

Abstract
A number of disease states including WHIM syndrome, HIV infection and cancer have been linked to the chemokine receptor CXCR4. High-affinity CXCR4 antagonist transition metal complexes of configurationally restricted bis-tetraazamacrocyclic ligands have been identified in previous studies. Recently synthesised and structurally characterised Co2+/Co3+ and Ni2+ acetate complexes of mono-macrocycle cross-bridged ligands have been used to mimic their known coordination interaction with the aspartate side chains on binding to CXCR4. Here, X-ray crystal structures for three Co2+/Co3+ acetate complexes and five Ni2+ acetate complexes are presented and demonstrate flexibility in the mode of binding to the acetate ligand concomitantly with the requisite cis-V-configured cross-bridged tetraazamacrocyle. Complexes of the smaller Co3+ metal ion exclusively bind acetate by chelating both oxygens of acetate. Larger Co2+ and Ni2+ metal ions in cross-bridged tetraazamacrocycles show a clear tendency to coordinate acetate in a monodentate fashion with a coordinated water molecule completing the octahedral coordination sphere. However, in unbridged tetraazamacrocycle acetate structures reported in the literature, the coordination preference is to chelate both acetate oxygens. We conclude that the short ethylene cross-bridge restricts the equatorial bulk of the macrocycle, prompting the metal ion to fill the equator with the larger monodentate acetate plus water ligand set. In unbridged ligand examples, the flexible macrocycle expands equatorially and generally only allows chelation of the sterically smaller acetate alone. These results provide insight for generation of optimised bis-macrocyclic CXCR4 antagonists utilising cobalt and nickel ions.
Introduction
The topological complexity of cross-bridged tetraazamacrocyles (Fig. 1) imparts rigidity and kinetic stability to their transition metal complexes.1 For this reason, these complexes have been utilised in applications where complex stability is paramount, such as aqueous oxidation catalysis2–11 and medical imaging.12,13 Another important property of these ligands is that the short cross-bridge restricts the configuration of the complex to a folded, cis geometry where the macrocycle takes up axial and cis-equatorial positions of octahedral,4 square-pyramidal,14 or trigonal bipyramidal15 coordination geometries (Fig. 1). Open coordination sites must be located cis to each other, which is important in oxidation applications2,11 and has been exploited more recently in producing optimised protein-binding complexes.16,17
Fig. 1.

A generic cross-bridged ligand structure and its metal complex showing a potential geometry.
We have taken advantage of these properties by designing bis-linked cross-bridged tetraazamacrocycle metal complexes (Fig. 2) that have remarkably efficient binding16,18–20 to the aspartate side chains of the CXCR4 chemokine receptor, a trans-membrane receptor important to the fusion process of the HIV virus to leukocytes,17 the metastasis of cancer cells,21 and other biological processes.22 Most relevant to this work, we have shown that a dinickel complex, the meta analogue of ligand 7 (Fig. 3) was similarly efficient as AMD3100 at binding CXCR4, with an IC50 of 14 nM.19 As part of our CXCR4 antagonist program, we have attempted to probe the aspartate-metal ion interaction by synthesizing acetate salts of cross-bridged complexes. The main aim of this work is to study physico-chemical parameters of components of compounds that are likely to be of relevance to CXCR4 antagonist design. Our goal was to produce single crystals suitable for X-ray diffraction that contain acetate ligands bound to the metal ion as a model for the aspartate-metal ion interaction occurring in the biological system. From these structures, we hoped to gain an understanding of the geometric and electronic requirements for producing strong-binding CXCR4 antagonists.
Fig. 2.

Topologically constrained CXCR4 antagonist complex bound to the CXCR4 chemokine receptor with potential coordination interactions shown.
Fig. 3.

Ligands used to form the compounds for analysis and testing in this work.
Because of the significant challenges in production of X-ray quality bis-linked tetraazamacrocycle complex crystals,18,23 single-macrocycle transition metal complexes are often used as models.16,18–20,23 To provide the most accurate model for our bis-macrocycle antagonists, which are linked through a xylene linker, we have synthesised a number of monobenzyl and dibenzyl4 pendant arm containing cross-bridged tetraazamacrocycles (Fig. 3). These ligands provide the same cross-bridged macrocycle geometric requirement around the metal ion, including the bulky benzyl group attached to one (or two) of the coordinated nitrogen atoms. In this work, we present the synthesis, characterization, and structural study of these ligands complexed to cobalt and nickel ions, which we are also evaluating in our research to determine the optimal combination of chelator and metal ion for CXCR4 antagonism. Additionally, we report here for the first time the synthesis and CXCR4 binding ability of a dicobalt bis-macrocyclic antagonist (Co2+)27, for comparison with the mono-macrocyclic model complexes, AMD3100, and our known dinickel and dicopper antagonists.
Experimental
General
Elemental analyses were performed by Quantitative Technologies Inc. Electrospray Mass spectra were collected at the Oklahoma University Health Sciences Center Laboratory for Molecular Biology and Cytometry Research on a Bruker-Daltonics HCT Ultra ion trap mass spectrometer. NMR spectra were obtained on a Varian Bruker AVANCE II 300 MHz NMR Spectrometer. Electronic spectra were recorded using a Beckman Coulter DU640 UV-Vis Spectrometer. Electrochemical experiments were performed on a BAS100B Electrochemical Analyzer. A button Pt electrode was used as the working electrode with a Pt-wire counter electrode and an Ag-wire pseudo-reference electrode. Scans were taken at 200 mV s−1. Acetonitrile solutions of the complexes (1 mM) with tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte were used. The measured potentials were referenced to SHE using ferrocene (+0.400 V versus SHE) as an internal standard. All electrochemical measurements were carried out under N2.
Synthesis
Anhydrous CoCl2, Co(OAc)2, and NiCl2 were purchased from Aldrich and used as received. Anhydrous Ni(OAc)2 was prepared from Ni(OAc)2·4H2O (Fluka) dried under vacuum over refluxing ethanol in an Abderhalden drying pistol until a constant weight was reached, which corresponded to the loss of four equivalents of water.
4,11-Dibenzyl-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (1),24 4,10-dibenzyl-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane (2),24 4-benzyl-11-methyl-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (3),16 and 4-benzyl-10-methyl-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane (4),20 4,11-dimethyl-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (5),24 4,10-dimethyl-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane (6),25 and 1,4-bis((11-methyl-1,4,8,11-tetraazabicyclo[6.6.2] hexadecan-4-yl)methyl)benzene (7).16
General complexation procedure for chloride complexes
1.00 mmol of the ligand (1–2) and 1.00 mmol of the anhydrous metal(II) chloride salt (Ni or Co) were added to 20 ml of dry DMF in an inert atmosphere glovebox. The reaction was stirred at room temperature for 18 h. Product MLCl2 precipitated over the course of the reaction. The reaction mixture was removed from the glovebox and the solid was isolated by filtration, washed with DMF, then ether, and dried under vacuum. Due to lack of solubility (the compounds are only slightly soluble in DMF and water) characterization was limited to elemental analysis and X-ray crystallography. The acetate salts were synthesised to allow complete characterisation (see below). [Co6Cl2]PF626 and [Ni6Cl2]27 were made according to literature procedures.
Co1Cl2: Purple powder. Yield: 0.223 g (42%). X-ray quality crystals were grown by ether diffusion into the mother liquor. Elemental analysis (%) calcd CoC26H38N4Cl2·H2O: C 56.32, H 7.27, N 10.10; Found C 56.23, H 6.96, N 10.00. MS (ES) m/z 500.2 [CoLCl]+.
Ni1Cl2: Reaction was refluxed overnight under nitrogen after removal from glovebox due to undissolved NiCl2. Green X-ray quality crystals formed upon cooling to room temperature. Yield: 0.256 g (48%). Elemental analysis (%) calcd NiC26H38N4Cl2: C 58.23, H 7.14, N 10.45; Found C 58.28, H 7.11, N 10.31. MS (ES) m/z 499.2 [NiLCl]+.
[Co2Cl2]Cl: Light purple powder. Yield: 0.375 g (59% based on elemental analysis formula). No X-ray quality crystals were obtained. Elemental analysis (%) calcd [CoC24H34N4Cl2] Cl·1.5H2O: C 50.50, H 6.53, N 9.80; Found C 50.14, H 6.34, N 10.21. MS (ES) m/z 471.2 [CoLCl]+, 509.1 [CoLCl2]+.
Ni2Cl2: Green-blue powder. Yield: 0.314 g (62%). X-ray quality crystals were grown by ether diffusion into a DMF solution. Elemental analysis (%) calcd NiC24H34N4Cl2: C 56.73, H 6.74, N 11.03; Found C 56.46, H 6.87, N 10.97. MS (ES) m/z 471.2 [NiLCl]+.
General complexation procedure for mononuclear acetate complexes
1.00 mmol of the ligand (1–6) and 1.00 mmol of the anhydrous metal(II) acetate salt (Ni or Co) were added to 25 ml of dry DMF in an inert atmosphere glovebox. The reaction was stirred at room temperature for 18 h. The crude [ML(OAc)][(OAc)] solution was removed from the glovebox, filtered to remove any trace solids, and evaporated to dryness. These crude products were dissolved in 10 ml of dry methanol, to which was added dropwise a 5 ml dry methanol solution of 5 equivalents (0.815 g, 5.00 mmol) of NH4PF6. Powders of the [ML(OAc)]PF6 salts precipitated, were collected, washed with cold methanol and ether, and dried under vacuum. Samples of [Ni1(OAc)]PF6 and [Ni5(OAc)]PF6 were synthesised as previously reported.19
[Co1(OAc)]PF6:28 Pale pink powder. Yield: 0.416 g (64%). No X-ray quality crystals were obtained. Elemental analysis (%) calcd [CoC26H38N4(C2H3O2)]PF6·H2O (687.572 g mol−1): C 48.91, H 6.30, N 8.15; Found C 49.19, H 6.50, N 8.29. MS (ES) m/z 524.2 [CoL(OAc)]+.
[Co2(OAc)](PF6)2: Purple powder. Oxidation to the Co3+ compound was again observed for the cyclen-based ligand. Yield: 0.416 g (64%). X-ray quality crystals were obtained from a cooled methanol solution. Elemental analysis (%) calcd [CoC24H34N4(C2H3O2)](C2H3O2)0.30(PF6)1.70·0.6 H2O (760.691 g mol−1): C 41.41, H 5.11, N 7.26; Found C 41.06, H 5.47, N 7.66. MS (ES) m/z 641.2 [CoL(OAc)][PF6]+, 496.3 [ML(OAc)]+.
[Ni2(OAc)]PF6:28 Pale purple powder. Yield: 0.414 g (67%). X-ray quality crystals were obtained from a cooled methanol solution. Elemental analysis (%) calcd [NiC24H34N4(C2H3O2)] PF6 (641.260 g mol−1): C 48.70, H 5.82, N 8.74; Found C 48.58, H 6.00, N 8.79. MS (ES) m/z 495.2 [NiL(OAc)]+.
[Co3(OAc)]PF6: Purple powder. Yield: 0.207 g (35%). No X-ray quality crystals were obtained. Elemental analysis (%) calcd [CoC20H34N4(C2H3O2)]PF6·0.4NH4PF6·1.2 H2O (680.278 g mol−1): C 38.84, H 6.08, N 9.06; Found C 39.14, H 5.78, N 8.67. MS (ES) m/z 448.2 [CoL(OAc)]+, 465.2 [CoL(OAc)(OH)]+.
[Ni3(OAc)](PF6): Pale purple powder. Yield: 0.356 g (60%). No X-ray quality crystals were obtained. Elemental analysis (%) calcd [NiC20H34N4(C2H3O2)]PF6·0.35NH4PF6 (650.266 g mol−1): C 40.64, H 5.95, N 9.37; Found C 40.46, H 5.70, N 9.59. MS (ES) m/z 447.2 [NiL(OAc)]+.
[Co4(OAc)](PF6)2: Bright pink powder. Oxidation to the Co3+ compound was again observed for the cyclen-based ligand. Yield: 0.350 g (49%). No X-ray quality crystals were obtained. Elemental analysis (%) calcd [CoC18H30N4(C2H3O2)](PF6)2 (710.369 g mol−1): C 33.82, H 4.68, N 7.89; Found C 33.82, H 4.61, N 8.00. MS (ES) m/z 421.2 [CoL(OAc)]+, 437.2 [CoL(OAc)OH]+.
[Ni4(OAc)]PF6: Pale purple powder. Yield: 0.488 g (86%). X-ray quality crystals were obtained from the evaporation of a methanol solution. Elemental analysis (%) calcd [NiC18H30N4 (C2H3O2)]PF6 (565.162 g mol−1): C 42.50, H 5.89, N 9.91; Found C 42.11, H 5.77, N 9.82. MS (ES) m/z 419.2 [NiL(OAc)]+.
[Co5(OAc)]PF6: Pink powder. Yield: 0.184 g (31%). X-ray quality crystals were obtained from diffusion of ether into an acetone solution. Elemental analysis (%) calcd [CoC14H30N4(C2H3O2)]PF6·1.8 H2O (544.38 g mol−1): C 35.30, H 6.71, N 10.29; Found C 35.46, H 6.68, N 10.10. MS (ES) m/z 372.2 [CoL(OAc)]+.
[Co6(OAc)](PF6)2: Bright pink powder. Yield: 0.386 g (61%). X-ray quality crystals were obtained from diffusion of ether into an acetonitrile solution. Elemental analysis (%) calcd [CoC12H26N4(C2H3O2)](PF6)2 (634.27 g mol−1): C 26.51, H 4.61, N 8.83; Found C 26.77, H 4.48, N 8.81. MS (ES) m/z 344.2 [CoL(OAc)]+.
[Ni6(OAc)]PF6: Pale purple powder. Yield: 0.104 g (24%). X-ray quality crystals were obtained from ether diffusion into dichloromethane solution. Elemental analysis (%) calcd [NiC12H26N4(C2H3O2)]PF6 (489.06 g mol−1): C 34.38, H 5.98, N 11.46; Found C 34.15, H 5.96, N 11.37. MS (ES) m/z 343.2 [NiL(OAc)]+.
[Co27(OAc)2](PF6)2: 1.00 mmol (0.583 g) of the ligand (7) and 2.00 mmol (0.360 g) of the anhydrous cobalt(II) acetate salt were added to 30 ml of dry CH3CN in an inert atmosphere glovebox. The reaction was stirred at room temperature for 18 h. The crude [Co2L(OAc)2](OAc)2 solution was removed from the glovebox, filtered to remove any trace solids, and evaporated to dryness. These crude products were dissolved in 10 ml of dry methanol, to which was added dropwise a 5 ml dry methanol solution of 5 equivalents (0.815 g, 5.00 mmol) of NH4PF6. A pink powder of the [Co27(OAc)](PF6)2 complex precipitated, was collected, washed with cold methanol and ether, and dried under vacuum. Yield: 0.115 g (10%). Elemental analysis (%) calcd [Co2C34H62N8(C2H3O2)2](PF6)2·0.4NH4PF6·4H2O (1246.05 g mol−1): C 36.63, H 6.28, N 9.44; Found C 36.89, H 6.21, N 9.07. MS (ES) m/z 410.0 [Co2L(OAc)2]2+.
X-ray crystallography
The sets of X-ray diffraction intensity data from all samples were collected in series of ω-scans using a Stoe IPDS2 image plate diffractometer operating with MoKα radiation. Crystals were mounted at the end of a glass fiber and cooled to 150(2) K in an Oxford Cryosystems nitrogen gas cryostream. Data were scaled and merged and a multi-scan method was applied for the absorption corrections of the collected data.29 The structures were solved using dual-space methods within SHELXT and full-matrix least squares refinement was carried out within SHELXL-2014 via the WinGX program interface.30 All non-hydrogen positions were located in the direct and difference Fourier maps and refined using anisotropic displacement parameters.
The structure of [Co1Cl2] was twinned by 180° rotation about the 1 0 4 reciprocal direction. The twin fraction was 0.946 : 0.054(4). The crystal of [Ni1Cl2] was refined as an inversion twin with twin fraction 0.52 : 0.48(2). The structure of [Ni4(OAc)]PF6 was twinned by 180° rotation about the 0 0 1 reciprocal direction (Twin fraction 0.8420 : 0.1580(17)). A small number of reflections suspected of being partially overlapped between two twin domains were omitted from the final refinement. The crystal of [Co6(OAc)](PF6)2 was refined as an inversion twin with twin fraction 0.54 : 0.46(4).
Anti-viral assays
Anti-HIV activity and cytotoxicity measurements in MT-4 and other cells were based on the viability of cells that had been infected or not infected with the HIV-1 strain IIIB and exposed to various concentrations of the test compound. After the cells were allowed to proliferate for 5 days, the number of viable cells was quantified by a tetrazolium-based colorimetric method as described by Pauwels et al.31,32 The metal complexes were dissolved in water or phosphate buffer prior to addition. Initial dissolution in DMSO was required for the hexafluorophosphate salt compounds followed by dilution into aqueous solution.
Results and discussion
Preparation of metal complexes
The initial complex formation reactions utilised anhydrous chloride salts of Ni2+ and Co2+, following procedures previously used for dimethyl cross-bridged cyclam and cyclen ligands26,27 or more recently cross-bridged homocyclen.25 The resulting complexes, although pure and amenable to crystallisation from the reaction solution by simply cooling them, or diffusing in ether, were not comprehensively characterised for two reasons. First, they were only slightly soluble in solvents such as acetonitrile and methanol. Lack of solubility hindered the ability to obtain solution phase data such as electronic spectra and cyclic voltammetry. It had been observed in previous studies in our group that making acetate complexes rather than chloride complexes increased the solubility of the resulting complexes significantly.16,18–20 Secondly, the complexes synthesised from acetate salts are of high interest to characterise the coordination interaction of the metal centres with carboxylate functional groups, which occurs when complexes of this type bind to the aspartate side chains of the CXCR4 chemokine receptor.20,33 Therefore, we decided to synthesise and comprehensively characterise complexes with all six ligands starting from acetate salts of Ni2+ and Co2+.
Complexation of the ligands with the acetate salts were carried out in an inert atmosphere glovebox, primarily to protect the ligands from exposure to water, which tends to protonate these highly basic ligands and inhibit complex formation.2,15,34 After visible colour changes and stirring overnight to complete the complexation reactions, the reaction mixtures were removed from the glovebox to work up in air. Interestingly, all cobalt complexes with cyclen ligands air-oxidized to give Co3+ products, while the cyclam-based ligand complexes were air stable and gave only Co2+ products. This is consistent with prior work on cobalt complexes of cross-bridged cyclen ligands.26 It appears the smaller cyclen ligand cavity favours the smaller Co3+ ion. Cyclic voltammetry studies examining the redox behaviour of these complexes is discussed below.
Crystallography
Tables S1 and S2† contains crystallographic data for the new crystal structures presented here. Table S3† contains selected bond lengths and bond angles for these structures.
Macrocycle-metal ion interactions
Due to their relevance to this work, two closely related crystal structures from one of our previous publications19 are included in this discussion: [Ni1(OAc)(H2O)]+ and [Ni5(OAc)(H2O)]+. Crystallographic details for these new structures, along with selected bond lengths and angles, are presented in Tables S1–S3 in the ESI.†
Prior to focussing on the detailed structural parameters, some general observations can be made. First, as constrained by the ligand cross-bridge, all complexes are found in the cis-V configuration.35 Fig. 4 shows the three chloride examples characterised, using both metal ions and both macrocycle ring sizes. Fig. 4a is Co1Cl2; Fig. 4b is Ni1Cl2; and Fig. 4c is Ni2Cl2 all with six-coordinate octahedral coordination geometries. Consistent with prior work and all of the other structures presented below, changing the identity of the metal ion, the alkyl substituent, benzyl in both cases of Fig. 4, or the labile additional equatorial ligands does not alter this configuration, which is a fixed feature of ethylene cross-bridged transition metal complexes.
Fig. 4.

Structures of (a) Co(1)Cl2 (b) Ni(1)Cl2 and (c) Ni(2)Cl2 demonstrating the cis-V configuration for both cyclam and cyclen based ligands.
Second, how fully engulfed the metal ion is by the ligand is dependent on the parent macrocycle ring size. Fig. 5 demonstrates this tendency using the [Ni6(OAc)]+ complex from a cyclen macrocycle and the [Ni5(OAc)(H2O)]+ complex from a cyclam macrocycle. We have found that the Nax–M–Nax bond angle is a convenient measure of how far into the folded macrocyclic cavity the metal ion is found. As shown in Fig. 5a, this bond angle is 163.82(14)° for the smaller cyclen parent macrocycle ring, indicating reduced ability of the complex to achieve an undistorted octahedral structure where this angle would be 180°. Fig. 5b, illustrates that the same Ni2+ metal ion in the larger cyclam parent ring ligand is much closer to linear for this bond angle at 173.41(11)°.
Fig. 5.

Structures of (a) [Ni(6)(OAc)]+ with N4–Ni1–N2 bond angle of 163.82(14)° and (b) [Ni(5)(OAc)(H2O)]+ with N4–Ni1–N2 bond angle of 173.41(11)°, showing the extent of engulfment of the same Ni2+ metal ion by two different macrocycle ring sizes.
Third, the ionic radius of the metal ion also plays a role in the deviation from regular octahedral geometry for the complex. Fig. 6 illustrates this trend with the three different metal ions present in the complexes discussed: low spin Co3+ (69 pm ionic radius); high spin Ni2+ (83 pm); and high spin Co2+ (89 pm). Fig. 6a and b shows the comparison of low spin Co3+ and high spin Ni2+ in same coordination sphere of ligand 7, and an iso-bidentate acetate. The smaller cross-bridged cyclen ligand is more complementary for the small, low spin Co3+ ion, having an Nax–M–Nax bond angle of 171.06(19)°, while the larger high spin Ni2+ ion is not as well accommodated with a 163.82(14)° Nax–M–Nax bond angle. The significant difference in ionic radius (14 pm) results in a ~7° bond angle difference. A much smaller difference is discernible in Fig. 6c and d in the larger cross-bridged cyclam system where two cations of much more similar ionic radius (high spin Co2+, 89 pm; high spin Ni2+, 83 pm) are similarly situated within the ligand 5 cavity and bind monodentate acetate anions and water molecules to complete their octahedral coordination geometries. The Nax–M–Nax bond angles are 173.0(2)° (hs Co2+)
Fig. 6.

Structures of (a) [Co(6)(OAc)]2+ with N2–Co1–N4 171.06(19)°, (b) [Ni(6)(OAc)]+ with N2–Ni1–N4 163.82(14)°, (c) [Co(5)(OAc)(H2O)]+ with N2–Co–N4 173.0(2)°, and (d) [Ni(5)(OAc)(H2O)]+ with N2–Ni1–N4 173.41 (11)° demonstrating the effect of metal ionic radius.
Together, these three trends echo those seen for other cross-bridge tetraazamacrocycle transition metal complexes and most usefully compiled in our previous work.25
Acetate binding
In this work, we are using acetate as a model to better understand the binding of these cobalt and nickel complexes to the aspartate carboxylate side chain on the surface of the CXCR4 chemokine receptor.16,18,20 In a recent study of Cu2+ and Zn2+ CXCR4 chemokine receptor antagonists,23 we were able to discern several trends based on similar acetate-as-model crystal structures that shed light on the likely coordination environment in the antagonist/receptor interaction and rationalised our antagonist binding affinities and residence time data.23 The aim of this study is to learn similar information about our cobalt and nickel antagonists. Fig. 7 shows all of the crystal structures of Ni/Co cross-bridged ligands 1–6 containing acetate bound to the metal ions. The acetate binding mode is briefly described along with the coordination sphere of these complexes prior to drawing conclusions from the structural study. Fig. 8 contains additional Ni/Co complex crystal structures from the literature, where the metal ion is bound to an unbridged tetraazamacrocycle derived from cyclam or cyclen and coordinated in a cis configuration where each metal ion is also bound to an acetate ligand. Table 3 provides the geometrical parameters for all discussed structures.
Fig. 7.

Nickel and cobalt cross-bridged tetraazamacrocycle acetate complexes discussed in the paper: (a) [Co(2)(OAc)]2+ (b) [Co6(OAc)]2+ (c) [Co(5)(OAc)(H2O)]+ (d) [Ni5(OAc)(H2O)]+ (e) [Ni1(OAc)(H2O)]+ (f) [Ni2(OAc)(H2O)]+ (g) [Ni4(OAc)]+ (h) [Ni6(OAc)]+.
Fig. 8.

Nickel and cobalt un-bridged tetraazamacrocycle acetate complexes from the literature for comparison to the cross-bridged complexes: (a) 37[Co(Bn2Cyclam)(OAc)]+ (b) 37[Ni(Bn2Cyclen)(OAc)]+ (c) 37[Ni (Bn2Cyclen)(OAc)(H2O)]+ (d) 38[Ni(Me4Cyclen)(OAc)]+ (e) 39[Ni(Cyclen) (OAc)]+ (f) 40[Ni(Bn1Cyclam)(OAc)]+. H atoms have been omitted for clarity.
Table 3.
Redox potentials (vs. SHE) with peak separations for nickel acetate complexes
| Complex | Eox (V) Ni2+/Ni3+ | E1/2 (V) Ni2+/Ni3+ | (Ea–Ec), mV |
|---|---|---|---|
| [Ni1(OAc)]+ | +1.255 | — | — |
| [Ni2(OAc)]+ | — | +1.117 | 106 |
| [Ni3(OAc)]+ | +1.277 | — | |
| [Ni4(OAc)]+ | — | +1.077 | 106 |
| [Ni5(OAc)]+ | +1.193 | 78 | |
| [Ni6(OAc)]+ | +1.062 | 92 |
For the cross-bridged ligand 1–6 acetate complexes, there are three groups of related structures. Fig. 7a [Co2(OAc)]2+ and Fig. 7b [Co6(OAc)]2+ contain the first type of observed complex. These are slightly distorted octahedral complexes of Co3+ ions with four nitrogens from the cross-bridged ligand occupying the two axial and two cis-equatorial positions. The acetate ligands in these complexes are acting as iso-bidentate chelates at the remaining cis-equatorial sites. Even though Co2+ salts were used for complexation, aerobic workup of the formed complexes leads to oxidation to Co3+. Both ligand 2 and 7 are derived from the smaller 12-membered cyclen ring, which, in our hands, always results in isolation of the smaller Co3+ ion, which is more complementary to the smaller ring size.26
The second group of related structures are shown in Fig. 7c–f. Fig. 7c contains [Co(5)(OAc)(H2O)]+, which features the larger Co2+ ion in a larger cyclam-derived ring system. Aerobic workup does not lead to oxidation, as the larger ring system is more complementary for the larger Co2+ ion.26 This Co2+ complex has common features with Fig. 7d–f featuring Ni2+ ions, Fig. 7d [Ni(5)(OAc)(H2O)]+, Fig. 7e [Ni(1)(OAc) (H2O)]+, Fig. 7f [Ni(2)(OAc)(H2O)]+. All of these M2+ complexes have slightly distorted octahedral geometries with the macrocycle nitrogens again occupying both axial and two cis-equatorial sites. However, in each of these four cases, the acetate ligand is bound equatorially in a monodentate fashion, with the uncoordinated acetate oxygen acting as a hydrogen bond acceptor from a water molecule coordinated in the final equatorial position. Only one of these complexes incorporates a cyclen-derived ligand.
The final two structures also contain cyclen-derived ligands: Fig. 7g [Ni4(OAc)]+ and Fig. 7h [Ni6(OAc)]+. In both cases, the Ni2+ ions are in distorted octahedral geometries with the acetate ligands bound in an isobidentate manner equatorially, and the remaining coordination sites occupied by the cross-bridged ligand nitrogen donors.
Among these eight cross-bridged tetraazamacrocycle acetate complexes, the coordination of water accompanying the monodentate coordination of the acetate ligand was an unexpected result, and could play a significant role in understanding the coordination of our cross-bridged CXCR4 antagonists to the aspartate carboxylate side-chains where they bind. Thermodynamically, bidentate coordination of the acetate chelate should be favoured over two monodentate ligands.1 However, similar behaviour observed for our Zn2+ cross-bridged complexes, as examined by crystallography and DFT calculations, revealed that the acetate-water ligand pair interacting through hydrogen bonding was energetically more favourable than the bidentate coordination of acetate alone.20 The cross-bridge plays an important role in dictating the acetate coordination mode.20
To characterise the influence of the cross-bridge for cobalt and nickel, we required examples of unbridged cis-coordinated tetraazamacrocycle complexes having similar acetate ligands. Fig. 8 shows six such cyclam and cyclen derived examples that were found in the literature for comparison to our cross-bridged complexes. Table 1 lists geometrical parameters for all 14 complexes in Fig. 7 and 8.
Table 1.
X-ray structural parameters determining acetate binding mode in Co/Ni complexes
| Complex | Mn+ | r (pm)36 | Nax–M–Nax angle (°) | Neq–M–Neq angle (°) | O–M–O angle (°) | OAc binding mode | M–N bond distance (Å) | M–O bond distance (Å) |
|---|---|---|---|---|---|---|---|---|
| [Co2(OAc)]2+ | Co3+ | 69 | 170.77(16) | 90.42(17) | 68.16(18) | Iso-bidentate | Co–Nax = 2.033(3) Co–Neq = 1.898(3) |
Co–O = 1.939(3) |
| [Co6(OAc)]2+ | Co3+ | 69 | 171.06(19) | 90.85(19) | 67.57(19) | Iso-bidentate | Co–Nax = 2.006 (avg) Co–Neq = 1.903 (avg) |
Co–O = 1.933 (avg) |
| [Co5(OAc)(H2O)]+ | Co2+ | 89 | 173.0(2) | 83.39(19) | 88.18(18) | Monodentate/H2O | Co–Nax = 2.179 (avg) Co–Neq = 2.126 (avg) |
Co–O = 2.042(4) (OAc) Co–O = 2.111(5) (H2O) |
| 19[Ni5(OAc)(H2O)]+ | Ni2+ | 83 | 173.41(11) | 84.02(10) | 88.35(10) | Monodentate/H2O | Ni–Nax = 2.192 (avg) Ni–Neq = 2.097 (avg) |
Ni–O = 2.064(2) (OAc) Ni–O = 2.077(3) (H2O) |
| 19[Ni1(OAc)(H2O)]+ | Ni2+ | 83 | 175.41(10) | 85.50(12) | 88.18(11) | Monodentate/H2O | Ni–Nax = 2.183 (avg) Ni–Neq = 2.132 (avg) |
Ni–O = 2.052(2) (OAc) Ni–O = 2.109(3) (H2O) |
| [Ni2(OAc)(H2O)]+ | Ni2+ | 83 | 163.52(6) | 85.59(7) | 87.61(6) | Monodentate/H2O | Ni–Nax = 2.1532 (avg) Ni–Neq = 2.0580 (avg) |
Ni–O = 2.0639(14) (OAc) Ni–O = 2.0770(15) (H2O) |
| [Ni4(OAc)]+ | Ni2+ | 83 | 163.71(13) | 87.69(14) | 63.01(12) | Iso-bidentate | Ni–Nax = 2.150 (avg) Ni–Neq = 2.025 (avg) |
Ni–O = 2.106 (avg) |
| [Ni6(OAc)]+ | Ni2+ | 83 | 163.82(14) | 87.08(14) | 62.78(13) | Iso-bidentate | Ni–Nax = 2.145 (avg) Ni–Neq = 2.023 (avg) |
Ni–O = 2.094 (avg) |
| 37[Co(Bn2Cyclam)(OAc)]+ | Co2+ | 89 | 171.54(10) | 97.16(11) | 61.51(9) | Iso-bidentate | Co–Nax = 2.254 (avg) Co–Neq = 2.107 (avg) |
Co–O = 2.134 (avg) |
| 37[Ni(Bn2Cyclen)(OAc)]+ | Ni2+ | 83 | 160.84(9) | 101.83(10) | 62.31(8) | Iso-bidentate | Ni–Nax = 2.180 (avg) Ni–Neq = 2.033 (avg) |
Ni–O = 2.115 (avg) |
| 37[Ni(Bn2Cyclen)(OAc)(H2O)]+ | Ni2+ | 83 | 160.29(7) | 96.99(7) | 88.19(6) | Monodentate/H2O | Ni–Nax = 2.161 (avg) Ni–Neq = 2.066 (avg) |
Ni–O = 2.0525(15) (OAc) Ni–O = 2.0750(15) (H2O) |
| 38[Ni(Me4Cyclen)(OAc)]+FODTAV | Ni2+ | 83 | 158.44(16) | 108.64(17) | 61.92(14) | Iso-bidentate | Ni–Nax = 2.154 (avg) Ni–Neq = 2.104 (avg) |
Ni–O = 2.114 (avg) |
| 39[Ni(Cyclen)(OAc)]+XADMAR | Ni2+ | 83 | 160.5(2) | 102.1(2) | 61.47(15) | Iso-bidentate | Ni–Nax = 2.098(4) Ni–Neq = 2.050 (avg) |
Ni–O = 2.121 (avg) |
| 40[Ni(Bn1Cyclam)(OAc)]+NEXQEN | Ni2+ | 83 | 173.54(6) | 100.36(6) | 62.01(6) | Iso-bidentate | Ni–Nax = 2.149 (avg) Ni–Neq = 2.073 (avg) |
Ni–O = 2.133 (avg) |
Analysis of the geometric parameters
The identity of the metal ion is not a good predictor of the acetate binding mode for these metals ions. Both coordination modes, isobidentate and monodentate with water binding, were characterised for the divalent ions, Co2+ and Ni2. However, oxidation state was more predictive; for Co3+, only isobidentate coordinated acetate was observed. This observation may be most related to the size of the metal ion. Co3+ in an octahedral geometry has a 69 pm ionic radius, while the Ni2+ and Co2+ ionic radii are much larger, at 83 pm and 89 pm, respectively. The small Co3+ ion has short Co–Neq bonds (~1.90 Å) in both complexes, which contributes to the large 90.42(17)° and 90.85(19)° Neq–Co–Neq bond angles, the largest of any of the cross-bridged complexes. Below, we discuss how this latter parameter is the most accurate predictor of coordination mode.
Nax–M–Nax bond angles varied significantly, from 158.44(16)° for unbridged [Ni(Me4Cyclen)(OAc)]+ to 175.41(10)° for cross-bridged cyclam complex [Ni1(OAc)(H2O)]+. However, the value of this parameter does not correlate well with the acetate binding mode. For example, both observed coordination modes are found for Nax–M–Nax bond angles near both extremes for this parameter: largest iso-bidentate value 173.54(6)° for [Ni(Bn1Cyclam)(OAc)]+; smallest iso-bidentate value 158.44(16)° for [Ni(Me4Cyclen)(OAc)]+; largest monodentate/H2O value 175.41(10)° for [Ni1(OAc)(H2O)]+; smallest monodentate/H2O value 160.29(7)° for [Ni(Bn2Cyclen)(OAc)(H2O)]+.
Neq–M–Neq bond angles varied significantly as well, from 83.39(19)° for [Co(5)(OAc)(H2O)]+ to 108.64(17)° for [Ni(Me4Cyclen)(OAc)]+. As a general rule, if a complex had an Neq–M–Neq angle >85.59°, its coordination mode was iso-bidentate, and if the Neq–M–Neq angle <85.59°, the coordination mode was monodentate/H2O. This result is similar to what we found in our recent study of Cu2+ and Zn2+ complexes of related ligands.20 An explanation for this trend is that the short ethylene cross-bridge often restricts the Neq–M–Neq angle to less than the ideal 90°, which causes an abundance of space on the opposite equatorial side, which can best be filled by the more sterically demanding pair of cis ligands consisting of a monodentate acetate hydrogen bonded to a water molecule, which demonstrate O–M–O bond angles all near 88° (Table 1). Even a modest increase in the Neq–M–Neq angle begins to restrict this space, allowing the smaller iso-bidentate coordination mode (O–M–O bond angles 61.47°–68.16°, Table 1) on the opposite equatorial side to adequately fill the smaller equatorial space.
From Table 1, it is apparent that the monodentate/H2O coordination mode is much more prevalent among the cross-bridged ligand complexes. Excluding the small Co3+ ion complexes, only two complexes of cross-bridged ligands bind acetate in the iso-bidentate mode: [Ni4(OAc)]+ and [Ni6(OAc)]+. Both ligands are cyclen-derived, but this is not a general rule as 2 forms a Ni2+ complex which coordinates acetate in the monodentate plus H2O mode. Evidently, although the ethylene cross-bridge favours monodentate acetate/H2O coordination, it does not dictate it in all cases.
From the previously published unbridged ligand structures used for comparison, five out of six bind acetate in the iso-bidentate coordination mode. The lack of an ethylene crossbridge allows much more flexibility in the macrocycle, most clearly represented by the variation in the Neq–M–Neq angles, ranging from 96.99° to 108.64°. In contrast to the cross-bridged complexes discussed above, the larger Neq–M–Neq angles restrict the equatorial space available on the opposite equatorial side, so that coordination of the iso-bidentate acetate is then favoured. However, even in this group, there is one example of the monodentate/H2O coordination mode [Ni (Bn2Cyclen)(OAc)(H2O)]+ (Fig. 8c).
Interestingly, for these chelators, the same metal–ligand combination can produce both coordination modes: [Ni (Bn2Cyclen)(OAc)]+ (Fig. 8b) is iso-bidentate with Neq–M–Neq angle 101.83(10)° and O–Ni–O angle 62.31(8)°, values clearly in line with the other iso-bidentate complexes of unbridged ligands. However, [Ni(Bn2Cyclen)(OAc)(H2O)]+ (Fig. 8c) is monodentate/H2O with Neq–M–Neq angle 96.99(7)° and O–Ni– O angle 88.19(6)°. This latter complex is the greatest outlier in Table 3, with Neq–M–Neq angle much greater than the 85.59° cut-off identified above for this coordination mode. Yet, this complex still manages an O–Ni–O angle of 88.19(6)°, in line with the monodentate/H2O structures of the cross-bridged ligand complexes. This may indicate that precise prediction is not warranted, or may be a feature of the more flexible non-bridged ligands. In general terms, cross-bridged ligand complexes appear to favour monodentate acetate/H2O coordination, while non-bridged ligand complexes appear to favour iso-bidentate acetate coordination in their Ni2+ and Co2+ complexes.
Electronic properties
The electronic spectra of nickel and cobalt complexes have been used to compare the properties of their ligands with those currently in the literature. In addition to their use as structural models for the binding of cross-bridged metal complexes to aspartate in the CXCR4 receptor protein, these new Ni2+, Co2+, and Co3+ complexes can provide insight into the general properties of cross-bridged tetraazamacrocyclic ligand complexes through comparison with other related bridged derivatives and to unbridged macrocyclic ligands.
In addition, transition metal complexes introduced into biological systems are challenged by a number of redox active compounds that may oxidize or reduce the metal ion, which may result in inactivation or metal ion release. Characterising the redox behaviour of a potential inorganic medicinal compounds is therefore important for understanding their biological stability.41,42
Electronic spectra of nickel(II) complexes
The electronic spectra of the six Ni2+ acetate complexes exhibit classic octahedral Ni2+ electronic spectra, with three major absorbances in the range of 300–1100 nm, as exemplified by the spectrum of [Ni3(OAc)]+ in acetonitrile (Fig. 9) and tabulated in Table 2. The electronic spectra of octahedral nickel(II) complexes are useful for determining ligand field strengths.27,43,44 Δo is given directly by the energy of the lowest energy absorption band. For the six octahedral acetate complexes, this gives the following results: Δo = 10 215 cm−1 for [Ni1(OAc)]+; Δo = 10 515 cm−1 for [Ni2(OAc)]+; Δo = 10 604 cm−1 for [Ni3(OAc)]+; Δo = 10 638 cm−1 for [Ni4(OAc)]+; Δo = 11 236 cm−1 for [Ni5(OAc)]+; and Δo = 11 403 cm−1 for [Ni6(OAc)]+.
Fig. 9.

Electronic spectrum of [Ni3(OAc)]+ in acetonitrile.
Table 2.
Electronic spectra of nickel and cobalt acetate complexes in acetonitrile
| Complex | Metal ion | Absorption in nm (extinction coefficient M−1 cm−1) | Calculated Δo (cm−1) | |||
|---|---|---|---|---|---|---|
| [Ni1(C2H3O2)]+ | Ni2+ | 354 (15) | 570 (7) | 829sh (5) | 979 (12) | 10 215 |
| [Ni2(C2H3O2)]+ | Ni2+ | 334 (37) | 559 (10) | 845sh (28) | 951 (36) | 10 515 |
| [Ni3(C2H3O2)]+ | Ni2+ | 354 (12) | 555 (9) | 821sh (9) | 943 (15) | 10 604 |
| [Ni4(C2H3O2)]+ | Ni2+ | 334 (21) | 556 (10) | 836sh (32) | 940 (38) | 10 638 |
| [Ni5(C2H3O2)]+ | Ni2+ | 337 (15) | 526 (12) | 777 (10) | 890 (17) | 11 236 |
| [Ni6(C2H3O2)]+ | Ni2+ | 318 (26) | 513 (11) | 794 (35) | 877 (38) | 11 403 |
| [Co1(C2H3O2)]+ | Co2+ | 464sh (17) | 510 (20) | 547sh (15) | 1063 (12) | |
| [Co2(C2H3O2)]2+ | Co3+ | 380 (235) | 523 (356) | 22 920 | ||
| [Co3(C2H3O2)]+ | Co2+ | 550 (74) | ||||
| Co(4)(C2H3O2)2+ | Co3+ | 370 (195) | 523 (249) | 22 920 | ||
| Co(5)(C2H3O2)+ | Co2+ | 453sh (19) | 501 (22) | 542sh (18) | 1008 (11) | |
| Co(6)(C2H3O2)2+ | Co3+ | 364 (151) | 507 (239) | 23 524 | ||
Of note are the higher extinction coefficients for the cyclen-based complexes (~10–40 M−1 cm−1) compared to the cyclam-based complexes (~5–17 M−1 cm−1). This behaviour is likely due to greater distortion away from octahedral for the smaller ligand ring, which can’t engulf the metal ion as fully. This greater distortion would make the transitions that are forbidden in the truly octahedral geometry, more likely to occur, giving the higher extinction coefficients observed.27
Interestingly, the trend of increasing ligand field strength in these complexes is with a decrease in macrocycle size, from the 14-membered cyclam-based ligands to the 12-membered cyclen-based ligands. This is the opposite of the trend that was observed for similar Ni2+ dichloride complexes with dimethyl cross-bridged ligands.27 In that series of octahedral dichloro complexes, the observation was decreasing ligand field strength with decreasing macrocycle ring size, which was attributed to poorer orbital overlap as the octahedron became more distorted with the ligand size decrease. The change from methyl substituents to at least one benzyl group, as well as the change from two chloro ligands to one acetate ligand, appears to sufficiently effect the electronic properties to reverse the trend. For example, Δo = 11 236 cm−1 for cyclam-based [Ni5(OAc)]+; and Δo = 11 403 cm−1 for cyclen-based [Ni6(OAc)]+. The smaller ring system appears to enforce a stronger ligand field on the Ni2+ cation in these acetate complexes.
What did not change, however, is the similarity in Δo between these cross-bridged ligand complexes and those of cis-binding unbridged macrocycles. For example, the value for cis-Ni(13[ane]N4)Cl2 is Δo = 11 111 cm−1 (ref. 45) and cis-Ni (TACD)(NO3)2 is Δo = 9756 cm−1 (TACD = 1,4,7,10-tetrabenzyl1,4,7,10-tetraazacyclododecane).46 The ethylene cross-bridge does not greatly change the ligand field strength of the macrocyclic ligand if both are bound in a cis configuration. However, the cross-bridge does topologically prohibit trans configurations, which have much higher ligand field strengths for Ni2+. The value of Dqxy used to measure the ligand field strength of such tetragonally distorted complexes47 can be significantly larger. For example, for the unbridged cyclam ligand, [Ni(cyclam)Cl2] is in a trans configuration and the value of Dqxy = 14870 cm−1.47
Finally, the effect of N-substitution on the cross-bridged ligands can be considered. The smooth increase in Δo from ligand 1 to ligand 6 indicates the presence of benzyl groups lessens the ligand field strength as, for both cyclam (ligands 1, 3, and 5) and cyclen (ligands 2, 4, and 6) series: dibenzyl < monobenzyl < dimethyl. The steric requirements of the benzyl pendant arms may disrupt the preferred ligand conformation, resulting in a weaker ligand field strength.
Electronic spectra of cobalt complexes
As noted above, the cyclen-based complexes oxidised upon workup in air, resulting in Co3+ complexes of ligands 2, 4, and 6. However, the cyclam-based complexes were air stable and to allow Co2+ complexes to be isolated. Therefore, some of the comparisons that could be made for the Ni2+ complexes aren’t possible. However, CoLCl2 (Co2+) and [CoLCl2]+ (Co3+) complexes with ligands 5 and 6 have all been previously synthesised and spectroscopically characterised,26 so several meaningful comparisons can be made. Fig. 10 shows representative spectra and Table 2 lists the relevant numerical parameters.
Fig. 10.

Electronic spectra in acetonitrile of (a) [Co1(OAc)]+ and (b) [Co2(OAc)]2+.
The electronic spectra in acetonitrile of the cyclam-based, d7 Co2+ complexes are typical of high spin Co2+ complexes, having a single major absorption band centred between 500 and 600 nm and low extinction coefficients.44 Interestingly, the spectrum for the [Co3(OAc)]+ complex (not pictured, λmax = 550 nm, ε = 74 M−1 cm−1) has only a single smooth peak with no fine structure but an increased ε, while in the spectrum for each of the [Co1(OAc)]+ and [Co5(OAc)]+ complexes (see Fig. 10a) the major absorption peak is split with one two shoulders on the maximum absorption peak and (ε ~ 15–20 M−1 cm−1). These latter spectra are similar to those observed for the CoLCl2 complexes with ligands 5 and 6 previously published, which all have this major peak split in the same way and similarly small extinction coefficients.26 For Co5Cl2 λmax = 540 (24 M−1 cm−1) and 558 nm (21 M−1 cm−1) and for Co(6)Cl2 λmax = 546 (34 M−1 cm−1) and 568 nm (35 M−1 cm−1). The change in splitting pattern and extinction coefficient for [Co3(OAc)]+ indicate that the asymmetry of the single benzyl group of [Co3(OAc)]+ results in a different structure from all of the other Co2+ complexes. Unfortunately, we were unable to produce X-ray quality crystals of this sample to better understand what this structural change is.
The cyclen-based Co3+ complexes are confirmed as the expected low spin d6 according to sharp proton and carbon NMR spectra. Their electronic spectra are typical of octahedral Co3+ amine complexes.48 These spectra show the usual two absorption bands between 300 and 700 nm (with much higher extinction coefficients than the Co2+ complexes) that are generally associated with cis configuration CoN4 complexes.44 Fig. 9b shows the spectrum for [Co2(OAc)]2+, which is representative of all three Co3+ complexes.
As with Ni2+, the electronic spectra of octahedral Co3+ complexes can be used to estimate the ligand field strengths of the ligands, expressed as Δo.49,50 In this method, the energy of the lowest energy absorption band plus the Racah parameter (3800 cm−1 for Co3+)49–51 equals Δo. Since both [Co2(OAc)]2+ and [Co4(OAc)]2+ have their lowest energy absorption at 523 nm, they have the identical Δo = 22 920 cm−1. This similarity in Δo for both of these cyclen-based ligands was not quite as apparent in the Ni2+ complexes above, where the values of Δo differed by 123 cm−1. However, the value of Δo for [Co6(OAc)]2+, Δo = 23 524 cm−1, is somewhat larger, as was observed for the replacement of benzyl with methyl substituents for the Ni2+ complexes, above. Again, disruption of the preferred ligand configuration by the bulky benzyl groups may be explain the lower values of Δo in benzylated ligands.
Comparison of Δo for these three complexes is appropriate with the [CoLCl2]+ complexes of ligands 5 and 6,26 Δo = 19 430 cm−1 for Co(5)Cl2+; and Δo = 21 130 cm−1 for Co(6)Cl2+. Of course, the most appropriate comparison is the latter one, because all three of these ligands are based on the 12-membered cyclen ring. The increase in Δo in the present ligand 2, 4, and 6 cases may be due to two factors. The first is the change of the equatorial ligands from Cl to O donors; these O donors should be slightly stronger field ligands.52 The unbridged cyclen complex cis-[Co(cyclen)CO3]+ has been reported to have Δo = 22 670 cm−1.51 Here the macrocyclic ligand is forced to be cis by the chelating carbonate ligand. This complex has a very similar coordination environment to [Co2(OAc)]2+, [Co4(OAc)]2+, and [Co6(OAc)]2+.
The second reason for the higher Δo values for the present acetate complexes is the fact that the lowest energy band in the present ligand 2, 4, and 6 complexes is actually an overlapping pair E(1T1g) and A(1T1g) absorbances, which is why cis CoN4X2+ complexes only appear to have two total absorbance bands.44,48 [Co6Cl2] +, surprisingly, exhibits all three bands in a cis configuration complex, most likely due to larger-than-normal distortion of the octahedral geometry.26 Changing the two chloro ligands to an acetate ligand together with the addition of the one or two benzyl groups at the macrocycle N-donors leads to less severe distortion and a return to the usual two absorption bands. In terms of Δo calculations, the values for [Co2(OAc)]2+, [Co4(OAc)]2+, and [Co6(OAc)]2+ will be high compared to [Co6Cl2]+, because the Δo values are calculated for a broad peak which mixes in a higher energy absorbance, while the unique lowest energy absorbance peak was used for [Co6Cl2]+.
Electrochemical studies of nickel complexes
The cyclic voltammograms in acetonitrile of (a) [Ni4(OAc)]+ (representative also of [Ni2(OAc)]+, [Ni5(OAc)]+, and [Ni6(OAc)]+) and (b) [Ni1(OAc)]+ (representative also of [Ni3(OAc)]+) and are shown in Fig. 11. The redox potentials and peak separations of all six Ni2+ acetate complexes are listed in Table 4. The cyclam ligands 1 and 3 surprisingly give only irreversible oxidations to Ni3+ (Table 3 and Fig. 11b). This was unexpected since all three of the NiLCl2 complexes where L = 5–6 have reversible Ni2+/3+ couples as well as irreversible reductions assigned to Ni2+/+.27 The substitution of benzyl for methyl groups (for example changing ligand 5 to ligand 1) on cross-bridged ligand complexes of iron and manganese had minimal effects on the cyclic voltammetry of those complexes,2,4 so significant changes were not expected for these nickel complexes. However, all of the iron and manganese complexes referred to were dichloro complexes and so there may be some influence of the acetato ligand on the nickel complex properties in this work. Perhaps there is reactivity of the bound acetate ligand upon oxidation of the mono- and di-benzyl-cyclam nickel complexes that leads to the irreversible behaviour. In support of this idea is the known oxidation catalyst behaviour of the iron53 and manganese2,9 complexes of cross-bridged cyclams. Oxidative Ni3+ coordination to carbon ligands is also well-known,54–59 so perhaps modification of the benzyl pendant arms is occurring. The anodic peaks are nearly identical, +1.255 V for [Ni1(OAc)]+ and +1.277 V for [Ni3(OAc)]+, suggesting that the presence of either one or two benzyl groups has little effect on the ability of the metal ion to be oxidized.
Fig. 11.

Cyclic voltammograms for (a) [Ni4(OAc)]+ and (b) [Ni1(OAc)]+.
Table 4.
Redox potentials (vs. SHE) with peak separations for cobalt acetate complexes
| Co2+ complex | Eox (V) unassigned | E1/2 (V) Co2+/Co3+ | (Ea–Ec), mV | E1/2 (V) Co2+/Co3+ | (Ea–Ec), mV |
| [Co1(OAc)]+ | +1.226 | +0.648 | 75 | +0.392 | 167 |
| [Co3(OAc)]+ | +1.159 | +0.564 | 100 | +0.293 | 177 |
| [Co5(OAc)]+ | +1.415 | +0.536 | 208 | +0.246 | 220 |
| Co3+ complex | E1/2 (V) Red Co3+/Co2+ | (Ea–Ec), mV | E1/2 (V) Red Co2+/Co+ | (Ea–Ec), mV | |
| [Co2(OAc)]2+ | +0.014 | 109 | −0.640 | 178 | |
| [Co4(OAc)]2+ | +0.040 | 185 | −0.758 | 104 | |
| [Co6(OAc)]2+ | −0.144 | 107 | — | — | |
In contrast, the cyclen-based complexes (ligands 2, 4, and 6) and the dimethyl cyclam complex (ligand 5) all give reversible oxidation cycles for Ni2+/3+ (Table 3 and Fig. 11a). Clearly, the smaller cyclen ring better stabilises the small Ni3+ ion in these complexes, as oxidation occurs more easily than for the cyclam-based complexes. Apparently, the lower oxidation potential does not activate the process that leads to the irreversible nature of the cyclam-based benzyl-containing complexes. Among cyclen-based ligand complexes, the difference in oxidation potential between the mono- and di-benzyl ligands and the dimethyl ligand is minimal. Even though reversible oxidation is present, the irreversible reduction common to all three of the NiLCl2 complexes where L = 5–6 27 are not seen. An explanation could be that the hard oxygen donors of the acetate ligand do not stabilise soft Ni+ as well as chloride does.
Electrochemical studies of cobalt complexes
The cyclic voltammograms in acetonitrile of (a) [Co2(OAc)]+ (representative also of [Co4(OAc)]+) and (b) [Co3(OAc)]+ (representative also of [Co1(OAc)]+) are shown in Fig. 12. The redox potentials and peak separations of all four cobalt acetate complexes can be found in Table 4. The cyclen-based compounds are initially in the Co3+ oxidation state, while the cyclam-based compounds are initially in the Co2+ oxidation state.
Fig. 12.

Cyclic voltammograms for (a) [Co2(OAc)]2+ and (b) [Co3(OAc)]+.
The Co3+ cyclen-based complex voltammograms are relatively simple, with quasi-reversible reductions. For [Co2(OAc)]2+ two such reductions occur at E1/2 = +0.014 V (ΔE = 109 mV) and E1/2 = −0.640 V (ΔE = 178 mV). For [Co4(OAc)]2+ the corresponding reductions occur at E1/2 = +0.040 V (ΔE = 185 mV) and E1/2 = −0.758 V (ΔE = 104 mV). For Co(6)(OAc)2+, only one reduction is observed at E1/2 = −0.144 V (ΔE = 107 mV). The reductions can be assigned as the Co3+/2+ and Co2+/+ couples, with the spacing between them in the order of 650–800 mV, which corresponds well to the spacing of MnLCl2 (L = 5 and 6) which both have reversible Mn2+/3+ and Mn3+/4+ couples nearly 750 mV apart.2 The Co3+/2+ reduction of [Co6(OAc)]2+ complex occurs at the most negative potential, −0.144 V, and uniquely among this set of cyclen complexes, a second reduction is not observed. The benzyl pendant arms of 2 and 4 help stabilise and/or enclose Co+, whereas this stabilization is not present in [Co6(OAc)]2+, which does not reach the Co+ oxidation state in our experiments. Interestingly, the CoLCl2 (L = 5–6) complexes all have similar Co2+/3+ redox couples with E1/2 values near 0.00 V vs. SHE. However, these dichloro complexes have only irreversible reductions to Co+ at much lower potentials, below −2.00 V.26 The presence of only one negatively charged acetate, rather that two negatively charged chlorides, as well as at least one benzyl pendant arm, makes the reduction to Co+ both more accessible and more reversible for the ligand 2 and 4 complexes.
The Co2+ cyclam-based complexes have quite different cyclic voltammograms (see Fig. 11). Starting from Co2+, the expected oxidation to Co3+ is seen for all three ligand complexes (1, 3, and 5). This initial oxidation is at E1/2 = +0.392 V (ΔE = 167 mV) for [Co1(OAc)]+; at E1/2 = +0.293 V (ΔE = 177 mV) for [Co3(OAc)]+; and at E1/2 = +0.246 V (ΔE = 220 mV) for [Co5(OAc)]+. These potentials occurring 200–300 mV more positive than the Co2+/3+ redox couples of the equivalent cyclen-based ligands (2, 4, and 6, see above) complexes makes sense, as the larger cyclam ring would not stabilise the smaller Co3+ ion as well, resulting in a less favoured oxidation.
The cobalt(II) cyclam-based complex voltammograms also contain two additional waves. First, there is an additional reversible oxidation approximately only ~250 mV more positive for all three complexes: E1/2 = +0.648 V (ΔE = 75 mV) for [Co1(OAc)]+; E1/2 = +0.564 V (ΔE = 100 mV) for [Co3(OAc)]+; and E1/2 = +0.536 V (ΔE = 208 mV) for [Co5(OAc)]+. The proximity to the initial oxidation of this additional redox couple makes it unlikely to be the result of a Co3+/4+ oxidation. More likely is the oxidation from Co2+ to Co3+ of a second species with a different ligand set in the same solution. This behaviour has been observed for the Co2+ complexes CoLCl2 (L = 5–6) in acetonitrile, where cyclic voltammetry in TBAPF6 supporting electrolyte gave complex features assigned to multiple species in solution which differ by the number of bound chloride ligands.26 It is possible that either the coordination mode of the acetate ligand is changing, producing two different species for the [Co1(OAc)]+, [Co3(OAc)]+, and [Co5(OAc)]+ complexes, or there is an equilibrium mixture of bound and free acetate ligand complexes. These different species would have different redox potentials and might give rise to the two closely spaced reversible redox couples observed. Finally, an additional feature for all three cyclam-based complexes is an irreversible oxidation at greater than 1.1 V vs. SHE. This feature has not been fully assigned, but might either be due to an oxidation to Co4+, or a ligand-based oxidation process.
Anti-HIV activity
AMD310017,60–62 and the high potency CXCR4 antagonists that we have developed16,18–20 are bismacrocyclic with an aryl (xylyl) linker. Our previously collected data indicates that monomacrocyclic compounds will also have affinity for the receptor which can result in anti-HIV activity, but this will be lower than for the bismacrocyclic derivatives. The discussion relates the anti-HIV activity to CXCR4 binding as this link has been borne out in all of our previous research. The main reason for synthesizing the monomacrocyclic compounds (metal complexes of 1–6) was to utilise them as simpler structural analogues to allow us to obtain X-ray structural data that models aspartate or glutamate coordination to the metal centre. We do not anticipate taking these compounds into further biological evaluation or in vivo studies as they have greater potential for off-target binding. However, it is still of interest to determine their antiviral activity and investigate the structure activity relationships for this subset of compounds.
Anti-HIV activity measurements in MT-4 cells were based on the viability of cells that had been infected or not infected with the HIV-1 (strain IIIB) and exposed to various concentrations of the test compound.63 Data are presented in Table 5. Viral strain IIIB is an X4 viral strain that solely uses the CXCR4 receptor as a co-receptor for cell entry, it does not use CCR5. Our previous studies show that activity in the anti-HIV assays usually indicates CXCR4 binding.
Table 5.
Anti-HIV IC50 values of the evaluated compounds using HIV-1 (viral strain IIIB)
| Complex | Ligand | Anti-HIV activity (IC50) [μM] |
|---|---|---|
| Co2+1 Ni2+1 |
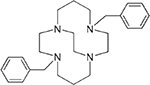 |
1.82 0.49 |
| Co3+2 Ni2+2 |
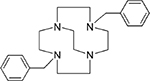 |
>100 0.59 |
| Co2+3 Ni2+3 |
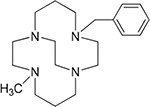 |
>100 0.80 |
| Co3+4 Ni2+4 |
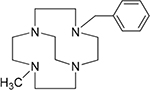 |
>100 0.55 |
| Co2+5 Ni2+5 |
 |
>100 3.48 |
| Co3+6 Ni2+6 |
 |
>100 7.69 |
| (Co2+)27 |  |
0.690 |
The free chelators showed IC50 values of greater than 100 μM indicating no measurable anti-HIV activity for these compounds.18 This is consistent with previously analysed free macrocyclic chelators in which the hydrogen-bonding potential of the chelator has been disrupted by alkylation and they are only activated on inclusion of the metal centre to give the potential for coordinate bond formation.
AMD3100 metal complexes have anti-HIV activity in this order: Zn2 > Ligand = Ni2 > Cu2 > Co2 ≫Pd2 according to the literature.62 In our study of cross-bridged analogues of AMD3100, we have shown that Cu2+, Zn2+, and Ni2+ complexes all have low nanomolar IC50 values against the HIV1 viral strain, with the exact ordering depending on how the specific ligand is designed.16,18–20 Here, we extend our studies to include cobalt. For the nickel(II) complexes previously studied, bis-macrocyclic bridged complexes with ethylene bridged structures were generally of lower anti-HIV potency than the AMD3100 complex.19 It was also shown that the binding of nickel(II) can be used to increase potency of unbridged AMD3100 derivatives that are functionalised at the linking xylyl group (increasing anti-HIV potency from 295 nM to 95 nM in one case).64
As described above, the smaller cyclen cross-bridged ligands select for Co3+, while the large cyclam cross-bridged ligands appear to stabilise the larger Co2+. Unlike Cu2+, Zn2+, and Ni2+ complexes,16,18–20 the Co2+/Co3+ complexes synthesised and screened here, do not appear to have very strong affinity for the CXCR4 receptor, by analogy with thethe X4 strain anti-HIV activity reported in Table 5. The only mononuclear cobalt complex with a measurable IC50 was [Co1(OAc)]+ with IC50 = 1.82 μM, which is not particularly potent compared to our previous Cu2+, Zn2+, and Ni2+ complexes.16,18–20 Of course, we do anticipate losing some potency as a result of having only one metal centre in the monomacrocyclic ligand, but all of the Ni2+ complexes show affinity in the low or sub micromolar range. Dinuclear (Co2+)27, as expected, shows a more potenti anti-HIV activity than any of the mononuclear cobalt complexes. Its IC50 = 0.690 μM, which is about 2.5-fold more efficient than [Co1(OAc)]+. This gain in efficiency for the dinuclear complex is actually somewhat lower than expected. For example, our published dinuclear nickel complex,19 most analogous with Ni2+3, exhibited an anti-HIV IC50 = 0.014 μM. This value is 57-fold more efficient than Ni2+3. Clearly, cobalt complexes, even in dinuclear compounds, are not as favourable for continued development of anti-HIV CXCR4 antagonists.
It is well-known that the substitution kinetics of low spin, octahedral Co3+ complexes are very slow, which would explain the poor binding of the Co3+ complexes. An explanation for the reduced activity of the Co2+ complexes may simply lie in the Irving–Williams series for the binding strength of first row transition metals that predict weaker binding from right to left on the first transition row.65
Consistent with the Irving–Williams series is the enhanced binding affinity of the Ni2+ complexes over those of Co2+. Each Ni2+ complex tested gave a measurable anti-HIV activity, with the ligand 1–4 complexes demonstrating sub-micromolar activity, with the ligand 5–6 complexes approximately an order of magnitude less potent. Perhaps this disparity can be attributed to the presence of at least one benzyl group in ligands 1–4, which has been shown to be crucial to the high CXCR4 affinity of AMD3100.17
Relating the anti-HIV activity of the Ni2+ complexes to the coordination modes observed in the crystal structures above, is not clear-cut, as ligand sets (1–4 and 5–6) give structures of both the iso-bidentate and monodentate/H2O coordination modes. However, we should keep in mind that water molecules and the aspartate carboxylate groups would be available at the site of CXCR4 binding, which would allow a given complex to select its most favourable binding mode in the protein environment.
Conclusions
Six mono-macrocyclic cross-bridged tetraazamacrocyclic ligands have been complexed to Co2+/Co3+ and Ni2+ concurrently with an acetate anion, which serves as a model carboxylate ligand for the aspartate side chains shown to interact with xylyl-bridged bis-cyclam CXCR4 antagonists on binding to the receptor. X-ray crystal structures of three of the Co2+/Co3+ and five of the Ni2+ complexes were obtained, to complement recently published analogues. All of these structures were examined to learn about preferences for Co2+/Co3+ and Ni2+ macrocycle complexes in binding carboxylate ligands, which could potentially be applied to CXCR4 antagonist design.
The cross-bridged Co3+ complexes studied were all complexed to the smaller cyclen-based ligands and were found only to coordinate to acetate in a chelating iso-bidentate manner, which is likely due the short M–N bond lengths and the resulting slightly distorted octahedral coordination geometries have near 90° Neq–M–Neq bond angles that allow only room for a single chelated acetate opposite the cross-bridge. As antagonists, these complexes are poor, potentially due to the slow substitution kinetics of Co3+. These complexes were diamagnetic and had electronic spectra typical of a low spin, octahedral Co3+ ion. Their cyclic volatammograms were simple, with quasi-reversible reductions to Co2+ and Co+.
The cross-bridged Co2+ complexes studied were all complexed to the larger cavity cyclam-based ligands and the only structurally characterised example, [Co5(OAc)(H2O)]+, binds acetate in a monodentate fashion with a water molecule completing the coordination sphere. The larger Co2+ cation formed a much smaller Neq–M–Neq bond angle of ~83°, which allowed the monodentate acetate/water ligands room to bind equatorially. The only measurable affinity to CXCR4 of any of the cobalt complexes was from this group as perhaps the faster substitution kinetics of high spin Co2+ allow binding. This high spin nature was confirmed for all three cyclam-based ligand complexes by electronic spectra typical of this species. Cyclic voltammetry revealed complex behaviour with multiple oxidations which is consistent with the high spin species leading to multiple different complexes in acetonitrile solution incorporating bound solvent molecule(s).
Stable Ni2+ complexes were formed with both cross-bridged cyclam- and cyclen-based ligands. Most of these complexes were found to include a monodentate acetate/water ligand pair equatorially opposite of the ligand cross-bridge, as was observed for the larger Co2+ ion. Here, the Neq–M–Neq bond angles were always <85.6°. However, coordination sphere flexibility was observed as two complexes, both of the smaller cyclen-based ligands 4 and 6, demonstrated iso-bidentate chelation of acetate and Neq–M–Neq bond angles >87°. In comparison to unbridged tetraazamacrocycle Ni2+ complexes of acetate, it was discovered that the more flexible non-bridged macrocycles can expand equatorially and produce larger Neq– M–Neq bond angles (up to ~108°) which strongly selected for the equatorial coordination of acetate in the bidentate chelating mode. Electronic spectra of all Ni2+ cross-bridged complexes gave typical distorted octahedral behaviour, allowing for the calculation of Δo, which was larger for cyclen-based ligands and was reduced as benzyl substituents were added. Cyclic voltammograms of the cross-bridged Ni2+ complexes mostly showed reversible Ni2+/Ni3+ redox couples, although cyclam-based ligands with at least one benzyl substituent made the oxidation irreversible.
All of the Ni2+ complexes showed micromolar activity as CXCR4 antagonists, showing that this ion is a better choice than Co2+/Co3+ for this application. Only one mononuclear cobalt(II) complex exhibited measurable CXCR4 activity. However, the mono-macrocyclic antagonists investigated in this work do not rival the bis-macrocyclic antagonists we have previously identified which can bind CXCR4 through interaction with two aspartate residue side chains.19 The first dicobalt(II) bis-macrocyclic complex we have prepared showed only a slight improvement over its mononuclear analogue, a surprisingly small improvement based on data from other metal ions. This result will be checked with the synthesis of other dicobalt analogues, but suggests that cobalt(II) will not be the metal of choice for out high-efficiency bismacrocyclic CXCR4 antagonist program. Our future work will involve further investigation of a wider range of Ni2+ bis-macrocyclic compounds as CXCR4 antagonists.
Supplementary Material
Acknowledgements
T. J. H. acknowledges the Health Research award for project number HR13-157, from the Oklahoma Center for the Advancement of Science and Technology. This project was supported by the National Center for Research Resources and the National Institute of General Medical Sciences of the National Institutes of Health through Grant Number 8P20M103447. T. J. H. acknowledges the Research Corporation (CC6505) for funding. T. J. H. also acknowledges the Henry Dreyfus Teacher-Scholar Awards Program for support of this work. We gratefully acknowledge the Daisy Appeal Charity for funding (Grant: DAhul0211) and fellowship funding for BPB, and the University of Hull for infrastructure support. This work, in part, was supported by funding of the KU Leuven (GOA/10/014, PF/10/018 and C22/17/008) and the Foundation of Scientific Research (FWO no. G.0485.08 and G.0528.12).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic supplementary information (ESI) available: Tables from X-ray crystallographic studies. CCDC 1566342, 1566343, 1566345, 1566346, 1567486–1567489, and 1567495. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04728f
Notes and references
- 1.Hubin TJ, Synthesis and coordination chemistry of topologically constrained azamacrocycles, Coord. Chem. Rev, 2003, 241, 27–46. [Google Scholar]
- 2.Hubin TJ, McCormick JM, Collinson SR, Buchalova M, Perkins CM, Alcock NW, Kahol PK, Raghunathan A and Busch DH, New iron(II) and manganese(II) complexes of two ultra-rigid, cross-bridged tetraazamacrocycles for catalysis and biomimicry, J. Am. Chem. Soc, 2000, 122(11), 2512–2522. [Google Scholar]
- 3.Collinson SR, Alcock NW, Hubin TJ and Busch DH, Synthesis and characterization of the novel bridged ligand 5,8-dimethyl-1,5,8,12-tetraazabicyclo 10.3.2 heptadecane and its complexes with iron(II) and manganese(II) ions, J. Coord. Chem, 2001, 52(4), 317–331. [Google Scholar]
- 4.Hubin TJ, McCormick JM, Collinson SR, Alcock NW, Clase HJ and Busch DH, Synthesis and X-ray crystal structures of iron(II) and manganese(II) complexes of unsubstituted and benzyl substituted cross-bridged tetraazamacrocycles, Inorg. Chim. Acta, 2003, 346, 76–86. [Google Scholar]
- 5.Wilson KR, Cannon-Smith DJ, Burke BP, Birdsong OC, Archibald SJ and Hubin TJ, Synthesis and structural studies of two pyridine-armed reinforced cyclen chelators and their transition metal complexes, Polyhedron, 2016, 114, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matz DL, Jones DG, Roewe KD, Gorbet MJ, Zhang Z, Chen ZQ, Prior TJ, Archibald SJ, Yin GC and Hubin TJ, Synthesis, structural studies, kinetic stability, and oxidation catalysis of the late first row transition metal complexes of 4,10-dimethyl-1,4,7,10-tetraazabicyclo[6.5.2] pentadecane, Dalton Trans, 2015, 44(27), 12210–12224. [DOI] [PubMed] [Google Scholar]
- 7.Shircliff AD, Wilson KR, Cannon-Smith DJ, Jones DG, Zhang Z, Chen ZQ, Yin GC, Prior TJ and Hubin TJ, Synthesis, structural studies, and oxidation catalysis of the manganese(II), iron(II), and copper(II) complexes of a 2-pyridylmethyl pendant armed side-bridged cyclam, Inorg. Chem. Commun, 2015, 59, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones DG, Wilson KR, Cannon-Smith DJ, Shircliff AD, Zhang Z, Chen ZQ, Prior TJ, Yin GC and Hubin TJ, Synthesis, Structural Studies, and Oxidation Catalysis of the Late-First-Row-Transition-Metal Complexes of a 2-Pyridylmethyl Pendant-Armed Ethylene Cross-Bridged Cyclam, Inorg. Chem, 2015, 54(5), 2221–2234. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Coats KL, Chen ZQ, Hubin TJ and Yin GC, Influence of Calcium(II) and Chloride on the Oxidative Reactivity of a Manganese(II) Complex of a Cross-Bridged Cyclen Ligand, Inorg. Chem, 2014, 53(22), 11937–11947. [DOI] [PubMed] [Google Scholar]
- 10.Busch DH, Collinson SR and Hubin TJ, Catalysts and methods for catalytic oxidation, US Pat, 6906189, 2005. [Google Scholar]
- 11.Brewer SM, Wilson KR, Jones DG, Reinheimer EW, Archibald SJ, Prior TJ, Ayala MA, Foster AL,Hubin TJ and Green KN, Increase of Direct C-C Coupling Reaction Yield by Identifying Structural and Electronic Properties of High-Spin Iron Tetraazamacrocyclic Complexes, Inorg. Chem, 2018, 57, 8890–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sprague JE, Peng Y, Fiamengo AL, Woodin KS, Southwick EA, Weisman GR, Wong EH, Golen JA, Rheingold AL and Anderson CJ, Synthesis, Characterization and In Vivo Studies of Cu(II)-64-Labeled Cross-Bridged Tetraazamacrocycle-amide Complexes as Models of Peptide Conjugate Imaging Agents, J. Med. Chem, 2007, 2527–2535. [DOI] [PubMed] [Google Scholar]
- 13.Miranda CS, Burke BP, Lee RE, Nigam S, Clemente G, Thompson JA, Ruest T, D’huys T, Schols D, Greenman J, Cawthorne C and Archibald SJ, CXCR4 chemokine receptor imaging: evaluation and validation of a new configurationally restricted tetraazamacrocyclic CXCR4 antagonist, 64Cu-CB-bicyclam, Eur. J. Nucl. Med. Mol. Imaging, 2016, 43(Suppl 1), S1–S734. [Google Scholar]
- 14.Hubin TJ, Alcock NW and Busch DH, The square-pyramidal Pd-II complex of a cross-bridged tetraazamacrocycle, Acta Crystallogr., Sect. C: Cryst. Struct. Commun, 1999, 55, 1404–1406. [Google Scholar]
- 15.Hubin TJ, McCormick JM, Alcock NW, Clase HJ and Busch DH, Crystallographic characterization of stepwise changes in ligand conformations as their internal topology changes and two novel cross-bridged tetraazamacrocyclic Copper(II) complexes, Inorg. Chem, 1999, 38(20), 4435–4446. [DOI] [PubMed] [Google Scholar]
- 16.Khan A, Nicholson G, Greenman J, Madden L, McRobbie G, Pannecouque C, De Clercq E, Silversides JD, Ullom R, Maples DL, Maples RD, Hubin TJ and Archibald SJ, Binding optimization through coordination chemistry: CXCR4 chemokine receptor antagonists from ultrarigid metal complexes, J. Am. Chem. Soc, 2009, 131, 3416–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Clercq E, The bicyclam AMD3100 story, Nat. Rev. Drug Discovery, 2003, 2, 581–587. [DOI] [PubMed] [Google Scholar]
- 18.Valks GC, McRobbie G, Lewis EA, Hubin TJ, Hunter TM, Sadler PJ, Pannecouque C, De Clercq E and Archibald SJ, Configurationally restricted bismacrocyclic CXCR4 receptor antagonists, J. Med. Chem, 2006, 49(21), 6162–6165. [DOI] [PubMed] [Google Scholar]
- 19.Smith R, Huskens D, Daelemans D, Mewis RE, Garcia CD, Cain AN, Carder Freeman TN, Pannecouque C, De Clercq E, Schols D, Hubin TJ and Archibald SJ, CXCR4 chemokine receptor antagonists: nickel(II) complexes of configurationally restricted macrocycles, Dalton Trans, 2012, 41, 11369–11377. [DOI] [PubMed] [Google Scholar]
- 20.Maples RD, Cain AN, Burke BP, Silversides JD, Mewis R, D′huys T, Schols D, Linder DP, Archibald SJ and Hubin TJ, Aspartate-based CXCR4 chemokine receptor binding of cross-bridged tetraazamacrocyclic copper(II) and zinc(II) complexes, Chem. – Eur. J, 2016, 22, 12916–12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.A. M. E. Fulton, Chemokine Receptors in Cancer, Humana Press, New York, 2009. [Google Scholar]
- 22.Khan A, Greenman J and Archibald SJ, Small Molecule CXCR4 Chemokine Receptor Antagonists: Developing Drug Candidates, Curr. Med. Chem, 2007, 14, 2257–2277. [DOI] [PubMed] [Google Scholar]
- 23.Soibinet M, Dechamps-Olivier I, Guillon E, Barbier J-P, Chuburu F, Le Baccon M and Handel H, XAS, ESR and Potentiometric Studies of Three Dinuclear N,N’-para-Xylylenebis(tetraazamacrocycle)copper(II) Complexes - X-ray Crystal Structure of [N,N’-p-Xylylenebis(cyclen)]copper(II), Eur. J. Inorg. Chem, 2003, 1984–1994. [Google Scholar]
- 24.Weisman GR, Wong EH, Hill DC, Rogers ME, Reed DP and Calabrese JC, Synthesis and transitionmetal complexes of new cross-bridged tetraamine ligands, Chem. Commun, 1996, 947–948. [Google Scholar]
- 25.Matz DL, Jones DG, Roewe KD, Gorbet MJ, Zhang Z, Chen ZQ, Prior TJ, Archibald SJ, Yin GC and Hubin TJ, Synthesis, structural studies, kinetic stability, and oxidation catalysis of the late first row transition metal complexes of 4,10-dimethyl-1,4,7,10-tetraazabicyclo 6.5.2 pentadecane, Dalton Trans, 2015, 44(27), 12210–12224. [DOI] [PubMed] [Google Scholar]
- 26.Hubin TJ, Alcock NW, Clase HJ, Seib LL and Busch DH, Synthesis, characterization, and X-ray crystal structures of cobalt(II) and cobalt(III) complexes of four topologically constrained tetraazamacrocycles, Inorg. Chim. Acta, 2002, 337, 91–102. [Google Scholar]
- 27.Hubin TJ, Alcock NW, Clase HJ and Busch DH, Potentiometric titrations and nickel(II) complexes of four topologically constrained tetraazamacrocycles, Supramol. Chem, 2001, 13(2), 261–276. [Google Scholar]
- 28.Hubin TJ, Walker AN, Davilla DJ, Carder Freeman TN, Epley BM, Hasley TR, Amoyaw PNA, Jain S, Archibald SJ, Prior TJ, Krause JA, Oliver AG,Tekwani BL and Khan MOF, Tetraazamacrocyclic derivatives and their metal complexes as antileishmanial leads, submitted 2018. [DOI] [PMC free article] [PubMed]
- 29.Blessing RH, An empirical correction for absorption anisotropy, Acta Crystallogr., Sect. A: Found. Crystallogr, 1995, 51, 33–38. [DOI] [PubMed] [Google Scholar]
- 30.Sheldrick GM, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv, 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J and De Clercq E, Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds, J. Virol. Methods, 1988, 20, 309–321. [DOI] [PubMed] [Google Scholar]
- 32.Pannecouque C, Daelemans D and De Clercq E, Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: revisited 20 years later, Nat. Protoc, 2008, 3, 427–434. [DOI] [PubMed] [Google Scholar]
- 33.Liang X, Parkinson JA, Weishaupl M, Gould RO, Paisey SJ, Park H-S, Hunter TM, Blindauer CA,Parsons S and Sadler PJ, Structure and dynamics of metallomacrocycles: recognition of zinc xylyl-bicyclam by an HIV coreceptor, J. Am. Chem. Soc, 2002, 124, 9105–9112. [DOI] [PubMed] [Google Scholar]
- 34.Lichty J, Allen SM, Grillo AI, Archibald SJ and Hubin TJ, Synthesis and characterization of the cobalt(III) complexes of two pendant-arm cross-bridged cyclams, Inorg. Chim. Acta, 2004, 357(2), 615–618. [Google Scholar]
- 35.Bosnich B, Poon CK and Tobe ML, Complexes of Cobalt(III) with a Cyclic Tetradentate Secondary Amine, Inorg. Chem, 1965, 4, 1102–1108. [Google Scholar]
- 36.Shannon RD, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr, 1976, 32(5), 751–767. [Google Scholar]
- 37.Hubin TJ, unpublished data. 2015.
- 38.Wallick JL, Riordan CG and Yap GPA, Acetate and acetamide complexes of [Ni(Me 4 [12]aneN 4)]PF 6 : a tale of two ligands, Acta Crystallogr., Sect. C: Struct. Chem, 2014, 70, 640–643. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Ren Y-W, Zhang J-H and Yang P, Crystal structure of a nickel(II) complex with macrocyclic tetraamine, J. Chem. Crystallogr, 2004, 34, 409–410. [Google Scholar]
- 40.Tina M, Hunter TM, McNae IW, Simpson DP, Smith AM, Moggach S, White F, Walkinshaw MD, Parsons S and Sadler PJ, Configurations of Nickel– Cyclam Antiviral Complexes and Protein Recognition, Chem. – Eur. J, 2007, 13, 40–50. [DOI] [PubMed] [Google Scholar]
- 41.Wadas TJ, Wong EH, Weisman GR and Anderson CJ, Copper Chelation Chemistry and its Role in Copper Radiopharmaceuticals, Curr. Pharm. Des, 2007, 13, 3–16. [DOI] [PubMed] [Google Scholar]
- 42.Woodin KS, Heroux KJ, Boswell CA, Wong EH, Weisman GR, Niu WJ, Tomellini SA, Anderson CJ, Zakharov LN and Rheingold AL, Kinetic inertness and electrochemical behavior of copper(II) tetraazamacrocyclic complexes: Possible implications for in vivo stability, Eur. J. Inorg. Chem, 2005, (23), 4829–4833. [Google Scholar]
- 43.Drago RS, Physical Methods for Chemists, Saunders College Publishing-Harcourt Brace Jovanovich, Ft. Worth, 2nd edn, 1992. [Google Scholar]
- 44.A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 2nd edn, 1984. [Google Scholar]
- 45.Martin LY, Ph.D. Thesis, The Ohio State University, 1974. [Google Scholar]
- 46.Kalligeros GA and Blinn EL, Strained five- and six-coordinated macrocyclic nickel(II) complexes, Inorg. Chem, 1972, 11, 1145–1148. [Google Scholar]
- 47.Martin LY, Sperati CR and Busch DH, The spectrochemical properties of tetragonal complexes of high spin nickel(II) containing macrocyclic ligands, J. Am. Chem. Soc, 1977, 99, 2968–2981. [Google Scholar]
- 48.Cotton FA and Wilkinson G, Advanced Inorganic Chemistry, Wiley & Sons, New York, 5th edn, 1988. [Google Scholar]
- 49.Wentworth RAD and Piper TS, A Crystal Field Model for the Spectral Relationships in Monoacidopentaammine and Diacidotetraammine Complexes of Cobalt(III), Inorg. Chem, 1965, 4, 709–714. [Google Scholar]
- 50.Wentworth RAD and Piper TS, An Empirical Molecular Orbital Treatment of Tetragonal Ammine Complexes of Cobalt(III), Inorg. Chem, 1965, 4, 1524–1526. [Google Scholar]
- 51.Hung Y, Martin LY, Jackels SC, Tait AM and Busch DH, Ring size effects among metal complexes with macrocyclic ligands: synthesis, stereochemistry, spectrochemistry, and electrochemistry of cobalt(III) complexes with unsubstituted, saturated tetraaza macrocycles, J. Am. Chem. Soc, 1977, 99, 4029–4039. [Google Scholar]
- 52.Miessler GL, Fischer PJ and Tarr DA, Inorganic Chemistry, Pearson Education, Inc., Upper Saddle River, NJ, 5th edn, 2014. [Google Scholar]
- 53.Feng Y, England J and Que L, Iron-Catalyzed Olefin Epoxidation and cis-Dihydroxylation by Tetraalkylcyclam Complexes: the Importance of cis-Labile Sites, ACS Catal, 2011, 1(9), 1035–1042. [Google Scholar]
- 54.Everson DA, Shrestha R and Weix DJ, Nickel-Catalyzed Reductive Cross-Coupling of Aryl Halides with Alkyl Halides, J. Am. Chem. Soc, 2010, 132, 920–921. [DOI] [PubMed] [Google Scholar]
- 55.Biswas S and Weix DJ, Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides, J. Am. Chem. Soc, 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joshi-Pangu A, Wang C-Y and Biscoe MR, Nickel-Catalyzed Kumada Cross-Coupling Reactions of Tertiary Alkylmagnesium Halides and Aryl Bromides/Triflates, J. Am. Chem. Soc, 2011, 133, 8478–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu X, Yang T, Wang S, Xu H and Gong H, Nickel-Catalyzed Reductive Cross-Coupling of Unactivated Alkyl Halides, Org. Lett, 2011, 13, 2138–2141. [DOI] [PubMed] [Google Scholar]
- 58.Dai Y, Zang Z, You H and Gong H, Ni-Catalyzed Reductive Allylation of Unactivated Alkyl Halides with Allylic Carbonates, Chem. – Eur. J, 2017, 18(3), 808–812. [DOI] [PubMed] [Google Scholar]
- 59.Xu H, Zhao C, Qian Q, Deng W and Gong H, Nickel-catalyzed cross-coupling of unactivated alkyl halides using bis (pinacolato)diboron as reductant, Chem. Sci, 2013, 4, 4022–4029. [Google Scholar]
- 60.Bridger GJ, Skerlj RT, Thornton D, Padmanabhan S, Martellucci SA, Henson GW, Abrams MJ, Yamamoto N, De Vreese K, Pauwels R and De Clercq E, Synthesis and structure-activity relationships of phenylenebis(methylene)-linked bis-tetraazamacrocycles that inhibit HIV replication–effects of macrocyclic ring size and substitution on the aromatic linker, J. Med. Chem, 1995, 38, 366–378. [DOI] [PubMed] [Google Scholar]
- 61.Gerlach LO, Jakobsen JS, Jensen KP, Rosenkilde MR, Skerlj RT, Ryde U, Bridger GJ and Schwartz TW, Metal ion enhanced binding of AMD3100 to Asp(262) in the CXCR4 receptor, Biochemistry, 2003, 42, 710–717. [DOI] [PubMed] [Google Scholar]
- 62.Este JA, Cabrera C, De Clercq E, Struyf S, Van Damme J, Bridger G, Skerlj RT, Abrams MJ, Henson G, Gutierrez A, Clotet B and Schols D, Activity of different bicyclam derivatives against human immunodeficiency virus depends on their interaction wiht the CXCR4 chemokine receptor, Mol. Pharmacol, 1999, 55, 67–73. [DOI] [PubMed] [Google Scholar]
- 63.Harada S, Koyanagi Y and Yamamoto N, Science, 1985, 229, 563–566. [DOI] [PubMed] [Google Scholar]
- 64.Poty S, Désogère P, Goze C, Boschetti F, D’huys T, Schols D, Cawthorne C, Archibald SJ, Maëckee HR and Denat F, New AMD3100 derivatives for CXCR4 chemokine receptor targeted molecular imaging studies: synthesis, anti-HIV-1 evaluation and binding affinities, Dalton Trans, 2015, 44, 5004–5016. [DOI] [PubMed] [Google Scholar]
- 65.Irving H and Williams RJP, The stability of transitionmetal complexes, J. Chem. Soc, 1953, 3192–3210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.