

Abstract

Apoptosis signal-regulating kinase 1 (ASK1) is a key mediator in the apoptotic and inflammatory cellular stress response. To investigate the therapeutic value of modulating this pathway in neurological disease, we have completed medicinal chemistry studies to identify novel CNS-penetrant ASK1 inhibitors starting from peripherally restricted compounds reported in the literature. This effort led to the discovery of 21, a novel ASK1 inhibitor with good potency (cell IC50 = 138 nM), low clearance (rat Cl/Clu = 0.36/6.7 L h–1 kg–1) and good CNS penetration (rat Kp,uu = 0.38).
Keywords: Apoptosis signal-regulating kinase 1, ASK1, rational design, CNS penetration, neurodegenerative diseases
Apoptosis signal-regulating kinase 1 (ASK1) is a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family that activates downstream MAP kinases (MAPKs), c-Jun N-terminal kinases (JNKs), and p38 MAPKs in response to stress.1 This pathway plays an essential role in the regulation of both apoptosis and inflammation,2 and genetic animal models of neurodegeneration have implicated ASK1 in a number of diseases including amyotrophic lateral sclerosis (ALS)3 and multiple sclerosis.4 Specifically, genetic deletion of ASK1 in Cu/Zn-superoxide dismutase (SOD1)mut mice extended the life span of these animals,5 and this event was found to be concurrent with reduced motor neuron loss in the spinal cord. These findings were partially recapitulated in SOD1 mice that had been administered with the ASK1 inhibitors K811 (1, Figure 1) or K812 (2, Figure 1)6 at disease onset (28 weeks), thus linking modulation of the kinase activity of ASK1 with disease progression in ALS. Despite the potential benefit associated with targeting this pathway in neurological disease,7 the only ASK1 inhibitor in the clinic is selonsertib (3, GS-4997, Figure 1), which is in phase II clinical trials8 for the treatment of liver fibrosis in combination with other agents.9
Figure 1.

ASK1 inhibitors reported in the literature.
To gain a better understanding of the impact of modulating the ASK1 pathway in neurological disease, we have recently disclosed the identification of macrocyclic inhibitors with good potency, pharmacokinetic (PK) profile, and moderate CNS penetration following a deconstruction–cyclization strategy (analog 4, Figure 2, top).10 Herein we describe an alternative approach to gain access to potent and brain penetrant ASK1 inhibitors that relied on the optimization of the phenyl group in the initially deconstructed analog 5 (Figure 2, bottom).
Figure 2.

Binding mode of compound 3 bound to ASK1 (PDB code 6OYT) and optimization strategy. Compound 3 occupies the ATP-binding pocket of ASK1 and interacts with the hinge binder residue (Val 757) and with the catalytic lysine residue (Lys 709). Compound 5 was predicted to retain these two key interactions and place the phenyl group in a solvent exposed region.
At the outset, the potency of 5 was determined using both a biochemical11 and an internally developed cellular assay,12−14 and this analog was found to be a weak ASK1 inhibitor (IC50 = 607 nM, cell IC50 > 20 μM, Table 1). The poor potency of this compound was rationalized by docking experiments that predicted a binding mode on which the lipophilic phenyl group in 5 would be placed in a solvent-exposed area (Figure 2). This insight led us to pursue the synthesis of compounds on which the phenyl group would be replaced with more polar five-member heterocyclic groups (Table 1).15,16 The first analog synthesized following this strategy was pyrazole 6, and it was found to be 20-fold more potent in the biochemical assay relative to 5 (6, IC50 = 29 nM vs 5, IC50 = 607 nM, Table 1). To evaluate the importance of the attachment point of the pyrazole to the aminopyridine fragment, the regioisomeric 4-substituted pyrazole 7 was synthesized, and this analog was found to have improved cellular potency relative to 6 (7, cell IC50 = 6.8 μM vs 6, cell IC50 > 20 μM). To mask the H-bond of the amide in 7, substituents with the ability to form an intramolecular H-bond were then introduced in the pyrazole. Among these, the methoxy group had the biggest impact in potency and the resulting analog 8 (cell IC50 = 90 nM) was also found to have low in vivo clearance (Cl/Clu = 1.6/4.0 L h–1 kg–1) in a rat PK experiment.17 An initial attempt to further improve the profile of this inhibitor by implementing the previously described macrocyclization strategy10 led to the identification of analog 9 (cell IC50 = 32 nM),18 but this compound had high in vivo clearance in a rat PK experiment (Cl/Clu = 4.7/21 L h–1 kg–1) and also suffered from high efflux ratio (ER) (9, Table 1, ER = 26).19 Given the shortcomings of this macrocyclization strategy, the effort shifted toward the synthesis of analogs with reduced polar surface area (PSA) in an attempt to reduce their high ER. Among these, analog 10, on which the central pyridine ring had been replaced with a phenyl group, was found to be a very weak ASK1 inhibitor in the biochemical assay (IC50 = 6.2 μM), and despite its reduced PSA, it was still found to be a P-gp substrate (ER = 104). In contrast, replacement of the triazole ring in 8 with an imidazole led to the identification of inhibitor 11, which had good potency (cell IC50 = 93 nM) and low ER (2.2), but it was found to have high clearance in a rat PK experiment (Cl/Clu = 3.4/42 L h–1 kg–1).
Table 1. In Vitro and in Vivo Data for 3 and Analogs 5–11.

Biochemical assay.
Inhibition of ASK1 autophosphorylation. IC50 results are the geometric mean of a minimum of two determinations.
MDCK-MDR1 human P-gp transfected cell line. Papp = apparent permeability, apical-to-basolateral (10–6 cm/s); ER = (B–A)/(A–B) efflux ratio.
Clu = Cl/fu.
DMSO/PG (1:1) used as vehicle. Vdss = 1.8 L/kg; t1/2 = 0.9 h.
DMSO/PG (1:1) used as vehicle. Vdss = 2.7 L/kg; t1/2 = 0.7 h.
DMA/EtOH/PG/water (1:1:3:5) used as vehicle. Vdss = 3.5 L/kg; t1/2 = 2.0 h.
Given the limited success of this optimization strategy, it was decided to focus the synthetic effort toward modification of the N-methyl substituent in the pyrazole analog 8. In an attempt to increase the structural diversity of these inhibitors, a small library of N-alkylated and N-arylated pyrazoles was synthesized (Table 2). These studies revealed that introduction of small alkyl groups in the pyrazole N (analogs 12–14) resulted in compounds with improved cellular potency. Specifically, analog 13 on which a cyclopropylmethyl group had been introduced was over 7-fold more potent relative to the parent methyl substituted analog 8 (13, cell IC50 = 12 nM vs 8, cell IC50 = 90 nM), but all the analogs synthesized with an alkyl group were found to have high ER (Table 2, analogs 12–14). In contrast, introduction of a 2-pyridyl group (analog 15) was found to have significantly lower ER (3.5) as well as an acceptable level of cellular potency (IC50 = 299 nM). Systematic synthesis of the other pyridyl rings was then carried out (analogs 16 and 17), and it was found that the analog containing a 3-pyridyl ring (17) had dramatically improved cellular potency relative to 15 (a 25-fold improvement in potency was observed), although this analog was a P-gp substrate (17, ER = 16). Subsequent introduction of a cyclopropyl substituent in 17 led to analog 18 which was found to have good potency but similarly high ER (16). In contrast, introduction of a dimethylamino group led to a compound with moderate ER (4.5), but its microsomal stability was poor (19, RLM, HLM = 136, 81 mL min–1 kg–1). Introduction of a pyrimidine substituent led to analog 20 which had good microsomal stability (RLM, HLM = 22, <5 mL min–1 kg–1) and cellular potency (74 nM) but high ER (19). Interestingly, analog 21, on which a pyrazine ring had been introduced, was found to have a good balance of potency (cell IC50 = 138 nM) and ER (5.0) and its good microsomal stability (RLM, HLM = <11, 6 mL min–1 kg–1) was found to correlate well with a low in vivo clearance in a rat PK experiment (Cl/Clu = 0.36/6.7 L h–1 kg–1).
Table 2. In Vitro and in Vivo Data for Pyrazole Analogs 8 and 12–21.
![]()

Biochemical assay.
Inhibition of ASK1 autophosphorylation. IC50 results are the geometric mean of a minimum of two determinations.
MDCK-MDR1 human P-gp transfected cell line. Papp = apparent permeability, apical-to-basolateral (10–6 cm/s); ER = (B–A)/(A–B) efflux ratio.
Clu = Cl/fu.
DMSO/PG (1:1) used as vehicle. Vdss = 1.8 L/kg; t1/2 = 0.9 h.
DMA/EtOH/PG/water (1:1:3:5) used as vehicle. Vdss = 1.3 L/kg; t1/2 = 1.8 h.
DMA/EtOH/PG/water (1:1:3:5) used as vehicle. Vdss = 2.3 L/kg; t1/2 = 5.2 h.
To confirm the binding mode of compound 21, an X-ray cocrystal structure with human ASK1 was elucidated (Figure 3). As expected, the same H-bond interactions of the carbonyl group of the amide to the backbone NH of Val 757 and the triazole N to the catalytic Lys observed in 3 were also present in the cocrystal structure of analog 21 with ASK1. As predicted, an intramolecular H-bond between the methoxy group and the amide was also observed.20
Figure 3.
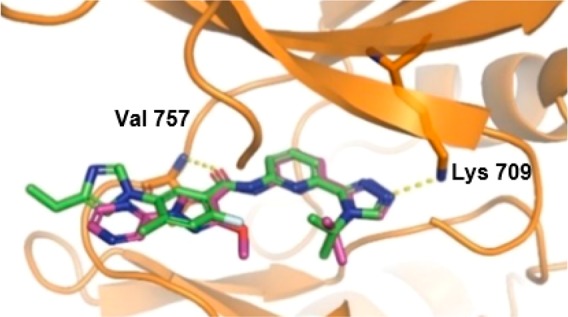
Overlay of the cocrystal structure of analogs 3 and 21 in the kinase domain (659–951) of human ASK1. Key interactions of the ligands with Val 757 and Lys 709 are represented with yellow dashes (PDB code 6VRE).
Compounds 11 and 21 displayed good potency and low ER and were selected for further in vivo characterization. Specifically, rat Kp,uu experiments (Table 3) revealed that both analogs had satisfactory CNS penetration (compound 11, Kp,uu = 0.61, compound 21, Kp,uu = 0.38).21 Further in vitro characterization showed that inhibitor 21 had superior CYP inhibition profile (relative to 11) and it was also inactive in a hERG assay (Table 3). On the basis of these data, analog 21 was selected for a kinase selectivity profile against a panel of 468 kinases, and it was found to be moderately selective (S-score (10) = 0.07).22
Table 3. Summary of Key in Vitro and in Vivo ADME Properties for Representative Compounds.
| analog | bioc IC50 (nM)a | cell IC50 (nM)b | RLM, HLM (mL min–1 kg–1) | MDCK-MDR1cPapp/ER | RPPB/HPPB (fu) | Cl/Clu (L h–1 kg–1)d | rat Kp,uue | CYP3A4/2C9,f IC50 (μM) | hERG IC50 (μM) |
|---|---|---|---|---|---|---|---|---|---|
| 8 | 3.9 | 90 | <11, 14 | 1.7/15 | 39/40 | 1.6/4.0 | >10/10 | 30 | |
| 11 | 2.4 | 93 | 72, 9 | 10/2.2 | 8/13 | 3.4/42 | 0.61 | 5.9/0.7 | 15 |
| 21 | 21 | 138 | <11, 6 | 5.0/5.0 | 5.3/5.4 | 0.36/6.7 | 0.38 | 8.8/>10 | >30 |
Biochemical assay.
Inhibition of ASK1 autophosphorylation. IC50 results are the geometric mean of a minimum of two determinations.
MDCK-MDR1 human P-gp transfected cell line. Papp = apparent permeability, apical-to-basolateral (10–6 cm/s); ER = (B–A)/(A–B) efflux ratio.
Clu=Cl/fu.
DMA/EtOH/PG/water (1:1:3:5) was used as the vehicle.
Midazolam and tolbutamide were used as substrates.
To predict the target coverage upon oral dosing of 21, a mouse PK experiment on which this compound was administered at 10 mg/kg (po) was also carried out (Figure 4). This experiment revealed that the projected dose to cover the cellular IC50, IC70, IC90 at trough in the brain for 12 h was 50, 115, 435 mg/kg, respectively, making this compound a suitable candidate for further in vivo studies.23
Figure 4.

Mouse PK experiment with analog 21 (10 mg/kg, po) and projected dose to cover the cellular IC50 in the brain for 12 h.
In summary, systematic optimization of the solvent-exposed substituent of a peripherally restricted ASK1 inhibitor led to the identification of the CNS penetrant analog 21. This compound was found to have good biochemical and cellular potency, low clearance, and good brain penetration in rodents, making it a suitable candidate for more advanced in vivo pharmacological studies.
Acknowledgments
We thank Kevin Barry for chiral separation support, Michael Dechantsreiter for parallel synthesis support, Sam Cziria for compound handling, Josh Johnson for support with collection of analytical data, and Jay Chodaparambil for X-ray support. We also thank Xiaoqing Wu, Xiaoping Wang, and Wenkui Zhao for efforts coordinating the synthesis of some of the compounds included in this manuscript and Janaky Coomaraswamy and Eric Tien for helpful discussions.
Glossary
Abbreviations
- ALS
amyotrophic lateral sclerosis
- ASK1
apoptosis signal-regulating kinase 1
- Cl
clearance
- CNS
central nervous system
- ER
efflux ratio
- HLM
human liver microsome
- HPPB
human plasma protein binding
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated kinase
- MAP3K
mitogen-activated kinases kinase kinase
- MDCK
Madin–Darby canine kidney
- MDR1
multidrug resistance 1
- MS
multiple sclerosis
- PG
propylene glycol
- P-gp
P-glycoprotein
- PK
pharmacokinetics
- RLM
rat liver microsome
- RPPB
rat plasma protein binding
- SOD1
superoxide dismutase 1
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00611.
(i) Experimental procedures for the synthesis of all the compounds described in this manuscript, (ii) experimental conditions for cocrystallization, collection, and refinement statistics for compound 21, and (iii) kinase selectivity profile for compound 21 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kawarazaki Y.; Ichijo H.; Naguro I. Apoptosis Signal-Regulating Kinase 1 as a Therapeutic Target. Expert Opin. Ther. Targets 2014, 18, 651–664. 10.1517/14728222.2014.896903. [DOI] [PubMed] [Google Scholar]
- Matsukawa J.; Matsuzawa A.; Takeda K.; Ichijo H. The ASK1-MAP Kinase Cascades in Mammalian Stress Response. J. Biochem. 2004, 136, 261–265. 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- Nishitoh H.; Kadowaki H.; Nagai A.; Maruyama T.; Yokota T.; Fukutomi H.; Noguchi T.; Matsuzawa A.; Takeda K.; Ichijo H. ALS-Linked Mutant SOD1 Induces ER Stress- and ASK1-Dependent Motor Neuron Death by Targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X.; Harada C.; Namekata K.; Matsuzawa A.; Camps M.; Ji H.; Swinnen D.; Jorand-Lebrun C.; Muzerelle M.; Vitte P.-A.; Rückle T.; Kimura A.; Kohyama K.; Matsumoto Y.; Ichijo H.; Harada T. Regulation of the Severity of Neuroinflammation and Demyelination by TLR-ASK1-p38 pathways. EMBO Mol. Med. 2010, 2, 504–515. 10.1002/emmm.201000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relative to their ASK1 wild type littermates.
- Fujisawa T.; Takahashi M.; Tsukamoto Y.; Yamaguchi N.; Nakoji M.; Endo M.; Kodaira H.; Hayashi Y.; Nishitoh H.; Naguro I.; Homma K.; Ichijo H. The ASK1-Specific Inhibitors K811 and K812 Prolong Survival in a Mouse Model of Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2016, 25 (2), 245–253. 10.1093/hmg/ddv467. [DOI] [PubMed] [Google Scholar]
- Fujisawa T. Therapeutic Application of Apoptosis Signal-Regulating Kinase 1 Inhibitors. Adv. Biol. Regul. 2017, 66, 85–90. and references therein 10.1016/j.jbior.2017.10.004. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov. Search result page. https://clinicaltrials.gov/ct2/results?cond=&term=NCT02781584+&cntry=&state=&city=&dist= (accessed 2020-2-11).
- Bansal R.; Nagorniewicz B.; Prakash J. Clinical Advancements in the Targeted Therapies Against Liver Fibrosis. Mediators Inflammation 2016, 2016, 1–26. 10.1155/2016/7629724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelbauer M. K.; Xin Z.; Jones J. H.; Enyedy I.; King K.; Marcotte D. J.; Murugan P.; Santoro J. C.; Hesson T.; Spilker K.; Johnson J. L.; Luzzio M. J.; Gilfillan R.; Gonzalez-Lopez de Turiso F. Rational Design and Optimization of a Novel Class of Macrocyclic Apoptosis Signal-Regulating Kinase 1 Inhibitors. J. Med. Chem. 2019, 62, 10740–10756. and references therein 10.1021/acs.jmedchem.9b01206. [DOI] [PubMed] [Google Scholar]
- The biochemical potency was determined at Reaction Biology Corporation using the assay conditions described in the Supporting Information of ref (10).
- This assay quantified the level of ASK1 phosphorylation at residue T848 (see also the Supporting Information of ref (10)).
- Prediction of the ability of these analogs to enter the CNS was performed using an in vitro MDCK-MDR1 assay as previously described. In these experiments, an MDCK NIH cell line transfected with MDR1 was used as an in vitro predictor of CNS penetration. Good correlation between the ER in this assay and brain exposure has been reported when using this cell line (see ref (14)).
- Kikuchi R.; de Morais S. M.; Kalvass J. C. In Vitro P-Glycoprotein Efflux Ratio Can Predict the In Vivo Brain Penetration Regardless of Biopharmaceutics Drug Disposition Classification System Class. Drug Metab. Dispos. 2013, 41 (12), 2012–2017. 10.1124/dmd.113.053868. [DOI] [PubMed] [Google Scholar]
- Pyrazole-containing ASK1 inhibitors have been recently reported in the literature although these compounds were not optimized for CNS-penetration (see ref (16)).
- Gibson T. S.; Johnson B.; Fanjul A.; Halkowycz P.; Dougan D. R.; Cole D.; Swann S. Structure-Based Drug Design of Novel ASK1 Inhibitors Using an Integrated Lead Optimization Strategy. Bioorg. Med. Chem. Lett. 2017, 27, 1709–1713. 10.1016/j.bmcl.2017.02.079. [DOI] [PubMed] [Google Scholar]
- All in vivo PK studies were conducted in accordance with Biogen Institutional Animal Care and Use Committee (IACUC) guidelines.
- This compound was synthesized as the racemate, and the enantiomers were separated using chiral SFC (the absolute stereochemistry of 9 was arbitrarily assigned). See Supporting Information.
- This finding, in combination with the difficulty associated with synthesizing these analogs, led us to the deprioritization of this approach.
- In addition to the highlighted interactions, the isopropyl group of 3 also interacts with the side chain of Val 694. The pyrazine ring forms a carbonyl−π interaction with the carbonyl of Gly 759 and the pyrazole ring stacks against Gly 760. A nonconventional CH–C=O hydrogen bond of the pyrazole with the hinge amino acid residue: Val757 can also be invoked.
- In these experiments the compounds were administered via an iv infusion (using DMA/EtOH/PG:water in a 1:1:3:5 ratio as the vehicle) in the carotid artery for a period of 4 h (1 mg/kg, 0.1 mg/mL) to reach steady state. After this time the plasma and brain concentration levels were quantified, and the values were adjusted by the measured protein binding in plasma and brain homogenate to calculate the Kp, uu.
- See Supporting Information.
- This dose compares favorably to that projected for compound 2 (internal in vitro and in vivo data suggest that the estimated dose to cover the cellular IC50 at trough in the brain for 12 h for compound 2 would be >100-fold higher relative to compound 21). Additional in vivo experiments with 21 and other ASK1 inhibitors will be reported in the future.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.