 163, 121–133 10.1016/S0002-9440(10)63636-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang A., Midura R. J., Vasanji A., Wang A. J., and Hascall V. C. (2014) Hyperglycemia diverts dividing osteoblastic precursor cells to an adipogenic pathway and induces synthesis of a hyaluronan matrix that is adhesive for monocytes. J. Biol. Chem. 289, 11410–11420 10.1074/jbc.M113.541458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhattacharjee A., Shukla M., Yakubenko V. P., Mulya A., Kundu S., and Cathcart M. K. (2013) IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic. Biol. Med. 54, 1–16 10.1016/j.freeradbiomed.2012.10.553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eguchi J., Kong X., Tenta M., Wang X., Kang S., and Rosen E. D. (2013) Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes 62, 3394–3403 10.2337/db12-1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshioka M., Kayo T., Ikeda T., and Koizumi A. (1997) A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 46, 887–894 10.2337/diab.46.5.887 [DOI] [PubMed] [Google Scholar]
- 21. Wang J., Takeuchi T., Tanaka S., Kubo S. K., Kayo T., Lu D., Takata K., Koizumi A., and Izumi T. (1999) A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody mouse. J. Clin. Invest. 103, 27–37 10.1172/JCI4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mathews C. E., Langley S. H., and Leiter E. H. (2002) New mouse model to study islet transplantation in insulin-dependent diabetes mellitus. Transplantation 73, 1333–1336 10.1097/00007890-200204270-00024 [DOI] [PubMed] [Google Scholar]
- 23. Ron D. (2002) Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J. Clin. Invest. 109, 443–445 10.1172/JCI0215020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gurley S. B., Clare S. E., Snow K. P., Hu A., Meyer T. W., and Coffman T. M. (2006) Impact of genetic background on nephropathy in diabetic mice. Am. J. Physiol. Renal Physiol. 290, F214–F222 10.1152/ajprenal.00204.2005 [DOI] [PubMed] [Google Scholar]
- 25. Szkudelski T. (2001) The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 50, 537–546 [PubMed] [Google Scholar]
- 26. Gordon S. (2003) Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 10.1038/nri978 [DOI] [PubMed] [Google Scholar]
- 27. Gordon S., and Taylor P. R. (2005) Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964 10.1038/nri1733 [DOI] [PubMed] [Google Scholar]
- 28. Gordon S., and Martinez F. O. (2010) Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604 10.1016/j.immuni.2010.05.007 [DOI] [PubMed] [Google Scholar]
- 29. Lawrence T., and Natoli G. (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 10.1038/nri3088 [DOI] [PubMed] [Google Scholar]
- 30. Ahyi A. N., Chang H. C., Dent A. L., Nutt S. L., and Kaplan M. H. (2009) IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J. Immunol. 183, 1598–1606 10.4049/jimmunol.0803302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El Chartouni C., Schwarzfischer L., and Rehli M. (2010) Interleukin-4 induced interferon regulatory factor (Irf) 4 participates in the regulation of alternative macrophage priming. Immunobiology 215, 821–825 10.1016/j.imbio.2010.05.031 [DOI] [PubMed] [Google Scholar]
- 32. Cipolletta C., Ryan K. E., Hanna E. V., and Trimble E. R. (2005) Activation of peripheral blood CD14+ monocytes occurs in diabetes. Diabetes 54, 2779–2786 10.2337/diabetes.54.9.2779 [DOI] [PubMed] [Google Scholar]
- 33. Szablewski L., and Sulima A. (2017) The structural and functional changes of blood cells and molecular components in diabetes mellitus. Biol. Chem. 398, 411–423 10.1515/hsz-2016-0196 [DOI] [PubMed] [Google Scholar]
- 34. Gerhardt T., and Ley K. (2015) Monocyte trafficking across the vessel wall. Cardiovasc. Res. 107, 321–330 10.1093/cvr/cvv147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parsa R., Andresen P., Gillett A., Mia S., Zhang X. M., Mayans S., Holmberg D., and Harris R. A. (2012) Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes 61, 2881–2892 10.2337/db11-1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang X., Goncalves R., and Mosser D. M. (2008) The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter 14, Unit 14.11 10.1002/0471142735.im1401s83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oyadomari S., Koizumi A., Takeda K., Gotoh T., Akira S., Araki E., and Mori M. (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109, 525–532 10.1172/JCI0214550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Calabro A., Benavides M., Tammi M., Hascall V. C., and Midura R. J. (2000) Microanalysis of enzyme digests of hyaluronan and chondroitin/dermatan sulfate by fluorophore-assisted carbohydrate electrophoresis (FACE). Glycobiology 10, 273–281 10.1093/glycob/10.3.273 [DOI] [PubMed] [Google Scholar]