

Abstract
ChIP-Seq, a technique that allows for quantification of DNA sequences bound by transcription factors or histones, has been widely used to characterize genome-wide DNA-protein binding at baseline and induced by specific exposures. Integrating results of multiple ChIP-Seq datasets is a convenient approach to identify robust DNA- protein binding sites and determine their cell-type specificity. We developed brocade, a computational pipeline for reproducible analysis of publicly available ChIP-Seq data that creates R markdown reports containing information on datasets downloaded, quality control metrics, and differential binding results. Glucocorticoids are commonly used anti-inflammatory drugs with tissue-specific effects that are not fully understood. We demonstrate the utility of brocade via the analysis of five ChIP-Seq datasets involving glucocorticoid receptor (GR), a transcription factor that mediates glucocorticoid response, to identify cell type-specific and shared GR binding sites across the five cell types. Our results show that brocade facilitates analysis of individual ChIP-Seq datasets and comparative studies involving multiple datasets.
Introduction
Transcription factors play a key role in the regulation of gene expression and the re-organization of chromatin by binding to specific genomic loci in a cell type-specific manner1,2. Chromatin immunoprecipitation sequencing (ChIP-Seq) is a popular genomic approach to identify binding sites in the genome for a protein of interest3. The technique involves reverse cross-linking of a protein of interest to DNA, followed by fragmentation, immunoprecipitation and parallel sequencing, which permits identification of DNA sequences bound by transcription factors or histones across the genome4. ChIP-Seq has been widely used, with over 40,000 ChIP-Seq assays of various DNA-binding proteins available in public repositories, such as the Gene Expression Omnibus (GEO) and Sequence Read Archive (SØ)5. The availability of these datasets enables researchers to explore novel questions related to transcriptional response by comparing results of multiple ChIP-Seq datasets.
Analysis of ChIP-Seq data is a multi-step process that includes obtaining quality control measures, mapping of sequencing reads to a reference genome, peak calling, performing differential binding analysis, annotating sites, and identifying motifs6. Several specialized informatics tools have been developed to perform these tasks7. The ENCODE project produced many ChIP-Seq datasets along with published standards and guidelines that recommend use of biological replicates, specific antibody and input control characteristics, and uniform processing pipelines to analyze ChIP-Seq data of different protein classes8,9. For example, to determine whether transcription factor binding sites are associated with corresponding gene transcription, it is recommended that ChIP-Seq experiments include measures of RNA polymerase II (RNAP2) occupancy along with the transcription factor of interest10. In the absence of RNAP2 ChIP-Seq data, genome-wide gene expression results derived from microarray or RNA-Seq studies with similar treatment conditions as the ChIP-Seq study of interest can be used to link transcription factor binding sites with transcriptomic changes.
Software tools to analyze ChIP-Seq data include the R packages chipseq11 and CSAR12, which perform binding site identification based on aligned data obtained via command-line tools, while Homer13, seqMINER14, and Sole- Search15 are popular tools that focus solely on aspects of downstream analysis. Pipelines that combine existing informatics tools to perform comprehensive ChIP-Seq analyses include ChiLin16 and the ENCODE pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2), which consist of Python scripts that integrate steps for sequence data mapping, peak calling, and differential binding analysis (only available in ChiLin). The Galaxy-based platforms Cistrome17 and Nebula18 are similar cloud-based options, but their computational speed and customization of commands are comparatively limited. None of the existing pipelines, however, facilitate analysis of publicly available ChIP-Seq data via automated retrieval of raw sequencing files and phenotype information from public repositories. Thus, there is a need for an end-to-end ChIP-Seq data analysis pipeline that includes direct access to the growing resource of public datasets in a sophisticated yet straightforward manner.
Here, we present brocade, an efficient and customizable ChIP-Seq data analysis pipeline that can be used to analyze publicly available ChIP-Seq data. To demonstrate the utility of brocade, we applied it to study the tissue-specificity of the glucocorticoid receptor (GR). Glucocorticoids are anti-inflammatory drugs commonly used to treat diseases such as asthma19. At a cellular level, glucocorticoids act by diffusing across the cell membrane and binding to GRs that then translocate to cell nuclei and modulate transcription of various genes in a tissue-dependent fashion20, including the upregulation of anti-inflammatory genes21. Although glucocorticoids are known to directly modulate gene transcription via GR binding to glucocorticoid response elements (GREs) and other transcription factors, the cell-specificity of GR-mediated gene transcription is not fully understood. We used brocade to analyze five publicly available GR ChIP-Seq datasets corresponding to five cell types to identify cell type-specific direct transcription targets of the GR.
Methods
Reproducible Analysis of ChIP-Seq Data
We created brocade, a set of Python scripts that generate bash and R markdown scripts that invoke various functions to facilitate the nearly automated analysis of any ChIP-Seq dataset, with human decision-making required for key steps such as selection of relevant phenotype data (Figure 1). A high-performance computing (HPC) environment is required to run the pipeline efficiently; bash scripts generated by brocade are currently in Platform Load Sharing Facility (LSF) format, a common workload management platform used in IBM clusters. Bash scripts are submitted to an HPC environment to perform GEO/SRA file downloads, read alignment and mapping, peak calling, quality control and differential binding analysis. The data download step can be skipped if a user would like to analyze local ChIP-Seq files. R markdown scripts produced by brocade are used to generate three html reports that (1) describe publicly downloaded datasets, (2) provide quality control metrics, and (3) report differential binding analysis results. Full instructions and code are available on GitHub at https://github.com/HimesGroup/brocade (individual Pythons scripts are in the pipeline_scripts/ directory; R markdown template files are in the template_files/ directory). We keep separate Python scripts for each of these steps rather than consolidating them into a master script because user input is required at key points during analysis. Additionally, separating steps allows users to more easily understand and customize scripts to suit their needs.
Figure 1.
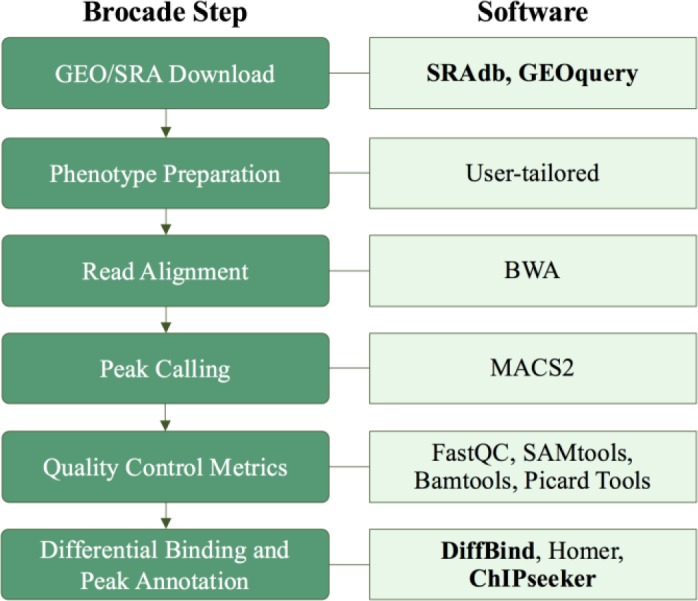
Steps followed by brocade to analyze ChIP-Seq data along with corresponding R packages (in bold) and other software used.
Publicly Available Data Download and Phenotype Preparation. Brocade downloads ChIP-Seq raw sequence data in fastq format by retrieving files from an ftp address corresponding to a user-provided GEO accession number using the SRAdb R package. A phenotype file is generated based on information extracted from the phenoData object of the GSE Matrix file that users must confirm is appropriate for desired analyses. Alternatively, users can provide their own fastq and phenotype files.
Read Alignment, Peak Calling, and Quality Control. Adapters, if specified, are trimmed using Trimmomatic22 and overall quality control metrics of fastq files are obtained using FastQC23. Trimmed reads are aligned to the reference genome (currently hg38) using BWA24. Bamtools25 is used to count and summarize the number of mapped reads, while Picard Tools25 is used to compute the insert size of paired-end libraries. Aligned read files are converted to bigwig format, which can be uploaded to the UCSC Genome Brower for peak visualization. DNA-protein binding sites are identified using MACS226 where broad or narrow peak calling is assigned to histone modifications as needed.
Differential Binding and Peak Annotation. Differential binding analysis is performed with the R package DiffBind27, which has built-in functions to retrieve read counts by Rsamtools and GenomicAlignments, and uses DESeq2 to compare binding differences between conditions of interest (e.g. treatment vs. control vehicle)28. The Benjamini- Hochberg method is used to adjust for multiple comparisons and calculate adjusted p-values (q-values). Binding sites are annotated with ChIPseeker29 and binding motif identification is performed with Homer13 and visualized with the R package seqLogo.
Primary GR-Mediated Transcriptomic Changes
ChIP-Seq Datasets. Publicly available ChIP-Seq datasets that measured GR-binding induced by glucocorticoid exposure were obtained from GEO/SRA by searching for the term “glucocorticoid receptor” with Homo sapiens selected as organism. To identify genes with active transcription in/near GR binding sites, we utilized RNAP2 ChIP- Seq datasets provided with GR ChIP-Seq datasets when possible. For GR ChIP-Seq datasets with no available RNAP2 ChIP-Seq profiles, we sought microarray datasets obtained for equivalent cell types under similar treatment conditions in GEO. We utilized brocade to analyze the GR and RNAP2 ChIP-Seq datasets and identify differential binding sites for samples exposed to glucocorticoids vs. control. GR and RNAP2 binding sites were considered to be significantly changed by glucocorticoids if q-values were <0.05. Differential gene expression results for microarray samples were obtained using RAVED (https://github.com/HimesGroup/raved)30. Genes were considered to be significantly differentially expressed with glucocorticoid exposure if q-values were <0.05.
Identification of Shared and Cell Type-Specific Primary GR Target Genes. We defined primary GR target genes as those genes with a GR-binding site within ±20 kb distance of the gene’s transcription start site (TSS), while also having either (1) an RNAP2-binding site within ±3kb of the gene’s TSS (i.e. promoter region), or (2) a significant differential expression result in the corresponding microarray dataset. We identified shared and cell type-specific primary GR target genes by comparing results obtained across all available cell types.
Results
Overview of Brocade Reports
Brocade output html reports include quality control checks and a summary of annotated binding sites for each study of interest. The quality control report has tables and plots illustrating the summary read counts, percentage of mapped and unmapped reads (Figure 2A) and a principal component analysis (PCA) plot based on mapped reads per sample. The differential binding report provides volcano plots (Figure 2B), a PCA plot based on differential binding results, heatmaps, and boxplots of log2 normalized counts (Figure 2C) for significant binding sites identified (i.e., those with q-value <0.05). The report includes select plots from the R package ChIPseeker: frequency of read counts within ±3 kb of the TSS (Figure 2D) and distribution of binding site distance relative to TSS (Figure 2E), as well as sequence logos of top motifs (Figure 2F).
Figure 2.

Sample figures included in brocade quality control and differential binding analysis reports.
Publicly Available ChIP-Seq Datasets
Our GEO/SRA search yielded glucocorticoid response ChIP-Seq datasets with dexamethasone exposure for five cell types: airway smooth muscle, bronchial epithelial (Beas-2B), adenocarcinomic alveolar basal epithelial (A549), acute lymphoblastic leukemia (RS4;11) and lymphoblastoid (Table 1). RNAP2 ChIP-Seq profiles were available for the three structural cell types (i.e., airway smooth muscle31, Beas-2B32 and A54933). We used gene expression microarray datasets GSE71615 (10nM dexamethasone, 24 hr) and GSE44248 (1000nM dexamethasone, 8 hr) for the RS4;1134 and lymphoblastoid cells35, respectively. Following analysis of ChIP-Seq datasets with brocade, we retained all significant differential binding sites (q-value < 0.05) for the three structural cell types. For the RS4;11 and lymphoblastoid cell datasets, we retained glucocorticoid-induced GR-binding sites with log2-fold change ≥1, as the RS4;11 dataset lacked replicates and no significant GR-binding sites (q-value < 0.05) were identified for the lymphoblastoid cell line. Significant genes (q-value <0.05) from the microarray differential expression analysis were also retained.
Table 1.
ChIP-Seq datasets selected to study cell-specific GR-binding differences.
| GEO ID | Cell Type | Control Treatment | Dexamethasone Treatment | GR | RNAP2 | ||
|---|---|---|---|---|---|---|---|
| Control Samples (N) | Treated Samples (N) | Control Samples (N) | Treated Samples (N) | ||||
| GSE95632 | Airway smooth muscle | Ethanol, 1 hr | 100 nM, 1 hr | 2 | 2 | 2 | 2 |
| GSE79803 | Beas-2B, airway epithelial cells | Ethanol, 1 hr | 100 nM, 1 hr | 2 | 2 | 2 | 2 |
| SRP000762 | A549, adenocarcinomic alveolar epithelial cells | Ethanol, 1 hr | 100 nM, 1 hr | 2 | 2 | 2 | 2 |
| GSE71616 | RS4;11, acute lymphoblastic leukemia | None | 10 nM, 1 hr | 1 | 1 | - | - |
| GSE45638 | lymphoblastoid cell lines | Ethanol | 1000 nM, 1 hr | 2 | 2 | - | - |
ChIP-Seq Results
We found 39,328 differential GR-binding sites for airway smooth muscle, 20,620 for Beas-2B, 7,818 for A549, 5,113 for RS4;11 and 999 for lymphoblastoid cell line, which corresponded to 6,421, 4,130, 2,297, 1,391, and 137 gene targets, respectively. Most differential GR-binding sites lied beyond 20kb of TSSs (Figure 3A), with a correspondingly large proportion of GR-binding sites located in intronic and distal intergenic regions (Figure 3B). Compared to the large number of GR-binding sites, only 3,220, 1,845, and 2,173 differential RNAP2-binding sites were identified for airway smooth muscle, Beas-2B, and A549, respectively. 8,449 differentially expressed genes for RS4;11, and 262 for lymphoblastoid cell line were identified. Thus, the number of primary GR target genes, based on overlap of GR-binding-associated genes with evidence of active transcription (i.e., RNAP2 bound genes or differentially expressed genes obtained via microarray), was 710 for airway smooth muscle, 405 for Beas-2B, 249 for A549, 685 for RS4;11, and 1 for lymphoblastoid cell line (Figure 3C).
Figure 3.

Characteristics of GR-binding sites for each cell type. (A) Number of GR binding sites within and beyond 20kb of TSS of annotated genes. (B) Proportion of GR binding sites according to type of genomic region. (C) Genes with GR-binding that have (green) vs. do not have (blue) active transcription in response to glucocorticoid exposure as determined by RNAP2 or microarray results. ASM: airway smooth muscle; Beas-2B: airway epithelial cells; A549: adenocarcinomic alveolar epithelial cells; RS4;11: acute lymphoblastic leukemia cells; LCL: lymphoblastoid cell lines.
Shared and Cell Type-Specific Primary GR Target Genes
The majority of primary GR target genes were cell type-specific, consistent with our previous transcriptomic findings for genes that were differentially expressed in response to glucocorticoid exposure (Figure 4)30. Because the lymphoblastoid cells only had one primary GR target gene (C16orf87) that did not overlap with the primary GR target genes of any of the other cell types, its results were excluded from Figure 4. Twelve primary GR gene targets, (i.e., FKBP5, MAP3K6, ST3GAL4, IRAK3, NFKBIA, IER2, STX10, ZFP36, TIPARP, IGF2BP3, NBN, DECR1) were shared across airway smooth muscle, Beas-2B, A549 and RS4;11, which included the well-known glucocorticoid- responsive gene FKBP536 and IRAK3, a gene that is highly expressed in response to glucocorticoid exposure in airway smooth muscle36,37 and whose variants have been associated with asthma in European populations38, 39. Thirty-eight additional genes were shared across structural cells, including the known glucocorticoid-responsive genes PER136,37, CEBPD36,37 and DUSP136, 37, 40. Although Beas-2B and A549 originate from bronchial epithelium, Beas-2B and airway smooth muscle shared more primary GR target genes than Beas-2B and A549 (195 vs. 90).
Figure 4.
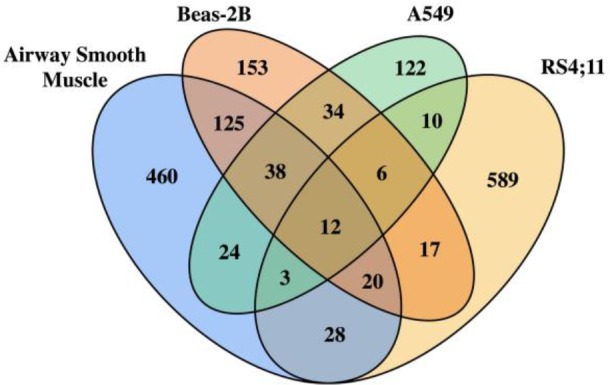
Overlap of primary GR target genes among four cell types.
To confirm shared and cell type-specific results of some primary GR target genes at the level of mapped reads, we checked plots of bigwig files created by brocade. Specifically, we confirmed that there was differential GR-binding induced by glucocorticoids in three cell types for FKBP5 and CRISPLD2, a gene known to be differentially expressed in response to glucocorticoid exposure in airway smooth muscle.36,37 FKBP5 had 4 significant differential GR-binding sites in airway smooth muscle, 6 in Beas-2B, and 6 in A549 (Figure 5A), while CRISPLD2, had 5 in airway smooth muscle, 4 in Beas-2B, and one in A549 (Figure 5B). Differential RNAP2-binding sites for FKBP5 were observed in all cell types (Figure 5A), whereas for CRISPLD2, they were only observed in airway smooth muscle cells after glucocorticoid exposure (Figure 5B). To confirm that ChIP-Seq results were consistent with transcriptomic results more broadly, we obtained gene expression data for each gene available in our web application REALGAR41, and confirmed that FKBP5 was differentially expressed with glucocorticoid exposure vs. control in all three cell types, while CRISPLD2 was differentially expressed only in airway smooth muscle.
Figure 5.

Differential GR- and RNAP2-binding sites near genes (A) FKBP5 and (B) CRISPLD2 in airway smooth muscle, Beas-2B, and A549 cells treated with dexamethasone (green) and control vehicle (orange). Asterisk (*) indicates most significant GR or RNAP2 binding site.
Discussion
With the growing interest in leveraging publicly available data, we present brocade, a pipeline that facilitates automated ChIP-Seq data analysis spanning data download from GEO/SRA to differential binding site identification and annotation. As integration of multiple datasets relies on having accurate and consistently obtained results for individual studies, brocade was designed to streamline and standardize ChIP-Seq data analysis. It produces well- documented yet concise html reports that are easily customizable to allow users to include alternative methods. Although brocade is nearly automatic, researchers are required to manually inspect quality control metrics and plots to identify sample outliers and define phenotypes of interest prior to performing differential binding analyses. To maximize use of all publicly available ChIP-Seq datasets, brocade can process standard ChIP-Seq experimental designs that include input DNA and biological replicates to control for genomic artifacts and individual sample variation, respectively3, but also datasets that lack such controls.
There is no consensus on how to assign a specific GR-binding site to a potential gene target on a genomic scale, especially when GR-binding occurs distant to TSS regions. The majority of GR-binding sites have been found to be outside of promoter regions but enriched within 100kb of glucocorticoid-responsive genes42. We restricted our definition of GR-binding sites to include those within 20kb of a target gene’s TSS, a plausible distance that is used in many studies31,32 and one supported by the fact that among the hundreds of primary target genes we identified, many were well-known glucocorticoid-responsive genes. However, we may have excluded GR bindings that occur in distal enhancers, as the ability to accurately link distal binding events to transcriptional regulation is limited42.
Previous studies have compared protein-DNA binding regions across cell types to identify shared and unique transcription factor binding sites1,43. However, transcription factor binding events alone do not guarantee their role in mediating gene transcription. To identify GR-binding sites that were more likely to mediate glucocorticoid- induced gene transcription, we combined GR-binding site data with evidence of gene transcription provided by RNAP2-binding or differential gene expression results. Previous studies identified glucocorticoid-responsive genes such as FKBP5 across multiple cell types, and PER1 and CRISPLD2 as specific to airway smooth muscle36,37. Our results found that the transcriptional response of CRISPLD2 to glucocorticoids occurs via promoter-proximal GR binding and is specific to airway smooth muscle, but these cell type-specific findings require further experimental validation. Future studies could compare DNA sequences of the GR-binding sites identified in CRISPLD2 or other genes to determine whether shared and cell type-specific transcriptomic changes involve direct GR binding to GREs or GR-tethering to transcription factors.
Ideally, ChIP-Seq experiments include controls such as RNAP2 ChIP-Seq that help to determine whether transcriptional response accompanies transcription factor binding, but some ChIP-Seq studies use gene expression changes measured by microarray or RNA-Seq as a proxy for changes in transcriptomic activity10. Our results were limited by the design of individual experiments, including different assays used to infer active transcription of genes (i.e., RNAP2 ChIP-Seq for airway smooth muscle, Beas-2B, and A549; microarray for RS4;11 and lymphoblastoid cell line), which likely introduced bias. Other limitations when comparing results across cell types are the heterogeneous dosages of dexamethasone administered, and the fact that one dataset lacked any replicates, which decreases the reliability of its results. Nonetheless, comparison of the GR ChIP-Seq datasets yielded promising results that stand to improve as more publicly available data is generated.
Future versions of brocade will include automated approaches to identify and process publicly available datasets using phenotypes and meta-data extracted from GEO/SRA entries and linked publications to increase its automaticity. For example, for datasets without RNAP2 ChIP-Seq, an automated search through GEO/SRA could identify an appropriately matched gene expression dataset. Future versions of brocade will also include analysis of more recent DNA-protein binding techniques such as ATAC-Seq, DNase-Seq, and STARR-Seq, as these become more widely used.
Conclusion
We developed brocade, a reproducible ChIP-Seq analysis pipeline, that facilitates analysis of publicly available datasets in GEO/SRA. We applied brocade to explore the cell-type specificity of primary GR target genes in five cell types (i.e., airway smooth muscle, Beas-2B, A549, RS4;11, lymphoblastoid). Most primary GR target genes showed cell type-specific activity (e.g., CRISPLD2), but twelve genes were shared across four cell types (e.g., FKBP5). These results indicate potential cell-specific mechanisms of glucocorticoid action that can be explored in future studies.
Figures & Table
References
- 1.Gertz J, Savic D, Varley KE, Partridge EC, Safi A, Jain P, et al. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol Cell. 2013;52((1)):25–36. doi: 10.1016/j.molcel.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie D, Boyle AP, Wu L, Zhai J, Kawli T, Snyder M. Dynamic trans-acting factor colocalization in human cells. Cell. 2013;155((3)):713–24. doi: 10.1016/j.cell.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10((10)):669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13((12)):840–52. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clough E, Barrett T. The Gene Expression Omnibus Database. Methods Mol Biol. 2016;1418:93–110. doi: 10.1007/978-1-4939-3578-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, et al. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput Biol. 2013;9((11)):e1003326. doi: 10.1371/journal.pcbi.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shin H, Liu T, Duan X, Zhang Y, Liu XS. Computational methodology for ChIP-seq analysis. Quant Biol. 2013;1((1)):54–70. doi: 10.1007/s40484-013-0006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489((7414)):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22((9)):1813–31. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mokry M, Hatzis P, Schuijers J, Lansu N, Ruzius FP, Clevers H, et al. Integrated genome-wide analysis of transcription factor occupancy, RNA polymerase II binding and steady-state RNA levels identify differentially regulated functional gene classes. Nucleic Acids Res. 2012;40((1)):148–58. doi: 10.1093/nar/gkr720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarkar D, Gentleman R, Lawrence M, Yao Z. chipseq: chipseq: A package for analyzing chipseq data. R package version 1.32.0. [Google Scholar]
- 12.Muino JM, Kaufmann K, van Ham RC, Angenent GC, Krajewski P. ChIP-seq Analysis in R (CSAR): An R package for the statistical detection of protein-bound genomic regions. Plant Methods. 2011:7–11. doi: 10.1186/1746-4811-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage- determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38((4)):576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye T, Ravens S, Krebs AR, Tora L. Interpreting and visualizing ChIP-seq data with the seqMINER software. Methods Mol Biol. 2014;1150:141–52. doi: 10.1007/978-1-4939-0512-6_8. [DOI] [PubMed] [Google Scholar]
- 15.Blahnik KR, Dou L, O’Geen H, McPhillips T, Xu X, Cao AR, et al. Sole-Search: an integrated analysis program for peak detection and functional annotation using ChIP-seq data. Nucleic Acids Res. 2010;38((3)):e13. doi: 10.1093/nar/gkp1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, et al. ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics. 2016;17((1)):404. doi: 10.1186/s12859-016-1274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12((8)):R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boeva V, Lermine A, Barette C, Guillouf C, Barillot E. Nebula--a web-server for advanced ChIP-seq data analysis. Bioinformatics. 2012;28((19)):2517–9. doi: 10.1093/bioinformatics/bts463. [DOI] [PubMed] [Google Scholar]
- 19.Fanta CH. Asthma. N Engl J Med. 2009;360((10)):1002–14. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- 20.Pratt WB, Morishima Y, Murphy M, Harrell M. Chaperoning of glucocorticoid receptors. Handb Exp Pharmacol. 2006;(172):111–38. doi: 10.1007/3-540-29717-0_5. [DOI] [PubMed] [Google Scholar]
- 21.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 1998;94((6)):557–72. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 22.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30((15)):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrews S. FastQC A Quality Control tool for High Throughput Sequence Data. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25((14)):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27((12)):1691–2. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP- Seq (MACS) Genome Biol. 2008;9((9)):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark R, Brown GD. DiffBind: differential binding analysis of ChIP-seq peak data. Bioconductor. http://bioconductor.org/packages/release/bioc/html/DiffBind.html. [Google Scholar]
- 28.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15((12)):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31((14)):2382–3. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 30.Kan M, Shumyatcher M, Diwadkar A, Soliman G, Himes BE. Integration of Transcriptomic Data Identifies Global and Cell-Specific Asthma-Related Gene Expression Signatures. AMIA Annu Symp Proc. 2018;2018:1338–47. [PMC free article] [PubMed] [Google Scholar]
- 31.Sasse SK, Kadiyala V, Danhorn T, Panettieri RA, Jr., Phang TL, Gerber AN. Glucocorticoid Receptor ChIP-Seq Identifies PLCD1 as a KLF15 Target that Represses Airway Smooth Muscle Hypertrophy. Am J Respir Cell Mol Biol. 2017;57((2)):226–37. doi: 10.1165/rcmb.2016-0357OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadiyala V, Sasse SK, Altonsy MO, Berman R, Chu HW, Phang TL, et al. Cistrome-based Cooperation between Airway Epithelial Glucocorticoid Receptor and NF-kappaB Orchestrates Anti-inflammatory Effects. J Biol Chem. 2016;291((24)):12673–87. doi: 10.1074/jbc.M116.721217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19((12)):2163–71. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu JN, Pinello L, Yissachar E, Wischhusen JW, Yuan GC, Roberts CWM. Functionally distinct patterns of nucleosome remodeling at enhancers in glucocorticoid-treated acute lymphoblastic leukemia. Epigenetics Chromatin. 2015:8–53. doi: 10.1186/s13072-015-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luca F, Maranville JC, Richards AL, Witonsky DB, Stephens M, Di Rienzo A. Genetic, functional and molecular features of glucocorticoid receptor binding. PLoS One. 2013;8((4)):e61654. doi: 10.1371/journal.pone.0061654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kan M, Koziol-White C, Shumyatcher M, Johnson M, Jester W, Panettieri RA, Jr., et al. Airway Smooth Muscle-Specific Transcriptomic Signatures of Glucocorticoid Exposure. Am J Respir Cell Mol Biol. 2019;61((1)):110–20. doi: 10.1165/rcmb.2018-0385OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B, et al. RNA-Seq transcriptome profiling identifies CRISPLD2 as a glucocorticoid responsive gene that modulates cytokine function in airway smooth muscle cells. PLoS One. 2014;9((6)):e99625. doi: 10.1371/journal.pone.0099625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balaci L, Spada MC, Olla N, Sole G, Loddo L, Anedda F, et al. IRAK-M is involved in the pathogenesis of early-onset persistent asthma. Am J Hum Genet. 2007;80((6)):1103–14. doi: 10.1086/518259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pino-Yanes M, Sanchez-Machin I, Cumplido J, Figueroa J, Torres-Galvan MJ, Gonzalez R, et al. IL-1 receptor-associated kinase 3 gene (IRAK3) variants associate with asthma in a replication study in the Spanish population. J Allergy Clin Immunol. 2012;129((2)):573–5. doi: 10.1016/j.jaci.2011.10.001. 5 e1-10. [DOI] [PubMed] [Google Scholar]
- 40.Shipp LE, Lee JV, Yu CY, Pufall M, Zhang P, Scott DK, et al. Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS One. 2010;5((10)):e13754. doi: 10.1371/journal.pone.0013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shumyatcher M, Hong R, Levin J, Himes BE. Disease-Specific Integration of Omics Data to Guide Functional Validation of Genetic Associations. AMIA Annu Symp Proc. 2017;2017:1589–96. [PMC free article] [PubMed] [Google Scholar]
- 42.Burd CJ, Archer TK. Chromatin architecture defines the glucocorticoid response. Mol Cell Endocrinol. 2013;380((1-2)):25–31. doi: 10.1016/j.mce.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Polman JA, Welten JE, Bosch DS, de Jonge RT, Balog J, van der Maarel SM, et al. A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci. 2012:13–118. doi: 10.1186/1471-2202-13-118. [DOI] [PMC free article] [PubMed] [Google Scholar]