

Abstract
Infectious diseases are caused by microorganisms belonging to the class of bacteria, viruses, fungi, or parasites. These pathogens are transmitted, directly or indirectly, and can lead to epidemics or even pandemics. The resulting infection may lead to mild-to-severe symptoms such as life-threatening fever or diarrhea. Infectious diseases may be asymptomatic in some individuals but may lead to disastrous effects in others. Despite the advances in medicine, infectious diseases are a leading cause of death worldwide, especially in low-income countries. With the advent of mathematical tools, scientists are now able to better predict epidemics, understand the specificity of each pathogen, and identify potential targets for drug development. Artificial intelligence and its components have been widely publicized for their ability to better diagnose certain types of cancer from imaging data. This chapter aims at identifying potential applications of machine learning in the field of infectious diseases. We are deliberately focusing on key aspects of infection: diagnosis, transmission, response to treatment, and resistance. We are proposing the use of extreme values as an avenue of interest for future developments in the field of infectious diseases. This chapter covers a series of applications selectively chosen to showcase how artificial intelligence is moving the field of infectious disease further and how it helps institutions to better tackles them, especially in low-income countries.
Keywords: Infectious diseases, Machine learning, Epidemics, Antibiotic resistance, Prediction, Transmission
Preamble on infectious diseases
Infectious diseases are caused by pathogenic microorganisms, such as bacteria, viruses, parasites, or fungi. The diseases can be symptomatic or asymptomatic. Certain infectious diseases such as human immunodeficiency virus (HIV) can be fairly asymptomatic but can lead to disastrous consequences after few years if uncontrolled (https://www.who.int/topics/infectious_diseases/en/). The spread of infectious diseases varies from microorganisms to microorganisms. For instance, certain viruses such as HIV are only transmitted upon close physical contacts (sexual transmission or blood contact) while influenza virus infection is transmitted by emitted droplets following sneezing, coughing, or speaking, within few meters of distance. Zoonotic diseases are infectious diseases of animals that can cause disease when transmitted to humans.
In the 20th-century infectious diseases were responsible for the largest number of premature death and disability worldwide. The Spanish flu occurred in the beginning of the previous century (Taubenberger and Morens, 2006; https://www.cdc.gov/features/1918-flu-pandemic/index.html). It is estimated that one-third of the world’s population (500 million individuals) was infected and has symptoms during the 1918–19 pandemic (Fig. 1A). The disease was one of the deadliest of all influenza pandemics. It was estimated that at least 50 million individuals died following the infection. The impact of this pandemic was not restricted to the first quarter of the 20th century since almost all cases of influenza A were caused by mutated versions of the 1918 virus. While we will not cover the virologic or immunological aspect of influenza infection, it is important to understand the purpose of this chapter why the pandemic occurred. The 1918 flu pandemic happened during World War I where proximity, bad hygiene, and unusual mass movement (troops and population) helped the spread of the virus. Even the United Stated reported more than 600,000 death in its country despite the distance. Many of the countries involved in the war “failed” to communicate on the death toll caused by influenza. This was purposely kept silence in order to sustain public morale. While this could be understood on a military aspect, it has deadly consequences as the virus would come in other waves. At that time, viruses were not known yet and diagnostic, prevention and treatments were very limited. As such, people would suffer from influenza virus itself (flu illness) and its consequences such as lung infection by bacteria (pneumonia) in susceptible individuals. This shows how poor communication and wrong usage of pandemics data could affect millions of lives. Since then, progresses have been made in order to follow influenza A pandemics. Since 1952, the World Health Organization’s Global Influenza Surveillance and Response System (GISRS) have been monitoring the evolution of influenza viruses. It also serves as a global alert mechanism for emerging viruses with pandemic potential as observed in 1918. We now better understand the factors that influence transmission (Fig. 1B). Influenza is just one of the various pandemics we have been through. In fact, besides influenza, smallpox, tuberculosis, and cholera are constant threats (Holmes et al., 2017). Improving the hygiene conditions and vaccination campaigns have been very effective means to reduce the spread of infections. There are different cases of viral spread, for instance, there is constant follow-up on polio cases as three countries still report cases while WHO has the mission to eradicate it completely. The 21st century has already seen emerging pandemic infectious such as SARS (severe acute respiratory syndrome), MERS (Middle East respiratory syndrome), Ebola, and Zika viruses. By controlling infections, we can reduce premature death as well as infection-driven diseases such as cirrhosis (hepatitis B), liver cancer (hepatitis C), stomach cancer (Helicobacter pylori), or worsening of conditions such as cardiovascular and respiratory (influenza A). Because we cannot always rely on medicine to develop rapidly vaccines or other treatments, the best prevention is to detect early possible pandemics and stop the transmission. By blocking transmission, we could eventually also reduce the mutation of the viruses and thus keep the virus in a stage that vaccines could help fight.
Fig. 1.
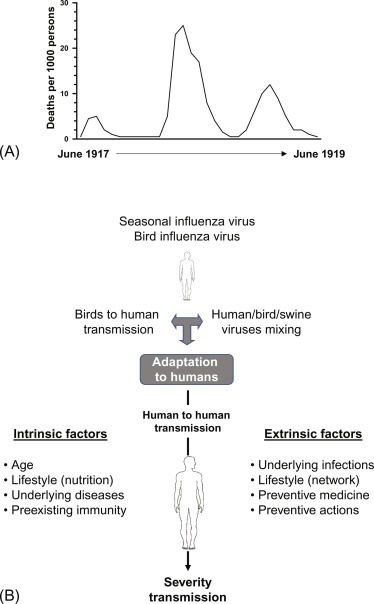
Lessons from the 1918 “Spanish” flu. (A) Graph representing the number of deaths during the peak of the 1918 influenza pandemic. (B) Since the “Spanish” flu, much knowledge has been acquired in the mechanisms of influenza transmission and factors influencing it.
Artificial intelligence in health care
Among the existing analytical tools artificial intelligence (AI) has been identified as the most powerful and promising for mankind (Silver et al., 2017). AI is the output from the input resource: big data that needs to be cleaned, structured, and integrated. What we refer as big data can be defined by volume, velocity, variety, variability, veracity, and complexity. These terms refer to the amount of data, the speed of data in and out, the range of data types and sources, and accuracy and correctness, respectively. However, most of the volume and velocity of data in health care as of today are not high enough to require big data. Most health-related studies do not require the support of data scientists but of bioinformaticians and statisticians. However, in a context of omics generating hundreds of thousands of data points for gene polymorphism, gene expression, metabolomics, lipidomics, and proteomics, there is a need to develop better tools to identify specific cases from the overall orientation of the mass of data.
Detection of weak signals enables the early identification of trends before they become significant and important. This is highly used in the field of cybersecurity. Translated to health care, this would mean identifying a signature in few individuals or a cluster of individuals and predicting the clinical trajectory of the rest of the population. Various sets of data have been elegantly used to predict infectious diseases epidemics. The problem with infectious diseases, as introduced above is their unpredictability as well as the multiple factors that affect the process of infection and transmission. AI is the form of computing that allows machines to act or react to input, similar to the way humans do, by performing cognitive functions. On the contrary, traditional computing also react to data but the output has been necessarily hand coded to react that way. There is no cognitive function performed, as such the independent intelligence is missing. If an unexpected situation is encountered, traditional computing cannot react. In short, AI platforms are constantly adapting their behavior to changes and modify their reactions accordingly. In an AI approach machines are designed to analyze, interpret, and solve a problem. In one of its leading application, machine learning, the computer learns once how to act or react to a certain result and knows in the future to act in the same way.
Recent reports have shown the added value of machine learning for image processing where classical tools could not identify early signs of diseases (Chen and Asch, 2017). This is particularly true for cancer (Boon et al., 2018) which diagnosis and treatment are often assisted by AI approaches. Even in developing countries where the resources, health-care cost, and other limitations prevent from providing optimal care, this is applicable. A group has recently shown the possibility to develop a low-cost point of care for lymphoma diagnosis based on basic imaging and deep learning (Im et al., 2018). Several reports suggested the use of Bayesian network (BN) for representing statistical dependencies (Xu et al., 2016). A BN is a graph-based model of joint multivariate probability distributions that captures properties of conditional independence between variables (Belle et al., 2013). In the era of systems biology and personalized medicine, the development of appropriate analytical is growing. A new class of data, often referred to as recreational data, will become more and more relevant in the context of health care: Internet of things (IoT). The IoT is a growing network of devices and objects that we use in our daily life and that can collect information. Smartphones with their numerous applications and wearables are the typical example of devices that generate continuous streams of data and this can be used to better understand our lifestyle. It is estimated that > 7 billion connected things are currently in use worldwide and making use of this would magnify the possibilities to improve our life. Such datasets and classical health-care datasets are being used to better understand infectious diseases, the mechanisms of infections, resistance to treatment, transmission as well as to improve vaccine designs (Fig. 2 ).
Fig. 2.
Essential principles in the control of infectious diseases. In the sequence, the important aspects to control transmission and improve control by preventive measures (vaccination and hygiene) are presented. The role of the AI ecosystem in this endeavor is central.
Figures with designs by Freepik from www.flaticon.com.
The utilization of AI in infectious diseases
Improved diagnosis and blocking transmission
Diagnosis
The fear of infectious disease transmission has led authorities to set up processes to detect individuals at risk. As such, in Singapore airport terminals, temperature checks are performed systematically using a thermal camera to identify individuals with high temperature. This minimal check is one piece of the multiple steps taken forward to block transmission of infections. Recent approaches using mathematical modeling are improving this type of surveillance. A similar system was developed to detect infected patients by classification using vital signs (Sun et al., 2015). Hence, respiration rate, heart rate, and facial temperature were used to successfully classify individuals at higher risk for influenza using neural network and fuzzy clustering method. Fuzzy clustering methods differ from the k-means clustering because of the addition of the membership values (degree of belongness to a cluster based on edge/centroid position in the said cluster) and the fuzzifier. As such, each point can belong to multiple clusters contrary to the nonfuzzy clustering methods. This demonstrates the ability to develop effective methods for identifying populations at risk. This triage is necessary and part of the process, even in the case of emergent infectious diseases, where efforts have to be prioritized. The use of machine learning methods can be used in more sophisticated contexts. For instance, a combination of support vector machine (SVM) learning algorithm, Matlab, leave one out cross-validation (LOOCV) method, and nested one-versus-one (OVO) SVM were used to better separate gene sequences from bacteria over other methods such as high-resolution melt (HRM). The combination of SVM and HRM could identify with high accuracy (100%) isolated bacteria (Fraley et al., 2016). In real-life biological samples, blood samples from patients, the accuracy was affected which shows the limitation of developing tools from data generated in a controlled environment (laboratory). Whether this was due to poor quality of the biological samples or inherent to the interactions of the bacteria in a nonartificial environment is not known. Still, this shows that the mathematical tools developed should consider certain practicalities such as quality of the samples or duration of the laboratory process. This was tackled in the case of tuberculosis diagnosis, the second leading cause of death from infection in the world (Saybani et al., 2015). Because of the lengthy process to have a final decision regarding diagnosis, early indicators of the infectious were sought (Fig. 1). Existing systems exist such as the artificial immune recognition system (AIRS) for various diseases diagnosis. AIRS was developed using the immune system’s feature. The role of the immune system is to recognize threats and keep these in memory. Immunological memory is probably the most important feature of immunity as it allows us to better respond when the threat (infectious agent) is encountered a second time. This is in line with developing AI tools based on human cognitive function, the only difference here is the fact intelligence is decentralized to the periphery (blood). AIRS use k-nearest neighbor (kNN) as a classifier. Few issues with kNN in machine learning (i) it identifies patterns of data without demanding for an exact match to known patters which provides low accuracy and (ii) if k is too small or large there may be issues with noise and loose neighborhood, respectively. The AIRS that uses supervised machine learning methods (Watkins and Boggess, 2002) has shown good accuracy (Cuevas et al., 2012). Saybani et al. have improved the accuracy of such a classification aid by using SVM instead of kNN as classifier. SVM is a much more robust classifier and was applied to a tuberculosis cohort. With an accuracy of 100%, sensitivity of 100%, specificity of 100%, Youden’s Index of 1, area under the curve (AUC) of 1, and root mean squared error (RMSE) of 0, the new AIRS method was able to successfully classify tuberculosis patients. Another life threatening and pandemic infection, malaria, has been under intense research to develop novel, easily implementable, and cost-effective methods for diagnosis. Malaria diagnosis is time consuming and may require the intervention of several health services. Machine learning algorithms were developed to detect red blood cells (RBCs) infected with malaria from digital in-line holographic microscopy data, a fairly cheap technology (Go et al., 2018). Segmented holograms from individual RBC were tagged with several parameters and 10 of these were statistically different between healthy and infected RBCs. Several machine learning algorithms were applied to improve the malaria diagnostic capacity and the model trained by the SVM showed the best accuracy in separating healthy from infected RBCs for training (n = 280, 96.78%) and testing sets (n = 120, 97.50%). This DIHM-based AI methodology is simple and does not require complex blood sample processing.
Epidemiology and transmission
Epidemiological studies can be performed at the population level or at the patient’s bed (clinical epidemiology). Epidemiological studies should be acquired along a specific timeline, with infection-related data collected in a longitudinal manner. When done properly, mathematical models can predict the size of emerging infectious diseases. Large datasets and prediction models exist for noncommunicable diseases (NCD). A recent study collated the data from 50 American states for a series of NCD such as diabetes, cardiovascular diseases, hypertension, and others over a period of 5 years (Luo et al., 2015). Data from 30 states were used for training and tested in the remaining 20 states. This colossal amount of data and machine learning modeling enabled to reach near-reality output. However, what defined NCD is the lack of transmission from patient to patient because of proximity or shared environment in a short period of time. This is what defines infectious diseases, because they can be transmitted from one individual to the other within very short periods of time. The severity of symptoms and mortality associated with the infection are driving the urgency of predict future size and location of the epidemic. Many of the machine learning methods presented in this chapter beside giving indications on size and location, which is often the communicated information, are primarily used by mathematicians to estimate variables related to infection (e.g., incubation time, transmission mode, symptoms, resistance to treatments). The input data for epidemiological studies is very diverse and enables it to utilize various assets of AI (Fig. 1). Epidemiological studies enable us to predict an epidemic from very small foyer as shown in a recent work on Kyasanur forest disease which is a tick-borne viral infectious disease (Majumdar et al., 2018). Using extremal optimization tuned neural network, the team of scientists showed the high prediction rate and proposed localization data to be implemented in future databases in order to better control transmission. Recent life-threatening outbreaks, such as Ebola, have pushed the community to innovate in the field of prediction. Using machine learning, a single-layer artificial neural network (ANN), logistic regression (LR), decision tree (DT), and SVM classifiers scientists generated an ensemble of predictors that could be applied to different combinations of Ebola-related data (Colubri et al., 2016). An important issue in such health crisis is often the lack of immediate response and poor quality of the data from initial foyer of infection. As in forensic science, the history of transmission relies significantly on the early steps. Colubri et al., showed how missing information and/or small sample size issues can be tackled when utilizing machine learning approaches: a composite of machine learning approaches rather than a single model.
Several teams from United States (Kane et al., 2014), China, New Zealand (Zhang et al., 2014), and South Africa (Adeboye et al., 2016) have utilized autoregressive integrated moving average (ARIMA) for predicting infectious diseases. The ARIMA model was originally developed for economic applications but has been used in other domains such as for infections that occur in cyclic or repeating patterns. The time series models such as ARIMA are used to predict future outbreaks as they filter out high-frequency noise in the data to detect local trends based on linear dependence in observations in the series. The ARIMA model can integrate dynamic relationships and update the model based on recent events. Hence, ARIMA models have been widely used for epidemic time series forecasting including hemorrhagic fever, dengue fever, and tuberculosis. For the later and other infectious diseases, seasonality is an important aspect (Mohammed et al., 2018). Using seasonal ARIMA (SARIMA) and neural network auto-regression (SARIMA-NNAR) tuberculosis incidence and seasonality was analyzed in South Africa. This machine learning approach indicated the need to tackle coinfection issues, especially HIV, and also festival peak periods to be risk factors driving transmission. SARIMA-NNAR outputs with the best model based on the simulation performance: the Akaike information criterion (AIC), second-order AIC (AICc), and Bayesian information criterion (BIC) which were lower than SARIMA alone. Other methods than ARIMA can indicate a potential risk associated with infection outbreak. In a study on Rift Valley Fever (RFV) emergence in Africa and the Arabic peninsula, maximum entropy machine learning methods have identified the key determinants associated with infectious risk. RVF is a vector-borne viral zoonosis and the ML output identified intermittent wetland, wild Bovidae richness, and sheep density as the main associations with landscape suitability for RVF outbreaks (Walsh et al., 2017). A comparative analysis of H5N1 outbreak in Egypt identified random forest as a more robust method for prediction than ARIMA (Kane et al., 2014). Derivatives of ARIMA such as an optimized ARIMA-generalized regression neural network (GRNN) model were used for forecasting and control of tuberculosis in a far from ideal environment of high population movement and coinfection history (HIV) in the Heng county (China). This shows a superior performance that the previous models to predict future incidence of tuberculosis (Wei et al., 2017). This suggests again that several strategies should be run in parallel and adapted to the local environment and context. This can be exemplified by a study showing how modeling [using three-step floating catchment area (3SFCA)] can help optimize health-care utilization by reallocating health-care resources to sites where the ratio of demand/supply is increasing dramatically (Chu et al., 2016).
A group of experts in game has recently used AI approaches (disease simulation model) to demonstrate success in using AI algorithms to search for the optimal Malaria intervention strategies (Wilder et al., 2018). The variety and volume of existing data for malaria control is quite dense (e.g., Malaria Atlas Project, Malaria Immunology Database, Mapping Malaria Risk in Africa projects, PlasmoDB) and enabled such an approach (Wong et al., 2018; Okell et al., 2008). Very often, it is not the quantity but the quality and specificity of the input data that will influence the accuracy of the predictive model. Following inoculation of the dengue virus by mosquitoes, dengue hemorrhagic fever can occur (5% of cases). Applying SVM with the radial basis function (RBF) kernel, scientists were able to forecast the high morbidity rate and take precautions to prevent such cases to happen. The parameter that was able to reach a high level of accuracy was not linked to climate but to the infection rate of the mosquitoes that transmit dengue virus (Kesorn et al., 2015). It most cases of infectious diseases the success transmission blockade is usually linked to outreach (Saybani et al., 2016). Strategies should then identify the best way to communicate and reach the various categories based on age, gender, and other socioeconomic variables. The use of AI for predicting infectious diseases pandemics should also integrate pipelines for adapted solutions. The high-level AI analytical approach presented above is possible when each of the various database has a high level of veracity.
Treatments and antimicrobial drug resistance
Despite a good ability to diagnose malaria and probably with improved diagnosis in the near future, there is a strong problematic with antibacterial and antiparasitic drugs: resistance (Blasco et al., 2017). The adoption of artemisinin-based combination therapies 20 years ago is now being challenged by the emergence of Plasmodium falciparum malaria parasites with decreased susceptibility to artemisinin-based combination therapies. Mathematical modeling using intrahost parasite stage-specific pharmacokinetic-pharmacodynamic relationships predicted that ART resistance was a result of ring stages becoming refractory to drug action (Saralamba et al., 2011). Antibiotic resistance can be better tackled with the existence of databases (Jia et al., 2017) reflecting this phenomenon. The comprehensive antibiotic resistance database (CARD) contains high-quality reference data on the molecular basis of antimicrobial resistance (http://arpcard.mcmaster.ca). CARD is ontologically structured, model centric, and spans the breadth of antimicrobial resistance drug classes and mechanisms. The database is an interconnected and hierarchical structure allowing optimized data sharing and organization. This highlights the importance of the right architecture for the database (big data architecture). Recent studies have also shown the use of machine learning in effectively identifying the potential antimicrobial capacity of candidate compounds (Wang et al., 2016). In a more systematic way, Ekins et al. have used a series of machine learning approaches to predict responsiveness to tuberculosis infection in mice (Ekins et al., 2016). This includes Laplacian-corrected naïve Bayesian classifier models and SVM models using Discovery Studio 4.1. Computational models were validated using leave-one-out cross-validation, in which each sample was left out one at a time, a model was built using the remaining samples, and that model was utilized to predict the left-out sample. As in many studies the receiver operator characteristic (ROC) plots and the areas under the cross-validated ROC curves are useful validation tools. Bayesian model with SVM, recursive partitioning forest (RP forest), and RP single tree models were compared. For each tree, a bootstrap sample of the original data is taken, and this sample is used to grow the tree. A bootstrap sample is a data set of the same total size as the original one, but a subset of the data records can be included multiple times. Their data clearly suggest that Bayesian models constructed with data generated by different laboratories in various mouse models can have predictive value and can be used in conjunction with other datasets for the selection of the most-fit antimicrobial compound. The same mathematical approaches can be performed either on a very specific target for potential drugs (Djaout et al., 2016) or for a more systematic analysis such as performed for the known inhibitors of fructose bisphosphate aldolase, an enzyme central to the glycolysis pathway in M. tuberculosis, in short, an essential player in the bacteria metabolism (Tiwari et al., 2016). Naïve Bayes, random forest, and C4.5 J48 algorithms were used with an approach to improve models by avoiding over fitting and generating faster and cost-effective models. Overall, this and previous studies (Zhanga and Amin, 2016) suggest that machine learning provides good accuracy confirming other studies validating in silico methods to be used for screening of large datasets to identify potential antiinfectious candidates. In line with this, Shen et al. have clearly shown how treatments can be assisted using mathematical models (Shen et al., 2018). Shen et al. proposed a decision support system can propose an antibiotic therapy adapted to the patient based on factors such as the body temperature, infection sites, symptoms/signs, complications, antibacterial spectrum, and even contraindications and drug-drug interactions. This was possible thanks to the impressive array of data used to construct the model: “the ontology contains 1,267,004 classes, 7,608,725 axioms, and 1,266,993 members of “SubClassOf” that pertain to infectious diseases, bacteria, syndromes, anti-bacterial drugs and other relevant components. The system includes 507 infectious diseases and their therapy methods in combination with 332 different infection sites, 936 relevant symptoms of the digestive, reproductive, neurological and other systems, 371 types of complications, 838,407 types of bacteria, 341 types of antibiotics, 1504 pairs of reaction rates (antibacterial spectrum) between antibiotics and bacteria, 431 pairs of drug interaction relationships and 86 pairs of antibiotic-specific population contraindicated relationships.” Another study has developed models in order to reduce the utilization of antibiotics. Infants can experience symptoms that are cause by pathogens (sepsis) or by noninfectious agents: systemic inflammatory response syndrome (SIRS). As it is difficult to have a clear diagnosis rapidly using classical laboratory tests, it was shown that applying a random forest approach could identify the best set of predictors out of laboratory variables measured at onset (Lamping et al., 2018). Beside antibiotics, antibodies are very effective in protecting against viral infection. The basis of vaccination is to mount an effective memory response when the vaccine will encounter the virus later. This happens via the production of antibodies that play a role in blocking the replication of the virus. Studies have shown that the use of machine learning was very important to identify best candidate vaccines (Choi et al., 2015). Using unsupervised learning as well as unsupervised learning: (i) penalized LR: LR incorporating into the model a lasso penalty term λ || β ||1, (ii) regularized random forest: DT-based method that generates multiple DTs over bootstrap replicates of the data, and (iii) SVM: kernel-based nonlinear classifier that finds a separating hyperplane between the classes to minimize the risk of classification error, Choi et al. model associations between antibody features and immune functions. The antibody features can predict qualitative and quantitative functional outcomes which provide with a novel and objective approach to assess immune correlates to antibody features. All the aspects of response to treatment depend on an essential parameter: compliance. There are very few systems to verify adherence to infection-related treatments in a large-scale manner. In the case of the HIV, a way to follow the evolution of the infection is to test the blood level of HIV RNA. This is very efficient to adjust the therapy but rarely affordable in poor resources settings. As the evolution of HIV infectivity highly depend on the antiretroviral therapy (Petersen et al., 2008), it becomes important to ensure the therapy is taken appropriately and that viral loads are not affected because of loose adherence to treatments. In populations at risk for virological failure, the implementation of a sensitive approach may enable us to detect individuals with irregular therapy adherence and help improve their health status, reduce costs of repetitive testing, and raise awareness on necessity to adhere to therapy regimens. The self-reported and questionnaire type data is usually of low sensitivity (50% or less). One way to better track adherence to antiretroviral therapy is by analyzing data from pharmacy. The classification of failure to comply with medication regimen was significantly better with pharmacy refill data than with self-reported data (Bisson et al., 2008). This has been the focus of a recent study (Petersen et al., 2015) in which adherence was followed by a monitoring system that provides the highest quality of data compared with self-reported, pharmacy data, or others. The device is a cap that fits on standard medicine bottles and records the time and date each time the bottle is opened and closed. The aim was to improve accuracy of data in order to have a better estimate of adherence to treatments and how this impacts virologic failure. The data generated by this type of device is analyzed with Super Learner (van der Laan et al., 2007), a data-adaptive algorithm based on cross-validation via multiple internal data splits. Again, the use of machine learning was very efficient and could improve classification of virological failure in a cohort of > 1000 patients in the United States. The combination of machine learning with the standard HIV follow-up (CD4 + T cell count) and antiretroviral therapy regimen significantly improved classification (ROC ≈ 0.8) compared to the electronic monitoring system alone. The contribution of machine learning in this context is reducing the necessity to perform viral load tests (by 1/3) with a sensitivity for virological failure detection at > 95%: saving time, resources and lives without low compromise. Altogether, those applications of machine learning, summarized in Fig. 3 have greatly improved the management of infectious diseases. While this shows the enormous potential of AI, there are still many aspects that seek adjustments in order to fully utilize the capacity of AI to help eradicate unwanted pathogens, reduce the burden of seasonal viruses, and better understand the interactions between pathogens and humans.
Fig. 3.
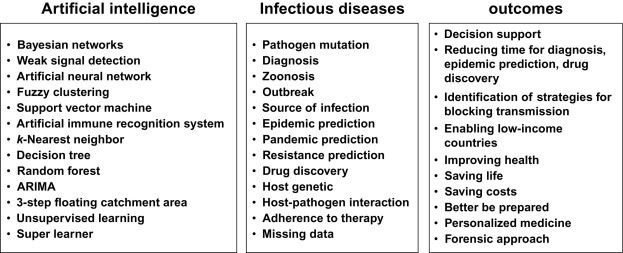
AI tools, their use and potential outcomes. The series (nonexhaustive) of machine learning tools used in the field of infectious diseases and the aspects they target. Expected outcomes from the contribution of AI are presented.
Improving the process
On the technical aspects
In this with the need to improve the utilization of AI in the context of infectious diseases, we believe that identification of earliest signs of transmission: the combination of extreme value theory and robust statistical methods-based analysis should be applied (Ferro and Segers, 2003; Mikosch and Wintenberger, 2014; Deheuvels, 1991; Smith, 1989). The first two steps of correlation/event analysis framework should filter most of the data and only let a small fraction pass to the last step. The goal is to scrutinize the unfiltered data in order to detecting suspicious observations. Here again, the main issue lies in the learning of the dynamic BN. For this purpose, the first step is to store dataset in a convenient way, often in a specific database (e.g., not only structured query language: NoSQL) that will enable fast extraction. The second problem that must be handled lies in the scores used by learning algorithms to determine the conditional independences required to define the graphical structure of the BN. Actually, those are closely related to statistical independence tests (essentially to cross entropy) (Gonzales and Wuillemin, 2011). While this is meaningful in many contexts, it seems that this kind of test is inappropriate for learning anomalies because the latter should be related to rare events whereas the aforementioned statistical tests are not. Therefore, the focus should be in the rare events and to apply statistical methods suited for this context. This paradigm shift is important because, in our opinion, it is one of the keys to better detect outliers in medical database (Barnett and Lewis, 1995; Filmoser et al., 2008). An important aspect of uncertainty discovery is the focus on rare events, which characterize precisely what the solution is looking for. For this purpose, we need to integrate into algorithms the most recent statistical techniques for the study of large multimodal datasets.
The potential of extreme value theory
Existing medical tests are effective to find the “known bad,” aka diseases, but can be ineffective in detecting “unknown bad.” This is often the case as medical tests are specific for one disease, often under specific conditions (age, gender, genetic, stage of disease, type of treatment). Hence there is a need to develop tools for the unknown bad such as new infectious diseases or increased burden of existing infections due to mutation of existing strains. The parallel between cybersecurity and infectious diseases is on the spread following an “attack.” In the medical field, majority of the data corresponds to normal behaviors and known suspicious observations. Extreme values and rare events are of utmost importance for extreme risk evaluation (Japan’s recent events show the need for such methods). In the context of our problematic, infectious diseases, the low-frequency risk makes extreme value theory and rare event analysis models of choice for representing appropriately infectious risk-related uncertainties. It is obvious that having a sophisticated uncertainty representation that best fits data is of utmost importance for correctly detecting infectious risk (extreme values). The study and application of the extreme value theory coupled with the copula allows us to detect weak signals in large structured and unstructured data. It should be known that when we use dimension reduction methods, the weak signal at the extreme may be lost. With the era of AI and distributed computing we can partially overcome the curse of dimensionality. It should not be confusing with classical AI tools as extreme value theory is a branch of statistics that seeks to assess, from a given ordered sample the probability of events that are more extreme than any previously observed. It is then a high-dimensional statistical approach. There were several attempts to use extreme value theory in machine algorithms and deep learning such as the extreme value machine method by Rudd et al. (2018). Other algorithms mining on extreme regions and values were developed (Brownlees et al., 2015; Cai et al., 2011; Goix et al., 2015, Goix et al., 2016; Mendelson, 2018; Ohannessian and Dahleh, 2012). By combining high-dimensional statistics and AI, there should be enough synergy to convince stakeholders and decision-makers to test their potential for predicting and modeling rare events such as pandemics, with a level of certainty (Reiss and Thomas, 2007).
Basics on the concept of extreme values
The development of extreme value theory originated from the needs in industrial applications (risk, finance, natural events, etc.) where the study of “high” values was a major concern because, in these applications, those may incur important costs. Basically, from a set of events X 1, …, X n, indexed by their observation time, 1, …, n the key idea is to look for the maximum M n = max {X 1, …, X n} and, as such, modeling the probability distributions ℙ(M n ≤ x), as such, the probability that M n is lower or equal to x , this being dependent on x and time n. Fréchet, Gumbel, and Weibull have contributed significantly to the definition of the distributions showcased here: .
In practice, the methods of determination of extreme values is done using the “Block Maxima” method where data are divided into blocks corresponding to separate periods of time. Alternatively, extreme value analysis rely on extracting from a continuous record, the peak values reached for any period during which values exceed or fall below a certain threshold (Fig. 4 ). This method is generally referred to as the “Peak Over Threshold” (POT) method. Of course, the choice of this threshold is, in itself, a problem, and this conditions the estimates of the parameters. On top of that, there are cluster phenomena, mainly when the time between two consecutive observations is small. In this case, it is common to observe “clusters” of extreme values, excluding being treated as if they were independent observations. Finally, in fine, it is often necessary to base the analysis of extreme values on small samples, even when they come from sets containing thousands of observations. For POT data, the analysis may involve fitting two distributions: one for the number of events in a known period of time (Poisson distribution) and the other for the size of the exceedances (Pareto distribution).
Fig. 4.
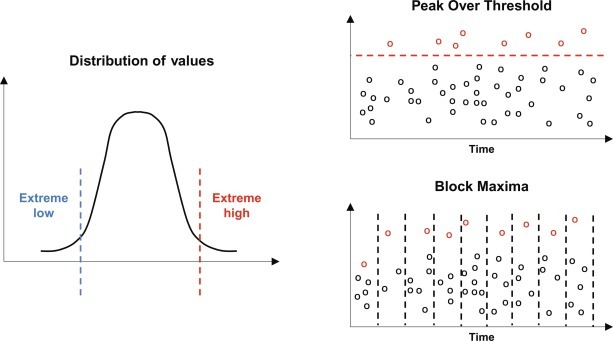
Concept of extreme value theories. The distribution of the data often shows the presence of extreme low and/or high values. These values are identified using two methods: Block Maxima which consider defined and repeated periods of time (e.g., by year and waves of infection) or Peak Over Threshold which consider any data beyond a set threshold over a continuous period of time.
Very often, a single observation is from various sets of numerical data. Additionally, rare events are often associated. Recently it was understood that dependency of extreme values could be viewed under the copulas. In statistics, a copula is a multivariate probability distribution for which the marginal probability distribution of each variable is uniform. They describe the dependence between random variables and have already been used in neurosciences (Eban et al., 2013). The copula of a random vector (U, V) is defined by C(u, v) as the joint cumulative distribution function: ℙ(U ≤ x, V ≤ y) = C(ℙ(U ≤ x), ℙ(V ≤ y)). Many of the existing methods should integrate the notion of copulas in order to solve problems related to high-dimensional statistics and this is extended to deep and machine learning (Deheuvels, 1991, Deheuvels, 2009).
On the design of data collection
The emergence of the IoT is probably the biggest opportunity in the utilization of data for human kind safety and progress. The IoT-derived data, if obtained with the highest standards of ethics and secured with the highest standards of security, will enable us to better track and control infectious diseases globally. In most studies, the parameters that show high dependence for dissemination of infections are the role played by the individuals and their close environment. As such, social interactions are very important drivers of infectious diseases transmission. Social interactions are probably even more important for primates and other group-living animals (Rushmore et al., 2017). Network analysis becomes a must not only to better control zoonosis but also to understand how in a group of individuals, be it animals, the social interactions shape the infectious disease dynamics. Human studies have shown how social media data (Lim et al., 2017) could be used to map infectious diseases that have not yet been identified by public health institutions. Using unsupervised machine learning model (i) no name of disease and (ii) no symptoms as a bottom-up approach. Our interpretation and review of the method is that by avoiding the integration of the name of the infection and the symptoms, there is more change to cover diseases and unknown diseases and to also enrich the databased related to known infectious diseases. Because most of the terms used in social media (symptoms, body parts, and pain locations) are not the scientific/medical level often reported by health-care institution it is more likely to cover a wide range of individuals and as such outputs with a denser geographical area. Although the study was limited in time (8 months), the validation was performed with accurate (electronic medical records) from a small group of individuals (n = 104), and the model was able to predict with a high precision, recall and F values on average above 0.7 (Lim et al., 2017). This suggests that sentiments expressed in social media is still valuable information, despite their heterogeneity and better classification of sentiments should be a priority for the next generation of social media-related studies. This approach of mass data to identify specific signals may enable us to enter into the era of personalized medicine. In line with this, patient similarity using electronic medical record has been successfully used to recommend treatments (Wang et al., 2015). The diagnoses, demographic data, vital signs, and structures laboratory results were used for similarity testing. The addition of an intelligence to the electronic medical record may enables us to reach a precision > 0.8. This again shows that dynamic learning models can be used as assistive technology for decision-making.
On the integration of AI in health-care institutions
The IoT gather a myriad of information on our habits. We can predict that the field of medicine will also highly benefit from IoT. More and more clinical laboratory tests are automated and the complexity of data generated can be more complex. Strategies to implement AI in health-care institutions still need to be developed (Beeler et al., 2018). The first aim is to setup a state-of-the-art data management system. While most hospitals and clinics have such systems in place, these are often obsolete as they are not adapted for the type of data we generate nowadays. In the constant chase against infectious diseases hospitals must have in place a systematic way to predict the emergence of nosocomial infections (Fig. 5 ). Such intelligent systems should consider multiple parameters including emergence of infectious diseases but also any particular change in the hospital’s routine. This was effectively performed using random forests and could bring attention to the hospital staff for possible gaps (Valleron, 2017). To reach such a level of predictivity there are several items to consider (i) upgrade the expertise in AI, (ii) develop synergies between mathematicians, biologists, and clinicians, and (iii) develop the culture around AI as done for previous technological developments. Of course, this implementation is not without some sacrifices. However, while AI is widely seen as a threat for “common” jobs, it should be seen as an opportunity. Hence, hospitals, clinics, and other surveillance institutions should turn it as a chance. Recent works have shown the benefit of integrating AI approaches for improved diagnosis. Ultrasound has proved to be a useful tool to validate diagnosis of lung infection: pneumonia. This diagnosis depends on two factors: the expertise of the operator and the potential bias during interpretation by the medical personal. Using pattern recognition and image analysis was used for automatic classification of pneumonia (Correa et al., 2018). The neural network trained correctly identified pneumonia infiltrates (> 90% sensitivity and 100% specificity). Moreover, the geographic information related to infectious diseases should be matched with the patients’ medical record and history (Hay et al., 2013). This is very important to immediately understand the relationship between location and other characteristics of the patients such as professional activity, family environment, housing type, contact with animals, etc. This will enable hospitals, which are often the first site where patients are in contact with professionals, to become experts in forensic science, where the culprit is a microorganism. Recent advances were proposed such as building on simulation experiences (Hogan et al., 2016). Preparedness for epidemic is mostly tested in hospital regarding number of beds, and activation of specific measures to avoid transmission of infection. Here, the Apollo structured vocabulary (XML Schema Document-based syntax) was developed to represent infectious disease scenarios and enable its utilization in independent simulators.
Fig. 5.

Major sources of nosocomial infections. The development of algorithms for the early identification of nosocomial infections is taking into account the sources for transmission.
Several systems are already in use in health care. But there is far more to develop to benefit significantly from the amount of available data. Progress has been fairly slow in general and mostly due to the fragmentation or nonexistence of data repositories. The contribution of major players in the field of technology (including the GAFA) may help fasten this process. Recently Catalia Health developed a humanoid robot named Mabu, a personal health-care assistant for patients suffering from congestive heart failure. Beside doing an accurate follow-up of medication compliance, it supports an adaptive conversation with the patients regarding the overall health status. Such systematic follow-up in intensive care unit, however, combining conversation with the medical personnel should enable them to better track and reduce the transmission of infections (Fig. 5). The University of Iowa Hospitals & Clinics have used machine learning to reduce surgical site infections by 74% over the past 3 years. Using DASH analytics systems, the hospitals is using the high-definition care platform (HDCP), which integrates with the hospital’s electronic health records to assess the risk for individual patients. Focusing on surgery-derived infections, the system uses the WHO surgical safety checklist. The checklist comprises three phases of the surgery: (1) before the induction of anesthesia, (2) before the first incision of the skin, and (3) before the patient leaves the operating room. This should ensure the correct care protocol has been followed. Once this data and that from the existing electronic health records is collected the DASH system inputs it all into a prediction model and identified potential risks. This enables us to provide costly treatments only to patients who need them. This is a precision medicine approach. In another hospital, a new algorithm could predict a patient’s risk of contracting Clostridium difficile, a life-threatening nosocomial infection. Every year in the United States, > 400,000 people contract a C. difficile infection following hospitalization and about 30,000 of them will die because of the infection. Medical doctors and computer scientist from the Massachusetts General Hospital worked together to use AI for prediction. Using a dataset from > 300,000 patients admitted and the knowledge that C. difficile spreads through physical contacts, every possible interaction between patients was taken into account when developing the algorithm. Another tool developed by Royal Phillips (Connected Care) enables to reduce by 87% the time for identification of nosocomial infection in hospitals, suggesting that wider utilization of such application could significantly reduce hospital-acquired morbidity and mortality.
Conclusions and future perspectives
The utilization of AI and ML are very promising as shown with examples discussed in this chapter. There are a few issues for the full integration of AI in our daily health-care lives. How are regulatory institutions such as the Food and Drug Administration (Harrington and Johnson, 2018) going to help AI integration? Guidelines will need to be developed and implemented. This raises another important question: how can we harmonize AI approaches across institution? The Hippocratic oath refers to equity in treatment and the aim to deliver the best treatment with the best of existing knowledge. Harmonizing the integration of AI (Collins and Tabak, 2014) will enable us to enforce its use for infectious pandemics prediction, better understanding of infections, and reduce time for drug discovery. Some tools exist to warranty the reproducibility of analysis (https://rmarkdown.rstudio.com) and such initiatives should be. While access to medication is still a (financial) limitation in developing countries, the use of AI to break transmission may become the best long-term affordable strategy. Risk analysis will also depend on how we are able to integrate and verify the quality of input data, especially based on IoT. All this will be for a global benefit (Velsko and Bates, 2016).
Bio-surveillance still remains largely uncoordinated systems. There is a need to take advantage of the rapid progress made in the past decades in data processing, analytics, and utilization. Existing structures such as the MOH-driven global influenza surveillance could be used as examples to develop long-term capabilities in preventing infectious disease pandemics and their deleterious effects. Even intracountry organizations often fail to communicate information properly and to group this in a timely manner. As discussed in this chapter, the architecture of the data is an essential part that should be defined upstream to enable such data sharing, merging, and analysis. With increasing product exchanges and travels the risks for dissemination of infectious diseases would not be reduced unless strategic decisions are made at the global level regarding implementation of big data architectures and their integration for AI-driven solutions. There is also a temptation not to wait for policies to be implemented. For instance, personalized approaches for infectious diseases risk have been proposed (Vinarti and Hederman, 2018). This is taking three essential parameters of infectious diseases: (i) pathogen’s availability, (ii) transmission method, and (iii) susceptible host. By testing three transmission types of infectious diseases, tuberculosis (air borne), Dengue (vector borne), and Cholera-India (water/food borne) the authors showed the effective and automatic generation of BN (risk probabilities), highly influenced by the person and the environment. Both population-wide and personalized approaches should be developed as infectious diseases have different infectivity, incubation time, transmission mode, and will lead to different symptoms depending on the host.
Acknowledgments
AL is supported by the Singapore Immunology Network, Agency for Science Technology and Research and JCO Development Program (Grant #1434 m00115). AL and SA are cofounders of Yobitrust, a Data Science company.
References
- Adeboye A., Obaromi D., Odeyemi A., Ndege J., Muntabayi R. Seasonality and trend forecasting of tuberculosis prevalence data in Eastern Cape, South Africa, using a hybrid model. Int. J. Environ. Res. Public Health. 2016;13(8):757. doi: 10.3390/ijerph13080757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett V., Lewis T. third ed. Wiley; New York: 1995. Outliers in Statistical Data. [Google Scholar]
- Beeler C., Dbeibo L., Kelley K., Thatcher L., Webb D., Bah A., Monahan P., Fowler N.R., Nicol S., Judy-Malcolm A., Azar J. Assessing patient risk of central line-associated bacteremia via machine learning. Am. J. Infect. Control. 2018;46(9):986–991. doi: 10.1016/j.ajic.2018.02.021. [DOI] [PubMed] [Google Scholar]
- Belle A., Kon M.A., Najarian K. Biomedical informatics for computer-aided decision support systems: a survey. Sci. World J. 2013;2013 doi: 10.1155/2013/769639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson G.P., Gross R., Bellamy S., Chittams J., Hislop M., Regensberg L., Frank I., Maartens G., Nachega J.B. Pharmacy refill adherence compared with CD4 count changes for monitoring HIV-infected adults on antiretroviral therapy. PLoS Med. 2008;5(5) doi: 10.1371/journal.pmed.0050109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco B., Leroy D., Fidock D.A. Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017;23(8):917–928. doi: 10.1038/nm.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon I.S., Yong T.P.T.A., Boon C.S. Assessing the role of artificial intelligence (AI) in clinical oncology: utility of machine learning in radiotherapy target volume delineation. Medicines (Basel) 2018;5(4) doi: 10.3390/medicines5040131. pii: E131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlees C., Joly E., Lugosi G. Empirical risk minimization for heavy-tailed losses. Ann. Stat. 2015;43(6):2507–2536. [Google Scholar]
- Cai J.J., Einmahl J.H.J., DeHaan L. Estimation of extreme risk regions under multivariate regular variation. Ann. Stat. 2011:1803–1826. [Google Scholar]
- Chen J.H., Asch S.M. Machine learning and prediction in medicine—beyond the peak of inflated expectations. N. Engl. J. Med. 2017;376(26):2507–2509. doi: 10.1056/NEJMp1702071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I., Chung A.W., Suscovich T.J., Rerks-Ngarm S., Pitisuttithum P., Nitayaphan S., Kaewkungwal J., O’Connell R.J., Francis D., Robb M.L., Michael N.L., Kim J.H., Alter G., Ackerman M.E., Bailey-Kellogg C. Machine learning methods enable predictive modeling of antibody feature:function relationships in RV144 vaccinees. PLoS Comput. Biol. 2015;11(4) doi: 10.1371/journal.pcbi.1004185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H.J., Lin B.C., Yu M.R., Chan T.C. Minimizing spatial variability of healthcare spatial accessibility—the case of a dengue fever outbreak. Int. J. Environ. Res. Public Health. 2016;13(12):1235. doi: 10.3390/ijerph13121235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F.S., Tabak L.A. Policy: NIH plans to enhance reproducibility. Nature. 2014;505(7485):612–613. doi: 10.1038/505612a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colubri A., Silver T., Fradet T., Retzepi K., Fry B., Sabeti P. Transforming clinical data into actionable prognosis models: machine-learning framework and field-deployable app to predict outcome of Ebola patients. PLoS Negl. Trop. Dis. 2016;10(3) doi: 10.1371/journal.pntd.0004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa M., Zimic M., Barrientos F., Barrientos R., Román-Gonzalez A., Pajuelo M.J., Anticona C., Mayta H., Alva A., Solis-Vasquez L., Figueroa D.A., Chavez M.A., Lavarello R., Castañeda B., Paz-Soldán V.A., Checkley W., Gilman R.H., Oberhelman R. Automatic classification of pediatric pneumonia based on lung ultrasound pattern recognition. PLoS One. 2018;13(12) doi: 10.1371/journal.pone.0206410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas E., Osuna-Enciso V., Zaldivar D., Perez-Cisneros M., Sossa H. Multi-threshold segmentation based on artificial immune systems. Math. Probl. Eng. 2012;2012 20 pages. [Google Scholar]
- Deheuvels P. On the limiting behavior of the Pickands estimator for bivariate extreme-value distributions. Statist. Probab. Lett. 1991;12:429–439. [Google Scholar]
- Deheuvels P. A multivariate Bahadur-Kiefer representation for the empirical copula process. J. Math. Sci. 2009;163(4):382–398. [Google Scholar]
- Djaout K., Singh V., Boum Y., Katawera V., Becker H.F., Bush N.G., Hearnshaw S.J., Pritchard J.E., Bourbon P., Madrid P.B., Maxwell A., Mizrahi V., Myllykallio H., Ekins S. Predictive modeling targets thymidylate synthase ThyX in Mycobacterium tuberculosis. Sci. Rep. 2016;6 doi: 10.1038/srep27792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eban E., Rothschild R., Mizrahi A., Nelken I., Elidan G. Dynamic copula networks for modeling real-valued time series. J. Mach. Learn. Res. 2013:31. Carvalho, C., Ravikumar, P. (Eds.) [Google Scholar]
- Ekins S., Perryman A.L., Clark A.M., Reynolds R.C., Freundlich J.S. Machine learning model analysis and data visualization with small molecules tested in a mouse model of Mycobacterium tuberculosis infection (2014–2015) J. Chem. Inf. Model. 2016;56(7):1332–1343. doi: 10.1021/acs.jcim.6b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro C.A., Segers J. Inference for clusters of extreme values. J. R. Stat. Soc. Ser. B: Stat Methodol. 2003;65(2):545–556. [Google Scholar]
- Filmoser P., Maronna R., Werner M. Outlier identification in high dimensions. Comput. Stat. Data Anal. 2008;52:1694–1711. [Google Scholar]
- Fraley S.I., Athamanolap P., Masek B.J., Hardick J., Carroll K.C., Hsieh Y.H., Rothman R.E., Gaydos C.A., Wang T.H., Yang S. Nested machine learning facilitates increased sequence content for large-scale automated high resolution melt genotyping. Sci. Rep. 2016;6 doi: 10.1038/srep19218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go T., Kim J.H., Byeon H., Lee S.J. Machine learning-based in-line holographic sensing of unstained malaria-infected red blood cells. J. Biophotonics. 2018;11(9) doi: 10.1002/jbio.201800101. [DOI] [PubMed] [Google Scholar]
- Goix N., Sabourin A., Clemencon S. Learning the dependence structure of rare events: a non-asymptotic study. Conference on Learning Theory; 2015. pp. 843–860. [Google Scholar]
- Goix N., Sabourin A., Clemencon S. Sparse representation of multivariate extremes with applications to anomaly ranking. Artificial Intelligence and Statistics; 2016. pp. 75–83. [Google Scholar]
- Gonzales C., Wuillemin P.H. PRM inference using Jaffray and Faÿ’s Local Conditioning. Theor. Decis. 2011;71(1):33–62. [Google Scholar]
- Harrington S.G., Johnson M.K. The FDA and artificial intelligence in radiology: defining new boundaries. J. Am. Coll. Radiol. 2018;S1546-1440(18):31343–31347. doi: 10.1016/j.jacr.2018.09.057. [DOI] [PubMed] [Google Scholar]
- Hay S.I., Battle K.E., Pigott D.M., Smith D.L., Moyes C.L., Bhatt S., Brownstein J.S., Collier N., Myers M.F., George D.B., Gething P.W. Global mapping of infectious disease. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 2013;368(1614) doi: 10.1098/rstb.2012.0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan W.R., Wagner M.M., Brochhausen M., Levander J., Brown S.T., Millett N., DePasse J., Hanna J. The Apollo Structured Vocabulary: an OWL2 ontology of phenomena in infectious disease epidemiology and population biology for use in epidemic simulation. J. Biomed. Semant. 2016;7:50. doi: 10.1186/s13326-016-0092-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes K.K., Bertozzi S., Bloom B.R., Jha P., Gelband H., DeMaria L.M., Horton S. Major infectious diseases: key messages from disease control priorities. In: Holmes K.K., Bertozzi S., Bloom B.R., Jha P., editors. Major Infectious Diseases. third ed. The International Bank for Reconstruction and Development/The World Bank; Washington, DC: 2017. (Chapter 1) [PubMed] [Google Scholar]
- Im H., Pathania D., McFarland P.J., Sohani A.R., Degani I., Allen M., Coble B., Kilcoyne A., Hong S., Rohrer L., Abramson J.S., Dryden-Peterson S., Fexon L., Pivovarov M., Chabner B., Lee H., Castro C.M., Weissleder R. Design and clinical validation of a point-of-care device for the diagnosis of lymphoma via contrast-enhanced microholography and machine learning. Nat. Biomed. Eng. 2018;2(9):666–674. doi: 10.1038/s41551-018-0265-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B., Raphenya A.R., Alcock B., Waglechner N., Guo P., Tsang K.K., Lago B.A., Dave B.M., Pereira S., Sharma A.N., Doshi S., Courtot M., Lo R., Williams L.E., Frye J.G., Elsayegh T., Sardar D., Westman E.L., Pawlowski A.C., Johnson T.A., Brinkman F.S., Wright G.D., McArthur A.G. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(Database issue):D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M.J., Price N., Scotch M., Rabinowitz P. Comparison of ARIMA and Random Forest time series models for prediction of avian influenza H5N1 outbreaks. BMC Bioinformatics. 2014;13(15):276. doi: 10.1186/1471-2105-15-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesorn K., Ongruk P., Chompoosri J., Phumee A., Thavara U., Tawatsin A., Siriyasatien P. Morbidity rate prediction of dengue hemorrhagic fever (DHF) using the support vector machine and the Aedes aegypti infection rate in similar climates and geographical areas. PLoS One. 2015;10(5) doi: 10.1371/journal.pone.0125049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamping F., Jack T., Rübsamen N., Sasse M., Beerbaum P., Mikolajczyk R.T., Boehne M., Karch A. Development and validation of a diagnostic model for early differentiation of sepsis and non-infectious SIRS in critically ill children—a data-driven approach using machine-learning algorithms. BMC Pediatr. 2018;18:112. doi: 10.1186/s12887-018-1082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S., Tucker C.S., Kumara S. An unsupervised machine learning model for discovering latent infectious diseases using social media data. J. Biomed. Inform. 2017;66:82–94. doi: 10.1016/j.jbi.2016.12.007. [DOI] [PubMed] [Google Scholar]
- Luo W., Nguyen T., Nichols M., Tran T., Rana S., Gupta S., Phung D., Venkatesh S., Allender S. Is demography destiny? Application of machine learning techniques to accurately predict population health outcomes from a minimal demographic dataset. PLoS One. 2015;10(5) doi: 10.1371/journal.pone.0125602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A., Debnath T., Sood S.K., Baishnab K.L. Kyasanur forest disease classification framework using novel extremal optimization tuned neural network in fog computing environment. J. Med. Syst. 2018;42:187. doi: 10.1007/s10916-018-1041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson S. Learning without concentration for general loss functions. Probab. Theory Relat. Fields. 2018;171(1):459–502. [Google Scholar]
- Mikosch T., Wintenberger O. The cluster index of regularly varying sequences with applications to limit theory for functions of multivariate Markov chains. Probab. Theory Relat. Fields. 2014;159:157–196. [Google Scholar]
- Mohammed S.H., Ahmed M.M., Al-Mousawi A.M., Azeez A. Seasonal behavior and forecasting trends of tuberculosis incidence in Holy Kerbala, Iraq. Int. J. Mycobacteriol. 2018;7(4):361–367. doi: 10.4103/ijmy.ijmy_109_18. [DOI] [PubMed] [Google Scholar]
- Ohannessian I.M., Dahleh M.A. Rare probability estimation under regularly varying heavy tails. Conference on Learning Theory; 2012. pp. 1–21. [Google Scholar]
- Okell L.C., Drakele C.J., Bousema T., Whitty C.J., Ghani A.C. Modelling the impact of artemisinin combination therapy and long-acting treatments on malaria transmission intensity. PLoS Med. 2008;5(11) doi: 10.1371/journal.pmed.0050226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M.L., van der Laan M.J., Napravnik S., Eron J.J., Moore R.D., Deeks S.G. Long-term consequences of the delay between virologic failure of highly active antiretroviral therapy and regimen modification. AIDS. 2008;22(16):2097–2106. doi: 10.1097/QAD.0b013e32830f97e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M.L., LeDell E., Schwab J., Sarovar V., Gross R., Reynolds N., Haberer J.E., Goggin K., Golin C., Arnsten J., Rosen M., Remien R., Etoori D., Wilson I., Simoni J.M., Erlen J.A., van der Laan M.J., Liu H., Bangsberg D.R. Super learner analysis of electronic adherence data improves viral prediction and may provide strategies for selective HIV RNA monitoring. J. Acquir. Immune Defic. Syndr. 2015;69(1):109–118. doi: 10.1097/QAI.0000000000000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss R.D., Thomas M. Birkhäuser; 2007. Statistical Analysis of Extreme Values With Applications to Insurance, Finance, Hydrology and Other Fields. [Google Scholar]
- Rudd E.M., Jain P.L., Scheirer W.J. The extreme value machine. IEEE Trans. Pattern Anal. Mach. Intell. 2018;40:3. doi: 10.1109/TPAMI.2017.2707495. [DOI] [PubMed] [Google Scholar]
- Rushmore J., Bisanzio D., Gillespie T.R. Making new connections: insights from primate-parasite networks. Trends Parasitol. 2017;33(7):547–560. doi: 10.1016/j.pt.2017.01.013. [DOI] [PubMed] [Google Scholar]
- Saralamba S., Pan-Ngum W., Maude R.J., Lee S.J., Tarning J., Lindegårdh N., Chotivanich K., Nosten F., Day N.P., Socheat D., White N.J., Dondorp A.M., White L.J. Intrahost modeling of artemisinin resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 2011;108(1):397–402. doi: 10.1073/pnas.1006113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saybani M.R., Shamshirband S., Hormozi S.G., Wah T.Y., Aghabozorgi S., Pourhoseingholi M.A., Olariu T. Diagnosing tuberculosis with a novel support vector machine-based artificial immune recognition system. Iran. Red Crescent Med. J. 2015;17(4):e24557. doi: 10.5812/ircmj.17(4)2015.24557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saybani M.R., Shamshirband S., Golzari S., Wah T.Y., Saeed A., Kiah L.M., Balas V.E. RAIRS2 a new expert system for diagnosing tuberculosis with real-world tournament selection mechanism inside artificial immune recognition system. Med. Biol. Eng. Comput. 2016;54:385. doi: 10.1007/s11517-015-1323-6. [DOI] [PubMed] [Google Scholar]
- Shen Y., Yuan K., Chen D., Colloc J., Yang M., Li Y., Lei K. An ontology-driven clinical decision support system (IDDAP) for infectious disease diagnosis and antibiotic prescription. Artif. Intell. Med. 2018;86:20–32. doi: 10.1016/j.artmed.2018.01.003. [DOI] [PubMed] [Google Scholar]
- Silver D., Schrittwieser J., Simonyan K., Antonoglou I., Huang A., Guez A., Hubert T., Baker L., Lai M., Bolton A., Chen Y., Lillicrap T., Hui F., Sifre L., van den Driessche G., Graepel T., Hassabis D. Mastering the game of Go without human knowledge. Nature. 2017;550:354–359. doi: 10.1038/nature24270. [DOI] [PubMed] [Google Scholar]
- Smith R.L. Extreme value analysis of environmental time series: an application to trend detection in ground-level ozone. Stat. Sci. 1989;4(4):367–377. [Google Scholar]
- Sun G., Matsui T., Hakozaki Y., Abe S. An infectious disease/fever screening radar system which stratifies higher-risk patients within ten seconds using a neural network and the fuzzy grouping method. J. Infect. 2015;70(3):230–236. doi: 10.1016/j.jinf.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger J.K., Morens D.M. 1918 influenza: the mother of all pandemics. Emerg. Infect. Dis. 2006;12(1):15–22. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari K., Jamal S., Grover S., Goyal S., Singh A., Grover A. Cheminformatics based machine learning approaches for assessing glycolytic pathway antagonists of Mycobacterium tuberculosis. Comb. Chem. High Throughput Screen. 2016;19(8):667–675. doi: 10.2174/1386207319666160610080716. [DOI] [PubMed] [Google Scholar]
- Valleron A.J. Data science priorities for a university hospital-based institute of infectious diseases: a viewpoint. Clin. Infect. Dis. 2017;65(Suppl. 1):S84–S88. doi: 10.1093/cid/cix351. [DOI] [PubMed] [Google Scholar]
- van der Laan M.J., Polley E.C., Hubbard A.E. Super learner. Stat. Appl. Genet. Mol. Biol. 2007;6:25. doi: 10.2202/1544-6115.1309. [DOI] [PubMed] [Google Scholar]
- Velsko S., Bates T. A conceptual architecture for national biosurveillance: moving beyond situational awareness to enable digital detection of emerging threats. Health Secur. 2016;14(3) doi: 10.1089/hs.2015.0063. [DOI] [PubMed] [Google Scholar]
- Vinarti R., Hederman L. A knowledge-base for a personalized infectious disease risk prediction system. Stud. Health Technol. Inform. 2018;247:531–535. [PubMed] [Google Scholar]
- Walsh M.G., de Smalen A.W., Mor S.M. Wetlands, wild Bovidae species richness and sheep density delineate risk of Rift Valley fever outbreaks in the African continent and Arabian Peninsula. PLoS Negl. Trop. Dis. 2017;11(7) doi: 10.1371/journal.pntd.0005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Tian Y., Tian L.L., Qian Y.M., Li J.S. An electronic medical record system with treatment recommendations based on patient similarity. J. Med. Syst. 2015;39:55. doi: 10.1007/s10916-015-0237-z. [DOI] [PubMed] [Google Scholar]
- Wang Y., Yang Y.J., Chen Y.N., Zhao H.Y., Zhang S. Computer-aided design, structural dynamics analysis, and in vitro susceptibility test of antibacterial peptides incorporating unnatural amino acids against microbial infections. Comput. Methods Prog. Biomed. 2016;134:215–223. doi: 10.1016/j.cmpb.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Watkins A., Boggess L.C. A new classifier based on resource limited artificial immune systems. Proceedings of Congress on Evolutionary Computation, IEEE World Congress on Computational Intelligence Honolulu; 2002. [Google Scholar]
- Wei W., Jiang J., Gao L., Liang B., Huang J., Zang N., Ning C., Liao Y., Lai J., Yu J., Qin F., Chen H., Su J., Ye L., Liang H. A new hybrid model using an autoregressive integrated moving average and a generalized regression neural network for the incidence of tuberculosis in Heng County, China. Am. J. Trop. Med. Hyg. 2017;97(3):799–805. doi: 10.4269/ajtmh.16-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder B., Tambe M., Suen S.C. Preventing infectious disease in dynamic populations under uncertainty. AAAI Conference on Artificial Intelligence; 2018. [Google Scholar]
- Wong Z.S.Y., Zhou J., Zhang Q. Artificial intelligence for infectious disease Big Data Analytics. Infect Dis. Health. 2018 doi: 10.1016/j.idh.2018.10.002. pii: S2468-0451(18)30144-5. [DOI] [PubMed] [Google Scholar]
- Xu J., Wickramarathne T.L., Chawla N.V. Representing higher-order dependencies in networks. Sci. Adv. 2016;2(5) doi: 10.1126/sciadv.1600028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhang T., Young A.A., Li X. Applications and comparisons of four time series models in epidemiological surveillance data. PLoS One. 2014;9(2) doi: 10.1371/journal.pone.0088075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanga X., Amin E.A. Highly predictive support vector machine (SVM) models for anthrax toxin lethal factor (LF) inhibitors. J. Mol. Graph. Model. 2016;63:22–28. doi: 10.1016/j.jmgm.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]