

Abstract
Purpose
Triple negative breast cancer (TNBC)/ basal-like breast cancer (BLBC) is a highly aggressive form of breast cancer. We previously reported that a small molecule agonist ligand for the orphan nuclear receptor estrogen-related receptor beta (ERRβ or ESRRB) has growth inhibitory and anti-mitotic activity in TNBC cell lines. In this study, we evaluate the association of ESRRB mRNA, copy number levels, and protein expression with demographic, clinicopathological, and gene expression features in breast tumor clinical specimens.
Methods
ESRRB mRNA level expression and clinical associations were analyzed using RNAseq data. Array-based comparative genomic hybridization determined ESRRB copy number in AA and Caucasian women. Transcription factor activity was measured using promoter-reporter luciferase assays in TNBC cell lines. Semi-automatic quantification of immunohistochemistry measured ERRβ protein expression on a 150-patient tissue microarray series.
Results
ESRRB mRNA expression is significantly lower in TNBC/BLBC vs. other breast cancer subtypes. There is no evidence of ESRRB copy number loss. ESRRB mRNA expression is correlated with the expression of genes associated with neuroactive ligand-receptor interaction, metabolic pathways, and deafness. These genes contain G/C-rich transcription factor binding motifs. The ESRRB message is alternatively spliced into three isoforms, which we show have different transcription factor activity in basal-like vs. other TNBC cell lines. We further show that the ERRβ2 and ERRβsf isoforms are broadly expressed in breast tumors at the protein level.
Conclusions
Decreased ESRRB mRNA expression, and distinct patterns of ERRβ isoform subcellular localization and transcription factor activity are key features in TNBC/BLBC.
Keywords: Triple negative breast cancer, Basal-like breast cancer, Molecular subtypes, Estrogen-related receptor beta
INTRODUCTION
Breast cancer is the most commonly diagnosed cancer in women and is the number two cause of cancer-related death [1]. Breast cancer subtypes are predominantly classified by two methods: immunohistochemistry (IHC) or gene expression. IHC tests for three proteins: estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor two (HER2), and based on the expression of these three receptors, patients are classified as having ER+, HER2 overexpressing, or triple negative breast cancer (TNBC). Patients who are diagnosed with TNBC are clinically defined as lacking ER, PR, and HER2 [2]. Gene expression profiling uses a 50-gene panel to determine if a breast cancer is one of 5 intrinsic, or Pam50 subtypes: luminal A, luminal B, HER2-enriched, normal-like and basal-like (BLBC) [3, 4]. In clinic, TNBC and BLBC largely converge [5], with ≥ 80% of patients with TNBC also categorized as having BLBC. TNBC/BLBC is a biologically aggressive subtype of breast cancer with characteristic high genomic instability [6]. It is diagnosed more frequently in African-American (AA) women with much worse prognosis than Caucasian/White (CW) women [7].
Since patients diagnosed with TNBC have tumors that lack ER and HER2, they are unresponsive to ER and HER2–targeted therapies. Patients must instead be given systemic chemotherapy, which is accompanied by toxic side effects [8]. Due to the aggressive nature of TNBC, it is important to identify new prognostic marker genes capable of defining targets and conversely subtypes within TNBC. Historically, nuclear receptors (NR) have been great targets for cancer treatment, such as the ER in ER+ breast cancer. The NR superfamily consists of many members including orphan nuclear receptors (ONRs) [9], which are defined as lacking any known endogenous ligands. One ONR subgroup contains what are known as the estrogen related receptors (ERRs), named for their resemblance to the ER, although they do not bind or respond to estrogen [9]. ERR beta (ESRRB, ERRβ), which is alternatively spliced, is one of the first discovered ONRs and is known to have important functions in development [10]. Our lab and others, have previously shown the function of ERRβ in cancer. Our 2016 publication showed that patients with BLBC whose tumors have high ESRRB mRNA expression have significantly improved distant-metastases free, and recurrence- free survival in comparison to patients with low expression [11]. Previous publications have shown that patients with BLBC overall have tumors with significantly lower ESRRB mRNA expression in comparison to other breast cancer subtypes [12, 13].
The goal of the present study is to: (1) define ESRRB expression levels in breast cancer, specifically in BLBC and TNBC; (2) determine how DNA copy number and protein levels are modulated in various data sets to determine if the previously observed decrease in ESRRB is caused by copy number loss, and the effects of decreased mRNA on protein expression and function; (3) characterize clinical correlations found in breast cancer. We show that ESRRB mRNA expression is significantly lower in patients with BLBC/ TNBC and that there is no observed decrease in DNA copy number. ERRβ splice variants have differential transcription factor activity in TNBC cell lines. Lastly, we also show that ERRβ splice variant protein levels are different in breast cancer patient samples depending on the IHC subtype. Patients with HER2 or TNBC have similar ERRβ protein expression and cellular localization vs. patients with ER+. Our study supports further investigation into the establishment of ERRβ as a TNBC/BLBC therapeutic target or prognostic marker, and as a tool to provide more insights into this aggressive cancer and its mechanisms of progression.
Materials and Methods
In silico analyses
Illumina HT 12 gene array data from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) study was analyzed via KMplottter [14] using a subset of patients who did not receive systemic treatment. A total of 300 patients (145 luminal A, 85 luminal B, 22 HER2, 48 basal-like) were included in the hazard ratio analysis.
RNA-sequencing (RNAseq) data was obtained from two publically-available datasets sets: the Sweden Cancerome Analysis Network – Breast (SCAN-B) [15, 16] and The Cancer Genome Atlas (TCGA) [17]. Gene expression profiles of GSE96058 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96058) were downloaded from Gene Expression Omnibus (GEO) database for the SCAN-B data analyses. This SCAN-B cohort contained 3273 samples (136 were replicates) analyzed by Illumina paired-end RNAseq and expression estimation. This data comes from Sweden and country law states that the submitter cannot provide raw sequence data in a public repository as sequencing may contain personally-identifiable information and hereditary mutations. Data was processed as previously described [15, 16] and gene expression data was generated as FPKM (expression measurement +0.1 FPKM followed by log2 transformation). Clinical information was publically available on GEO Accession Viewer and kindly provided in a comprehensive, table format by Dr. Lao Saal (Lund University, Sweden). TCGA raw, processed and clinical data were obtained from the GDC legacy archive (https://portal.gdc.cancer.gov) and accessed using TCGABiolinks.
Gene expression analysis was performed using Rstudio (version 3.6.0) and Bioconductor. Gene expression levels, measured as FPKM (fragments per kilobase of transcript per million), were determined for all breast cancer patient tumors as two datasets: Pam50 subtypes reported by the authors, or IHC subtypes parsed out using clinical information. Overall survival was determined using survminer. Differentially expressed genes (DEGs) were detected using edgeR. Genes with p<0.05 and fold change ≥2 were considered significant (Table S1). Pathway and represented disease analyses were performed on overexpressed DEGs using KEGG pathway analysis [18, 19].TNBC subtypes were determined using Vanderbilt’s TNBCtype tool [20, 21].
For SCAN-B data, cox regression was used to determine correlations between ESRRB expression and clinical characteristics. For promoter analysis, Bioconductor was used to isolate DEGs found within annotated genome UCSC, hg19 and search the promoter region of these genes, −4000 to +500 bp from the transcription start site for enriched short, ungapped, redundant sequences of up to 8 base pairs using Discriminative Regular Expression Motif Elicitation (DREME, [22]). The primary set of sequences was shuffled to create a control set. Significantly overrepresented motifs were determined using Fisher’s Exact test. We then used TomTom [23] to compare our overrepresented motifs to the publically-available JASPAR CORE database [24]which provides the binding preferences of a large database of known transcription factors. Source code for SCAN-B and TCGA analyses can be found at https://github.com/RigginsLabGU/Rmarkdown/blob/master/SCANB%20analysis.Rmd and https://github.com/RigginsLabGU/Rmarkdown/blob/master/TCGA%20analysis.Rmd.
Array comparative genomic hybridization
The 106-patient cohort of 37 Caucasian (CW) and 69 African American (AA) patients with TNBC and non TNBC (NTN) was collected and processed as previously described by Sugita, et al [25],: 17 CW/TNBC; 20 CW/NTN; 39 AA/TNBC; 30 AA/NTN. Copy number was determined from ESRRB probes on the Agilent SurePrint aCGH platform. Log2 intensity >3 was defined as amplification, ≥/ ≤ 0.25 were defined as copy number gain and loss respectively, and ≥3 was defined as deletion. PRISM 8.0 (Graphpad, San Diego, CA) was used for all statistical analyses of copy number and clinical demographic information.
Cell Culture
HCC1806 breast cancer cells were purchased from ATCC (Manassas, VA). MDA-MB-453 breast cancer cells were a gift from Dr. Anna Riegel (Lombardi Comprehensive Cancer Center (LCCC). BT549, MCF7, and MCF10A breast cancer cells were obtained from the LCCC Tissue Culture Shared Resource. Cells were routinely tested for Mycoplasma spp. and tested negative and fingerprinted using 9 standard STR loci and Y chromosome- specific amelogenin to verify authenticity. All cells were maintained in a humid carbon dioxide (CO2) incubator, 95% air: 5% CO2. HCC1806 and MDA-MB-453 cells were grown in improved minimal essential media (IMEM; Life Technologies, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS, purchased from LCCC Tissue Culture Shared Resource). BT549 cells were cultured in IMEM with 10% FBS and 10 μg/mL insulin (purchased from Life Technologies, Grand Island, NY).
Plasmids and transfection
The psG5 empty vector, ERRβsf (murine ERRβ, >90% homology to human ERRβ, Addgene #52188), ERRβ2 (Addgene #52186), and ERRβ-Δ10 (Addgene #52187) constructs have all been published previously [11, 26]. The ERRE-luciferase (Addgene #37851) and p21-luciferase (Addgene #21723) have been previously described [26]. Plasmids were introduced using Mirus TransIT-X2 Dynamic Delivery System (Mirus Bio LLC; MIR600Q) according to manufacturer’s instructions.
Dual-luciferase promoter-reporter assays
Cells were seeded into 24-well plastic tissue culture dishes at 150,000 cells per well on day 0. On day 1, cells were transfected using 500 ng DNA/ well (137 ng receptor; 360 ng luciferase reporter plasmid; 3 ng Renilla control) for 24 hours. On day 2, media containing the transfection complexes was removed and fresh media was added to the cells. On day 3, at 48 hours, cells were harvested for luciferase assay (https://www.promega.com/products/reporter-assays-and-transfection/reporter-assays/dual_luciferase-reporter-assay-system/) according to the manufacturer’s instructions. Luciferase activity was normalized to Renilla. All experiments were performed 3-5 times.
Western blotting and antibodies
Lysate collected for dual-luciferase activity assay were run on 4-12% poly acrylamide gels using electrophoresis for 90 minutes. After protein was transferred to nitrocellulose membranes, the membranes were blocked for one hour in 5% nonfat dry milk in Tris-Buffered Saline with Tween-20 (TBST) and probed overnight at 4°C with ERRβ #PP-H6707-00 (cl.07) 1:500. All membranes were then re-probed with loading control β-tubulin at 1:10:000 for 1 hour at room temperature or overnight at 4°C.
Horseradish peroxidase enzyme-conjugated anti-mouse whole immunoglobulin (IgG) secondary antibody (GE #NXA931 Buckinghamshire, U.K.) was used the following day at 1:5000 for 1 hour at room temperature. The membranes were then imaged for enhanced chemiluminescence (ECL, Denville Scientific, Holliston, MA) on the Amersham Imager 600 (GE Life Sciences).
Image analysis and statistical analysis of in vitro work
Images and figures were composed using Adobe Photoshop, Illustrator and InDesign. FIJI was used to perform densitometry on imaged blots. Statistical analyses of dual-luciferase activity assay and western blot densitometry were done using PRISM 8.0 (Graphpad, San Diego, CA).
Invasive Ductal Carcinoma Breast Cancer TMA series
The Histopathology and Tissue Shared Resource (HTSR) at Georgetown University Medical Center’s Lombardi Comprehensive Cancer Center (LCCC) constructed the Invasive Ductal Carcinoma Tissue Microarrays (TMA) series. The cohort consists of 150 patients with breast cancer distributed as 50 patients each on a series of 3 TMAs. All patients are research-consented through the HTSR, the Survey, Recruitment and Biospecimen Shared Resource (SRBSR), and/or Indivumed groups under the following respective Georgetown University Medical Center IRB protocols: 1992-048, Pr0000007, and 2007-345. The 50 patients for each TMA were grouped based on the following molecular subtypes of their tumors: ER alpha positive TMA (>10% ER alpha positivity, PR+/−, HER2 negative), HER2 positive TMA (ER+/−, PR+/−) and Triple Negative Breast Cancer (TNBC) TMA. The TMAs were stained for ER alpha, PR, HER2, Ki67 and panCytokeratin in a single multiplexed assay. High resolution images are available for all stained cores as well as quantification of percentage positive for ER, PR, and Ki67 and threshold analysis for HER2 positivity. Two cores for each patient are available side by side on the TMA. Each TMA has matching biological and immunological controls including tonsil, spleen, testis, reduction mammoplasty (benign breast), placenta and 6 well characterized breast cancer cell lines. Inclusion criteria for the TMA were female patients diagnosed with invasive ductal carcinoma with at least 3 years clinical follow up (majority with 5-year follow up, or otherwise deceased) with a primary breast cancer surgical resection at MedStar Georgetown University Hospital (MGUH) between 2004-2014. Exclusion criteria were male patients, patients diagnosed with ductal carcinoma in situ only or lobular carcinoma, known BRCA or familial mutation carriers, or evidence of neoadjuvant therapy. Racial distribution of the patients was 64% White/Caucasian, 26% Black/African American, 10% other or unknown, allowing for support for projects analyzing racial disparities. Clinical, treatment and follow up data were retrieved by the Innovation Center for Biomedical Informatics (ICBI) from the MGUH Cancer Registry. Pathology data was manually extracted from the original surgical pathology reports. All demographic, clinical, pathology, and available follow up data were de-identified and uploaded into a REDCap database for query and analysis. High resolution images of hematoxylin and eosin stained cores are available for all cores on the TMA.
Immunohistochemical staining
IHC staining of breast cancer tissue was performed for ERRβ. Five micron sections from formalin fixed paraffin embedded tissues were de-paraffinized with xylenes and rehydrated through a graded alcohol series. Heat induced epitope retrieval (HIER) was performed by immersing the tissue sections in Target Retrieval Solution, Low pH (DAKO) in the PT Link (DAKO). IHC staining was performed using the VectaStain Kit from Vector Labs according to manufacturer’s instructions. Briefly, slides were treated with 3% hydrogen peroxide, avidin/biotin blocking, and 10% normal goat serum and independently exposed to primary antibodies for ERRβ2- cl .07, 1:150, 1:240 (R&D systems, #PP-H6707-00) and ERRβsf- cl .05, 1:150 (R&D systems, #PP-H6705-00) for 1 hour at room temperature. Slides were exposed to appropriate biotin-conjugated secondary antibodies (Vector Labs), Vectastain ABC reagent and DAB chromagen (Dako). Slides were counterstained with Hematoxylin (Fisher, Harris Modified Hematoxylin), blued in 1% ammonium hydroxide, dehydrated, and mounted with Acrymount. Control tissues with the primary antibody omitted were used as negative controls. Images of the full TMA slide stained for of hematoxylin and eosin, ERRβsf- cl .05, and ERRβ2- cl .07 are available on figshare (https://doi.org/10.6084/m9.figshare.9992891.v1).
Scanning and Analysis using Vectra3
Stained slides were scanned using the Vectra3 Multi-Spectral Imaging Microscope with Vectra and Phenochart software (Perkin Elmer). Every available TMA core was imaged as a 3x3 composite image to capture almost the entire core in one image. The scanned images were analyzed in inForm software version 2.3 (Akoya). The inForm software is trained to identify individual cells within the TMA core by hematoxylin staining and intensity, average cell size, and cell splitting algorithms. Brown DAB staining that overlapped with hematoxylin staining was identified by the software as being a nuclear staining, while staining without overlap with the nuclear signal was considered cytoplasmic. In this way, nuclear versus cytoplasmic staining was differentiated. An average of 11,000 cells were counted per TMA core.
Statistical analysis of tissue microarray data
To prepare for statistical analysis, individual cores were manually examined and matched to their corresponding coordinates defined by Vecta3 software. Any cores missing more that 50% of tissue due to adipose tissue or folding were omitted from final analysis. The two cores from each patient were individually scored then averaged to determining the 50 scores per TMA slide used in the analyses.
ERRβ-clone 07 (recognizes ERRβ2), ERRβ-clone 05 (recognizes ERRβsf), and their ratio, as well as demographic variables were summarized by using mean (standard deviation, sd) and median (interquartile range, IQR) for continuous variables and frequency and percentage for categorical variables. Kruskal-Wallis tests were used to test whether median ERRβ2 and ERRβsf expression, and their ratio, were significantly different among the three IHC receptor subtypes. To assess whether the expression of each isoform was significantly different among the three IHC subtypes while considering lymph node status, race, and age, and if there was any interaction between IHC subtype and demographic variables, we first made a logit transformation of ERRβ– clone 07 and ERRβ– clone 05 which achieved approximate normality. Then two-way ANOVA was used for analysis following ANOVA procedure. If a significant difference was observed, pairwise comparisons were performed by Dwass, Steel, Critchlow-Fligner multiple comparison procedure (DSCF).
Nuclear staining and cytoplasmic staining were summarized by IHC subtype using mean (sd) and median (IQR). Spearman’s correlation coefficient was calculated to measure the association between the nuclear and cytoplasmic staining in each IHC subtype.
All tests were two-sided at a significant level 0.05. No method was used for adjusting multiple comparisons. All analyses were performed using SAS 9.4 and Rstudio (Version 0.99.902) software.
RESULTS
Low ESRRB mRNA expression is associated with shorter overall survival
We previously published that high expression of ESRRB mRNA is associated with improved recurrence-free and distant-metastasis free survival in a merged cohort of patients with BLBC from multiple independent studies [11,27]. Here, we analyzed the association of ESRRB mRNA expression with overall survival by analyzing Illumina HT 12 gene array data from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) study [28]. Importantly, the subset of patients selected for this analysis did not receive systemic treatment, providing a more compelling link between ESRRB expression and clinical outcome that is not confounded by treatment effect. We found that low ESRRB expression (defined as below median) is associated with significantly shorter overall survival (OS) only in patients with BLBC (Fig. 1).
Fig. 1. ESRRB association with overall survival (OS) in women with systemically untreated breast cancer.

Hazard ratios, 95% confidence intervals, and log-rank p value for low ESRRB expression (METABRIC RNAseq data, below median) relative to OS.
ESRRB mRNA expression levels are significantly decreased in BLBC and TNBC, but not associated with age, grade, or lymph node status
We used two large, publicly-available RNAseq data sets –SCAN-B [15, 16] and The Cancer Genome Atlas (TCGA) [17] – to analyze ESRRB mRNA expression in association with demographic, clinicopathologic, and gene expression features. ESRRB expression as FPKM was measured in both data sets in patients stratified by either IHC or Pam50 breast cancer subtypes for analysis (Fig. 2). In SCAN-B, ESRRB expression was significantly lower in patients with BLBC compared to patients with luminal A or normal-like breast cancer (Fig. 2a). ESRRB expression was also significantly lower in patients with TNBC vs. patients with ER+ and ER+/HER2+ breast cancer (Fig. 2b). In TCGA data, we confirmed the previously published findings of Garattini, et al. [13] and found that patients with BLBC had significantly lower ESRRB expression compared to patients with luminal A breast cancer (Fig. 2c). Like the SCANB cohort, patients classified as TCGA-TNBC also had significantly lower ESRRB expression compared to patients with ER+ and ER+/HER2+ breast cancer (Fig. 2d).
Fig. 2. ESRRB mRNA expression in SCAN-B and TCGA data sets.

Analysis of RNAseq data from SCAN-B (a, b, e, f, g) and TCGA (c, d) data. Kruskal-Wallis one-way ANOVA and Tukey multiple comparisons of means with 95% family-wise confidence level. a-d. ESRRB mRNA FPKM levels by PAM50 subtype in SCAN-B (a) and TCGA (c) data. ESRRB mRNA levels by IHC subtype in SCAN-B (b) and TCGA (d) data. Shown as mean ± standard deviation, p < 0.0001 **** (e- g.) Correlations in SCAN-B dataset. e. Correlation of ESR1 to ESRRB mRNA expression f. ESRRB mRNA expression by age g. Correlation of ESRRB high and low expression, frequency of node status, age at diagnosis, NHG, endocrine treatment, and chemotherapy in all breast cancer patients in dataset. Spearman correlation. White spaces were not significantly different. Light to dark colors, correlation observed and statistically significant, p< 0.001
SCAN-B is a 3273-patient data set collected in Sweden starting in 2010 [15, 16]. Biospecimens were collected and RNAseq was performed on biopsy cores as previously described [15]. Comprehensive clinical and demographic information was also collected, allowing for analysis of ESRRB association with these data for both the Pam50 and IHC subtypes. Analysis of Pam50 subtype revealed patients with BLBC were significantly younger than patients with luminal A, luminal B, and HER2 breast cancer (Fig. S1a), a characteristic commonly found in patients with BLBC, while analysis by IHC subtype showed that patients with HER2 were significantly younger than patients with ER+ breast cancer (Fig. S1b).
We also assessed the correlation between ESRRB high and low mRNA expression and clinical characteristics. Across the entire SCAN-B cohort, there was a weak but statistically significant positive correlation between estrogen receptor (ESR1) and ESRRB mRNA expression (p = 8.1 e-05, Fig. 2e). There was no significant correlation between ESRRB expression and age (Fig. 2f). Filtered data sets were split into tertiles with the top third considered ESRRB “high” expression and the lower third considered “low” expression. Analysis within the Pam50 and IHC subtypes showed that there was no significant difference in age between patients with high versus low ESRRB within the Pam50 subtypes (not shown). There also were no significant differences observed in lymph node status, Nottingham grade (NHG), endocrine treatment or chemotherapy treatment between patients with high versus low ESRRB. However, receipt of chemotherapy was negatively correlated with age at diagnosis (p < 5e−107) and positively associated with Nottingham grade (NHG, p < 6.7e−127), which is broadly reflective of clinical management decisions made in the treatment of younger women and higher grade disease (Fig. 2g) [29].
OS was assessed comparing patients with ESRRB high versus ESRRB low breast cancer (upper vs. lower tertiles) of all subtypes, BLBC, and TNBC from TCGA and SCAN-B datasets. There was no statistically significant difference in OS found for patients with ESRRB high vs. low in SCAN-B or TCGA data (Fig. S1c-S1h). This is likely because in SCAN-B, >80% of the patient population remain alive, while in the TCGA data set filtering out patients with TNBC or BLBC leaves a low number of patients with an event (n = 123/1098), decreasing the power of these analyses. Though not statistically significant, patients with BLBC in the TCGA data set with high ESRRB expression did trend towards better OS than patients with ESRRB low (hazard ratio = 2.13). Overall, these data confirm lower ESRRB expression in patients with TNBC or BLBC from two large-scale cohorts but identify no statistically significant associations between ESRRB expression and age, grade, lymph node status, or treatment.
ESRRB does not exhibit copy number loss in TNBC
Next, we sought to determine if the observed decrease in ESRRB mRNA expression found in TNBC/BLBC may be due to copy number loss. We approached this by analyzing an independent, 106-patient cohort of Caucasian (CW) and African American (AA) patients with TNBC and non TNBC (NTN; all other breast cancer subtypes, i.e. ER+ and HER2-amplified) using array comparative genomic hybridization (aCGH) as previously described by Sugita, et al [25]. ESRRB copy number was calculated and defined as deletion, loss, gain, or amplification, and demographic information was analyzed for associations with ESRRB copy number changes (Fig. S2a-e). Fisher's exact test showed that in our cohort, patients with TNBC had significantly higher grade than non-triple negative (NTN) patients at diagnosis (p < 0.0001), indicating that our cohort is representative of the TNBC population (Fig. 3a). There were no significant differences observed between CW and AA patients with TNBC and NTN in relation to the total number of copy number alterations. However, > 75% of all patients showed copy number gain rather than loss at the ESRRB locus, 14q24.3 (Fig. 3b).
Fig. 3. ESRRB copy number in breast tumor specimens.

a. Women with TNBC have higher grade than women with nonTNBC, Fisher's exact test p<0.0001 **** b. Copy number determined from ESRRB probes on the Agilent SurePrint aCGH platform. Log2 intensity >3=amplification, >0.25=gain, ≤0.25=loss, ≥3=deletion. Shown by Caucasian (CA) vs. African American (AA) women, TNBC vs nonTNBC patients. Fisher’s exact test, all tumors, CA vs AA, n.s. c, d. Ancestrality. SNP chip Illumina Infinium QC Array (Illumina Inc., CA) looking at ~3,000 ancestral informative markers in a portion of the Caucasian American (CW) and African American (AA) patients included in the ESRRB copy number study. Four super populations were identified: European (EUR), African (AFR), Ad mixed American (AMR), and East Asian (ASN). d. ESRRB copy number in self-identified AA or CW redistributed as 1000s genomes project AFR descent or AMR descent. Mann-Whitney, n.s
Since self-reported race and ethnicity data are not always concordant with genotyping analyses, we also assessed ancestry informative markers (AIMs) and the distribution of ESRRB copy number in a small subset of our cohort [28, 30]. This 20-patient subset included 6 patients with TNBC and 14 patients with NTN and due to the small number is exploratory. Principal component analysis (PCA) of 3000 AIMs overlaid with self-reported ethnicity of our cases showed separation of distinct ancestral populations [25] when merged with two populations from the 1000 Genomes Project: African descent (AFR) and ad mixed American (AMR) (Fig. 3c). The mean rank for ESRRB was markedly lower in the AA, but there was no statistically significant difference in the average ESRRB copy number in breast tumors from the AA women (clustered with the AFR group) versus the CW (clustered with the AMR group) (Fig. 3d).
Differentially expressed genes in patients with tumors characterized by ESRRB high versus ESRRB low expression
We next identified DEGs in patients with tumors characterized by ESRRB mRNA-high versus ESRRB mRNA-low expression to determine what other genes are co-modulated with ESRRB expression. First, we compared tumors from patients that are ESRRB high and low (upper and lower tertiles, respectively) and assessed genes that were high in patients with ESRRB-high (left/ pink) and low in patients with ESRRB-high (right/ purple) (≥2-fold up- or down-regulated, p < 0.05, Fig. 4a-d; Table S1). We determined the overlap of DEGs between data sets and between BLBC and TNBC, finding the most overlap within datasets (Fig. 4e, Fig. S3a, b). Prior studies report that greater than 80% of patients with TNBC also have BLBC [5] and our analysis of the SCAN-B and TCGA data sets shows a similar overlap (Fig. S3c,d). KEGG pathway analysis [18, 19] found enrichment of genes associated with neuroactive ligand-receptor interaction (fold change ≥ 3.65) and metabolic pathways (fold change ≥ 2.34) overexpressed in patients with ESRRB high. Three fourths of the DEGs lists included deafness as one of the disease associations. Mutations in ESRRB are linked to a hereditary autosomal recessive hearing disorder [31, 32] (Fig. 4f).
Fig. 4. Differentially expressed genes (DEGs) associated with ESRRB expression in SCAN- and TCGA data sets.

DEG analysis in patients with BLBC (a, c) and TNBC (b, d). Shown are DEGs that are high in ESRRB high patients, and low in ESRRB high patients in SCANB data, p<0.01, fold change (FC) > 4. e. Heat map depicting DEG overlap between the four analyses shown in a-e. f. Top pathways and diseases represented by overexpressed DEGs in ESRRB high-versus- low patients.
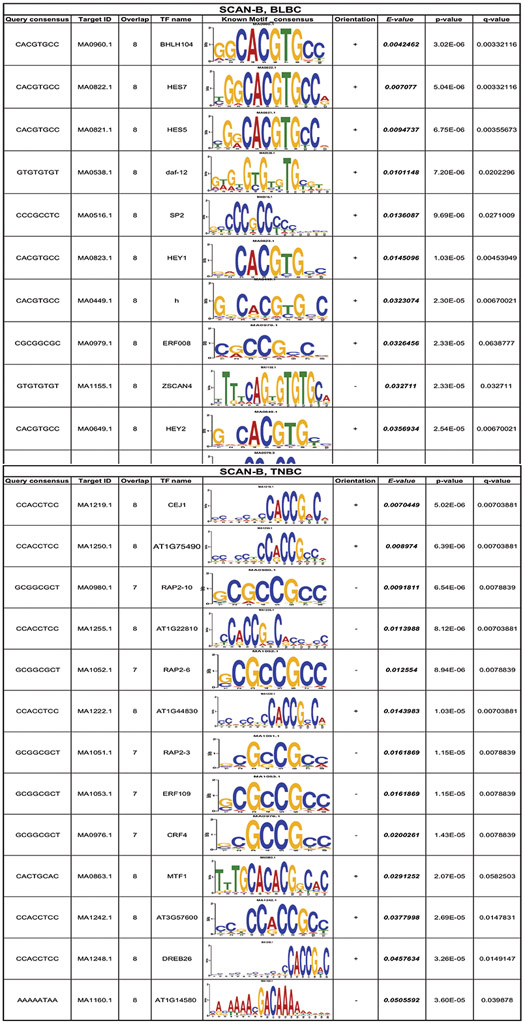
We then searched for transcription factor binding motifs of up to 8 base pairs within the promoter region (−4000 to +500 bp from the transcription start site) of the enriched DEGs in the SCAN-B dataset [22]. DEGs in BLBC samples had 39 enriched motifs which aligned to 320 known transcription factors TNBC samples had 40 enriched motifs which aligned to 611 known transcription factors (TOMTOM [23]; JASPAR [24], Fig. S3e). Top matches for both BLBC and TNBC in the SCAN-B dataset were extracted, E<0.05. The top motifs found in BLBC and TNBC were GGCACGTGCC and CCACCGACA, respectively. These motifs aligned to multiple known transcription factors, including basic helix-loop-helix (bHLH) and AP-2 motifs, both characterized as G/C-rich (Table 1).
Table 1. TOMTOM analysis of enriched transcription factor binding motifs.
Top motifs from DEGs aligned to JASPAR non-redundant DNA database. Corresponding motifs of known transcription factors are SCAN-B and TCGA data. Determined in BLBC and TNBC
 |
ERRβ isoform transcription factor activity differs across cellular models of TNBC molecular subtypes
A key challenge in defining bona fide ESRRB target genes amongst DEGs from high dimensional data sets is that ERRβ, at the protein level, is expressed as at least three distinct isoforms: ERRβsf, ERRβ2, ERRβ-Δ10 [33, 34]. These isoforms are produced via alternative splicing in which a genetic alteration at intron-exon boundaries, or splice sites, are altered at the mRNA level resulting in different proteins [35]. Two of these isoforms – ERRβsf and ERRβ2 – have opposing functions in regulating cell cycle progression [11], while less is known about ERRβ-Δ10 due to a lack of validated antibodies to detect this isoform at the protein level [36]. We first established basal level of two ERRβ isoforms – ERRβ2 and ERRβsf – in a panel of TNBC cell lines reflective of diverse TNBC subtypes. TNBC subtypes were initially defined as a step to further personalize medicine. There are 6 distinct TNBC subtypes based on gene expression and ontology [21, 37]. Cell lines representing 5 of the 6 subtypes, as well as non-transformed mammary epithelial cells MCF10A and estrogen-receptor positive cells MCF7, were probed for ERRβ2 and ERRβsf using two monoclonal antibodies we have previously characterized as selective for each isoform [11,26]. TNBC cell lines had a trend toward lower ERRβ protein levels in comparison to the MCF10As and MCF7s (Fig. 5a-c, Figure S4a, b). HCC1806s (basal-like 2, BL2) had the lowest ERRβ2:ERRβsf ratio while MDA-MB-453s (luminal androgen receptor, LAR) had the highest ratio, though these differences did not reach statistical significance (Fig. 5c). Consistent with this, assessment of TNBC subtypes, classified using TNBCtype [20, 21], at the mRNA level in SCAN-B tumor also showed no significant differences in ESRRB expression between the subtypes (Fig. S4c). Analysis of publicly available gene expression data in cell lines showed similar ERRβ2 mRNA expression (Fig. S4d, e).
Fig. 5. ERRβ transcription factor activity measured by heterologous promoter-reporter assays.

a,b. Endogenous expression of ERRβ2 (a) and ERRβsf (b) protein levels in cell lines representing TNBC subtypes. c. Ratio of ERRβ2 and ERRβsf expression in cell lines, determined by densitometry for each receptor and corrected by expression of the β-actin loading control. d. Measure of luciferase activity on sp1 and ERRE response elements in TNBC cell lines HCC1806, MDA-MB-453, and BT549. Kruskal-Wallis one-way ANOVA, ** p< 0.001, ***p<0.0001. (e-g) Western blot confirming overexpression of ERRβ splice variant plasmid DNA in cell lines.
To measure transcription factor function of ERRβ isoforms, we overexpressed cDNAs for each isoform in three different TNBC cell lines and measured their ability to induce transcription from heterologous promoter-reporter constructs. The three cell lines used were HCC1806, MDA-MB-453, and BT549, representing 3 of 6 TNBC subtypes [21]. The promoter-reporter constructs contained sequences corresponding to 2 established ERRβ response elements: estrogen related response element (ERRE, TNAAGGTCA) [38, 39] and specificity-protein-1 (SP1, (G/T)GGGCGG(G/A)(G/A)(C/T)) [40] (Fig. 5d). SP1 was also one of the top motifs identified in the BLBC subtype from the SCAN-B data set (Table 1). In HCC1806 (BL2) cells, there was significantly higher activity of both the ERRE and SP1 response elements induced by all three splice variants in comparison to empty vector. ERRβsf had significantly higher activity on the ERRE than ERRβ2 or ERRβ-Δ10, while ERRβ-Δ10 had significantly higher activity on the SP1 response element than ERRβsf or ERRβ2. In MDA-MB-453 (LAR) cells, ERRβsf had significantly higher activity on both the ERRE and SP1 response elements, while ERRβ-Δ10 had significantly higher activity on SP1 in comparison to empty vector. In BT549 (mesenchymal-like, ML) cells, ERRβsf and ERRβ-Δ10 had significantly higher activity on both the ERRE and SP1 response elements in comparison to empty vector. For both MDA-MB-453s and BT549s, ERRβ2 had no significant transcription factor activity on either ERRE or SP1. Western blot analysis confirmed overexpression of ERRβ isoforms in transfected cells (Fig. 5e-g). These data suggest that patterns of ERRβ isoform transcription factor activity may differ between basal-like and other TNBC molecular subtypes.
ERRβ isoform expression in breast tumor tissue varies by IHC subtype
We next assessed the protein expression of ERRβsf and ERRβ2 isoforms in patient tissues from a 150-patient TMA assembled by the HTSR at Georgetown University Medical Center’s LCCC. The TMA series consists of 50 invasive ductal carcinoma breast cancer patients each diagnosed with ER+, HER2 overexpressing, and TNBC IHC subtypes, with corresponding demographic and clinical information (race, pathological stage, age, treatment, Table 2). Post antibody optimization (Fig. S5a), consecutive sections of the TMA were stained for 1 of 2 antibodies: ERRβ-clone 05 (recognizes ERRβsf) or ERRβ-clone 07 (recognizes ERRβ2). Staining was quantified in a semi-automated fashion and nuclear versus cytoplasmic staining were differentiated using the Vectra3 Multi-Spectral Imaging Microscope. (Fig.6a-c, S5b). Representative image show the differential staining of the two antibodies (Fig. 6d). Statistical analyses were performed to determine the relationship of each isoform with lymph node status, race, and age.
Table 2.
Demographics of the 150-patient cohort included in the invasive ductal carcinoma (IDC) tissue microarray (TMA).
| ER+ | HER2+ | TNBC | ||||||
|---|---|---|---|---|---|---|---|---|
| Cohort Period | 2004-2011 | 2004-2014 | 2004-2012 | |||||
| Duration of follow up | Range (in years) | 1.05-11.14 | 0.84-13.96 | 1.24-22.63 | ||||
| Median (in years) | 6.39 | 5.48 | 6.49 | |||||
| Freq(%) ∣ Mean(sd), Median[IQR] | n | % | n | % | n | % | ||
| Sex | Female | 149 (99.33%) | 50 | 100% | 50 | 100% | 49 | 98% |
| Male | 1 (0.67%) | 0 | 0% | 0 | 0% | 1 | 2% | |
| Lymph Node Status | Positive | 47 (32.41%) | 18 | 36% | 17 | 34% | 12 | 24% |
| Negative | 98 (67.59%) | 32 | 64% | 32 | 64% | 34 | 68% | |
| Not documented | 5 (0.03%) | 0 | 0% | 1 | 2% | 4 | 8% | |
| Race | White/Caucasian | 96 (64%) | 31 | 62% | 33 | 66% | 32 | 64% |
| Black/African American | 40 (26.67%) | 15 | 30% | 12 | 24% | 13 | 26% | |
| Asian/Pacific Islander | 6 (4%) | 2 | 4% | 2 | 4% | 2 | 4% | |
| Other | 5 (3.33%) | 0 | 0% | 2 | 4% | 3 | 6% | |
| Unknown | 3 (2%) | 2 | 4% | 1 | 2% | 0 | 0% | |
| Age at diagnosis | Under 40 | 54.45(12.97), 55.05[43.89, 63.68] | 3 | 6% | 9 | 18% | 6 | 12% |
| 40-55 | 18 | 36% | 16 | 32% | 21 | 42% | ||
| Over 55 | 29 | 58% | 25 | 50% | 23 | 46% | ||
| Age Range | 30.7-88.23 | 27.63-88.32 | 28.06-88.52 | |||||
| Hispanic, Latino, Spanish Origin | Non-Spanish; non-Hispanic | 125 (83.33%) | 41 | 82% | 42 | 84% | 42 | 84% |
| Hispanic or Latino Origin | 1 (0.67%) | 0 | 0% | 0 | 0% | 1 | 2% | |
| Unknown | 24 (16%) | 9 | 18% | 8 | 16% | 7 | 14% | |
| Pathologic Stage Group | 0 | 1 (0.77%) | ||||||
| 1 | 38 (29.23%) | |||||||
| 2 | 1 (0.77%) | |||||||
| 4 | 2 (1.54%) | |||||||
| 1, 1 | 2 (1.54%) | |||||||
| 1, 1A | 1 (0.77%) | |||||||
| 1, 2A | 2 (1.54%) | |||||||
| 1A | 15 (11.54%) | |||||||
| 2A | 35 (26.92%) | |||||||
| 2A, 1A | 2 (1.54%) | |||||||
| 2A, 2B | 1 (0.77%) | |||||||
| 2B | 19 (14.62%) | |||||||
| 3A | 5 (3.85%) | |||||||
| 3B | 1 (0.77%) | |||||||
| 3C | 5 (3.85%) | |||||||
| Vital Status | Alive | 122 (81.33%) | 35 | 70% | 46 | 92% | 41 | 82% |
| Deceased | 28 (18.67%) | 15 | 30% | 4 | 8% | 9 | 18% | |
| Distant Metastasis | Yes | 7 (0.047%) | 3 | 6% | 0 | 0% | 4 | 8% |
| Recurrence | Yes | 11 (0.07%) | 5 | 10% | 2 | 4% | 4 | 8% |
| Breast Cancer Treatment | Surgery alone | 15 | 30% | 9 | 18% | 9 | 18% | |
| Surgery + radiation | 21 | 42% | 20 | 40% | 21 | 42% | ||
| Surgery + chemotherapy | 29 | 58% | 36 | 72% | 37 | 74% | ||
| Surgery + radiation + chemotherapy | 15 | 30% | 15 | 30% | 17 | 34% | ||
Fig. 6. ERRβ isoform expression by IHC.

A-D. Automatic scanning and semi-automatic quantification of ERRβ splice variant specific mouse monoclonal antibodies (ab) on tissue microarray (TMA). a. Blue/brown staining with ERRβ ab. Automatic detection of b. nuclear v cytoplasmic staining, and c. cells “positive”/ above threshold vs. “negative”/ below threshold. d. ERRβ protein expression in breast tumors. Representative images of ER+, HER2, and TNBC tumor tissues from 150 patient tissue microarray series stained with ERRβ ab
The median of ERRβ2, but not ERRβsf, expression was significantly different amongst the three IHC receptor subtypes: ER+, HER2, TNBC (p = 0.016). Post hoc analysis showed that ERRβ2 was significantly different between patients with ER+ and HER2 breast cancer (p = 0.017, Fig. 7a, Table S2.1). Median ERRβsf: ERRβ2 expression ratio was lowest in TNBC subtypes (indicating lower ERRβ2 or higher ERRβsf expression), however this ratio was not statistically significantly different amongst the three IHC receptor subtypes (Figure 7b).
Fig. 7. Semi-automated quantification of ERRβ IHC.

A. Total percent (%) of patients that are ERRβ positive, by IHC subtype, *p<0.01. b. Ratio of total ERRβ2:total ERRβsf determined for ER+, HER2+, and TNBC patients. c-e. ERRβ expression by race. Interactions between ERRβ2 receptor expression (c,d) and ERRβsf expression (e,f) and race. Two- way ANOVA, followed by DSCF pair-wise comparison *p < 0.05
We next assessed statistical interactions and main effects of three variables: IHC subtypes (variable 1), lymph node status, race or age (variable 2), and ERRβ status (variable 3). For ERRβ2 expression, there were no statistical interactions between the IHC subtypes and lymph node status, race or age that affected ERRβ status. There was no significant main effect on ERRβ2 expression by IHC subtypes with lymph node status, or, by IHC subtypes with age. However, race did show a significant main effect (p = 0.024) or difference in ERRβ2 expression amongst the different races (Fig. 7c, Table S3). Follow-up post-hoc Kruskal-Wallis found that the ERRβ2 median was significantly different amongst the three IHC subtypes in AA patients (p = 0.0312, Figure 7d). Further pairwise comparisons with DSCF method showed this observed difference was between patients with ER+ and TNBC (p = 0.0212, Fig. 7b, d).
There were no significant statistical interactions between the IHC subtypes and lymph node status or age that affected ERRβsf status, however we did see significant interaction effect with race (p = 0.009) indicating differential ERRβsf expression within the 5 races between the 3 IHC subtypes (Table S3, Fig. 7e). This significant interaction was followed up with a Kruskal-Wallis post hoc analysis which showed that median ERRβsf expression was significantly different among 5 races in the HER2 IHC receptor subtype (p = 0.0102). Follow-up pairwise comparisons showed that this significant difference was between AA and CW patients (p = 0.0282, Fig. 7f).
We next analyzed the subcellular localization of ERRβsf and ERRβ2 isoforms amongst the different IHC subtypes. Previous studies have shown that ERRβsf and ERRβ2 have different localization and functions within cells [11], with ERRβsf predominantly nuclear and ERRβ2 expressed in both the nucleus and cytoplasm. ERRβ2 had higher cytoplasmic vs. nuclear staining in patients with ER+ compared to patients with HER2 or TNBC, while ERRβsf had both nuclear and cytoplasmic staining in all three IHC subtypes (Fig. 8a, 8b, Table S2.2). We performed Spearman rank analysis to measure the association between the nuclear and cytoplasmic staining of ERRβ2 and ERRβsf in each IHC subtype. In the ER+ group, both ERRβ2 and ERRβsf nuclear staining were significantly positively correlated with cytoplasmic staining (r= 0.945, p < 0.0001 and r=0.488, p = 0.00038, respectively) meaning the expression of nuclear ERRβ2/ERRβsf increases when the value of cytoplasmic ERRβ2/ERRβsf increases. In the HER2 group, ERRβ2 nuclear staining was significantly negatively correlated with cytoplasmic staining (r= −0.922, p <0.0001), while ERRβsf nuclear staining was positively correlated with cytoplasmic staining (r=0.928, p <0.0001). In the TNBC group, ERRβ2 nuclear staining was also significantly negatively correlated with cytoplasmic staining (r= −0.729, p <0.0001), while ERRβsf nuclear staining was positively correlated with cytoplasmic staining, (r=0.93, p <0.0001). (Fig. 8c).
Fig. 8. Subcellular localization of ERRβ2 and ERRβsf in TMA cores.

Cytoplasmic and nuclear localization of ERRβ2 (a) and ERRβsf (b) determined for the three IHC subtypes. c. Correlation of ERRβ2 and ERRβsf between nuclear and cytoplasmic staining, by IHC subtype
DISCUSSION
In this study, we characterize ONR ESRRB/ERRβ copy number and expression and its clinical correlations in breast cancer. Patients with low ESRRB mRNA expression have significantly shorter overall survival. ESRRB mRNA expression is significantly lower in TNBC/BLBC vs. other breast cancer subtypes, but not significantly associated with age, lymph node status, or grade. In an independent dataset, we find no evidence of ESRRB copy number loss in patients with TNBC, suggesting that reduced mRNA expression is driven by other mechanisms. We further find that ESRRB expression is correlated with that of genes associated with neuroactive ligand-receptor interaction, metabolic pathways, and deafness, and that these genes contain G/C-rich transcription factor binding motifs. We show that the ESRRB message, which is alternatively spliced into three distinct isoforms, leads to different isoform transcription factor activity in TNBC cell lines that are characterized as basal-like vs. the mesenchymal or luminal androgen receptor subtype. Finally, we show in clinical samples that the ERRβ2 and ERRβsf isoforms are broadly expressed at the protein level.
We found that the loss of ESRRB mRNA in patients with TNBC/BLBC was not due to a loss of copy number, and in fact there was a gain in ESRRB copy number in all breast cancer subtypes. Though the copy number gain was not significant, it does suggest that there is a mechanistic variance occurring during transcription of this gene in breast cancer patients that drives the loss of mRNA that is consistently and significantly observed especially in TNBC/BLBC. While our ancestrality marker results were not significant, in the subset which had a small number of patients, we saw a trend toward reduced ESRRB copy number in AA versus CW patients which aligned with 1000 genome projects-defined AFR and AMR patients, respectively. This addresses a clinical diagnosis issue frequently encountered in which demographic information is misreported [41]. In this case, we looked at self-reported race in comparison to refined race and ethnic information from genotyping, and saw a difference in copy number that was not seen with only the self-reported race. The trend suggests that with a statistically-powered analysis (more patients) we may see more definitive results. Our results suggest that there is value to the addition of ancestrality markers to future analyses. It may also address an issue with accessibility to race data. For example, because of the limited amount of clinical data on race, when we filter for AA patients with TNBC with expression of ESRRB there are < 29 out of the 1098 patients in TCGA.
When looking at transcription factor activity we found that the three known splice variants ERRβsf, ERRβ2, ERRβ–Δ10 when exogenously expressed, have differential activity on known ESRRB response elements. In basal-like TNBC cell line, BL2, ERRβsf had higher activity on the ERRE while ERRβ–Δ10 had higher activity on the sp1 response element. Meanwhile in LAR and ML TNBC cell lines, ERRβsf and ERRβ–Δ10 had higher activity on both response elements, while ERRβ2 had no change in activity. The LAR and ML subtypes are consistent with our previous publication in which we showed that in a ER+ setting ERRβsf has higher activity on the ERRE while ERRβ2 shows no change in activity [11]. The difference in activity may be explained by the differing C-terminus of the splice variants. While all three isoforms share exons 3 - 9, ERRβsf is truncated at exon 9, ERRβ2 has exon 10 and part of 11 and ERRβ–Δ10 skips exon 10, but contains exons 11 and 12. Due to the different exon combinations, the 3 variants have different functional domains which may cause steric hindrance and thus may alter the binding of each variant to DNA. To further validate these findings, stable cell lines with inducible overexpression of ESRRB isoforms should be established. Transient overexpression limits the time frame in which experiments can be carried out to 24-72 hours, with optimal expression ~48 hours in the TNBC cell lines. Stable overexpression would allow us to further evaluate other features such as proliferation, cell death, migration, and invasion in association with overexpression of specific ESRRB isoforms. Non-transformed mammary epithelial cells MCF10As should also be established with both stable overexpression and knockdown/knockout with rescue.
To date, the small molecule agonist DY131 (DY) is one of the more promising ESRRB – targeting drugs [42]. Treatment with DY decreases cancer cell, but not non-transformed mammary epithelial cell, proliferation, induces apoptosis and causes spindle polarity defects. This, paired with the knowledge that ERRβ2 mediates mitotic arrest, as well as localization of the ERRβ2 splice variant [11] leads to the hypothesis that treatment with DY may target ERRβ2 preferentially in the cancer setting. Our finding in this work showing higher amounts of ERRβ2 compared to ERRβsf supports this hypothesis. This drug needs to be further studied and refined for use in humans. Using DY in conjunction with stable overexpression cell lines will strengthen our findings.
This is the first reported comparative quantification of ERRβsf and ERRβ2 protein expression in IHC breast cancer subtypes. Our 150-patient TMA revealed that in all three IHC subtypes (ER+, HER2, TNBC) a higher percentage of tumor cells are ERRβ2+ than ERRβsf+. We established that ERRβ2 was significantly different between patients with ER+ or HER2, and that the ratio of ERRβ2:ERRβsf was lower in patients with TNBC that it was in patients with ER+ or HER2. Though this difference was not statistically significant, it did suggest that in the more aggressive breast cancer subtype there is either an increase in expression of the ERRβsf splice variant, or a decrease in expression of the ERRβ2 splice variant. Our previous publications have led to the hypothesis that ERRβ-specific, ligand-mediated mitotic arrest is due to the ERRβ2 splice variant, and that the loss of this splice variant is an effect of the malignant transformation associated with TNBC/BLBC [11].
We have previously published that in breast cancer cell lines ERRβ2 localizes to the cytoplasm and centrosomes in the nucleus while ERRβsf localizes in the nucleus [11]. We further looked at this localization to see if there were differences amongst the three IHC subtypes. We established that while in ER+ tumors there was a positive correlation between nuclear and cytoplasmic staining for ERRβ2 and ERRβsf, both HER2 and TNBC - clinically aggressive subtypes - showed a positive correlation for ERRβsf and a negative correlation for ERRβ2. Furthermore, ERRβ2 localized almost exclusively in the nucleus in both of those settings. The inconsistency from our previous findings points to the question of heterogeneity and how to best treat cancer patients based on the different cells within a tumor.
This study has provided insight into the expression of ERRβ and its implications in TNBC/BLBC, an aggressive subtype of breast cancer which, to date, does not have any targeted therapies. Limitations of our study include the underrepresentation of AA patients, a group that is disproportionally affected by this breast cancer subtype. Though the SCAN-B data set is very large, it has limited racial and ethnic diversity. Our TMA had ~1/3 AA patients, which is an improvement over past studies, some of which did not include any AA patients. Additionally, we have shown the value ancestrality information can provide in lieu of self-reported race.
Another limitation of our analyses includes the inability to properly examine splice variant – specific ESRRB at the mRNA level. Due to the overall loss of ESRRB observed in the cancer setting, it is extremely difficult to look at individual isoform expression. To properly quantify splice variants at the RNA level, we would need to use ultra-deep RNAseq [43, 44]. This increases genome coverage and improves sequencing accuracy, providing assurance that we are accurately looking at isoform mRNA expression.
Future steps include in vitro validation of our DEG, pathway, and promoter analyses to further determine ESRRB co-regulatory partners. As previously stated, the establishment of stable cell lines will be instrumental in these validation studies. With this information ESRRB/ERRβ could serve as a future therapeutic target or as a biomarker in TNBC.
Supplementary Material
Supplemental Fig. 1. Age at diagnosis and association of ESRRB with OS in SCAN-B and TCGA data sets. a, b. Average age of patients in SCAN-B data set by Pam50 (a) and IHC (b) subtypes. ANOVA with multiple comparisons ***p<0.001, **** p<0.0001. c-h. KM plots showing overall survival of all breast cancer patients (c,f), BLBC patients (d,g), and TNBC patients (e,h) in SCAN-B (c-e) and TCGA (f-h) data
Supplemental Fig. 2. Demographics of aCGH cohort. A-E. Distribution of (a) age, (b) tumor size and ESRRB copy number, (c) pathology, (d) lymph node status and (e) metastasis
Supplemental Table 1. DEGs in ESRRB high and low patients. List of DEGs found in BLBC and TNBC patients from SCAN-B and TCGA data sets.
Supplemental Fig. 3. Overlap of BLBC patients and TNBC patients. a, b. Heat map representing differential expression of overlapping genes in SCAN-B versus TCGA data sets. c, d. Venn diagram of patients in SCAN-B (c) and TCGA (d) data. e. List of overrepresented motifs in promoter region DEGs in SCAN-B data sets
Supplemental Fig. 4. Protein and mRNA levels of ERRβ / ESRRB. Densitometry quantifying ERRβ splice variants ERRβ2 (a) and ERRβsf (b) protein levels in cell lines c. ESRRB mRNA levels in SCAN-B patients, sorted into TNBC subtypes. d, e. ESRRB mRNA levels in cell lines representing the TNBC cell lines
Supplemental Fig. 5. IHC optimization and localization. a. Negative and positive controls used for antibody optimization of ERRβsf ERRβ2. Scale bar = 100μm b. High magnification view representing subcellular staining of tumor cores staining for ERRβsf or ERRβ2. Selected panels for ERRβsf from tumor cores identified by Vectra3 as >90% nuclear and cytoplasmic staining, or >90% cytoplasmic staining. Arrows in left panels show positive nuclei.
Supplemental Table 2. ERRβ isoform expression and subcellular localization. Mean (sd) and median (IQR) expression of ERRβ2 receptor, ERRβsf receptor, and total ERRβ2:total ERRβsf expression (S2.1) and nuclear/ cytoplasmic localization of ERRβ2 receptor and ERRβsf (S2.2) in three IHC breast cancer subtypes
Supplemental Table 3. ERRβ isoform expression and clinical features. Analysis of ERRβ2 and ERRβsf receptor expression in the IHC subtypes and lymph node status, race, and age, with and without interaction
Acknowledgments
We are grateful to Allison Fitzgerald, Dr. Hillary Stires, Ayodeji Olukoya, and Sonali Persaud for their insights and/or critical reading of the manuscript. We would like to thank Drs. Bassem Haddad, Filipa Lynce, and Michael Johnson for their guidance in developing the HTSR’s invasive ductal carcinoma breast cancer tissue microarray series. We thank Henry Cho and Gaelle Palmer for their contributions to the TMA-associated REDCap database. Thank you to the Survey, Recruitment, and Biospecimen collection Shared Resource (SRBSR) for their support of research recruitment at MedStar Georgetown University Hospital. Thank you to Dr. Lao Saal of Lund University, Sweden for kindly providing the clinical information of SCAN-B data. We thank Garrett Graham for his guidance on all computational studies and Max Kushner for his aid with the DREME analysis. The results shown here are based in part upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding
These studies were supported in part by Department of Defense Breast Cancer Research Program award W81XWH-17-1-0615 to RBR. Georgetown University Medical Center Shared Resources are supported in part by P30 CA051008 (Lombardi Comprehensive Cancer Center Support Grant; Principal Investigator Dr. Louis Weiner). Fellowship funding for AIF was provided by the LCCC’s Graduate Training in Breast Cancer Health Disparities Research grant from Susan G. Komen for the Cure (GTDR15330383; Principal Investigator Lucile L. Adams-Campbell).
ABBREVIATIONS
- TNBC
Triple negative breast cancer
- BLBC
Basal-like breast cancer
- AA
African-American
- ESRRB
Estrogen related receptor beta
- IHC
Immunohistochemistry
- ER
Estrogen receptor
- PR
progesterone receptor
- HER2
human epidermal growth factor two
- CW
Caucasian/White
- NR
Nuclear receptor(s)
- ONR
Orphan nuclear receptor
- ERR
Estrogen related receptors
- OS
Overall survival
- SCAN-B
Sweden Cancerome Analysis Network – Breast
- FPKM
Fragments Per Kilobase of transcript per Million mapped reads
- ESR1
Estrogen receptor
- NHG
Nottingham grade
- NTN
Non triple negative breast cancer
- aCGH
Array comparative genomic hybridization
- AFR
African descent
- AMR
Ad mixed American
- DEGs
Differentially expressed genes
- BL2
Basal-like 2
- LAR
Luminal Androgen Receptor
- ML
Mesenchymal-like
- ERRE
Estrogen related response element
- SP1
Specificity-protein-1
- TMA
Tissue microarray
Footnotes
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
Ethical approval: This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
This article does not contain any studies with human participants performed by any of the authors.
References
- 1.Society AC: Breast Cancer Facts & Figures 2017-2018. In. Edited by Atlanta: American Cancer Society I; 2017. [Google Scholar]
- 2.Anders CK, Abramson V, Tan T, Dent R: The Evolution of Triple-Negative Breast Cancer: From Biology to Novel Therapeutics. Am Soc Clin Oncol Educ Book 2016, 35:34–42. [DOI] [PubMed] [Google Scholar]
- 3.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z et al. : Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009, 27(8): 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallden B, Storhoff J, Nielsen T, Dowidar N, Schaper C, Ferree S, Liu S, Leung S, Geiss G, Snider J et al. : Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics 2015, 8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO: Prognostic markers in triple-negative breast cancer. Cancer 2007, 109(1):25–32. [DOI] [PubMed] [Google Scholar]
- 6.Feng Y, Spezia M, Huang S, Yuan C, Zeng Z, Zhang L, Ji X, Liu W, Huang B, Luo W et al. : Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis 2018, 5(2):77–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott LC, Mobley LR, Kuo TM, Il'yasova D: Update on triple-negative breast cancer disparities for the United States: A population-based study from the United States Cancer Statistics database, 2010 through 2014. Cancer 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa RLB, Gradishar WJ: Triple-Negative Breast Cancer: Current Practice and Future Directions. J Oncol Pract 2017, 13(5):301–303. [DOI] [PubMed] [Google Scholar]
- 9.Mullican SE, Dispirito JR, Lazar MA: The orphan nuclear receptors at their 25-year reunion. J Mol Endocrinol 2013, 51(3):T115–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giguère V, Yang N, Segui P, Evans RM: Identification of a new class of steroid hormone receptors. Nature 1988, 331(6151):91–94. [DOI] [PubMed] [Google Scholar]
- 11.Heckler MM, Zeleke TZ, Divekar SD, Fernandez AI, Tiek DM, Woodrick J, Farzanegan A, Roy R, Üren A, Mueller SC et al. : Antimitotic activity of DY131 and the estrogen-related receptor beta 2 (ERRβ2) splice variant in breast cancer. Oncotarget 2016, 7(30):47201–47220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long MD, Campbell MJ: Pan-cancer analyses of the nuclear receptor superfamily. Nucl Receptor Res 2015, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garattini E, Bolis M, Gianni' M, Paroni G, Fratelli M, Terao M: Lipid-sensors, enigmatic-orphan and orphan nuclear receptors as therapeutic targets in breast-cancer. Oncotarget 2016, 7(27):42661–42682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z: An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment 2010, 123(3):725–731. [DOI] [PubMed] [Google Scholar]
- 15.Saal LH, Vallon-Christersson J, Häkkinen J, Hegardt C, Grabau D, Winter C, Brueffer C, Tang MH, Reuterswärd C, Schulz R et al. : The Sweden Cancerome Analysis Network - Breast (SCAN-B) Initiative: a large-scale multicenter infrastructure towards implementation of breast cancer genomic analyses in the clinical routine. Genome Med 2015, 7(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brueffer C, Vallon-Christersson J, Grabau D, Ehinger A, Häkkinen J, Hegardt C, Malina J, Chen Y, Bendahl P-O, Manjer J et al. : Clinical Value of RNA Sequencing-Based Classifiers for Prediction of the Five Conventional Breast Cancer Biomarkers: A Report From the Population-Based Multicenter Sweden Cancerome Analysis Network—Breast Initiative. JCO Precision Oncology 2018(2):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Network CGA: Comprehensive molecular portraits of human breast tumours. Nature 2012, 490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M: New approach for understanding genome variations in KEGG. Nucleic Acids Res 2019, 47(D1):D590–D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Goto S: KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 2000, 28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, Pietenpol JA: TNBCtype: a subtyping tool for triple-negative breast cancer. Cancer informatics 2012, 11: CIN. S9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA: Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011, 121(7):2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey TL: DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011, 27(12):1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS: Quantifying similarity between motifs. Genome Biol 2007, 8(2):R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018, 46(D1):D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugita B, Gill M, Mahajan A, Duttargi A, Kirolikar S, Almeida R, Regis K, Oluwasanmi OL, Marchi F, Marian C et al. : Differentially expressed miRNAs in triple negative breast cancer between African-American and non-Hispanic white women. Oncotarget 2016, 7(48):79274–79291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heckler MM, Riggins RB: ERRβ splice variants differentially regulate cell cycle progression. Cell Cycle 2015, 14(1):31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z: An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010, 123(3):725–731. [DOI] [PubMed] [Google Scholar]
- 28.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al. : The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486(7403):346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tesarova P: Breast cancer in the elderly-Should it be treated differently? Rep Pract Oncol Radiother 2012, 18(1):26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR et al. : A global reference for human genetic variation. Nature 2015, 526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collin RW, Kalay E, Tariq M, Peters T, van der Zwaag B, Venselaar H, Oostrik J, Lee K, Ahmed ZM, Caylan R et al. : Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet 2008, 82(1): 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ben Saïd M, Ayedi L, Mnejja M, Hakim B, Khalfallah A, Charfeddine I, Khifagi C, Turki K, Ayadi H, Benzina Z et al. : A novel missense mutation in the ESRRB gene causes DFNB35 hearing loss in a Tunisian family. Eur J Med Genet 2011, 54(6):e535–541. [DOI] [PubMed] [Google Scholar]
- 33.Chen F, Zhang Q, McDonald T, Davidoff MJ, Bailey W, Bai C, Liu Q, Caskey CT: Identification of two hERR2-related novel nuclear receptors utilizing bioinformatics and inverse PCR. Gene 1999, 228(1-2): 101–109. [DOI] [PubMed] [Google Scholar]
- 34.Zhou W, Liu Z, Wu J, Liu JH, Hyder SM, Antoniou E, Lubahn DB: Identification and characterization of two novel splicing isoforms of human estrogen-related receptor beta. J Clin Endocrinol Metab 2006, 91(2):569–579. [DOI] [PubMed] [Google Scholar]
- 35.Roy B, Haupt LM, Griffiths LR: Review: Alternative Splicing (AS) of Genes As An Approach for Generating Protein Complexity. Curr Genomics 2013, 14(3):182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Divekar SD, Tiek DM, Fernandez A, Riggins RB: Estrogen-related receptor β (ERRβ) - renaissance receptor or receptor renaissance? Nucl Recept Signal 2016, 14:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, Pietenpol JA: Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One 2016, 11(6):e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sem DS, Casimiro DR, Kliewer SA, Provencal J, Evans RM, Wright PE: NMR Spectroscopic Studies of the DNA-binding Domain of the Monomer-binding Nuclear Orphan Receptor, Human Estrogen Related Receptor-2 THE CARBOXYL-TERMINAL EXTENSION TO THE ZINC-FINGER REGION IS UNSTRUCTURED IN THE FREE FORM OF THE PROTEIN. Journal of Biological Chemistry 1997, 272(29):18038–18043. [DOI] [PubMed] [Google Scholar]
- 39.Gearhart MD, Holmbeck SM, Evans RM, Dyson HJ, Wright PE: Monomeric complex of human orphan estrogen related receptor-2 with DNA: a pseudo-dimer interface mediates extended half-site recognition. Journal of molecular biology 2003, 327(4):819–832. [DOI] [PubMed] [Google Scholar]
- 40.Castet A, Herledan A, Bonnet S, Jalaguier Sp, Vanacker J-M, Cavaillès V: Receptor-interacting protein 140 differentially regulates estrogen receptor-related receptor transactivation depending on target genes. Molecular Endocrinology 2006, 20(5): 1035–1047. [DOI] [PubMed] [Google Scholar]
- 41.Data NRCUPoDCoRaE: Improving Racial and Ethnic Data on Health: Report of a Workshop In: Improving racial and ethnic data on health. Edited by Melnick D, Perrin E. Washington (DC):National Acadamies Press (US); 2003. [PubMed] [Google Scholar]
- 42.Donna DY, Forman BM: Identification of an agonist ligand for estrogen-related receptors ERRβ/γ. Bioorganic & medicinal chemistry letters 2005, 15(5): 1311–1313. [DOI] [PubMed] [Google Scholar]
- 43.Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH: Accurate and comprehensive sequencing of personal genomes. Genome Res 2011, 21(9): 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirebrahim H, Close TJ, Lonardi S: De novo meta-assembly of ultra-deep sequencing data. Bioinformatics 2015, 31(12):i9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Age at diagnosis and association of ESRRB with OS in SCAN-B and TCGA data sets. a, b. Average age of patients in SCAN-B data set by Pam50 (a) and IHC (b) subtypes. ANOVA with multiple comparisons ***p<0.001, **** p<0.0001. c-h. KM plots showing overall survival of all breast cancer patients (c,f), BLBC patients (d,g), and TNBC patients (e,h) in SCAN-B (c-e) and TCGA (f-h) data
Supplemental Fig. 2. Demographics of aCGH cohort. A-E. Distribution of (a) age, (b) tumor size and ESRRB copy number, (c) pathology, (d) lymph node status and (e) metastasis
Supplemental Table 1. DEGs in ESRRB high and low patients. List of DEGs found in BLBC and TNBC patients from SCAN-B and TCGA data sets.
Supplemental Fig. 3. Overlap of BLBC patients and TNBC patients. a, b. Heat map representing differential expression of overlapping genes in SCAN-B versus TCGA data sets. c, d. Venn diagram of patients in SCAN-B (c) and TCGA (d) data. e. List of overrepresented motifs in promoter region DEGs in SCAN-B data sets
Supplemental Fig. 4. Protein and mRNA levels of ERRβ / ESRRB. Densitometry quantifying ERRβ splice variants ERRβ2 (a) and ERRβsf (b) protein levels in cell lines c. ESRRB mRNA levels in SCAN-B patients, sorted into TNBC subtypes. d, e. ESRRB mRNA levels in cell lines representing the TNBC cell lines
Supplemental Fig. 5. IHC optimization and localization. a. Negative and positive controls used for antibody optimization of ERRβsf ERRβ2. Scale bar = 100μm b. High magnification view representing subcellular staining of tumor cores staining for ERRβsf or ERRβ2. Selected panels for ERRβsf from tumor cores identified by Vectra3 as >90% nuclear and cytoplasmic staining, or >90% cytoplasmic staining. Arrows in left panels show positive nuclei.
Supplemental Table 2. ERRβ isoform expression and subcellular localization. Mean (sd) and median (IQR) expression of ERRβ2 receptor, ERRβsf receptor, and total ERRβ2:total ERRβsf expression (S2.1) and nuclear/ cytoplasmic localization of ERRβ2 receptor and ERRβsf (S2.2) in three IHC breast cancer subtypes
Supplemental Table 3. ERRβ isoform expression and clinical features. Analysis of ERRβ2 and ERRβsf receptor expression in the IHC subtypes and lymph node status, race, and age, with and without interaction