

Abstract
NLRP3 inflammasome mediated release of interleukin-1β (IL-1β) has been implicated in various diseases. In this study, rationally designed mimics of sulfonylurea moiety were investigated as NLRP3 inhibitors. Our results culminated into discovery of series of unprecedented N-cyano sulfoximineurea derivatives as potent NLRP3 inflammasome inhibitors. Compound 15 (IC50 = 7 nM) and analogues were found to be highly potent and selective NLRP3 inflammasome inhibitor with good pharmacokinetic profile. These effects translate in vivo, as 15, 29, and 34 significantly inhibit NLRP3 dependent IL-1β secretion in mice.
Keywords: NLRP3, NLRP3 inflammasome, Interleukin-1β (IL-1β), Inflammation, Sulfonylurea, N-cyano-sulfoximineurea derivatives
Inflammasomes are multiprotein complexes formed by innate immune sensors, including nucleotide oligomerization domain (NOD)-like receptor protein (NLR) family members NLRP1, NLRP3, and NLRC4, along with other non-NLR receptors such as AIM2 and IFI16.1 NLRP3 inflammasome activation is dependent on two successive signals. The first step comprises an initiating signal (priming) in which many danger associated molecular patterns (DAMPs) and pathogen associated molecular patterns (PAMPs) are recognized by TLRs, which in turn up-regulates transcription of inflammasome related components, including inactive NLRP3, proIL-1β, and proIL-18.2,3 In the second step of inflammasome activation, the oligomerization of NLRP3 and subsequent assembly of NLRP3, ASC, and procaspase-1 results in formation of a complex. This triggers the transformation of procaspase-1 to caspase-1, leading to the release of active pro-inflammatory cytokines IL-1β and IL-18.4
The NLRP3 inflammasome is the most well understood and widely studied owing to its role in host defense and innate immunity.5 The NLRP3 inflammasome is key component of the inflammatory response. The inappropriate activation of NLRP3 is implicated in a wide range of diseases, including rare periodic fever syndrome, CAPS, TRAPS, and a variety of human complex diseases such as multiple sclerosis, atherosclerosis, alzheimer’s disease, diabetes, asthma, gouty arthritis, inflammatory bowel disease (IBD), juvenile arthritis, and neurodegenerative and autoimmune diseases.6,7 Hence, the NLPR3 inflammasome might be a potential target for the treatment of these complex diseases.8,9
Additionally, advantage of blocking NLRP3 over simply using an immunosuppressant is that it targets this inflammatory response while leaving the rest of the immune system to operate as normal.10 Therefore, NLRP3 is emerging as a promising target to develop novel and specific compounds for the treatment of anti-inflammatory and autoimmune diseases.11 Current treatments for these diseases include biologic drugs that target IL-1β such as anakinra, canakinumab, and rilonacept.12 However, they also have major limitations such as inconvenient treatment routes and high cost. Hence, an oral small molecule inhibitor of NLRP3 is desirable. This has attracted a great deal of attention in the pharmaceutical industry and in academia.13−15
Several structurally diverse small molecule modulators of NLRP3 have been described (Figure 1). MCC950 is reported to be a small molecule inhibitor of NLRP3 inflammasome with an early promise for treatment of inflammatory diseases.16 In addition, novel boron compound (NBC-6),17 sulfonamide (JC-171),18 and compounds like CY0914 and OLT117719 have also been reported as NLRP3 inflammasome inhibitors. Oridonin20 and Tranilast21 have recently also been reported to block the NLRP3 inflammasome pathway.
Figure 1.

Selected known NLRP3 inflammasome inhibitors.
This communication describes the discovery of a series of unprecedented N-cyano sulfoximineurea derivatives as highly potent, orally bioavailable, and selective NLRP3 inflammasome inhibitors. We hypothesized that rationally designed mimics of sulfonylurea moiety in the MCC950 would result in structurally novel NLRP3 inflammasome inhibitors with improved properties (Figure 2). Accordingly, we focused our SAR efforts on exploring this region of the molecule.
Figure 2.

Schematic representation of ligand optimization.
A series of novel compounds have been synthesized by rational and systematic bioisosteric replacement of sulfonylurea (Figure 2). Drug-like properties such as molecular weight (<500) and clogP of these NCEs are generally within the acceptable range of oral drugs.22 The ability of test compounds to inhibit NLRP3 inflammasome was measured in IL-1β assay using THP1 cells.16Table 1 describes the structure–activity relationship (SAR) of replacement of sulfonylurea group. The most studied NLRP3 inhibitor, MCC950 (also called CRID3 and CP-456,773), was found to be potent in our hands as well (IC50 = 8 nM), and its corresponding phenyl (2) and tosyl (3) analogues were also potent, with IC50 of 35 and 15 nM, respectively. Introduction of cyanomethyl urea or 1-(2,2,2-trifluoroethyl) urea in place of sulfonylurea (as exemplified by analogues 4 and 5) resulted in loss of activity with less than 50% inhibition of IL-1β release at 1 μM. Changing the topography by introducing oxetane or squaric acid moiety also resulted in dramatic potency loss (6 and 7). Further, various other rationally designed mimics of sulfonylurea moiety, like 2-oxo-N-(arylsulfonyl)acetamide derivative 8, corresponding hydroxy derivative 9 and acetamide 10 also lowered the activity. Moreover, sulfonamide derivative 11, oxalamide derivative 12, phosphonamidic acid analogue 13, and cyanoguanidine derivative 14 were also synthesized and explored as NLRP3 inflammasome inhibitors. However, all these performed modulations lowered the activity obtaining less than 50% inhibition of IL-1β release at 1 μM.
Table 1. Core Modification on the Sulfonylurea Moiety: In Vitro Activity of MCC950 (1) and 2–20a.
![]()

IC50 values given are expressed as mean ± SEM of three independent experiments.
Calculated from Schrödinger Release 2018-3: QikProp, Schrödinger, LLC, New York, NY, 2018.
These results indicate that any modification of the sulfonylurea motif in 4–14 resulted in loss of IL-1β inhibition, showing the critical importance of the sulfonylurea hydrogen bond acceptor–donor pattern. Nevertheless, to our delight, N-cyano sulfoximineurea analogue 15 was found to be highly potent (IC50 = 7 nM), equipotent to MCC950. In addition, N-cyano sulfoximineurea derivative 16 was also found to be active (IC50 = 21 nM). Sulfoximine moiety, the monoaza analogues of sulfones, are stable pharmacophores which offer a rich and versatile tool in medicinal chemistry.23 This prompted us to explore additional N-substituted sulfoximineurea analogues. However, introduction of alkyl, methoxy, aryl, heteroaryl, amide, ester, or acyl group at sulfoximine nitrogen proved to be detrimental to activity (17–24). Overviews of all the compounds that have been synthesized and tested for IL-1β activity are listed in Table 1.
Notably, 15 and 16 were highly selective against TNF-α (IC50 > 10 μM). The production of TNF-α was not affected by 15 even at high concentrations (see Supporting Information), indicating its selective effect on the NLRP3 inflammasome. Moreover, 15 blocked the recruitment of ASC during NLRP3 activation, thus providing further evidence to the mechanistic hypothesis of our novel N-cyano sulfoximineurea derivatives as NLRP3 inflammasome inhibitors. Furthermore, this compound had no effect on the AIM2 inflammasome, demonstrating specificity for NLRP3 inflammasome. The results obtained suggested that incorporation of the N-cyano sulfoximineurea motif may open up new directions for further SAR development. Accordingly, we focused our investigation on this region of the molecule, and thus, in our optimization campaign, a series of novel substituted sulfoximineurea derivatives were synthesized and evaluated as NLRP3 inflammasome inhibitors.
An efficient synthesis allowed rapid access to 15, as outlined in Scheme 1. The key intermediate 25 was obtained from commercially available p-tosyl chloride using a known procedure.24 The another key intermediate, tricyclic amine (1,2,3,5,6,7-hexahydro-s-indacen-4-amine) (26), and its corresponding isocyanate 27, was synthesized via reported protocol on multigram scale.25 The coupling of sulfinimide 25 with 4-isocyanato-1,2,3,5,6,7-hexahydro-s-indacene (27) provided sulfinylurea 28 in high yields, using modified reaction conditions reported by Toth et al.26 Synthesis of N-substituted sulfoximines using alkylamines27 and cyanamide28,29 are reported in the literature. However, there are no reports for the synthesis of N-cyano sulfoximineurea, albeit synthesis of sulfinylurea followed by chlorination/amination to afford N-alkyl sulfoximineurea are documented in literature.26 Sulfinylurea 28 was converted to N-cyano sulfoximineurea derivative 15 using cyanamide with NCS as oxidant and potassium tert-butoxide as base in a acetonitrile mixture. Following the same reaction sequence with various sulfinimides resulted in test compounds 16 and 29–34 in good yield and high chemical purity. Synthesis of 2–24 is described in the Supporting Information.
Scheme 1. Synthesis of Compound 15.

Reagents and conditions: (a) 4-isocyanato-1,2,3,5,6,7-hexahydro-s-indacene (27), n-BuLi, dry THF, −78 °C to rt, 4 h (70%); (b) cyanamide, MeCN, t-BuOK, NCS, rt 5 h (22%).
Extensive SAR work on the left-hand side aryl ring fragment was also pursued.30 Key examples selected from a large set of modifications are presented in Table 2. Notably, the fluorinated compound 29 displayed excellent potency (IC50 = 5.3 nM) as well as good pharmacokinetic properties with AUC = 3.2 μg·h/mL at 3 mg/kg and mice oral bioavailability of 59%. Additional modifications on the left-hand side aryl ring, such as addition of electron-withdrawing groups (30 and 31), were made to potentially improve the pharmacokinetic profile. The 4-cyano analogue 30 displayed IL-1β inhibition IC50 of 34 nM. However, pharmacokinetic profile of this compound was comparatively inferior (AUC = 0.8 μg·h/mL at 3 mg/kg, t1/2 = 1 h, mice oral bioavailability 27%). The 3-cyano analogue 31 displayed IC50 of 12 nM, with similar pharmacokinetic profile (AUC = 1.2 μg·h/mL at 3 mg/kg, mice oral bioavailability 40%). Next, in a quest to explore heteroaryl analogues, the pyridyl compounds were investigated. The 4-pyridyl analogue 32 was found to be relatively less potent (IC50 = 112 nM). The regioisomer, 3-pyridyl compound 33 helped to regain in vitro potency but was accompanied by loss of plasma levels and bioavailability. In vitro potency was further improved for 2-pyridyl compound 34 (IC50 = 23 nM). In addition, the pharmacokinetic properties of 34 were also dramatically improved, with AUC = 8.2 μg·h/mL at 3 mg/kg, t1/2 = 5.2 h and mice oral bioavailability of 97%, perhaps due to good solubility. It is noteworthy to mention that all these compounds (15, 16 and 29–34) were highly selective against TNF-α (IC50 > 10 μM). In particular, 34 show a high oral bioavailability along with a long half-life and represent an improvement in pharmacokinetic properties with respect to regioisomers 32 and 33. This, combined with the potency of the compound, suggests potential for therapeutic efficacy in in vivo models.
Table 2. Modifications on Left-Hand Side of N-Cyano-sulfoximineurea Derivatives: In Vitro Activity and Pharmacokinetics Propertiesa of 15, 29–34.
![]()

Compounds dosed 1 mg/kg iv and 3 mg/kg po.
PO: Mouse PK data is mean data because of composite study design, n = 3/time point. Formulation: PO, 1% Tween 80 + 99% (0.5%) methyl cellulose in water; IV, 5% NMP + 5% solutol +90% normal saline.
Calculated from Schrödinger Release 2018-3: QikProp, Schrödinger, LLC, New York, NY, 2018. NC: Not calculated.
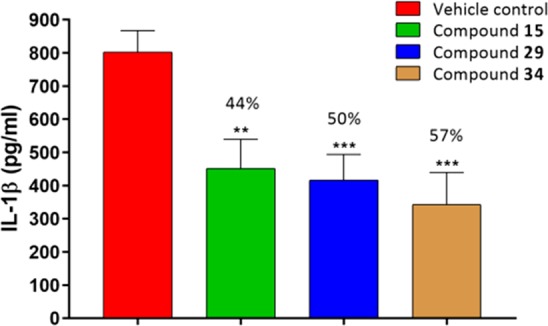
Next, having identified compounds with high potency and good pharmacokinetic profile, the effect of the lead compounds 15, 29 and 34, on the NLRP3 dependent release of IL-1β was evaluated in an acute in vivo LPS+ATP challenged model in female C57BL/6 mice.31 A single oral administration of test compounds at 10 mg/kg dose decreases the IL-1β levels by 44%, 50%, and 57% respectively, wrt vehicle control (Figure 3), indicating that even at this low dose, 15, 29, and 34 markedly reduced the IL-1β compared to the control.
Figure 3.
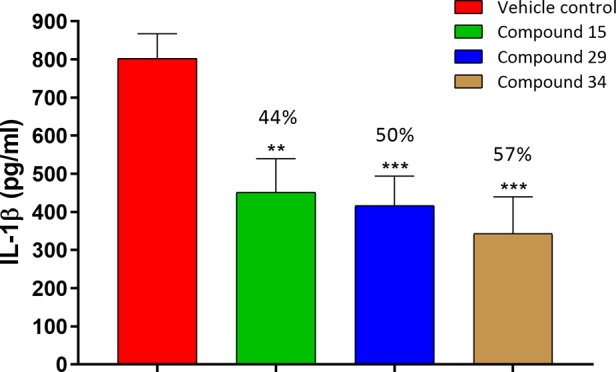
In vivo IL-1β inhibition of compounds 15, 29, and 34 in C57 mice (n = 6 animals/group; female C57 mice; po 10 mg/kg; formulation, 1% Tween 80 and 99% methyl cellulose (0.5%); ***P < 0.001 versus control, **P < 0.01 versus control; error bar indicates SEM).
In conclusion, through systematic exploration of rationally designed mimics of sulfonylurea moiety, we have identified a novel series of N-cyano sulfoximineurea derivatives as potent and selective NLRP3 inflammasome inhibitors. The in vitro inhibition potency of N-cyano sulfoximineurea analogue 15 on IL-1β release by LPS/ATP stimulation was found to be similar to the reference compound MCC950, thus indicating that structural modifications at the sulfonylurea moiety can be tolerated. Further 15, 29, and 34 exhibited significant IL-1β lowering efficacy in animal models with desired pharmacokinetic profile. These data also warrant further investigation of 15 and analogues for treatment of chronic inflammatory diseases mediated via NLRP3 inflammasome. These NLRP3 inflammasome inhibitors with novel chemical structure having high potency and desired PK/PD profile would be helpful in further investigations as potential clinical candidates.
Acknowledgments
We thank Jeevan Kumar for log P calculations using QikProp, Schrödinger Software.
Glossary
Abbreviations Used
- NLRP3
NOD-like receptor family, pyrin domain-containing protein 3
- ASC
apoptosis-associated speck-like protein containing a CARD
- AIM2
absent in melanoma 2
- DAMP
damage-associated molecular pattern
- PAMP
pathogen-associated molecular pattern
- TLR
Toll-like receptor
- IFI16
gamma-interferon-inducible protein 16
- IL-1R
interleukin-1 receptor
- LPS
lipopolysaccharide
- ATP
adenosine triphosphate
- CAPS
cryopyrin associated periodic syndromes
- TRAPS
tumor necrosis factor receptor-associated periodic syndrome
- NCE
New Chemical Entity
- NMP
N-methyl-2-pyrrolidone
- NCS
N-chlorosuccinimide
- PK/PD
pharmacokinetic/pharmacodynamics
- TNF-α
tumour necrosis factor alpha.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00433.
The authors declare no competing financial interest.
Notes
ZRC Communication No. 595.
Supplementary Material
References
- Guo H.; Callaway J. B.; Ting J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F.; Mayor A.; Tschopp J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- He Y.; Hara H.; Nunez G. Mechanism and regulation of nlrp3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao B. Z.; Xu Z. Q.; Han B. Z.; Su D. F.; Liu C. Nlrp3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262–271. 10.3389/fphar.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Shaw M. H.; Kim Y. G.; Nuñez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol.: Mech. Dis. 2009, 4, 365–398. 10.1146/annurev.pathol.4.110807.092239. [DOI] [PubMed] [Google Scholar]
- Strowig T.; Henao-Mejia J.; Elinav E.; Flavell R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- Zhen Y.; Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front. Immunol. 2019, 10, 276. 10.3389/fimmu.2019.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H.-H.; Yang Y.-X.; Meng X.; Luo X.-Y.; Li X. M.; Shuai Z.-W.; Ye D. Q.; Pan H. F. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun. Rev. 2018, 17, 694–702. 10.1016/j.autrev.2018.01.020. [DOI] [PubMed] [Google Scholar]
- Mangan M. S. J.; Olhava E. J.; Roush W. R.; Seidel H. M.; Glick G. D.; Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discovery 2018, 17, 588–606. 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- Dinarello C. A.; Simon A.; van der Meer J. W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discovery 2012, 11, 633–652. 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo-Castro E.; Faust D.; Fox S.; Baldwin A. G.; Osborne S.; Haley M. J.; Karran E.; Nuthall H.; Atkinson P. J.; Dawson L. A.; Routledge C.; Allan S. M.; Freeman S.; Brownlees J.; Brough D. Development of a characterized tool kit for the interrogation of NLRP3 inflammasome-dependent responses. Sci. Rep. 2018, 8, 5667. 10.1038/s41598-018-24029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachove I.; Chang C. Anakinra and related drugs targeting interleukin-1 in the treatment of cryopyrin-associated periodic syndromes. Open Access Rheumatol.: Res. Rev. 2014, 6, 15–25. 10.2147/OARRR.S46017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid A.; Li B.; Kombe A. J. K.; Jin T.; Tao J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. 10.3389/fimmu.2019.02538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; He H.; Chen Y.; Huang W.; Cheng J.; Ye J.; Wang A.; Tao J.; Wang C.; Liu Q.; Jin T.; Jiang W.; Deng X.; Zhou R. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. 10.1084/jem.20171419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Varadarajan S.; Munoz-Planillo R.; Burberry A.; Nakamura Y.; Nunez G. 3,4-Methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 2014, 289, 1142–1150. 10.1074/jbc.M113.515080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll R. C.; Robertson A. A.; Chae J. J.; Higgins S. C.; Munoz-Planillo R.; Inserra M. C.; Vetter I.; Dungan L. S.; Monks B. G.; Stutz A.; Croker D. E.; Butler M. S.; Haneklaus M.; Sutton C. E.; Nunez G.; Latz E.; Kastner D. L.; Mills K. H.; Masters S. L.; Schroder K.; Cooper M. A.; O’Neill L. A. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin A. G.; Rivers-Auty J.; Daniels M. J. D.; White C. S.; Schwalbe C. H.; Schilling T.; Hammadi H.; Jaiyong P.; Spencer N. G.; England H.; Luheshi N. M.; Kadirvel M.; Lawrence C. B.; Rothwell N. J.; Harte M. K.; Bryce R. A.; Allan S. M.; Eder C.; Freeman S.; Brough D. Boron-based inhibitors of the nlrp3 inflammasome. Cell Chem. Biol. 2017, 24, 1321–1335. e1325 10.1016/j.chembiol.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Fulp J. W.; Jiang Y.; Li X.; Chojnacki J. E.; Wu J.; Wang X. Y.; Zhang S. Development and characterization of a hydroxyl-sulfonamide analogue, 5-chloro-n-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel nlrp3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem. Neurosci. 2017, 8, 2194–2201. 10.1021/acschemneuro.7b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C.; Swartzwelter B.; Gamboni F.; Neff C. P.; Richter K.; Azam T.; Carta S.; Tengesdal I.; Nemkov T.; D’Alessandro A.; Henry C.; Jones G. S.; Goodrich S. A.; St. Laurent J. P.; Jones T. M.; Scribner C. L.; Barrow R. B.; Altman R. D.; Skouras D. B.; Gattorno M.; Grau V.; Janciauskiene S.; Rubartelli A.; Joosten L. A. B; Dinarello C. A. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E1530–E1539. 10.1073/pnas.1716095115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H.; Jiang H.; Chen Y.; Ye J.; Wang A.; Wang C.; Liu Q.; Liang G.; Deng X.; Jiang W.; Zhou R. Oridonin is a covalent nlrp3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. 10.1038/s41467-018-04947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Jiang H.; Chen Y.; Wang X.; Yang Y.; Tao J.; Deng X.; Liang G.; Zhang H.; Jiang W.; Zhou R. Tranilast directly targets nlrp3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. 10.15252/emmm.201708689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring M. J.; Arrowsmith J.; Leach A. R.; Leeson P. D.; Mandrell S.; Owen R. M.; Pairaudeau G.; Pennie W. D.; Pickett S. D.; Wang J.; Wallace O.; Weir A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discovery 2015, 14, 475–486. 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- Lücking U. Sulfoximines: A neglected opportunity in medicinal chemistry. Angew. Chem., Int. Ed. 2013, 52, 9399–9408. 10.1002/anie.201302209. [DOI] [PubMed] [Google Scholar]
- Krasnova L. B.; Yudin A. K. p-Tolylsulfinyl amides: reagents for facile electrophilic functionalization of olefins. J. Org. Chem. 2004, 69 (7), 2584–2587. 10.1021/jo035518a. [DOI] [PubMed] [Google Scholar]
- Urban F. J.; Jasys V. J.; Raggon J. W.; Buzon R. A.; Hill P. D.; Eggler J. F.; Weaver J. D. Novel synthesis of 1-(1,2,3,5,6,7-hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methyl-ethyl)-furan-2-sulfonyl]-urea, an anti-inflammatory agent. Synth. Commun. 2003, 33, 2029–2043. 10.1081/SCC-120021029. [DOI] [Google Scholar]
- Toth J. E.; Grindey G. B.; Ehlhardt W. J.; Ray J. E.; Boder G. B.; Bewley J. R.; Klingerman K. K.; Gates S. B.; Rinzel S. M.; Schultz R. M.; Weir L. C.; Worzalla J. F. Sulfonimidamide analogs of oncolytic sulfonylureas. J. Med. Chem. 1997, 40, 1018–1025. 10.1021/jm960673l. [DOI] [PubMed] [Google Scholar]
- Dannenberg C. A.; Bizet V.; Bolm C. Direct access to n-alkylsulfoximines from sulfides by a sequential imidation/oxidation procedure. Synthesis 2015, 47, 1951–1959. 10.1055/s-0034-1380536. [DOI] [Google Scholar]
- Lafitte G.; Parnet V.; Pierre R.; Raffin C.; Vatinel R.; Musicki B.; Tomas L.; Bouix-Peter C.; Ouvry G.; Daver S.; Arlabosse J.-M.; Boiteau J.-G.; Gerfaud T.; Harris C. S. Route scouting and optimization of a potent sulfoximine-based inverse agonist of RORγt. Tetrahedron 2018, 74, 5974–5986. 10.1016/j.tet.2018.08.043. [DOI] [Google Scholar]
- García Mancheño O. G.; Bolm C. Synthesis of sulfonimidamides from sulfinamides by oxidation with N-chlorosuccinimide. Beilstein J. Org. Chem. 2007, 3, 25. 10.1186/1860-5397-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R.; Iyer P.; Agarwal S.; Novel substituted sulfoximine compounds. WO 2018/225018, 2018.
- Perregaux D. G.; Mcniff P.; Laliberte R.; Hawryluk N.; Peurano H.; Stam E.; Eggler J.; Griffiths R.; Dombroski M. A.; Gabel C. A. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J. Pharmacol. Exp. Ther. 2001, 299, 187–197. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.