

Abstract
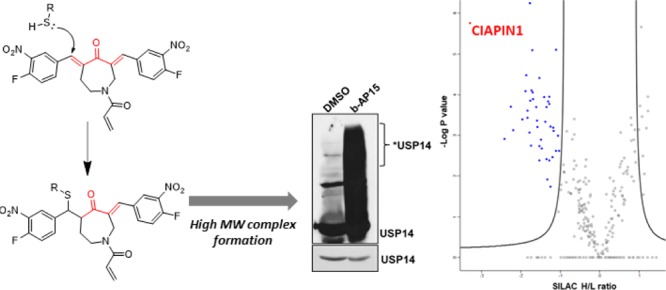
Deubiquitinating enzymes (DUBs) are a growing target class across multiple disease states, with several inhibitors now reported. b-AP15 and VLX1570 are two structurally related USP14/UCH-37 inhibitors. Through a proteomic approach, we demonstrate that these compounds target a diverse range of proteins, resulting in the formation of higher molecular weight (MW) complexes. Activity-based proteome profiling identified CIAPIN1 as a submicromolar covalent target of VLX1570, and further analysis demonstrated that high MW complex formation leads to aggregation of CIAPIN1 in intact cells. Our results suggest that in addition to DUB inhibition, these compounds induce nonspecific protein aggregation, providing molecular explanation for general cellular toxicity.
Introduction
Ubiquitination, the covalent addition of 76 amino acid protein ubiquitin (Ub) to protein substrates, is a widespread protein post-translational modification in eukaryotic cells.1 Because of its role in proteasomal degradation and a plethora of signaling pathways, ubiquitination is an emerging field of clinical interest in multiple disease states, including cancer.2−4 Several compounds have been reported in recent years targeting deubiquitinating enzymes (DUBs)4,5 which regulate the removal of Ub marks. However, the characterization of such inhibitors and clinical compounds from a target perspective is variable; although a DUB target is reported, compound selectivity and specificity across the proteome remained, in most cases, incompletely resolved. Comprehensive understanding of a compound’s targets facilitates interpretation of phenotypes in preclinical investigations and can identify mechanisms of toxicity6 and resistance at an early stage of testing. Only recently, and with the help of advanced activity-based protein profiling (ABPP)7−9 assays that allow the profiling of DUBs in a cellular context, highly selective DUB inhibitors have been reported.4,5,10 Here, we describe the proteomic investigation of two structurally related DUB inhibitors b-AP15 and VLX1570. These inhibitors share a reactive α,β-unsaturated carbonyl substructure motif capable of covalent interaction with nucleophilic residues. Initially taken forward into a phase I/II clinical trial for refractory multiple myeloma, VLX1570 has been put on full clinical hold because of dose-limiting toxicity. We demonstrate that these inhibitors target a diverse range of proteins beyond their reported targets, resulting in the formation of higher molecular weight (MW) complexes. Through a quantitative chemical proteomic approach, we identify CIAPIN1, also known as anamorsin, as a potent covalent target of VLX1570 which forms high MW complexes upon reaction with VLX1570, leading to aggregation of CIAPIN1 in intact cells.
Results and Discussion
b-AP15 has been previously described as a specific reversible inhibitor of the proteasomal DUBs USP14 and UCH-37 (also referred to as UCH-L5) with anti-cancer activities.11,12 Examination of the chemical structure of b-AP15, however, shows the presence of electrophilic Michael acceptor motifs (Figure 1A). Although the results obtained by D’Arcy and colleagues demonstrate that b-AP15 is an inhibitor of USP14/UCH-37,11 two unrelated cysteine protease enzymes from different DUB families, the chemical structure of b-AP15 suggests additional proteins may be targeted by this compound and its analogues. Indeed, b-AP15 possesses higher potency in intact cells than that in biophysical assays against USP14 and UCH-37.11 In support of compound promiscuity, another study describing the chemical synthesis of active-site-directed DUB probes showed data compatible with a nonspecific DUB inhibition profile upon increasing concentrations of b-AP15.13 In our hands, b-AP15 inhibits the cleavage of the DUB substrate Ub-AML (ubiquitin-aminoluciferin) by a number of purified recombinant DUBs (Figure 1B). Crude extracts of cells treated with increasing concentrations of b-AP15 also showed that b-AP15 is able to decrease the global DUB activity of these treated cells (Figure 1C). Furthermore, the b-AP15 treatment in both cancer cell lines and endothelial cells resulted in comparable cytotoxicity as observed by a cell viability assay, indicating the nonspecific toxicity of this chemotype (Figure S1).
Figure 1.

b-AP15 is a nonspecific DUB inhibitor. (A) Molecular structure of b-AP15 with Michael acceptors (black dot) and acrylamide (gray dot) motif indicated. (B) DUB activity measured by cleavage of the luminescent substrate Ub-AML in a panel of recombinant proteasomal (purified 19S proteasomes) and nonproteasomal DUBs treated with b-AP15 (100 μM) (n = 3–4). (C) DUB assay as described above using HeLa cell extracts (5 μg) incubated with the indicated b-AP15 concentrations (n = 3).
By immunoblot analysis, it was observed that b-AP15 induces the formation of high MW complexes with USP14 (Figure 2A,B) and UCH-37 (Figure S2) in both crude cell extracts and intact cells. The formation of these complexes is reduced by co-incubation with thiol containing reducing reagents dithiothreitol (DTT) or glutathione (GSH), in support of these higher MW complexes forming via the Michael acceptors. The protective effect of these reagents was hypothesized to be due to the blocking of these reactive sites. To test this, b-AP15 was reacted in vitro with an excess of GSH. As predicted, b-AP15 (GSH)2 could be observed by liquid chromatography–mass spectrometry (LC–MS) analysis after 30 min of incubation (Figure S3). A potential third addition via acrylamide to give b-AP15 (GSH)3 was not observed. In support of this finding, PYR-41, an ubiquitin-activating enzyme (E1) inhibitor (also containing Michael acceptors), has been reported to induce DTT-sensitive protein cross-linking and inhibition of DUBs and other cellular enzymes by formation of high MW complexes.14 In addition, high MW ubiquitylated proteins accumulated upon b-AP15 treatment, the levels of which were reduced by co-treatment with DTT and GSH (Figure 2C). The origin of these very high MW ubiquitylated proteins that accumulate upon treatment with b-AP15 in comparison with those that accumulate on application of proteasome inhibitors has been largely discussed.11,15 Interestingly, ubiquitylated protein complexes with very high MW are also reported by D’Arcy and other studies using α,β unsaturated carbonyl compounds.14,16−18
Figure 2.

b-AP15 inhibits DUBs through the formation of cytotoxic high MW protein complexes, which are averted in the presence of thiol-reducing agents. Immunoblot analysis of high MW complexes of USP14 in (A) HeLa crude cell extracts incubated with b-AP15 (10 μM) and in (B) HeLa cells treated with either vehicle, 10 μM b-AP15 or with 10 μM b-AP15, in combination with reducing agents (10 mM DTT or 10 mM GSH), both at 37 °C for 2 h. Shorter exposure times were used as a loading control (lower blots in the panel). (C) Immunoblot analysis of ubiquitin conjugates in HeLa cells treated with vehicle, 10 μM b-AP15, or with 10 μM b-AP15 in combination with reducing agents (10 mM DTT or 10 mM GSH) for 2 h. (D) Visualization of the b-AP15-induced high MW protein complexes in a SDS-PAGE gel, after Coomassie Blue staining, in HeLa cells treated as in (C).
The effects of b-AP15 on the global proteome were further evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Blue staining, revealing the formation of notably increased levels of high MW protein complexes in comparison to dimethyl sulfoxide (DMSO) control or cotreatment with DTT and GSH (Figure 2D). To better characterize the consequences of the global formation of high MW protein complexes on cell signaling, the status of mTOR was analyzed in crude extracts treated with b-AP15 by immunoblotting. Strikingly, a shift from mTOR in its native form toward a band with slower migration mobility is observed with nanomolar concentration of bAP-15 (Figure S4A,B). A similar observation was described by us in a previous study showing oxidation of proteins using a photosensitiser.19 A similar but less potent effect was observed on incubation with the natural product curcumin, which also possesses Michael acceptor groups (Figure S4C,D).
VLX1570 is a structural analogue of b-AP15 that shows higher potency and improved permeability.20,21 VLX1570 was taken forward into a phase I/II clinical trial for refractory multiple myeloma in combination with dexamethasone (NCT02372240) but was then put on full clinical hold in July 2017 because of dose-limiting toxicity. Based on our analysis of b-AP15, we postulated that this toxicity may be due to promiscuous covalent reaction with cellular nucleophiles and formation of high MW complexes. To test whether VLX1570 also forms high MW complexes, HeLa lysates were treated with varying concentrations of b-AP15 and VLX1570 and probed with the active-site-directed DUB probe HA-Ub-C2Br.22 Dose-dependent inhibition of probe labeling was observed for both compounds. Even on blotting for USP28, a DUB structurally unrelated to both USP14 and UCH-37, higher MW complex formation was observed (Figure S5). Co-incubation of both VLX1570 and b-AP15 with GSH in the multiple myeloma cell line KMS11 led to increased cell viability after 16 h, suggesting that the GSH is scavenging these compounds and reducing their active concentration (Figure S6).
GSH represents one of many nucleophiles accessible to VLX1570 within a biological context, and Michael acceptors are well known for their reactivity with cysteine residues.23 To comprehensively profile the covalent targets of VLX1570, ABPP was conducted. Accordingly, an alkyne-tagged analogue of VLX1570, compound 1 (Figure 3A) was designed and synthesized. The acrylamide position was selected for conversion to the alkyne, as acrylamide-lacking analogues had previously been reported in structure–activity relationship studies not to influence cell viability.20 Indeed, 1 retained equivalent cytotoxicity to VLX1570 in both KMS11 and U2OS cell lines, as observed with the CellTiter-Glo assay (Figures 3A and S7).
Figure 3.

Chemical proteomics reveals multiple covalent targets of VLX1570. (A) Structure of VLX1570 and probe derivative compound 1. EC50 values measured by CellTiter-Glo in U2OS and KMS11 cells are reported. (B) Volcano plot of 1-enriched proteome from U2OS cells, with targets significantly (FDR = 0.05, S0 = 0.2) competed by 20 μM VLX1570 indicated (blue). The most significant hit, CIAPIN1, is indicated. (C) Immunoblot analysis of VLX1570-treated KMS11 cells demonstrates a protective effect against high MW complex formation for CIAPIN1 on addition of GSH (10 mM, 2 h).
Before commencing proteomic-profiling experiments, suitable competitive conditions were determined. U2OS cells were pre-incubated with increasing concentrations of VLX1570 or bAP-15 for 30 min before addition of the affinity probe 1 at a final concentration of 5 μM. After 1 h, the cells were lysed. Subsequent ligation to AzTB,24 an azido-TAMRA-biotin capture reagent, via a copper catalyzed azide–alkyne cycloaddition (CuAAC) allowed visualization of probe-protein complexes by in-gel fluorescence. Both VLX1570 and b-AP15 pre-incubation caused reduction in labeling intensity for specific bands, most markedly affecting a band at approximately 37 kDa (Figure S8A). VLX1570 competitive conditions (5 and 20 μM) were taken forward for proteomic analysis. The spike-in SILAC methodology25 was employed as previously described.8,26 Briefly, cell lysates from competitive conditions were mixed in a 2:1 ratio with “spike”: lysate generated from 1-treated U2OS cells grown in R10K8 media. Subsequent CuAAC ligation, enrichment on NeutrAvidin-Agarose resin, tryptic digest, and LC–MS/MS analysis enabled proteome-wide in-cell target identification of 1-labeled proteins. Multiple covalent targets were identified using this method, with significant competition observed at both 1- and 4-fold competitor excess. Forty-four proteins were significantly competed by 20 μM VLX1570 (Figure 3B, Supporting Information Data set 1), 24 of which were also significantly competed by 5 μM of the compound (Figure S8B). On inspection of these protein sequences, all contained cysteine residues. Gene ontology cellular component analysis suggested that VLX1570 was covalently interacting with proteins across multiple cellular locations, including the cytoplasm, nucleus, and multiple organelles (Table S1). It should be noted that although we have demonstrated accumulation of higher MW bands for UCH-37, USP14, and USP28 by immunoblot analysis, none of these DUBs were identified in the ABPP experiment. As the ABPP experiment only identifies covalent interactors, it is possible that the high MW complex formation observed for DUBs is occurring through a noncovalent mechanism. This is in agreement with literature data reporting that although DTT does affect cytotoxicity of VLX1570,20 compound binding and enzymatic inhibition are both reversible.21 Alternatively, it is possible that the high MW complexes formed with these proteins sterically hinder the CuAAC ligation reaction, thereby preventing covalently 1-labeled proteins from being enriched and identified in this experiment.
The most significantly competed covalent target of VLX1570 identified was CIAPIN1. CIAPIN1, a 33 kDa protein, is a part of the electron transport chain that enables Fe–S cluster assembly.27 It possesses an N-methyltransferase domain at its N-terminus, although this has been reported to be catalytically inactive.28 Furthermore, CIAPIN1 exerts anti-apoptotic effects in cells, although it is unrelated to apoptosis regulatory molecules of the BCL2 or CASP families.29 In our hands, siRNA knockdown of CIAPIN1 lead to a reduction in cell viability of KMS11 cells (Figure S9). Knockdown of CIAPIN1 has been reported to induce apoptosis in several cancers30,31 and also non-cancer cell lines.32,33 In agreement with functional inhibition of CIAPIN1, both b-AP15 and VLX1570 are reported to exert cytotoxicity via an apoptotic mechanism that is insensitive to BCL2 overexpression.11,21
As observed for DUB targets of this chemotype, higher MW complex formation was observed on immunoblotting for CIAPIN1 in VLX1570-treated cells, an effect that was rescued on addition of GSH (Figure 3C). Incubation of VLX1570 with recombinant CIAPIN1 showed similar higher MW complex formation (Figure S10A). Formation of a covalent adduct was confirmed by electrospray MS analysis (Figure S10B), although covalently linked higher MW species were not observed. Further analysis by size exclusion chromatography demonstrated that these VLX1570 adducts result in the accumulation of protein aggregates (Figure 4A). LC–MS/MS analysis of the CIAPIN1–VLX1570 adduct following tryptic digest revealed that 7 out of a possible 10 cysteine residues were covalently modified by VLX1570, indicating that although VLX1570 modifies CIAPIN1 monomerically, it does so nonspecifically (Figure S11). Closer inspection of an unmodified peptide containing Cys249 showed the presence of an intrachain disulfide bond, which was disrupted in the equivalent VLX1570-modified peptide (Figure S12 and Table S2). This disrupted disulfide bond represents one potential mechanism by which VLX1570 adducts result in CIAPIN1 instability and ultimately aggregation. Finally, we examined the fate of CIAPIN1 in intact cells following the VLX1570 treatment. VLX1570 was titrated onto KMS11 cells and incubated for 1 or 6 h before lysis and immunoblot analysis (Figure 4B). Aggregation was observed with 100 nM of VLX1570 after 1 h, with extended higher MW complexes and depletion of stable CIAPIN1 observed after 6 h. This effect was conserved for b-AP15 (Figure S13).
Figure 4.
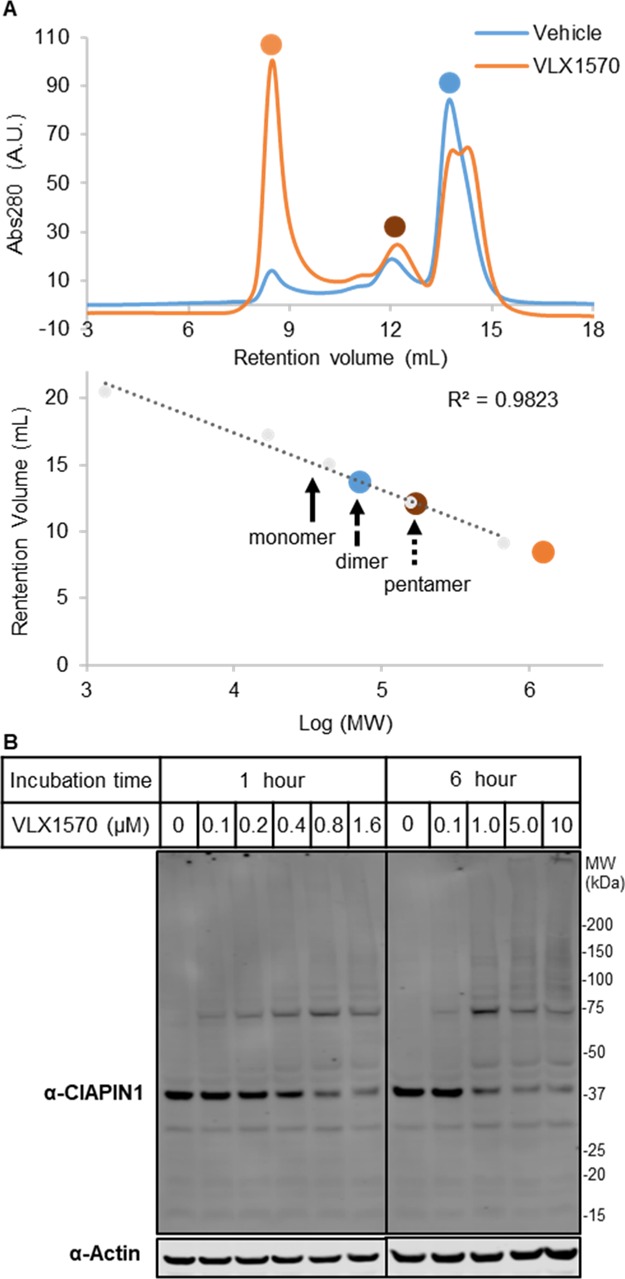
VLX1570 aggregates CIAPIN1. (A) Size exclusion chromatography of recombinant CIAPIN1 in the presence of VLX1570 or DMSO control indicates formation of an aggregation product. (B) Immunoblot analysis of high MW complex formation of CIAPIN1 following incubation with indicated concentrations of VLX1570 or DMSO.
Conclusions
In summary, our data indicate that b-AP15 and VLX1570 react with multiple cellular proteins in a nonspecific manner. Through a combination of immunoblot analysis and ABPP, we have identified several additional DUB targets and multiple proteins unrelated to ubiquitination machinery. Toxicity observed for drug candidates has been reported to be caused by off-target effects.34 In the case of b-AP15 and VLX1570, we show that it is linked to the formation of high MW complexes following compound treatment, an effect that is abated by co-incubation with GSH. Other molecules like curcumin18 and its derivative AC17,16 diterpene NSC-302979,18 synthetic compounds DBA18 and PYR-41,14 prostaglandin Δ12-PGJ2,18 and chalcone derivatives AM146,17 RA-9,17 and RA-1417 contain α,β-unsaturated ketones and have been already described to inhibit isopeptidases. The affinity probe 1 has enabled the comprehensive profiling of the covalent targets of VLX1570 in intact cells, leading to the identification of CIAPIN1 as a target. Further analysis of CIAPIN1 has identified protein depletion through aggregation as a mode of action of VLX1570, supporting our observations with b-AP15.
Collectively, the data demonstrate the power of target profiling to elucidate the mechanism of action and potential toxicity early in compound development. This strikes a cautionary note for the application of structurally similar compounds as tool compounds and the clinical investigation of compounds possessing this chemotype.
Experimental Section
General Methods and Reagents
Solvents were purchased from commercial sources at high-performance LC (HPLC) or analytical grade and used without purification. Analytical and preparative LC–MS were performed using a Waters system equipped with a Waters 2545 Binary Gradient Module, SecurityGuard ULTRA cartridges for EVO-C18 UHPLC HPLC, a Kinetex 5 μM EVO C18 100 Å 100 × 3.0 mm column, and a Waters SQ Detector 2. NMR spectra were recorded using a Bruker 400 MHz spectrometer using the deuterated solvent stated. Chemical shifts (δ) are quoted in parts per million (ppm) and referenced to the residual solvent peak. Reagents were purchased from Sigma-Aldrich unless otherwise stated and used without further purification. b-AP15 and VLX1570 were purchased from Selleckchem and used as supplied without further purification.
Synthesis of Compound 1 (Scheme 1)
Scheme 1. Synthesis of Compound 1.

To a solution of 4-perhydroazepinone hydrochloride (500 mg, 34 mmol, 1 equiv) and 4-fluoro-3-nitrobenzaldehyde (1.7 g, 101 mmol, 3 equiv) in acetic acid (50 mL), H2SO4 (10 mL) was added dropwise at room temperature. After stirring for 48 h, more H2SO4 (5 mL) was added. After a further 72 h, additional 4-fluoro-3-nitrobenzaldehyde (600 mg, 101 mmol, 1.06 equiv) was added (Scheme 1). After a further 48 h, the reaction was deemed complete by LC–MS analysis. The solution was diluted into 200 mL of H2O and extracted with EtOAc (2 × 150 mL). The combined layers were washed with brine (100 mL), dried over Na2SO4, and concentrated in vacuo. Crude intermediate A (1.4 g) was carried forward without purification. LC–MS 416 [M + H]+. To a solution of intermediate A (280 mg, 0.67 mmol, 1 equiv) and 3-butynoic acid (57 mg, 0.67 mmol, 1 equiv) in tetrahydrofuran (8 mL) at 0 °C, triethylamine (188 μL, 135 mmol, 2 equiv) was added, followed by dropwise addition of propylphosphonic anhydride (640 μL, 101 mmol, 1.5 equiv) as a 50% solution in EtOAc. The reaction was stirred at room temperature for 2 h. The reaction was then diluted with 75 mL of sat. NaHCO3 and extracted with ethyl acetate (2 × 75 mL). The combined layers were washed with brine (100 mL), dried over Na2SO4, and concentrated in vacuo. The compound was purified by preparative LC–MS. A 15 min gradient of 15–80% buffer B in buffer A was used (buffer A: acetonitrile/water = 5/95 with 20 mM ammonium acetate buffer, pH 6.0, buffer B: acetonitrile/water = 80/20 with 20 mM ammonium acetate buffer, pH 6.0) to provide compound 1 as a yellow solid (13 mg, 4%) in 98% purity. LC–MS: ES+ 482 [M + H]+, HRMS (ES+) found 482.1163. C24H18F2N3O6 requires 482.1158, 1H NMR (400 MHz, DMSO-d6): δ 8.36–8.19 (m, 2H), 8.05–7.88 (m, 2H), 7.76–7.54 (m, 3H), 7.51–7.36 (m, 1H), 6.50–5.80 (m, 1H), 5.36–5.11 (m, 2H), 4.99–4.85 (m, 1H), 4.85–4.65 (m, 1H), 3.90–3.74 (m, 2H), 3.22–2.92 (m, 2H), 13C NMR (101 MHz, DMSO): δ 214.12, 193.04, 166.94, 156.06, 153.39, 141.48, 138.21, 137.50, 137.41, 135.84, 134.02, 132.74, 127.74, 127.29, 119.39, 119.22, 87.08, 79.41, 74.86, 48.42, 48.24, 25.94.
Biological and Proteomic Methods
Detailed experimental procedures are included in the Supporting Information.
Acknowledgments
J.A.W. and K.V.M.H. are grateful for support by Myeloma UK (MUK). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. A.P.-F., O.F., and O.R.’s contributions to this study were supported by a grant from Innoviris (Région Bruxelles Capitale, Belgium). Initial work by J.A.W. was supported by a grant from the Institute of Chemical Biology EPSRC CDT.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00144.
Methods, IC50 values measured by the CellTiter Glo assay; immunoblot analysis; schematic of GSH addition to b-AP15; Gene ontology cellular component (GO CC) enrichment analysis; VLX1570-modified peptides identified by PEAKs analysis; and NMR spectra of 1 (PDF)
Spike-in SILAC proteomic analysis of VLX1570 (XLSX)
Molecular formula strings (CSV)
Author Contributions
# J.A.W. and A.P.-F. contributed equally to this work.
The authors declare no competing financial interest.
Notes
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE35 partner repository with the dataset identifier PXD015412.
Supplementary Material
References
- Bergink S.; Jentsch S. Principles of Ubiquitin and Sumo Modifications in DNA Repair. Nature 2009, 458, 461–467. 10.1038/nature07963. [DOI] [PubMed] [Google Scholar]
- Wertz I. E.; Wang X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. 10.1016/j.chembiol.2018.10.022. [DOI] [PubMed] [Google Scholar]
- Jacq X.; Kemp M.; Martin N. M. B.; Jackson S. P. Deubiquitylating Enzymes and DNA Damage Response Pathways. Cell Biochem. Biophys. 2013, 67, 25–43. 10.1007/s12013-013-9635-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kategaya L.; Di Lello P.; Rougé L.; Pastor R.; Clark K. R.; Drummond J.; Kleinheinz T.; Lin E.; Upton J.-P.; Prakash S.; Heideker J.; McCleland M.; Ritorto M. S.; Alessi D. R.; Trost M.; Bainbridge T. W.; Kwok M. C. M.; Ma T. P.; Stiffler Z.; Brasher B.; Tang Y.; Jaishankar P.; Hearn B. R.; Renslo A. R.; Arkin M. R.; Cohen F.; Yu K.; Peale F.; Gnad F.; Chang M. T.; Klijn C.; Blackwood E.; Martin S. E.; Forrest W. F.; Ernst J. A.; Ndubaku C.; Wang X.; Beresini M. H.; Tsui V.; Schwerdtfeger C.; Blake R. A.; Murray J.; Maurer T.; Wertz I. E. Usp7 Small-Molecule Inhibitors Interfere with Ubiquitin Binding. Nature 2017, 550, 534–538. 10.1038/nature24006. [DOI] [PubMed] [Google Scholar]
- Turnbull A. P.; Ioannidis S.; Krajewski W. W.; Pinto-Fernandez A.; Heride C.; Martin A. C. L.; Tonkin L. M.; Townsend E. C.; Buker S. M.; Lancia D. R.; Caravella J. A.; Toms A. V.; Charlton T. M.; Lahdenranta J.; Wilker E.; Follows B. C.; Evans N. J.; Stead L.; Alli C.; Zarayskiy V. V.; Talbot A. C.; Buckmelter A. J.; Wang M.; McKinnon C. L.; Saab F.; McGouran J. F.; Century H.; Gersch M.; Pittman M. S.; Marshall C. G.; Raynham T. M.; Simcox M.; Stewart L. M. D.; McLoughlin S. B.; Escobedo J. A.; Bair K. W.; Dinsmore C. J.; Hammonds T. R.; Kim S.; Urbé S.; Clague M. J.; Kessler B. M.; Komander D. Molecular Basis of Usp7 Inhibition by Selective Small-Molecule Inhibitors. Nature 2017, 550, 481–486. 10.1038/nature24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Esbroeck A. C. M.; Janssen A. P. A.; Cognetta A. B. 3rd; Ogasawara D.; Shpak G.; van der Kroeg M.; Kantae V.; Baggelaar M. P.; de Vrij F. M. S.; Deng H.; Allarà M.; Fezza F.; Lin Z.; van der Wel T.; Soethoudt M.; Mock E. D.; den Dulk H.; Baak I. L.; Florea B. I.; Hendriks G.; De Petrocellis L.; Overkleeft H. S.; Hankemeier T.; De Zeeuw C. I.; Di Marzo V.; Maccarrone M.; Cravatt B. F.; Kushner S. A.; van der Stelt M. Activity-Based Protein Profiling Reveals Off-Target Proteins of the Faah Inhibitor Bia 10-2474. Science 2017, 356, 1084–1087. 10.1126/science.aaf7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun M.; Kramer H. B.; Willems L. I.; McDermott J. L.; Leach C. A.; Goldenberg S. J.; Kumar K. G. S.; Konietzny R.; Fischer R.; Kogan E.; Mackeen M. M.; McGouran J.; Khoronenkova S. V.; Parsons J. L.; Dianov G. L.; Nicholson B.; Kessler B. M. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem. Biol. 2011, 18, 1401–1412. 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Ward J. A.; McLellan L.; Stockley M.; Gibson K. R.; Whitlock G. A.; Knights C.; Harrigan J. A.; Jacq X.; Tate E. W. Quantitative Chemical Proteomic Profiling of Ubiquitin Specific Proteases in Intact Cancer Cells. ACS Chem. Biol. 2016, 11, 3268–3272. 10.1021/acschembio.6b00766. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Flygare J. A.; Bogyo M.; Wertz I. E. Activity-Based Probes for the Ubiquitin Conjugation–Deconjugation Machinery: New Chemistries, New Tools, and New Insights. FEBS J. 2017, 284, 1555–1576. 10.1111/febs.14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberto I.; Liu X.; Seo H.-S.; Schauer N. J.; Iacob R. E.; Hu W.; Das D.; Mikhailova T.; Weisberg E. L.; Engen J. R.; Anderson K. C.; Chauhan D.; Dhe-Paganon S.; Buhrlage S. J. Structure-Guided Development of a Potent and Selective Non-Covalent Active-Site Inhibitor of Usp7. Cell Chem. Biol. 2017, 24, 1490–1500.e11. 10.1016/j.chembiol.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcy P.; Brnjic S.; Olofsson M. H.; Fryknäs M.; Lindsten K.; De Cesare M.; Perego P.; Sadeghi B.; Hassan M.; Larsson R.; Linder S. Inhibition of Proteasome Deubiquitinating Activity as a New Cancer Therapy. Nat. Med. 2011, 17, 1636–1640. 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- Tian Z.; D’Arcy P.; Wang X.; Ray A.; Tai Y.-T.; Hu Y.; Carrasco R. D.; Richardson P.; Linder S.; Chauhan D.; Anderson K. C. A Novel Small Molecule Inhibitor of Deubiquitylating Enzyme Usp14 and Uchl5 Induces Apoptosis in Multiple Myeloma and Overcomes Bortezomib Resistance. Blood 2014, 123, 706–716. 10.1182/blood-2013-05-500033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong A.; Merkx R.; Berlin I.; Rodenko B.; Wijdeven R. H. M.; El Atmioui D.; Yalçin Z.; Robson C. N.; Neefjes J. J.; Ovaa H. Ubiquitin-Based Probes Prepared by Total Synthesis to Profile the Activity of Deubiquitinating Enzymes. ChemBioChem 2012, 13, 2251–2258. 10.1002/cbic.201200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapuria V.; Peterson L. F.; Showalter H. D. H.; Kirchhoff P. D.; Talpaz M.; Donato N. J. Protein Cross-Linking as a Novel Mechanism of Action of a Ubiquitin-Activating Enzyme Inhibitor with Anti-Tumor Activity. Biochem. Pharmacol. 2011, 82, 341–349. 10.1016/j.bcp.2011.05.012. [DOI] [PubMed] [Google Scholar]
- D’Arcy P.; Linder S. Proteasome Deubiquitinases as Novel Targets for Cancer Therapy. Int. J. Biochem. Cell Biol. 2012, 44, 1729–1738. 10.1016/j.biocel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Zuo Y.; Li B.; Wang H.; Liu H.; Wang X.; Qiu X.; Hu Y.; Wen S.; Du J.; Bu X. Deubiquitinase Inhibition of 19s Regulatory Particles by 4-Arylidene Curcumin Analog Ac17 Causes Nf-Kappab Inhibition and P53 Reactivation in Human Lung Cancer Cells. Mol. Cancer Ther. 2013, 12, 1381–1392. 10.1158/1535-7163.mct-12-1057. [DOI] [PubMed] [Google Scholar]
- Issaenko O. A.; Amerik A. Y. Chalcone-Based Small-Molecule Inhibitors Attenuate Malignant Phenotype Via Targeting Deubiquitinating Enzymes. Cell Cycle 2012, 11, 1804–1817. 10.4161/cc.20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullally J. E.; Fitzpatrick F. A. Pharmacophore Model for Novel Inhibitors of Ubiquitin Isopeptidases That Induce P53-Independent Cell Death. Mol. Pharmacol. 2002, 62, 351–358. 10.1124/mol.62.2.351. [DOI] [PubMed] [Google Scholar]
- Pinto A.; Mace Y.; Drouet F.; Bony E.; Boidot R.; Draoui N.; Lobysheva I.; Corbet C.; Polet F.; Martherus R.; Deraedt Q.; Rodríguez J.; Lamy C.; Schicke O.; Delvaux D.; Louis C.; Kiss R.; Kriegsheim A. V.; Dessy C.; Elias B.; Quetin-Leclercq J.; Riant O.; Feron O. A New Er-Specific Photosensitizer Unravels (1)O2-Driven Protein Oxidation and Inhibition of Deubiquitinases as a Generic Mechanism for Cancer Pdt. Oncogene 2016, 35, 3976–3985. 10.1038/onc.2015.474. [DOI] [PubMed] [Google Scholar]
- Wang X.; D’Arcy P.; Caulfield T. R.; Paulus A.; Chitta K.; Mohanty C.; Gullbo J.; Chanan-Khan A.; Linder S. Synthesis and Evaluation of Derivatives of the Proteasome Deubiquitinase Inhibitor B-Ap15. Chem. Biol. Drug Des. 2015, 86, 1036–1048. 10.1111/cbdd.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Mazurkiewicz M.; Hillert E. K.; Olofsson M. H.; Pierrou S.; Hillertz P.; Gullbo J.; Selvaraju K.; Paulus A.; Akhtar S.; Bossler F.; Khan A. C.; Linder S.; D’Arcy P. The Proteasome Deubiquitinase Inhibitor Vlx1570 Shows Selectivity for Ubiquitin-Specific Protease-14 and Induces Apoptosis of Multiple Myeloma Cells. Sci. Rep. 2016, 6, 26979. 10.1038/srep26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A.; Ovaa H.; Kolli N.; Gan-Erdene T.; Wilkinson K. D.; Ploegh H. L.; Kessler B. M. Chemistry-Based Functional Proteomics Reveals Novel Members of the Deubiquitinating Enzyme Family. Chem. Biol. 2002, 9, 1149–1159. 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- Weerapana E.; Simon G. M.; Cravatt B. F. Disparate Proteome Reactivity Profiles of Carbon Electrophiles. Nat. Chem. Biol. 2008, 4, 405–407. 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heal W. P.; Jovanovic B.; Bessin S.; Wright M. H.; Magee A. I.; Tate E. W. Bioorthogonal Chemical Tagging of Protein Cholesterylation in Living Cells. Chem. Commun. 2011, 47, 4081–4083. 10.1039/c0cc04710d. [DOI] [PubMed] [Google Scholar]
- Geiger T.; Wisniewski J. R.; Cox J.; Zanivan S.; Kruger M.; Ishihama Y.; Mann M. Use of Stable Isotope Labeling by Amino Acids in Cell Culture as a Spike-in Standard in Quantitative Proteomics. Nat. Protoc. 2011, 6, 147–157. 10.1038/nprot.2010.192. [DOI] [PubMed] [Google Scholar]
- Sadaghiani A. M.; Verhelst S. H.; Bogyo M. Tagging and Detection Strategies for Activity-Based Proteomics. Curr. Opin. Chem. Biol. 2007, 11, 20–28. 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Calderone V.; Ciofi-Baffoni S.; Giachetti A.; Jaiswal D.; Mikolajczyk M.; Piccioli M.; Winkelmann J. Molecular View of an Electron Transfer Process Essential for Iron–Sulfur Protein Biogenesis. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7136–7141. 10.1073/pnas.1302378110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G.; Cheng C.; Li Y.; Shaw N.; Xiao Z.-C.; Liu Z.-J. Crystal Structure of the N-Terminal Methyltransferase-Like Domain of Anamorsin. Proteins: Struct., Funct., Bioinf. 2014, 82, 1066–1071. 10.1002/prot.24443. [DOI] [PubMed] [Google Scholar]
- Shibayama H.; Takai E.; Matsumura I.; Kouno M.; Morii E.; Kitamura Y.; Takeda J.; Kanakura Y. Identification of a Cytokine-Induced Antiapoptotic Molecule Anamorsin Essential for Definitive Hematopoiesis. J. Exp. Med. 2004, 199, 581–592. 10.1084/jem.20031858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Li Q.; Wang C.; Xiong Q.; Lin Y.; Sun Q.; Jin H.; Yang F.; Ren X.; Pang T. Knock-Down of Ciapin1 Sensitizes K562 Chronic Myeloid Leukemia Cells to Imatinib by Regulation of Cell Cycle and Apoptosis-Associated Members Via Nf-Kappab and Erk5 Signaling Pathway. Biochem. Pharmacol. 2016, 99, 132–145. 10.1016/j.bcp.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Li X.; Pan Y.; Fan R.; Jin H.; Han S.; Liu J.; Wu K.; Fan D. Adenovirus-Delivered Ciapin1 Small Interfering Rna Inhibits Hcc Growth in Vitro and in Vivo. Carcinogenesis 2008, 29, 1587–1593. 10.1093/carcin/bgn052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Wang W. E.; Zhang Q. Ciapin1 Sirna Inhibits Proliferation, Migration and Promotes Apoptosis of Vsmcs by Regulating Bcl-2 and Bax. Curr. Neurovasc. Res. 2013, 10, 4–10. 10.2174/156720213804805909. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Fang J.; Ma H. Inhibition of Mir-182-5p Protects Cardiomyocytes from Hypoxia-Induced Apoptosis by Targeting Ciapin1. Biochem. Cell Biol. 2018, 96, 646–654. 10.1139/bcb-2017-0224. [DOI] [PubMed] [Google Scholar]
- Lin A.; Giuliano C. J.; Palladino A.; John K. M.; Abramowicz C.; Yuan M. L.; Sausville E. L.; Lukow D. A.; Liu L.; Chait A. R.; Galluzzo Z. C.; Tucker C.; Sheltzer J. M. Off-Target Toxicity Is a Common Mechanism of Action of Cancer Drugs Undergoing Clinical Trials. Sci. Transl. Med. 2019, 11, eaaw8412 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yılmaz Ş.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The Pride Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.