

Abstract

The oxidative stress response, gated by the protein–protein interaction of KEAP1 and NRF2, has garnered significant interest in the past decade. Misregulation in this pathway has been implicated in disease states such as multiple sclerosis, rheumatoid arthritis, and diabetic chronic wounds. Many of the known activators of NRF2 are electrophilic in nature and may operate through several biological pathways rather than solely through the activation of the oxidative stress response. Recently, our lab has reported a nonelectrophilic, monoacidic, naphthalene-based NRF2 activator which exhibited good potency in vitro. Herein, we report a detailed structure–activity relationship of naphthalene-based NRF2 activators, an X-ray crystal structure of our monoacidic KEAP1 inhibitor, and identification of an underexplored area of the NRF2 binding pocket of KEAP1.
Keywords: KEAP1, NRF2, protein−protein interaction, oxidative stress
Chronic oxidative stress is implicated in a number of disease states, such as chronic obstructive pulmonary disease (COPD), multiple sclerosis, diabetic chronic wounds, and chronic kidney disease.1−6 Upregulating cellular defenses against oxidative stress may be a viable pathway for treatment or management of such diseases.7−9 NRF2 (nuclear factor (erythroid-derived 2)-like 2), a basic leucine zipper protein, regulates transcription of many antioxidant proteins. This oxidative stress response is gated primarily by the protein KEAP1 (Kelch-like ECH-associated protein 1), which sequesters NRF2 and, through a multiprotein assembly, polyubiquitinates it, marking it for proteosomal degradation.10 If the KEAP1-NRF2 protein–protein interaction is inhibited, NRF2 can no longer be sequestered and tagged for degradation. Inhibiting KEAP1 in this manner allows cytoplasmic NRF2 concentrations to increase, translocate into the nucleus, and promote the transcription of genes associated with the antioxidant response, such as NADPH quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), and glutamate cysteine ligases-C and -M (Figure 1).10−14
Figure 1.

Top: KEAP1-NRF2 interaction under basal conditions. Bottom: Mechanism of NRF2 via electrophilic and nonelectrophilic pathways.
The KEAP1-NRF2 interaction is inhibited in the presence of electrophiles, reactive oxygen species, or reactive nitrogen species, leading to a cytoprotective response in the cell.15 Some therapies that inhibit the KEAP1-NRF2 interaction utilize KEAP1’s sensitivity to electrophiles to increase cellular NRF2 levels. Some electrophiles may be promiscuous binders, and their lack of selectivity may make identification of mechanism of action more challenging.16,17
There have been multiple reports in recent years of nonelectrophilic KEAP1-NRF2 inhibitors with significant structural diversity, including various small molecules (1a–1j) and peptides (1k) (Chart 1). Most of these molecules possess anionic character at physiological pH. Due to the relative ease of modifying compounds such as naphthalene 1a, we and others have developed an SAR of these compounds via scaffold-hopping approaches and modification to the flanking benzenesulfonamide arms; however, comparatively little investigation has been done to probe variations in the regions that link the naphthalene core to the benzensulfonamides.20,28 In this Letter, we present structural modifications, informed by a crystal structure of monoacid inhibitor 1c (Figure 2), that provide valuable insights into the key interactions governing the potency and binding affinities of these 1,4-disubstituted naphthalene inhibitors.
Chart 1. Representative Examples of Known KEAP1 Inhibitors18−27.

Figure 2.

Structure of KEAP1 Kelch domain bound to compound 1c. (A, B) Diagram of interactions between KEAP1 Kelch residues (depicted as violet circles) and compound 1c. Of the four KEAP1 Kelch:1c complexes crystallized in the asymmetric unit, two subunits contain a formate ion (FMT, shown in teal) within hydrogen bonding distance of 1c (A) and two subunits contain a water molecule (B). 2fo – fc electron density of 1c and formate (A) and 1c and bridging water (B) is shown in blue mesh contoured at 1σ. (C) Superposition of KEAP1 Kelch:1c complex with the structures of KEAP1 bound to two other naphthalene-based compounds (1d, orange; 1e, teal) previously reported in the literature. Associated PDB codes (6V6Z, 4XMB, 4ZY3) are shown at right. Amino acids in close proximity to bound ligands are labeled on the protein surface.
Previously, we were unable to obtain a suitable cocrystal structure of 1c with the KEAP1 Kelch domain, so we analyzed the potential binding mode of monoacidic inhibitor 1cin silico.20 Docking experiments predicted that the carboxylate would likely interact with R483 and R415. We have now achieved success in cocrystallization of monoacidic inhibitor 1c with the Kelch domain of KEAP1 from a sodium formate solution. The cocrystal structure that we obtained contained a unit cell comprised of four Kelch domains, each possessing 1c in slightly different orientations. Two Kelch domains contained a formate ion interacting with the unsubstituted sulfonamide, while the remaining two displayed water molecules in this position. While these two variations contained slightly different orientations, the overall interactions between 1c and the Kelch domain remained similar (Figure 2A and Figure 2B). Interestingly, we observed that the carboxymethyl functionality is engaged in a hydrogen bond network and dipolar interactions with R415, N414, N382, S363, and a water molecule, which was contradictory with our docking experiments, which showed interactions with R415 and R483.20 In the crystal structure, key interactions appear to be made between the sulfonamide oxygen atoms and S363, S508, Y525, S555, S602 and two water molecules. With these data in hand, we set forth to determine which of these interactions are critical for inhibitor binding.
Our investigation into the structure–activity relationship began by probing the necessity of the bis-sulfonamide motif. In previous work, we showed that monoacidic analogs of 1b still maintained significant potency; however, no dicarboxylmonosulfonamide analogs were synthesized. We sought to determine if protein binding was driven more by the sulfonamide oxygens or the interactions of the carboxylates. A series of these monosulfonamide compounds were easily accessed through a Heck reaction of 1-bromo-4-aminonaphthalene (2) and subsequent derivatization (Scheme 1). Although these monosulfonamide compounds are structurally similar to reported monosulfonamide compound RA-839 (1f), none of these compounds retained nanomolar affinities for the Kelch-domain of KEAP1, as determined by fluorescence anisotropy (FA).21,29 This difference in affinity may be due to the rigidity of the acid in RA-839 versus the more freely rotating carboxylate groups in 3 and 4. Even though compounds 5 and 6 contained an electrophilic α,β-unsaturated carbonyl, the activity of these compounds in the FA assay would be solely dependent on nonelectrophilic inhibition since we used a truncated version of the KEAP1 protein which does not contain the previously mentioned electrophile-reactive cysteine residues.
Scheme 1. Synthesis of Monosulfonamide Analogs of 1b and 1c.

(a) Pd(OAc)2, P(o-tolyl)3, K2CO3, ethyl acrylate, dioxane, 100 °C 16 h; (b) 4-methoxybenzenesulfonyl chloride, pyridine; (c) ethyl bromoacetate, K2CO3, MeCN, rt, 16 h; (d) 15% NaOH(aq), MeOH, rt, 4 h; (e) 10 wt % Pd/C, H2 (40 psi), EtOH, rt.
Next, we sought to determine the extent to which the affinity of these compounds was driven by interactions stemming from the aryl groups appended to the sulfonamides. Alkynylsulfonamide 9 was obtained via a Sonogashira coupling of 1-bromo-4-aminonaphthalene (2) and 4-ethynylanisole, followed by sulfonylation of the amine. Hydrogenation of the alkyne yielded the saturated analog 10, and subsequent alkylation of the sulfonamide and saponification of the resulting ester produced phenethyl derivative 11 (Scheme 2). The affinity of this compound was determined by an FA assay, and it displayed reduced affinity for the Kelch-domain of KEAP1, which indicated that the benzenesulfonamide groups may be a major contributor to the activities of these compounds.
Scheme 2. Synthesis of Phenethyl Analog 11.

(a) Pd(PPh3)4, CuI, 4-ethynylanisole, NEt3, DMF, 80 °C, 20 h; (b) 4-methoxybenzenesulfonyl chloride, pyridine, rt, 18 h; (c) 5 wt % Pd/C, H2 (40 psi), EtOAc, rt, 18 h; (d) ethyl bromoacetate, K2CO3, MeCN, rt, 18 h; (e) 15% NaOH(aq), MeOH, rt, 5 h.
To test the hypothesis of the sulfonamide groups’ key interactions, we synthesized analog 12, in which a sulfone rather than a sulfonamide moiety was installed. Synthesis of these analogs began with a nitration of 1-methylnaphthalene to yield 1-nitro-4-methylnaphthalene 13. Benzylic bromination with azabisisobutyronitrile/N-bromosuccinimide, followed by substitution with 4-methoxybenzenethiol, yielded the thioether, which was oxidized with H2O2 to the corresponding sulfone 14. Reduction of the nitro group and sulfonylation produced the sulfone analog of 1a, which then underwent alkylation and saponification to yield the desired monoacidic sulfone analog 12 (Scheme 3).
Scheme 3. Synthesis of Sulfone Analog 12.

(a) N-Bromosuccinimide, azabisisobutyronitrile, MeCN, rt, 6 h; (b) 4-methoxybenzenethiol, 1 M NaOH(aq), dioxane, rt, 18 h; (c) 30% H2O2, AcOH, Ac2O, rt, 5 h; (d) 10 wt % Pd/C, H2 (40 psi), EtOAc; (e) 4-methyoxybenzenesulfonamide, pyridine, rt, 18 h; (f) ethyl bromoacetate, K2CO3, MeCN, rt, 18 h; (g) 15% NaOH(aq), MeOH, rt, 5 h.
Additionally, we attempted to synthesize analogs in which the carboxymethyl group was attached to the carbon α to the sulfone and where both sulfonamides were replaced with sulfones; however, we were unable to successfully isolate the desired products due to β-elimination of a sulfinate to give α,β-unsaturated compounds. Comparison of intermediates 15 and 16 to bis-sulfonamide 1a showed that monosulfone 15 had comparable activity to 1a, yet bis-sulfone 16 was inactive (Chart 2). When the sulfone monoacid 12 and diamino monoacid 1c were compared, sulfone monoacid 12 showed about a 10-fold drop in affinity. This loss of binding affinity may be due to an unfavorable increase of hydrophobicity in the Arg-rich area of the binding pocket.
Chart 2. Sulfone Analogs of Compounds 1a and 1c.

We were also interested in probing the ability of the binding pocket to accommodate substitutions α to the carboxylate group. Recently, a report by Lu et al. outlined an exploration of various α substituents based on amino acid side chains producing compounds of structure 17 (Chart 3).28 Due to the lack of activity of our monosulfonamide analogs, we opted to explore the activity of bis-sulfonamide compounds with structure 18. We hypothesized that the more congested nature of the substituents in 18 may constrain the three-dimensional structure, leading to enhanced interactions and binding affinity compared to the less constrained compounds with structure 17.
Chart 3. Structural Variations Explored by Lu et al. vs This Work.

Not only would α-substituents potentially lead to the development of an SAR for an underexplored area of these molecules, substitution in this position introduces chirality into this framework. Due to 12 being the best inhibitor in the series, we synthesized methyl analog 19 and phenyl analog 20 by substituting ethyl bromoacetate with ethyl-2-bromopropionate and benzyl-2-bromo-2-phenylacetate, respectively, for the alkylation step (Chart 4). Use of benzyl-2-bromo-2-phenylacetate was required because basic saponification of the α-phenylcarbonyl resulted in decomposition of the material.
Chart 4. Propionate and Phenylacetic Acid Analogs of 1c and 12.

The addition of an R group α to the carbonyl did indeed constrain the orientation of the substituents; the constraint is evident from this modification due to the formation of atropisomers, which displayed a 2:1 ratio of the major vs minor conformer. A variable temperature NMR experiment, from 20 to 70 °C, was performed in an effort to observe the interconversion of the atropisomers; however, no substantial shift or coalescence of the signals was observed. Attempts to separate the atropisomers were unsuccessful. Interestingly, the addition of the methyl group increased binding affinity of sulfone 19 5-fold compared to the unsubstituted sulfone 12, while addition of the phenyl ring severely diminished the activity. These results can be rationalized by examination of the crystal structure of 1c (Figure 1c), in which there is a small hydrophobic pocket where the methyl group may fit. It is likely that this pocket is unable to accommodate much larger substituents, like phenyl. When comparing the α-methyl compound 21 and unsubstituted monoacid 1c, we observe a 2-fold reduction in binding affinity. The reason for this slight decrease may be due to the methyl group inducing a less favorable binding pose for this series of compounds.
Because of the relatively high binding affinity of racemic propionate 19, we moved to determine whether the individual enantiomers of 19 displayed a preference for the binding pocket. To synthesize each of the enantiomers, it was necessary to preclude basic saponification to avoid racemization of the stereocenter (see Supporting Information). We utilized enantiopure benzyl esters, which were subsequently deprotected by Pd/C-catalyzed hydrogenation to afford the individual enantiomers with >95% ee (determined by chiral HPLC). Testing each of the enantiomers for their binding affinity in the FA assay indicated that there was a negligible eudysmic ratio (i.e., the ratio of the IC50 values of the high-affinity and low-affinity binder), indicating that further probing is warranted to determine the proper substituent for this area of the molecule. The results of the structural modifications detailed above are summarized in Table 1.
Table 1. IC50 ± Standard Deviation, Retention Times,a and cLogD7.0 of Tested Compounds.
| compd | IC50 ± standard deviation (nM)b | retention time (min) | cLogD7.0c |
|---|---|---|---|
| 1b | 29d | n.d. | 3.56 |
| 1c | 63d | n.d. | –3.81 |
| 3 | >25000 | 5.184 | –0.39 |
| 4 | 9300 ± 560 | 5.008 | –3.15 |
| 5 | >25000 | 5.254 | 0.25 |
| 6 | 13000 ± 3700 | 5.071 | –3.30 |
| 11 | 1500 ± 18 | 6.737 | 2.07 |
| 12 | 678 ± 29 | 5.886 | 0.13 |
| 15 | 3000 ± 120 | 6.072 | 3.07 |
| 16 | >25000 | 6.184 | 4.08 |
| 19 | 189 ± 9 | 5.995 | 1.19 |
| 20 | 14000 ± 1060 | 6.312 | 2.08 |
| 21 | 126 ± 20 | 5.910 | 0.44 |
| 22 | 151 ± 20 | 5.995 | 1.19 |
| 23 | 241 ± 8 | 5.995 | 1.19 |
See Supporting Information for HPLC method.
Standard deviation of two independent experiments.
Values calculated with ChemAxon’s LogD Predictor.30
Values were taken from Jain et al., and compounds were run in parallel as standards in the FA assay.20
Overall, using a combination of X-ray crystallographic analysis and experimental structure–activity relationship development, we have determined key features of naphthalene-based KEAP1-NRF2 inhibitors that are responsible for the binding affinity to the Kelch domain of KEAP1 (Chart 5).
Chart 5. Key Interactions and Sites for Optimization Identified.
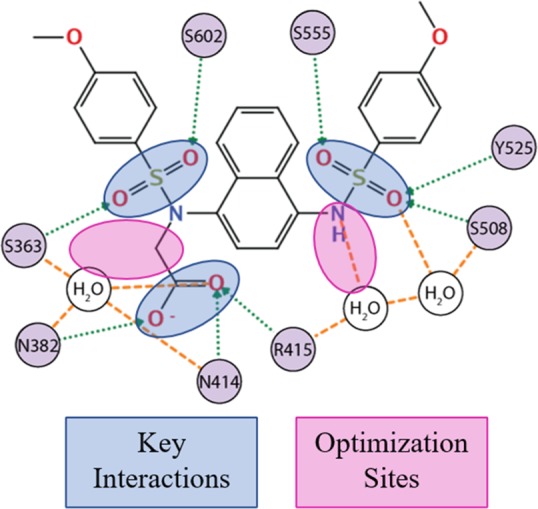
Key interactions are formed between serine residues within the Kelch domain and the oxygen atoms of either the sulfonamide or sulfone functionalities present within these inhibitors. Pairing the previously described serine–oxygen interactions with dipolar interactions between the carboxylate, asparagine, and arginine within the binding pocket appeared to be crucial for establishing strong protein–inhibitor interactions. Furthermore, we showed that one of the nitrogen atoms on the naphthalene ring could be replaced with a carbon atom and still maintain reasonable binding affinities. This substitution allows for the investigation of different geometries around this prochiral carbon instead of being confined to an achiral nitrogen linker. Lastly, our observations agree with Lu et al. in having identified a region α to the carbonyl of these inhibitors where additional exploration may yield inhibitors with higher affinity. Further experiments are underway to determine the role these α-substituents play in inhibitor binding.
Acknowledgments
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The authors thank the following funders for their support of this work: National Institute of Arthritis, Musculoskeletal and Skin Diseases to T.W.M. (Grant 1R01 AR069541-01A1).
Glossary
Abbreviations
- KEAP1
Kelch-like ECH-associated protein 1
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- FA
fluorescence anisotropy
- SAR
structure–activity relationship
- COPD
chronic obstructive pulmonary disorder
- DMF
dimethylformamide
- NQO1
NADPH quinone oxidoreductase 1
- HO-1
heme oxygenase 1
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00631.
Experimental procedures of syntheses, biochemical assay protocols, and crystal structure parameters (PDF)
Author Present Address
∥ A.D.J.: Northwestern Center for Molecular Innovation and Drug Design, 2145 Sheridan Road, Evanston, IL 60208, U.S.
Author Present Address
⊥ B.R.: Department of Chemistry, The Scripps Research Institute, 110 Scripps Way, Jupiter, FL 33458, U.S.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Long M.; Rojo de la Vega M.; Wen Q.; Bharara M.; Jiang T.; Zhang R.; Zhou S.; Wong P. K.; Wondrak G. T.; Zheng H.; Zhang D. D. An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes 2016, 65 (3), 780–793. 10.2337/db15-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani P. S.; Ellison T.; Waqas B.; Sultan D.; Abdou S.; David J. A.; Cohen J. M.; Gomez-Viso A.; Lam G.; Kim C.; Thomson J.; Ceradini D. J. Targeted Nrf2 activation therapy with RTA 408 enhances regenerative capacity of diabetic wounds. Diabetes Res. Clin. Pract. 2018, 139, 11–23. 10.1016/j.diabres.2018.02.021. [DOI] [PubMed] [Google Scholar]
- Gopal S.; Mikulskis A.; Gold R.; Fox R. J.; Dawson K. T.; Amaravadi L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the phase 3 DEFINE and CONFIRM studies. Multiple Sclerosis Journal 2017, 23, 1875–1883. 10.1177/1352458517690617. [DOI] [PubMed] [Google Scholar]
- Boutten A.; Goven D.; Artaud-Macari E.; Boczkowski J.; Bonay M. NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17 (7), 363–71. 10.1016/j.molmed.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Cui W.; Zhang Z.; Zhang P.; Qu J.; Zheng C.; Mo X.; Zhou W.; Xu L.; Yao H.; Gao J. Nrf2 attenuates inflammatory response in COPD/emphysema: Crosstalk with Wnt3a/beta-catenin and AMPK pathways. J. Cell. Mol. Med. 2018, 22 (7), 3514–3525. 10.1111/jcmm.13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezu M.; Souma T.; Yu L.; Suzuki T.; Saigusa D.; Ito S.; Suzuki N.; Yamamoto M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. 10.1016/j.kint.2016.08.023. [DOI] [PubMed] [Google Scholar]
- Magesh S.; Chen Y.; Hu L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrándiz M. L.; Nacher-Juan J.; Alcaraz M. J. Nrf2 as a therapeutic target for rheumatic diseases. Biochem. Pharmacol. 2018, 152, 338–346. 10.1016/j.bcp.2018.04.010. [DOI] [PubMed] [Google Scholar]
- Cuadrado A.; Rojo A. I.; Wells G.; Hayes J. D.; Cousin S. P.; Rumsey W. L.; Attucks O. C.; Franklin S.; Levonen A. L.; Kensler T. W.; Dinkova-Kostova A. T. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discovery 2019, 18, 295–317. 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- Kobayashi A.; Kang M.-I.; Okawa H.; Ohtsuji M.; Zenke Y.; Chiba T.; Igarashi K.; Yamamoto M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. M. U.; Luo L.; Namani A.; Wang X. J.; Tang X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta, Mol. Basis Dis. 2017, 1863, 585–597. 10.1016/j.bbadis.2016.11.005. [DOI] [PubMed] [Google Scholar]
- Cullinan S. B.; Gordan J. D.; Jin J.; Harper J. W.; Diehl J. A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.; Rojo A. I.; Salinas M.; Diaz R.; Gallardo G.; Alam J.; Ruiz De Galarreta C. M.; Cuadrado A. Regulation of Heme Oxygenase-1 Expression through the Phosphatidylinositol 3-Kinase/Akt Pathway and the Nrf2 Transcription Factor in Response to the Antioxidant Phytochemical Carnosol. J. Biol. Chem. 2004, 279, 8919–8929. 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- Itoh K.; Chiba T.; Takahashi S.; Oyake T.; Ishii T.; Yamamoto M.; Katoh Y.; Hayashi N.; Satoh K.; Igarashi K.; Hatayama I.; Nabeshima Y.-i. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Itoh K.; Tong K. I.; Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radical Biol. Med. 2004, 36, 1208–1213. 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- Clulow J. A.; Storck E. M.; Lanyon-Hogg T.; Kalesh K. A.; Jones L. H.; Tate E. W. Competition-based, quantitative chemical proteomics in breast cancer cells identifies new target profiles for sulforaphane. Chem. Commun. (Cambridge, U. K.) 2017, 53 (37), 5182–5185. 10.1039/C6CC08797C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yore M. M.; Kettenbach A. N.; Sporn M. B.; Gerber S. A.; Liby K. T. Proteomic analysis shows synthetic oleanane triterpenoid binds to mTOR. PLoS One 2011, 6 (7), e22862 10.1371/journal.pone.0022862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte D.; Zeng W.; Hus J. C.; McKenzie A.; Hession C.; Jin P.; Bergeron C.; Lugovskoy A.; Enyedy I.; Cuervo H.; Wang D.; Atmanene C.; Roecklin D.; Vecchi M.; Vivat V.; Kraemer J.; Winkler D.; Hong V.; Chao J.; Lukashev M.; Silvian L. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21 (14), 4011–9. 10.1016/j.bmc.2013.04.019. [DOI] [PubMed] [Google Scholar]
- Jiang Z. Y.; Lu M. C.; Xu L. L.; Yang T. T.; Xi M. Y.; Xu X. L.; Guo X. K.; Zhang X. J.; You Q. D.; Sun H. P. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- Jain A. D.; Potteti H.; Richardson B. G.; Kingsley L.; Luciano J. P.; Ryuzoji A. F.; Lee H.; Krunic A.; Mesecar A. D.; Reddy S. P.; Moore T. W. Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur. J. Med. Chem. 2015, 103, 252–268. 10.1016/j.ejmech.2015.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkel A. F.; Engel C. K.; Margerie D.; Kannt A.; Szillat H.; Glombik H.; Kallus C.; Ruf S.; Güssregen S.; Riedel J.; Herling A. W.; Von Knethen A.; Weigert A.; Brüne B.; Schmoll D. Characterization of RA839, a noncovalent small molecule binder to Keap1 and selective activator of Nrf2 signaling. J. Biol. Chem. 2015, 290, 28446–28455. 10.1074/jbc.M115.678136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies T. G.; Wixted W. E.; Coyle J. E.; Griffiths-Jones C.; Hearn K.; McMenamin R.; Norton D.; Rich S. J.; Richardson C.; Saxty G.; Willems H. M. G.; Woolford A. J. A.; Cottom J. E.; Kou J. P.; Yonchuk J. G.; Feldser H. G.; Sanchez Y.; Foley J. P.; Bolognese B. J.; Logan G.; Podolin P. L.; Yan H.; Callahan J. F.; Heightman T. D.; Kerns J. K. Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J. Med. Chem. 2016, 59, 3991–4006. 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- Callahan J. F.; Kerns J. K.; Li T.; Mccleland B. W.; Nie H.; Pero J. E.; Davies T. G.; Heightman T. D.; Griffiths-Jones C. M.; Howard S.; Norton D.; Verdonk M. L.; Woolford A. J. A.. Arylcyclohexyl pyrazoles as NRF2 regulators. Patent WO2017060855 A1, 2017.
- Lu M. C.; Jiao Q.; Liu T.; Tan S. J.; Zhou H. S.; You Q. D.; Jiang Z. Y. Discovery of a head-to-tail cyclic peptide as the Keap1-Nrf2 protein-protein interaction inhibitor with high cell potency. Eur. J. Med. Chem. 2018, 143, 1578–1589. 10.1016/j.ejmech.2017.10.052. [DOI] [PubMed] [Google Scholar]
- Yasuda D.; Nakajima M.; Yuasa A.; Obata R.; Takahashi K.; Ohe T.; Ichimura Y.; Komatsu M.; Yamamoto M.; Imamura R.; Kojima H.; Okabe T.; Nagano T.; Mashino T. Synthesis of Keap1-phosphorylated p62 and Keap1-Nrf2 protein-protein interaction inhibitors and their inhibitory activity. Bioorg. Med. Chem. Lett. 2016, 26 (24), 5956–5959. 10.1016/j.bmcl.2016.10.083. [DOI] [PubMed] [Google Scholar]
- Lazzara P. R.; David B. P.; Ankireddy A.; Richardson B.; Dye K.; Ratia K. M.; Reddy S. P.; Moore T. W. Isoquinoline Kelch-like ECH-Associated Protein 1-Nuclear Factor (Erythroid-derived 2)-like 2 (KEAP1-NRF2) Inhibitors with High Metabolic Stability. J. Med. Chem. 2019, 10.1021/acs.jmedchem.9b01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Lucas B.; Capacci A.; Lin E. Y.; Jones J. H.; Dechantsreiter M.; Enyedy I.; Marcotte D.; Xiao G.; Li B.; Richter K. Design, synthesis and identification of novel, orally bioavailable non-covalent Nrf2 activators. Bioorg. Med. Chem. Lett. 2020, 30 (4), 126852. 10.1016/j.bmcl.2019.126852. [DOI] [PubMed] [Google Scholar]
- Lu M. C.; Zhang X.; Wu F.; Tan S. J.; Zhao J.; You Q. D.; Jiang Z. Y. Discovery of a Potent Kelch-Like ECH-Associated Protein 1-Nuclear Factor Erythroid 2-Related Factor 2 (Keap1-Nrf2) Protein-Protein Interaction Inhibitor with Natural Proline Structure as a Cytoprotective Agent against Acetaminophen-Induced Hepatotoxicity. J. Med. Chem. 2019, 62 (14), 6796–6813. 10.1021/acs.jmedchem.9b00818. [DOI] [PubMed] [Google Scholar]
- Inoyama D.; Chen Y.; Huang X.; Beamer L. J.; Kong A. N.; Hu L. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. J. Biomol. Screening 2012, 17 (4), 435–447. 10.1177/1087057111430124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LogD Predictor. https://disco.chemaxon.com/apps/demos/logd/ (accessed Feb 2, 2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.