

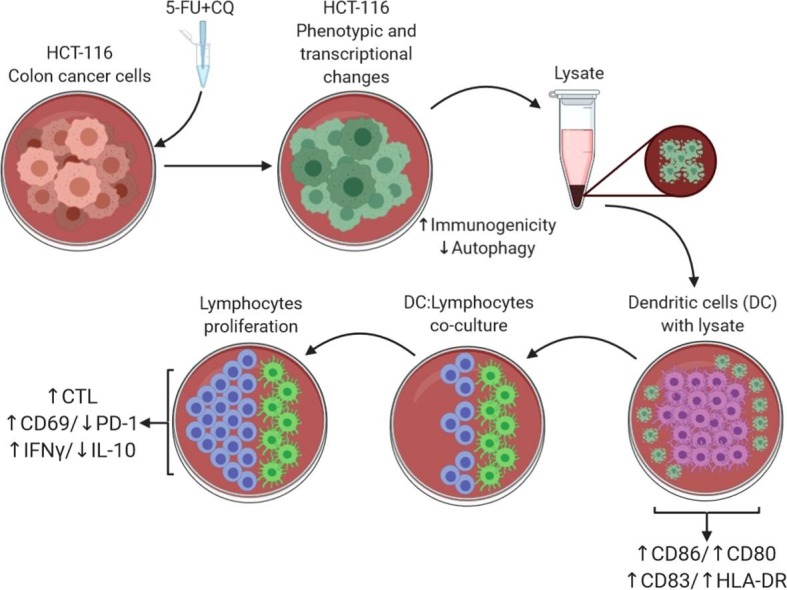
Graphical abstract
Keywords: Autophagy, Cancer, Colorectal, Chemotherapy, Dendritic cells, Cytotoxic T cells
Highlights
-
•
Autophagy helps tumor cells to face drug-induced damages, stimulating tolerance.
-
•
Autophagy inhibition improves the efficiency of antineoplastic agents.
-
•
Blockage of autophagy with Chloroquine increases synthesis of tumor antigens.
-
•
Blockage of autophagy changes the expression of ATG and tumor suppressor genes.
-
•
Lysate of drug-treated cells enhances maturation/activation of human dendritic cells.
-
•
Lysate of drug-treated cells induce Th1 alloresponse, and generation of tumor-specific CTL.
Abstract
Autophagy is an important mechanism for tumor escape, allowing tumor cells to recover from the damage induced by chemotherapy, radiation therapy, and immunotherapy and contributing to the development of resistance. The pharmacological inhibition of autophagy contributes to increase the efficacy of antineoplastic agents. Exposing tumor cells to low concentrations of select autophagy-inducing antineoplastic agents increases their immunogenicity and enhances their ability to stimulate dendritic cell (DC) maturation. We tested whether the application of an autophagy-inhibiting agent, chloroquine (CQ), in combination with low concentrations of 5-fluorouracil (5-FU) increases the ability of tumor cells to induce DC maturation. DCs sensitized with the lysate of HCT-116 cells previously exposed to such a combination enhanced the DC maturation/activation ability. These matured DCs also increased the allogeneic responsiveness of both CD4+ and CD8+ T cells, which showed a greater proliferative response than those from DCs sensitized with control lysates. The T cells expanded in such cocultures were CD69+ and PD-1- and produced higher levels of IFN-γ and lower levels of IL-10, consistent with the preferential activation of Th1 cells. Cocultures of autologous DCs and lymphocytes improved the generation of cytotoxic T lymphocytes, as assessed by the expression of CD107a, perforin, and granzyme B. The drug combination increased the expression of genes related to the CEACAM family (BECN1, ATGs, MAPLC3B, ULK1, SQSTM1) and tumor suppressors (PCBP1). Furthermore, the decreased expression of genes related to metastasis and tumor progression (BNIP3, BNIP3L, FOSL2, HES1, LAMB3, LOXL2, NDRG1, P4HA1, PIK3R2) was noted. The combination of 5-FU and CQ increases the ability of tumor cells to drive DC maturation and enhances the ability of DCs to stimulate T cell responses.
1. Introduction
Colorectal cancer (CRC) is the third most frequent cancer observed worldwide [1], with 40,990 new cases expected for 2020 in Brazil [2]. Conventional therapy for patients with CRC is based on total or partial colectomy, usually followed by neoadjuvant or adjuvant chemotherapy [3] with a fluoropyrimidine, oxaliplatin, irinotecan and/or taxanes [4]. The success of chemotherapy is often complicated by metastases and relapsing disease associated with the development of drug resistance [5]. One of the mechanisms responsible for drug resistance is autophagy, a selective cellular degradation process in which cytosolic proteins and organelles are sequestered into double-membrane autophagosomes that fuse with lysosome for the recycling of macromolecules [6], [7]. This process is also triggered to generate amino acids, nucleotides, and fatty acids under conditions of nutrient deprivation [8]. Other types of stress can also trigger autophagy, such as genomic stress, endoplasmic reticulum stress, intracellular infections, and exposure to drugs [9].
We have previously observed that certain chemotherapeutic agents at ultra-low concentrations can modulate signaling pathways and induce cytokines, such as IL-12, IL-10, IL-4, and TNF-α [10], without inducing apoptosis. Treatment of HCT-116 colorectal cancer cells with a nontoxic concentration of paclitaxel alters the expression of several genes, especially those responsible for the synthesis of heat shock proteins, components of the antigen processing machinery (APM) and tumor-associated antigens [11]. The increased immunogenicity of tumor cells induced by drug exposure is dependent on the onset of so-called immunogenic cell death, with increased expression of danger signals (e.g., danger-associated molecular patterns; DAMPs) such as calreticulin, heat shock proteins, ATP, and high mobility group box 1 (HMGB-1) [12], [13], [14], [15], [16]. Consistent with this notion, we also observed that exposing HCT-116 cells to a nontoxic concentration of paclitaxel or doxorubicin causes transcriptional alterations in several genes associated with the expression of tumor antigens [17]. In addition, dendritic cells (DCs) sensitized with the lysate of HCT-116 cells that were previously treated with low concentrations of paclitaxel induced cytotoxic T lymphocytes (CTLs) with higher lytic potential than did the DCs sensitized with untreated tumor cells [17].
More recently, combining antineoplastic agents with autophagy blockers as a therapeutic approach for treating cancer patients has been proposed. Exposing human colorectal cancer cells to 5-fluorouracil (5-FU) enhances autophagy in a considerable portion of these cells [18]. Exposure of these cells to the antimalarial drugs chloroquine (CQ), hydroxychloroquine, or mefloquine (quinolones) reduces autophagy and increases the susceptibility of cells to chemotherapy [19]. Quinolones function as weak bases that passively diffuse into the lysosomes where they are protonated and prevented from leaving this vesicle. Their presence increases the lysosome pH, interrupting its functions and preventing the end of autophagy [20], [21]. As anti-inflammatory drugs, quinolones have been used to treat some autoimmune diseases [22]. Currently, there are a number of clinical trials and experimental studies focusing on the feasibility of using CQ and hydroxychloroquine against SARS-CoV-2 (COVID-19) [23], [24] . In addition, the combination of CQ and antineoplastic drugs enhances the clinical response in patients with breast [25] and kidney cancer [26], showing a synergistic effect on the activation of mTOR (mammalian target of rapamycin), inhibiting autophagy, and thus increasing tumor cell death. No studies on the induction of autophagy in tumor cells treated with ultra-low doses of chemotherapeutic agents are available, and little is known about the effect of this phenomenon on the immunogenicity of tumor cells. Furthermore, there are no reports on the effects of using CQ combined with other drugs on the functions of immunocompetent cells such as lymphocytes or DCs.
In the present study, we tested the hypothesis that the application of CQ in combination with low concentrations of 5-FU blocks autophagy in tumor cells and promotes DC maturation, increasing their ability to enhance T lymphocyte cytotoxic granule expression.
We found that exposing HCT-116 cells to this combination induced transcriptional changes, and DCs treated with HCT-116 lysate showed an improved ability to stimulate the proliferation of allogeneic T cells and to enhance the generation of cytotoxic CD8+ T cells. These results lead us to conclude that antigenic and transcriptional changes induced in tumor cells by the combination of CQ and low concentrations of 5-FU can be used as a basis for developing better DC-based antitumor reactive T cells.
2. Materials and methods
2.1. Culture and treatments of colorectal cancer HCT-116 cells
The human colon cancer cell line HCT-116 was authenticated by DNA STR profiling using the GenePrint 10 commercial system (Promega, Madison, WI, USA) at the Viral Carcinogenesis and Cancer Biology Research Group, Institute of Biotechnology (IBTEC), Sao Paulo State University (UNESP). These cells were authenticated as mycoplasma-free and were cultured in 150 cm2 culture bottles in RMPI-1640 medium (Gibco) supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 10 mM HEPES, and 1% antibiotics/antimycotics solution (Life Technologies). The cells were cultured at 37˚C under a 5% CO2 atmosphere. After 24 h of culturing, the cells were treated with 80 µM CQ for 18 h, and then the minimum effective concentration of 5-fluorouracil (20 µM) (Eurofarma) was added and the cells were cultured for a further 24 h. The cells were then detached with trypsin and washed before subsequent analysis or were cryopreserved for the further preparation of cell lysates. The CQ and 5-FU concentrations and the incubation times were previously determined by MTT-based cytotoxicity assay. For this, 2x103 cells were plated in 96-well flat-bottomed culture plates and incubated with variable concentrations of CQ (80 to 200 µM) and 5-FU (0.06 to 100 µM). Increased optical density due to formazan reduction was read by a spectrophotometer to evaluate the toxic effect of the drug concentrations.
2.2. Analysis of cell viability and death
The concentration of HCT-116 cells was adjusted to 105 cells/mL and cultured overnight in a 24-well plate. The cells were treated with CQ overnight, followed by exposure to 5-FU for 24 h. As a positive control of cell death, cells were heated at 59 °C for 20 min. The cells were detached by trypsinization and centrifuged at 1200 rpm for 10 min. The pellet was suspended in 100 µl of cold PBS and centrifuged at 10,000 rpm for 30 sec. The cell pellet was resuspended in 100 μl of annexin buffer solution and incubated with annexin V at room temperature and protected from light. After incubation, 200 μl of the annexin buffer was added. Finally, the cells were stained with 7-amino-actinomycin D (7AAD) to assess viability and were analyzed by flow cytometry.
2.3. HCT-116 lysate
Cryopreserved aliquots were thawed in a 37 °C water bath for 3 min and then quickly refrozen in liquid nitrogen for 3 min (freeze/thaw cycle). This process was repeated five times. The samples were then centrifuged at 14,000 rpm for 15 min, and the supernatant was treated with 70 μl/mL of protease inhibitor (Halt ™ Protease Inhibitor Cocktail ThermoFisher), followed by storage at −80 °C. The total protein was quantified using a ThermoFisher BCA Protein Assay Kit, and samples were aliquoted (100 μg protein) and stored at −80 °C.
2.4. Western blot
Briefly, 40 μg of cell lysate proteins derived from each of the four treatment groups (WT, CQ, 5-FU, and 5-FU + CQ) were diluted in gel electrophoresis sample buffer and heated at 95 °C for 10 min. The proteins were separated on a polyacrylamide gel and then transferred to a polyvinylidene difluoride membrane. Nonspecific binding of proteins was blocked by incubating the membranes in 5% low-fat milk in PBS-Tween buffer for 1 h at room temperature. The membranes were subsequently incubated overnight with antigen-specific primary antibodies for autophagy markers: LC3-II (#2775 s Cell Signaling Technologies) and SQSTM1 (#pm045 MBL Intern. Corp.); secondary anti-rabbit (#A0545 Sigma) and anti-mouse (#A9044 Sigma) antibodies were applied, respectively. GAPDH (#g8795 Sigma) was used as a housekeeping gene. The membranes were washed with PBS-Tween buffer for 30 min and developed with enhanced chemoluminescence immunoreactive reagent and read on a UVITEC Cambridge photodocumentation system.
2.5. Ultrastructural analysis by transmission electron microscopy
Approximately 106 cells were cultured overnight in a 6-well culture plate and were treated with CQ and 5-FU according to the abovementioned protocol . After incubation, cell monolayers were trypsinized, and the collected cells were centrifuged for 10 min at 1,200 rpm at 20 °C. The samples were fixed in 1 mL of Karnovsky's solution, post-fixed in 1% osmium tetroxide in 0.1 M phosphate buffer at pH 7.3, incubated in 0.5% uranyl acetate in aqueous solution; dehydrated in a gradient series of acetone solutions, and embedded in Araldite glue. The ultrafine sections (50 nm) were contrasted with saturated uranyl acetate and lead citrate solutions and observed using a Tecnai Spirit (FEI Company) electron microscope at 80 kV.
2.6. Human monocyte-derived dendritic cells
DCs were differentiated in vitro from the peripheral blood adherent mononuclear cells (PBMCs) of six healthy donors. The PBMCs were obtained by centrifugation through a Ficoll-isopaque gradient, suspended in AIM-V culture medium (Invitrogen), and seeded in 6-well culture plates (1x106/per well). After incubating for 1 h at 37˚C, non-adherent cells were removed and adherent monocytes were cultured in complete culture medium containing 80 ng/ml recombinant human GM-CSF and IL-4 (PeproTech) for 5 days. They were then treated with HCT-116 lysate (100 µg per 106 DCs) and kept in culture for an additional 48 h. Immature DCs were submitted to seven different culture conditions: DCs (untreated immature DCs); WT (DCs exposed to the lysate of untreated HCT-116 cells); CQ (DCs exposed to the lysate of HCT-116 cells that were pretreated with 80 µM of CQ), 5-FU (DCs exposed to the lysate of HCT-116 cells that were pretreated with 20 µM of 5-FU), and 5-FU + CQ (DCs exposed to the lysate of HCT-116 cells that were pretreated with 20 µM of 5-FU and 80 µM of CQ). All procedures involving both normal and transformed human cells were approved by the Ethics Committee of the Botucatu School of Medicine – UNESP (CEP # 2.258.145).
2.7. DC phenotyping
The lysate-exposed and control DCs were incubated with fluorescent monoclonal antibodies for 30 min and washed with PBS containing 0.1% bovine serum albumin (BSA) and 0.1% sodium azide. The DCs were incubated with labeled antibodies (HLA-DR-PE, CD11c-APC, CD83-PE-Cy7, CD80-APC-H7, and CD86-FITC (BD Biosciences) for 20 min at 4 °C and then washed with PBS-BSA. The cells were suspended in 100 µl of PBS-BSA, and the samples were read in a FACSCanto II cytometer (Becton-Dickinson) and analyzed using FlowJo, version 7.2.4.
2.8. Mixed lymphocyte reaction assay (MLR)
The functional activity of DCs was first evaluated through their ability to stimulate the proliferation of normal allogeneic T lymphocytes. DCs from six different donors were cocultured with allogeneic T lymphocytes (previously marked with carboxyfluorescein succinyl ester (CFSE)) in flat-bottomed 96-well plates in a 1:10 (104:105) DCs: lymphocyte ratio. Cells were harvested five days later, and the lymphocyte proliferation was analyzed by flow cytometry based on the dilution of CFSE in the replicant cells. We also analyzed the expression of PD-1 and CD69 on CD3+ cells using anti-PD-1-PE and CD69-APC-H7 (BD Pharmingen).
2.9. IFN-γ and IL-10 detection
Supernatants of the MLR assay were collected and preserved at −80˚C. These samples were analyzed for the in vitro synthesis of IFN-γ and IL-10 using an ELISA kit according to the manufacturer's instructions (R&D Systems).
2.10. Generation of cytolytic T lymphocytes and antitumor cytotoxicity assay
To generate specific antitumor T cells, DCs were cocultured with an autologous T lymphocyte-rich suspension in a 1:10 DC:lymphocyte ratio (104:105) in complete culture medium supplemented with IL-7 (5 ng/ml) and IL-2 (40 IU/ml). The culture was pulsed with IL-2 every two days for 14 days. On day 14, the lymphocytes were harvested and evaluated for their cytotoxic activity against HCT-116 target cells. A lymphocytotoxicity assay was performed by adding the in vitro-generated lymphocytes to HCT-116 monolayer cells in different effector: target ratios (15:1, 7.5:1 and 3.25:1). Anti-CD107a-APC was added to these cocultures and incubated for 5 h at 37˚C in an atmosphere containing 5% CO2. The cells were then harvested and washed with 0.1% BSA in PBS and treated with the Cytofix/CytoPerm kit (BD Pharmingen). Finally, the cells were labeled with anti-perforin-Alexa 488 and anti-granzyme-PE antibodies and analyzed by flow cytometry.
2.11. Tumor cell transcription
To investigate how treatment with the combination of CQ and 5-FU changes tumor cell gene expression, we performed a transcription analysis of these cells. Total RNA was extracted using a QIAGEN RNeasy Plus Micro Kit. RNA was quantified using an RNA HS Assay Kit (Invitrogen) and a QBIT® system. The quality of RNA was analyzed using an Agilent RNA 6000 chip in a Bioanalyzer 2100 system. Only samples with an RNA integrity number higher than 8.0 (optimal quality) were processed.
All indications and steps of the Sure Select Strand-Specific RNA Library Preparation Kit were followed, and the dsDNA libraries of each treatment group with the adapters and index were analyzed on the Illumina Miseq platform. After obtaining the data, the reads were assembled based on the sequences of each transcript of interest using the CLC Genomics Workbench program. The effect of the treatments on gene expression was evaluated by comparing the fold change in RNA expression of the samples with that of the untreated control.
2.12. Statistical analysis
Homogeneity of variance was accessed by the Bartlett test, and the data were analyzed by analysis of variance (ANOVA) followed by the Tukey–Kramer test for multiple comparisons. Differences were considered significant when the error probability was less than or equal to 5% (p ≤ 0.05).
3. Results
3.1. Combination of chloroquine and 5-FU blocks autophagy
We first verified whether the working concentrations of CQ and 5-FU were able to block autophagy. Proteins extracted from untreated tumor cells (WT) or cells treated with either chloroquine (80 µM) or 5-fluorouracil (20 µM) or those treated with the combination of drugs (5-FU + CQ) were analyzed by western blot to evaluate the accumulation of LC3-II and SQSTM1 (both autophagic markers). We observed slightly increased LC3-II expression when treating with CQ alone, while the combination of CQ and 5-FU resulted in higher LC3-II expression. Interestingly, treatment with 5-FU alone induced a slight increase of LC3-II expression, indicating the induction of autophagy, as confirmed by the lower intensity of SQSTM1 in this group (Fig. 1 A).
Fig. 1.

Autophagy Inhibition prior to Chemotherapy Treatment of HCT-116 cells. Cells were treated with 80 μM CQ overnight and then treated with the minimum effective concentration of 5-FU (20 µM) for 24 h. A portion of these cells was lysed, and the total proteins were analyzed by western blot for LC3-II and SQSTM1 (A). GAPDH was used as a housekeeping gene (C). The remaining cells were analyzed by transmission electron microscopy (B-E). N = nucleus; green arrow: autophagosome; yellow arrow: autophagosome with undigested organelles; blue arrow: autophagolysosomes (vesicles with degraded material). Groups: Control (B), CQ (C), 5-FU (D), 5-FU + CQ (E).
3.2. Ultrastructural changes induced by 5-FU and CQ in HCT-116 cells
To reinforce the cytometric results of the induction and inhibition of autophagy and the induction of apoptosis, we used transmission electron microscopy to analyze the cells under different treatments. Cells treated with 5-FU showed double-membrane vesicles (autophagosomes) and single-membrane vesicles containing degraded material (autophagolysosomes) in the cytoplasm (Fig. 1D). By contrast, the control cells showed a very small number of autophagosomes containing flocculated and degraded material in addition to containing mitochondria and some rough endoplasmic reticulum cisterns (Fig. 1B). In the 5-FU + CQ combined treatment, the size of autophagosomes appeared to increase within the cytoplasm, and these vesicles exhibited different filler contents. Some vesicles had heterogeneous electron-dense material inside them, and others showed myelin figures and electron-lucent degraded material, indicating that some autophagosomes were not engaged in the digestion process and that the process of digestion was blocked. There was a clear accumulation of autophagosomes containing undigested organelles, demonstrating the failure or inhibition of autophagolysosome formation (Fig. 1E). This finding is in agreement with previous studies [27], [28], [29], [30].
3.3. Enhanced cytotoxic action of 5-FU by inhibiting autophagy with chloroquine
Table 1 shows the number of HCT-116 cells after incubation with CQ, 5-FU or the combination of both drugs (five independent assays). While CQ alone had a discrete effect on cell growth, 5-FU reduced the number of cells to approximately 50% of the control culture. The combination of 5-FU and CQ strongly inhibited cell growth in the cultures.
Table 1.
Number of HCT-116 cells following treatment with CQ, 5-FU, or the combination of both drugs. CTRL refers to untreated cells cultured in regular culture medium. Each column refers to an independent assay (cultured on different days).
| HC-T116 cell counting (x 107) | |||||
|---|---|---|---|---|---|
| Treatment | Assay 1 | Assay 2 | Assay 3 | Assay 4 | Assay 5 |
| CTRL | 6.7 | 6.5 | 6.3 | 6.4 | 6.4 |
| CQ | 5.7 | 5.1 | 5.6 | 5.2 | 5.3 |
| 5-FU | 3.0 | 3.4 | 3.5 | 3.1 | 3.3 |
| 5-FU + CQ | 1.2 | 1.5 | 1.3 | 1.2 | 1.4 |
Cells were cultured in 75 cm2 culture flasks until achieving 60% confluence and then either CQ 20 mM (24 h), 5-FU 20 mM (24 h), or the combination of both drugs (24 h + 24 h) was added. Cells were detached by trypsinization and counted. The cell viability was evaluated by the trypan blue exclusion test, and no differences of viability were observed among the cell suspensions (all greater than 94%). CTRL refers to untreated cells cultured in regular culture medium. Each column refers to an independent assay (cultured on different days).
Fig. 2 shows that early apoptosis (annexin V+) was the predominant type of cell death induced by the treatments. 5-FU alone (Fig. 2D) increased by four times the annexin V labeling compared to the control (Fig. 2B), while its combination with CQ increased early apoptosis in ten times compared with the same control (Fig. 2E), and around twice compared with 5-FU alone.
Fig. 2.

The combination of chloroquine and 5-FU induced apoptosis in HCT-116 cells. HCT-116 tumor cells were treated or not with 80 µM CQ overnight. Subsequently, they were treated or not with the minimum nontoxic or effective dose of 5-FU. After 24 h, the cells were trypsinized and stained with annexin V and 7AAD to assess apoptosis and necrosis (A). Pseudocolor showing cell size (FSC) and granularity (SSC), with analysis gate (B). Negative Control (C). Treatment with QC (D). Treatment with 5-FU (E). Treatment with QC + 5-FU (F).
3.4. Treatment of tumor cells with CQ plus 5-FU enhances their ability to induce DC maturation
Fig. 3 illustrates the effect of the HCT-116 lysates on the DC phenotype, highlighting the overall increase in the percentage of cells expressing maturation or activation markers CD83, CD80, CD86, and HLA-DR. The percentage of cells expressing the CD83 marker increased from 74 ± 3.4% in WT to more than 90% in the groups exposed to 5-FU + CQ. Cell markers associated with DC maturation (CD80, CD86, and HLA-DR) increased from 53.5 ± 2.7%; 74.5 ± 1.2%, and 65.9 ± 3.2% in the WT control to 63.7 ± 5.1%; 92.7 ± 1.5%, respectively, and to 68.7 ± 2.5%, 93.5 ± 1.12% and 86.3 ± 2.4% in cells treated with 5-FU + CQ, respectively. Although treating HCT-116 cells with CQ or 5-FU alone increase the immunogenic effect on DCs, this effect was not as intense as seen in the drug combination.
Fig. 3.

Lysates of HCT-116 exposed to 5-FU + CQ increase DC maturation and activation. DCs from six healthy donors were exposed to the lysates of HCT-116 cells previously treated with the minimal effective concentrations of 5-FU or its combination with CQ (5-FU + CQ). WT refers to DCs pulsed with the lysate of untreated HCT-116 cells, and CTRL refers to unstimulated DCs. Scatter plots illustrate the percentages of DCs (CD11c+) that co-express the markers CD83 (A), HLA-DR (B), CD80 (C), and CD86 (D). The mean and standard deviation of the six individual donors was analyzed by ANOVA. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.005.
3.5. DCs sensitized with lysates from HCT-116 cells pretreated with CQ and 5-FU showed an enhanced allogeneic response
Given the effect of HCT-116 lysates on the maturation and activation of DCs from healthy individuals, we analyzed whether phenotypic changes were accompanied by changes in DC function. The MLR assay was used to evaluate the effect of HCT-116 lysates on the ability of DCs to induce an allogeneic response. This study was performed only with the combination of CQ and 5-FU, which presented the most relevant phenotypic changes. Fig. 4 A shows the proliferative response of total CD3 lymphocytes, while Fig. 4B and C show the proliferation of CD4+ and CD8+ cells, respectively. Although the exposure of HCT-116 cells to CQ alone increased the DC maturation compared to WT, this effect was not as intense as the drug combination. Our results showed a marked effect on the proliferation of CD4+ T lymphocytes when DCs were sensitized with lysates from HCT-116 cells that were pretreated with 20 μM 5-FU + CQ.
Fig. 4.

Improved DC antigen-presenting function induced by lysates of HCT-116 treated with 5-FU + CQ. DCs were exposed to HCT-116 lysates and cocultured with allogeneic lymphocytes for 5 days. Growing total lymphocytes (CD3+ cells) were quantified by flow cytometry (A), as were CD4+ and CD8+ cells (B; C) (N = 9; * p ≤ 0.05; ** p ≤ 0.01). The CD4+ and CD8+ T lymphocytes were further analyzed separately for the expression of CD69 (activation marker) and PD-1 (regulatory molecule) (D-G; * p ≤ 0.05; ** p ≤ 0.02). MLR supernatants were collected and analyzed for the secretion of IFN-γ (H) and IL-10 (I) by ELISA (*** p ≤ 0.01).
3.6. Lymphocytes responding to allogeneic stimulation showed an activated cell profile
CD69, a marker of activated T lymphocytes, and PD-1, the checkpoint ligand of PD-L1 expressed by tumor cells, were assessed on DC-stimulated T cells. Fig. 4D-G indicates that lymphocytes generated through exposure to DC 5-FU + CQ had a high frequency of CD69+ expression, both among CD4+ T lymphocytes (30.7 ± 2.7%) and CD8+ lymphocytes (41.6 ± 3.2% and 42.5 ± 2.9%, respectively). In contrast, the expression of the regulatory marker PD-1 was lower in these lymphocytes, especially in CD4+ cells (5-FU + CQ: 16.5 ± 1.3%).
3.7. HCT-116 treated with 5-FU + CQ showed an increased ability to induce a Th1 response
As shown in Fig. 4H, DCs exposed to the lysates of HCT-116 cells that were treated with 5-FU + CQ had an increased ability to induce the production of IFN-γ via allogeneic lymphocytes. The levels of IFN-γ increased from 171.1 ± 11.4 pg/ml (WT) to 195.7 ± 16.5 pg/ml (CQ) and 389.3 ± 19.8 pg/ml (5-FU + CQ). Conversely, Fig. 4I shows that DC exposure to the lysates decreased the ability of DC-stimulated T cells to secrete IL-10, reducing the levels from 46.8 ± 6.3 pg/ml (WT) to 28.7 ± 7.2 pg/ml (CQ) and 8.2 ± 3.7 pg/ml (5-FU + CQ).
3.8. Dcs treated with lysate enhanced the in vitro generation of CTLs
Our analysis of cytotoxic T lymphocytes was restricted to the expression of perforin and granzyme B molecules. We tested the efficiency of HCT-116 lysate-treated DCs to generate autologous tumor-reactive T cells. We found that lymphocytes cultured with DCs exposed to lysates of HCT-116 cells treated with 5-FU + CQ induced the generation of lymphocytes with higher levels of perforin and granzyme B than in those cultured with control DCs (Fig. 5 ). No differences were observed upon labeling with anti-CD107a (data not shown).
Fig. 5.

In vitro generation of cytotoxic T lymphocytes (CTLs) is improved by DCs exposed to lysates of HCT-116 previously treated with 5-FU + CQ. Mean fluorescence intensity (MFI) of proliferating CD8+ cells (A) of four healthy donors at individual effector:target ratios (3.25:1, 7.5:1, and 15:1). These lymphocytes showed higher expression levels of the cytotoxicity markers perforin (MFI 15:1 ratio, 5-FU + CQ > WT (p < 0.05)) and granzyme B (MFI 7.5:1 ratio, 5-FU + CQ > WT (p < 0.05); MFI 15:1 ratio, 5-FU + CQ > WT (p < 0.05)) compared to the WT control group.
3.9. Transcriptional changes associated with autophagy blockade
To better understand the increase of DC maturation associated with blocking autophagy, we evaluated HCT-116 cells treated with 5-FU, CQ and their combination. The gene fold change was used to identify significant differences in gene expression among the groups (Table 2 ). CQ-treated cells showed a modest increase in the expression of the autophagy genes ATGs, SQSTM1, MAP1LC3B, and ULK1 and a considerable decrease in genes related to tumor progression (BNIP3, BNIP3L, FOSL2, HES1, LAMB3, LOXL2, NDRG1, P4HA1, and PIK3R2), as well as a decrease in nominal tumor antigens (members of the CEA family). Treatment with 5-FU induced an increase in autophagy genes. In contrast with the CQ group, we did not observe such an intense decrease in the genes related to tumor progression, while the expression of CEA genes was increased. Cells treated with 5-FU + CQ showed an increased expression of autophagy genes, as well as an increase in genes of the CEA family. Unlike the 5-FU group, the expression of genes associated with tumor progression was decreased.
Table 2.
Fold change expression of selected genes by tumor cells, comparing untreated cells (WT) with those treated with CQ, 5FU or CQ + 5FU.
| Gene functions | Gene name | WT vs CQ | WT vs 5-FU | WT vs 5-FU + CQ |
|---|---|---|---|---|
| Autophagy- | ATG12 | 1.7 | 3.2 | 7.1 |
| ATG5 | 1.9 | 4.5 | 6.4 | |
| MAP1LC3B | 1.8 | 2.3 | 5.7 | |
| ULK1 | 1.2 | 2.4 | 5.2 | |
| SQSTM1 | 2.1 | 1.9 | 4.8 | |
| BECN1 | 0.8 | 5.8 | 2.5 | |
| BNIP3 | −4.36 | −1.36 | −4.11 | |
| BNIP3L | −4.06 | −1.2 | −3.85 | |
| Cell adhesion | CEACAM5 | −0.9 | 1.5 | 8.4 |
| CEACAM6 | −1.3 | 2.4 | 6.5 | |
| CEACAM7 | −2.1 | 1.7 | 5.2 | |
| CEACAM1 | −1.2 | 2.1 | 5.7 | |
| HES1 | −3.16 | −0.5 | −7.1 | |
| FOSL2 | −4.72 | −0.72 | −3.6 | |
| NDRG1 | −10.43 | −1.43 | −6.11 | |
| Catalytic activity | PIK3R2 | −1.2 | −3.97 | −4.2 |
| PGK1 | −2.59 | −0.2 | −2.59 | |
| LOXL2 | −4.8 | −0.9 | −2.85 | |
| PGK1 | −2.59 | −0.2 | −2.59 | |
| ALDOC | −8.14 | −1.14 | −7.52 | |
| ANGPTL4 | −6.25 | −1.25 | −5.93 | |
| Structural molecule activity | LAMB3 | −4.26 | −0.76 | −3.59 |
| Heat shock proteins and HMGBs | HMGB1 | 1.02 | 1.9 | 1.12 |
| HMGB2 | 0.98 | 1.87 | 1.05 | |
| HMGB3 | 0.2 | 0.5 | 0.347 | |
| DNAJB1 (HSP40) | 1.17 | 1.5 | 0.85 | |
| HSP1A (HSP70) | 2.67 | 2.82 | 3.38 | |
| HSP90AB1 (HSP90) | 1.45 | 2.97 | 1.52 | |
| Oxidoreductase | P4HA1 | −6.53 | −1.1 | −6.89 |
| RNA binding | PCBP1 | 1.2 | 2.86 | 8.26 |
4. Discussion
The development of resistance to antitumor agents is one of the primary challenges of modern cancer therapy. Here, we investigated the effects of treating tumor cells with a combination of 5-FU and CQ. The concentration of 20 μM 5-FU was chosen because it was the lowest concentration capable of inhibiting tumor growth and may, therefore, represent a suitable in vitro model of the metronomic dose used in the clinic [31]. Although CQ has previously been applied in combination with several antineoplastic agents, a study using low concentrations of the drug has not yet been reported.
Increased LC3-II and SQSTM1 levels are currently used as suitable markers of effective autophagy inhibition. When autophagy is blocked by CQ, LC3-II accumulates [32]. SQSTM1 binds to ubiquitinated proteins, especially LC3-II, forming part of the autophagosome wall. Thus, if autophagy is enhanced, SQSTM1 is degraded. Inhibiting autophagy promotes the accumulation of SQSTM1, as well as LC3-II [33]. Low concentrations of 5-FU increased autophagy in HCT-116 cells, while CQ was able to inhibit the process, as evidenced by the accumulation of LC3-II. The analysis of SQSTM1 expression is consistent with constitutive autophagy in HCT-116 cells [34].
Autophagy induction by 5-FU was also confirmed by transcriptome analysis that showed an increase in expression of the autophagy-associated genes ATGs, SQSTM1, MAP1LC3B, BECN1, and ULK1. Their expression was enhanced when applying the combination of 5-FU + CQ. The increased expression of LC3B and BECN1 under stress conditions is consistent with previous reports [35], [36].
Ultrastructural analysis revealed that the untreated control cells had negligible expression of autophagosomes and autophagolysosomes, while those treated with CQ showed increased numbers of autophagosomes in the cytoplasm. Cells treated with 5-FU showed an evident increase in the number of autophagolysosomes containing degraded material, indicating the induction of autophagy and the completion of the process, consistent with prior reports [28], [29], [30], [37], [38]. The combined treatment of CQ and 20 µM 5-FU induced intense accumulation of cytoplasmic vesicles in several stages, especially autophagosomes containing undigested material. We also found cells with signs of autophagic and apoptotic death, such as cytoplasmic vacuolization, chromatin rarefaction (reduction of heterochromatic groups associated with the nuclear envelope), altered mitochondrial morphology (dilation of the cristae), and dilation of the nuclear envelope, which also agrees with previous reports [39], [40]. This phenomenon was confirmed by the observation that CQ + 5-FU treatment strongly reduced cell proliferation and induced early apoptosis in tumor cells, although CQ or 5-FU alone had no apparent effect on cell viability. 5-Fluorouracil induces apoptosis in cancer cells [41], and the inhibition of autophagy by CQ can potentiate the cytotoxicity of 5-FU. This occurs because the accumulation of vesicles in the cytoplasm due to incomplete autophagy can cause oxidative stress and lead to cell death. Based on this information, we aimed to confirm whether the toxicity of low 5-FU concentrations would be enhanced by CQ. In this study, the apoptosis and cell death results showed that 5-FU + CQ increased the levels of early apoptosis even though the drug concentrations had only a negligible effect if applied alone.
Analyzing the effects of CQ + 5-FU treatment on the immunogenicity of tumor cells revealed that DC exposure to the lysate of HCT-116 cells treated with drug enhanced the maturation of antigen-presenting cells. DC maturation is required for increased antigen-presenting ability and is accompanied by phenotypic changes, such as increased expression of MHC class II and co-stimulatory molecule expression. CD40, CD80, and CD86 expression are essential for T lymphocyte stimulation [42]. In addition to being a maturation marker, CD83 modulates the immune response by stimulating both naive and memory T cells [43]. CD83 expression was also enhanced by the drug combination. Although apoptotic cell death is considered non-immunogenic or weakly immunogenic, it is possible that apoptosis induced by the blockage of autophagy results in the expression of danger signals (e.g., DAMPs) that are usually not expressed during physiological apoptosis. In a parallel study, we observed that exposing HCT-116 cells to low concentrations of paclitaxel induces the expression of HSP-70 and 90 (unpublished data). In the present study, we observed a small increase in the expression of HSP-70 but irrelevant changes in the HSP-90 and HMGB genes. These findings indicated that the enhanced ability to sensitize DCs is mainly due to the increased expression of the tumor-associated genes of the CEA family (CEACAM 1, 5, 6, and 7).
In another study, we found that exposing HCT-116 cells to low doses of paclitaxel or doxorubicin increased the expression of several genes associated with enhanced tumor immunogenicity [11]. Total RNA extracted from HCT-116 colorectal cancer cells treated with 5-FU was more immunogenic when transfected into DCs [44]. Exposing tumor cells to other drugs such as paclitaxel and doxorubicin increases the expression of the antigen processing machinery via the cytosolic route, such as TAP1, tapasin, and calnexin [11]. This view is reinforced by the observation that chemical stress triggers the cell surface expression of calreticulin (CALR) [12], another molecule promoting the antigenic processing of tumor cells. Interestingly, CALR moves from the endoplasmic reticulum (where it functions as an APM component) to the cytoplasmic membrane, where it serves as a potent activating signal. CALR, together with HMGB1, plays an important role in the phenotypic maturation of DCs, increasing HLA-DR, CD80, and CD86 expression and functional maturation, as well as enhancing the secretion of IL-12 and INF-γ [45].
This enhancing effect on tumor cell immunogenicity was reinforced by our demonstration that the cell lysates had a functional effect on DCs. Indeed, the mixed lymphocyte reaction (MLR) allowed us to demonstrate that the phenotypic changes observed in DCs are accompanied by an improvement in their ability to stimulate allogeneic lymphocytes. Specifically, DCs sensitized with the lysates of HCT-116 cells treated with 5-FU + CQ promoted the enhanced proliferation of lymphocytes in the MLR.
The more intense proliferative effect on CD4+ T lymphocytes is also in agreement with their ability to interact with the HLA-DR molecules expressed by DCs. Furthermore, the increased proliferation of CD8+ T lymphocytes suggests the generation of cytotoxic T lymphocytes that are specific for tumor cells (since their proliferation was stimulated by DCs sensitized with HCT-116 lysates). Aiming to confirm that the combination of 5-FU and CQ can stimulate the proliferation of CD8+ lymphocytes, we performed a CTL assay, which, unlike the MLR, tests the ability of DCs to stimulate the proliferation of autologous CD8+ lymphocytes that target tumor cells [46]. Our main difficulty when evaluating the direct cytotoxicity of CTLs against target cells is that HCT-116 cells are adherent, but most available assays for this purpose work well for cells in suspension and are not reliable for adherent cell types. There are some chemiluminescence-based assays for this, but we were unable to acquire the appropriate kit. Therefore, we decided to evaluate the expression of perforin and granzyme B by CTLs challenged with tumor target cells, following the methodology proposed by Betts et al [47]. These proteins are considered to be functional markers in cells with cytotoxic capacity [48]; perforin polymerization perforates the target cell membrane, while granzyme B induces apoptosis [49]. As expected, we found increased expression (i.e., increased mean fluorescence intensity; MFI) of both perforin and granzyme B in the CD8+ population induced by DCs sensitized with 5-FU + CQ lysate.
We also observed that lymphocytes generated in response to sensitized DCs expressed higher levels of the ‘very early antigen’ CD69, which is associated with the effective stimulation of lymphocytes by antigen-presenting cells [50]. Consistently, lymphocytes stimulated with DCs exposed to 5-FU + CQ lysates induced earlier and more potent T cell activation. The findings also indicated that there was no selective activation of CD4+ or CD8+ T cells, since the percentage of CD69+ cells was increased in both subpopulations. Accordingly, these same treatments resulted in a reduced generation of PD-1+ cells among CD4+ T lymphocytes. PD-1 is a member of the CD28 family, and its primary function is to limit clustering of the TCR and costimulatory molecules within the immunologic synapse. PD-1 expression is increased in the T cells of patients with pancreatic cancer, lung cancer, and various leukemias and lymphomas [51]. Therefore, PD-1 expression in T cells and PD-L1 expression in tumor cells constitute a tumor escape mechanism. We observed reduced PD-1 expression on the expanding lymphocytes cocultured with DCs. This aspect is particularly interesting because the signals associated with PD-1 and PD-L1 are among the main checkpoint blockades of the antitumor response. Hence, in addition to the anti-CTLA-4 antibody, the anti-PD-1 antibodies [52] are mainstays of modern antitumor immunotherapy.
The lymphocytes cultured with DCs sensitized with drug-exposed tumor cells produced higher amounts of IFN-γ than controls while showing decreased production of IL-10. Naive CD4+ and CD8+ lymphocytes can differentiate into specific effector T cells with different functions that are attributed, in part, to the pattern of cytokines they produce [53]. CD4+ T cells can differentiate into Th1 cells that release IFN-γ and TNF-α to stimulate the generation of CD8+ cells; in this way, Th1 cells are involved in the development of the antitumor immune response. Interferon γ also recruits monocytes, inducing the differentiation of CD4+ T lymphocytes into Th1 lymphocytes [54], [55]. IL-10 is a DC-inhibiting cytokine that can be secreted by both Tregs and Th2, inhibiting antigen presentation, and decreasing the expression of MHC II and the co-stimulatory molecules CD80 and CD86 [56]. Thus, DCs under the combined treatment promoted preferential activation of Th1 lymphocytes, with little Th2 or Treg stimulation. An analysis of other cytokines, such as IL-12, IL-2, TNF, IL-4, IL-6, IL17, and IL-23, would be useful to provide a clearer view regarding how the different treatments affect the modulation of T cell subsets. The quantification of these cytokines should be considered in future studies.
Finally, the evidence that the combination of 5-FU and CQ can modulate tumor cell biology, as can the corresponding HCT-116 lysate, is reinforced by the increased expression of genes of the tumor-associated CEA family (CEACAM 5, 6, and 7), as previously reported [57], and the decreased expression of genes associated with tumor progression (BNIP3, BNIP3L, FOSL2, HES1, LAMB3, LOXL2, NDRG1, P4HA1, and PIK3R2).
5. Conclusion
Taken together, our results indicate that blocking autophagy with chloroquine increases the ability of tumor cells to mature DCs that can induce an antitumor response. In addition, these results confirm the clinical application of this protocol for improving the in vitro stimulation of DCs, as well as the use of combined anti-autophagic agents and conventional chemotherapeutic agents for the treatment of patients with colorectal cancer.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding sources
This research did not receive any specific grant from funding agencies in the public, commercial, or not for profit sectors. JAZR and CMG were recipients of doctorate scholarships from CAPES and the Brazilian Council for Research CNPq, Brazil. GGR was the recipient of a postdoctoral fellowship from CAPES. BFF and CSF were the recipients of scientific initiation scholarships from the Sao Paulo Research Foundation (FAPESP).
CRediT authorship contribution statement
Jofer Andree Zamame Ramirez: Methodology, Investigation, Formal analysis, Writing - original draft. Graziela Gorete Romagnoli: Conceptualization, Investigation, Validation. Bianca Francisco Falasco: Investigation. Carolina Mendonça Gorgulho: Investigation, Writing - review & editing. Carla Sanzochi Fogolin: Investigation. Daniela Carvalho dos Santos: Formal analysis, Writing - review & editing. João Pessoa Araújo Junior: Methodology, Formal analysis. Michael Thomas Lotze: Writing - review & editing. Rodrigo Portes Ureshino: Methodology. Ramon Kaneno: Conceptualization, Methodology, Writing - review & editing, Supervision.
Declaration of Competing Interest
The authors declare no potential conflicts of interest.
Acknowledgments
We thank Professor Deilson Elgui de Oliveira for the authentication of the cell line used in this study.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.intimp.2020.106495.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.R.L. Siegel, K.D. Miller, A. Jemal, Cancer statistics, 2020, CA: A Cancer J. Clinicians 70(1) (2020) 7–30. [DOI] [PubMed]
- 2.INCA, Estimativa 2020: incidência de câncer no Brasil, in: M.d. Saúde (Ed.) Estimativa, Instituto Nacional de Câncer José Alencar Gomes da Silva/ Ministério da Saúde., 2019, p. 120.
- 3.Andre T. An overview of adjuvant systemic chemotherapy for colon cancer. Clin. Colorectal Cancer. 2004;4(1):22–28. [PubMed] [Google Scholar]
- 4.Jonker Derek J., O'Callaghan Chris J., Karapetis Christos S., Zalcberg John R., Tu Dongsheng, Au Heather-Jane, Berry Scott R., Krahn Marianne, Price Timothy, Simes R. John, Tebbutt Niall C., van Hazel Guy, Wierzbicki Rafal, Langer Christiane, Moore Malcolm J. Cetuximab for the Treatment of Colorectal Cancer. N. Engl. J. Med. 2007;357(20):2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 5.Guan X. Cancer metastases: challenges and opportunities. Acta Pharmaceutica Sinica B. 2015;5(5):402–418. doi: 10.1016/j.apsb.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuervo A.M. Autophagy, sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Mehrpour M., Beau I. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 8.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 9.Eskelinen E.L. Autophagy, A lysosomal degradation pathway with a central role in health and disease. Biochimica et Biophysica Acta. 2009;1793:664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Shurin G.V., Kaneno R. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J. Immunol. 2009;183:137–144. doi: 10.4049/jimmunol.0900734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaneno R., Kaneno F.M. Chemotherapeutic agents in low noncytotoxic concentrations increase immunogenicity of human colon cancer cells. Cellular Oncol. (Dordrecht) 2011;34:97–106. doi: 10.1007/s13402-010-0005-5. [DOI] [PubMed] [Google Scholar]
- 12.Zitvogel L., Ghiringhelli F. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 13.Zitvogel L., Pequignot M.O. Immune response against dying tumor cells. Adv. Immunol. 2004;84:131–179. doi: 10.1016/S0065-2776(04)84004-5. [DOI] [PubMed] [Google Scholar]
- 14.Zitvogel L., Kroemer G. Anticancer immunochemotherapy using adjuvants with direct cytotoxic effects. J. Clin. Invest. 2009;119(8):2127–2130. doi: 10.1172/JCI39991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitt J.M., Kroemer G., Zitvogel L. Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment. In: Kalinski P., editor. Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy. Springer International Publishing; Cham: 2017. pp. 65–79. [Google Scholar]
- 16.Vanmeerbeek I., Sprooten J., De Ruysscher D., Tejpar S., Vandenberghe P., Fucikova J., Spisek R., Zitvogel L., Kroemer G., Galluzzi L., Garg A.D. Trial watch: chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology. 2020;9(1) doi: 10.1080/2162402X.2019.1703449. 1703449-1703449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaneno R., Shurin G.V., Tourkova I.L., Shurin M.R. Chemomodulation of human dendritic cell function by antineoplastic agents in low noncytotoxic concentrations. J. Transl. Med. 2009;7:58–66. doi: 10.1186/1479-5876-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sasaki K., Tsuno N.H., Sunami E. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10:370–375. doi: 10.1186/1471-2407-10-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amaravadi R.K., Yu D., Lum J.J. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrew F.G. Chloroquine, Mechanism of drug action and resistance in plasmodium falciparum. Pharmacol. Therapeut. 1993;57(2):203–235. doi: 10.1016/0163-7258(93)90056-j. [DOI] [PubMed] [Google Scholar]
- 21.Sharma N., Simmy T., Golden E.B. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012;326:143–154. doi: 10.1016/j.canlet.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 22.Ponticelli C., Moroni G. Hydroxychloroquine in systemic lupus erythematosus (SLE) Expert Opin. Drug Saf. 2017;16(3):411–419. doi: 10.1080/14740338.2017.1269168. [DOI] [PubMed] [Google Scholar]
- 23.Liu J., Cao R., Xu M., Wang X., Zhang H., Hu H., Li Y., Hu Z., Zhong W., Wang M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6(1):16. doi: 10.1038/s41421-020-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chinese Clinical Trial Register, 2020. http://www.chictr.org.cn.
- 25.GrohBrandt N., Beckmann I., Schrauder M.W., Loehberg M. Evaluation for synergistic suppression of breast cancer proliferation by Chloroquine in combination with the rapamycin derivative, Rad001. Geburtshilfe Und Frauenheilkunde. 2009;5(69):214–218. [Google Scholar]
- 26.Grimaldi A., Santini D., Zappavigna S. Antagonistic effects of chloroquine on autophagy occurrence potentiate the anticancer effects of everolimus on renal cancer cells. Cancer Biol. Therapy. 2015;4(16):567–579. doi: 10.1080/15384047.2015.1018494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eskelinen E.L., Prescott A.R., Cooper J., Brachmann S.M., Wang L., Tang X., Backer J.M., Lucocq J.M. Inhibition of autophagy in mitotic animal cells. Traffic. 2002;3(12):878–893. doi: 10.1034/j.1600-0854.2002.31204.x. [DOI] [PubMed] [Google Scholar]
- 28.Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia L., Dourmashkin R.R., Allen P.D., Gray A.B., Newland A.C., Kelsey S.M. Inhibition of autophagy abrogates tumour necrosis factor α induced apoptosis in human T-lymphoblastic leukaemic cells. Br. J. Haematol. 1997;98(3):673–685. doi: 10.1046/j.1365-2141.1997.2623081.x. [DOI] [PubMed] [Google Scholar]
- 30.Moon E.-K., Kim S.-H., Hong Y., Chung D.-I., Goo Y.-K., Kong H.-H. Autophagy inhibitors as a potential antiamoebic treatment for Acanthamoeba keratitis. Antimicrob Agents Chemother. 2015;59(7):4020–4025. doi: 10.1128/AAC.05165-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Longley D., Harkin D., Johnston P. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003;5(3):330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N., Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy. 2007;3(6):542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 33.Bjørkøy G., Lamark T., Pankiv S., Øvervatn A., Brech A., Johansen T. Academic Press; 2009. Chapter 12 Monitoring Autophagic Degradation of p62/SQSTM1, Methods in Enzymology; pp. 181–197. [DOI] [PubMed] [Google Scholar]
- 34.Kucharewicz K., Dudkowska M., Zawadzka A., Ogrodnik M., Szczepankiewicz A.A., Czarnocki Z., Sikora E. Simultaneous induction and blockade of autophagy by a single agent. Cell Death Dis. 2018;9(3):353. doi: 10.1038/s41419-018-0383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scherz-Shouval R., Weidberg H., Gonen C., Wilder S., Elazar Z., Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. PNAS. 2010;107(43):18511–18516. doi: 10.1073/pnas.1006124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J.-W., Zhang Q.-H., Liu T. Autophagy facilitates anticancer effect of 5-fluorouracil in HCT-116 cells. J. Cancer Res. Ther. 2018;14(12):1141–1147. doi: 10.4103/0973-1482.204898. [DOI] [PubMed] [Google Scholar]
- 37.Choi J.-H., Yoon J.S., Won Y.-W., Park B.-B., Lee Y.Y. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. APMIS. 2012;120(7):597–604. doi: 10.1111/j.1600-0463.2012.02876.x. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki K., Tsuno N.H., Sunami E., Tsurita G., Kawai K., Okaji Y., Nishikawa T., Shuno Y., Hongo K., Hiyoshi M., Kaneko M., Kitayama J., Takahashi K., Nagawa H. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10 doi: 10.1186/1471-2407-10-370. 370–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amaravadi R.K., Yu D., Lum J.J., Bui T., Christophorou M.A., Evan G.I., Thomas-Tikhonenko A., Thompson C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007;117(2):326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boya P., González-Polo R.-A., Casares N., Perfettini J.-L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005;25(3):1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mhaidat N.M., Bouklihacene M., Thorne R.F. 5-Fluorouracil-induced apoptosis in colorectal cancer cells is caspase-9-dependent and mediated by activation of protein kinase C-δ. Oncol Lett. 2014;8(2):699–704. doi: 10.3892/ol.2014.2211. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Adema G., Hartgers F., Verstraten R. A dendritic-cell-derived C-C chemokine that preferentially attracts naive T cells. Nature. 1997;387:713–717. doi: 10.1038/42716. [DOI] [PubMed] [Google Scholar]
- 43.Wang X., Ji J., Zhang H. Stimulation of dendritic cells by DAMPs in ALA-PDT treated SCC tumor cells. Oncotarget. 2015;42(6):688–702. doi: 10.18632/oncotarget.5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Almeida C.V., Zamame J.A., Romagnoli G.G., Rodrigues C.P., Magalhaes M.B., Amedei A., Kaneno R. Treatment of colon cancer cells with 5-fluorouracil can improve the effectiveness of RNA-transfected antitumor dendritic cell vaccine. Oncol. Rep. 2017;38(1):561–568. doi: 10.3892/or.2017.5692. [DOI] [PubMed] [Google Scholar]
- 45.Aarntzen E.H.J.G., Aarntzen I., Lesterhuis W.J., Schuurhuis D., Jacobs J.F.M., Bol K. Targeting CD4+ T-helper cells improves the induction of antitumor responses in dendritic cell–based vaccination. Cancer Res. 2013;1(73):19–29. doi: 10.1158/0008-5472.CAN-12-1127. [DOI] [PubMed] [Google Scholar]
- 46.Burleson G.R., Burleson F.G., Dietert R.R. The cytotoxic T lymphocyte assay for evaluating cell-mediated immune function. Methods Mol. Biol. 2010;598:195–205. doi: 10.1007/978-1-60761-401-2_14. [DOI] [PubMed] [Google Scholar]
- 47.Betts M.R., Brenchley J.M., Price D.A., De Rosa S.C., Douek D.C., Roederer M., Koup R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods. 2003;281(1):65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 48.Bryceson Y.T., Fauriat C., Nunes J.M., Wood S.M., Björkström N.K., Long E.O., Ljunggren H.-G. Functional Analysis of Human NK cells by Flow Cytometry. Methods Mol. Biol. (Clifton, N.J.) 2010;612:335–352. doi: 10.1007/978-1-60761-362-6_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapani J.A., Smyth M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002;2:735. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 50.Patsoukis N., Sari D., Boussiotis V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle. 2012;23(11):4305–4309. doi: 10.4161/cc.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cockerham L.R., Jain V., Sinclair E. Programmed death-1 expression on CD4+ and CD8+ T cells in treated and untreated HIV disease. AIDS. 2014;28(12):1749–1758. doi: 10.1097/QAD.0000000000000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sundar R., Cho B.C., Brahmer J.R., Soo R.A. Nivolumab in NSCLC: latest evidence and clinical potential. Therapeut. Adv. Med. Oncol. 2015;7(2):85–95. doi: 10.1177/1758834014567470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guermonprez P., Valladeau J., Zitvogel L. Antigen presentation and T cell stimulation by dendritic cells. Ann. Rev. Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 54.Guéry L., Hugues S. Th17 Cell Plasticity and Functions in Cancer Immunity. Biomed Res. Int. 2015;20:31–46. doi: 10.1155/2015/314620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knutson K.L., Disis M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol., Immunother. 2005;8(54):721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Margarida Saraiva A.O.G. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010;10(1):170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 57.Aquino A., Prete S.P., Guadagni F., Greiner J.W., Giuliani A., Orlando L., Masci G., De Santis S., Bonmassar E., Graziani G. Effect of 5-fluorouracil on carcinoembryonic antigen expression and shedding at clonal level in colon cancer cells. Anticancer Res. 2000;20(5B):3475–3484. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.