

Abstract
Hippocampal lesions including synaptic injury, neuroinflammation, and impaired neurogenesis are featured pathology closely associated with neuronal stress and cognitive impairment in Alzheimer’s disease (AD). Previous studies suggest that ghrelin and its receptor, growth hormone secretagogue receptor 1α (GHSR1α), promote hippocampal synaptic function and neurogenesis. GHSR1α activation thus holds the potential to be a therapeutic avenue for the treatment of hippocampal pathology in AD; however, a comprehensive study on the preventive effect of MK0677 on hippocampal lesions in AD-related conditions is still lacking. In this study, we treated a transgenic mouse model of AD-like amyloidosis (5xFAD mice) at the asymptomatic stage with MK0677, a potent ghrelin mimetic. We found that MK0677 fostered hippocampal neurogenesis in 5xFAD mice but observed little preventive function with regards to the development of hippocampal amyloid-β (Aβ) deposition, synaptic loss, microglial activation, or cognitive impairment. Furthermore, MK0677 at a dose of 3 mg/kg significantly increased 5xFAD mouse mortality. Despite enhanced hippocampal neurogenesis, MK0677 treatment has little beneficial effect to prevent hippocampal lesions or cognitive deficits against Aβ toxicity. This study, together with a failed large-scale clinical trial, suggests the ineffectiveness of MK0677 alone for AD prevention and treatment.
Keywords: Alzheimer’s disease, amyloid-β, growth hormone secretagogue receptor 1α, hippocampus, MK0677
INTRODUCTION
Alzheimer’s disease (AD) is characterized by progressive cognitive decline with featured pathology including brain amyloid-β (Aβ) deposition and tauopathy [1]. The hippocampus is an AD-sensitive brain region, and hippocampal lesions are closely associated with cognitive impairment accompanying AD patients [2, 3]. However, the etiopathogenetic mechanisms of hippocampal deficits in AD remain poorly understood. As a result, effective avenues to rescue hippocampal function in AD are still lacking.
Ghrelin is a neuropeptide predominantly produced in the stomach [4]. The abundance of the ghrelin receptor (growth hormone secretagogue receptor 1α (GHSR1α)) in the hippocampus suggests the relevance of the ghrelin/GHSR1α system to hippocampal function [5–7]. Indeed, in addition to its orexigenic function [8, 9], GHSR1α activation has been linked to the modulation of hippocampal neurogenesis and synaptic strength [10–13]. In this context, GHSR1α activation has the potential to exert protection on hippocampal function in AD-relevant pathological settings. A previous study reported promoting effects of GHSR1α activation by ghrelin on hippocampal neurogenesis in a mouse model of brain amyloidosis (5xFAD mice) [14]. Additionally, several studies have shown that GHSR1α’s natural ligand or agonists protect against Aβ toxicity-induced synaptic pathology and cognitive impairment in multiple lines of animal models mimicking AD pathology [15–18]. Notably, in a previous large-scale clinical trial, MK0677, a potent ghrelin mimetic, was found to be ineffective in mitigating cognitive impairment in AD patients [19]. It could be argued that the patients in the failed clinical trial were at “too” advanced stage for MK0677 to demonstrate its benefit. Therefore, it would be of great interest to examine the preventive effect of MK0677 at the “prodromal” stage of AD pathology.
In this study, we treated 5xFAD mice at the asymptomatic stage with MK0677. Although MK0677 promoted hippocampal neurogenesis in 5xFAD mice, the treatment failed to prevent the development of hippocampal Aβ accumulation, synaptic loss, and microglial activation. In addition, we did not observe the beneficial effect of MK0677 on 5xFAD mouse spatial learning and memory, which is in agreement with the lack of observed effect of MK0677 in mitigating cognitive impairment in AD patients [19]. Therefore, the most parsimonious interpretation of our study is that GHSR1α activation by MK0677 could enhance hippocampal neurogenesis in AD-related conditions but has limited translational potential for AD prevention and treatment.
MATERIALS AND METHODS
Mice and agonist treatment
5xFAD mice (B6SJL-Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799Vas/Mmjax) were obtained from Jackson Laboratory and crossed with B6SJL-Tg mice [20]. Ghsr null mice were originally from Dr. Jeffrey M. Zigman at UT Southwestern Medical Center and bred for experimental use [21]. Mouse breeding and usage was performed in accordance with the guidelines of the University of Texas at Dallas (UTD) Institutional Animal Care and Use Committee (IACUC) and National Institutes of Health (NIH). Mice were used in pairs of age-matched non-transgenic mice (nonTg). Genotypes of animals were confirmed using PCR and/or amyloid plaque staining. The number of mice used was determined by previous data and calculations to ensure that the minimal number of mice as required was used in these experiments. 3-month-old nonTg mice received daily intraperitoneal (i.p.) injections of saline (sterilized 0.9% NaCl) for one month. The age- and gender-matched 5xFAD mice and the littermates nonTg mice received daily i.p. injections of either saline (sterilized 0.9% NaCl) or MK0677 (Tocris, #5272, at doses of 1.5 or 3 mg/kg) diluted in saline for one month. Ghsr null mice received daily i.p. injections of saline (sterilized 0.9% NaCl) or MK0677 (Tocris, #5272, 1.5 mg/kg) diluted in saline for one month. Mouse body weight was recorded every five days until euthanasia. After the 1-month treatment, the mice were subjected to the Morris water maze behavioral test and then euthanized for tissue collection following IACUC-approved procedures.
Immunocytochemistry
Frozen sections were prepared as previously described [22]. Mouse brains were dissected and immediately fixed in 4% paraformaldehyde (PFA) (Sigma-Aldrich) for 24–26 h at 4°C followed by keeping in 30% sucrose for 24–26 h at 4°C. The slices were blocked with blocking buffer (5% goat or donkey serum as needed, 0.3% Triton X-100 in PBS, pH 7.4) for 1 h, then incubated with primary antibodies at room temperature overnight. The following antibodies were used: rabbit-anti-β-amyloid (CST, #8243, 1:1,000) for Aβ deposition detection, mouse-anti-Doublecortin (Santa Cruz Biotechnology, #sc271390, 1:100), rabbit-anti-PSD95 (CST, #3450, 1:400), guinea pig-anti-vGlut1 (Synaptic system, #135304, 1:400), goat-anti-Iba1 (abcam, #ab5076, 1:600). After washing with PBS, the slices were probed with appropriate cross-adsorbed secondary antibodies conjugated to Alexa Fluor 488 and/or Alexa Fluor 594 (Thermo Fisher Scientific, 1:400). Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific, #R37606, 1 drop/500 μl) as needed. Images were collected on a Nikon confocal microscope.
Amyloid plaque deposition analysis
For amyloid plaque deposition analysis, the amyloid plaque channel was subjected to “Binary” then “Define Threshold” in NIS element software. The threshold was set to include all the positive staining and the same setting was applied to all images. Volume measurement was then performed for all the plaques as well as each single plaque in both the hippocampal and cortical areas using the ROI selection option. Amyloid plaque-occupied percentage in the hippocampus or cortex and the average individual plaque volume was used to compare amyloid plaque deposition between 5xFAD saline and MK0677 treatment groups.
Neurogenesis analysis
Neurogenesis in the hippocampal dentate gyrus (DG) area was presented by performing doublecortin (DCX) staining in fixed brain slices as previously described [14]. DCX-positive (DCX+) cell number was counted manually. The DG area was measured using NIS element software. DCX+ cell density was calculated by dividing the DCX+ cell number by the DG area to evaluate neurogenesis.
Synaptic density analysis
For synaptic density analysis, vesicular glutamate transporter 1 (vGlut1)- and postsynaptic density 95 (PSD95)-stained channels were saved as two binary layers, the threshold for each channel defined separately and then applied to all the images. The overlapping areas were analyzed by using the “AND” operation in the “Binary Operation” dialogs of NIS element software [23]. Hippocampal volume was measured as described in the previous section (amyloid plaque deposition analysis). The overlapping dots represent synapses and average synapse number per selected area volume was calculated to represent synaptic density.
Microglia morphology and plaque-associated microglia density analysis
Analysis of microglia morphology was done by producing three-dimensional (3D) reconstructions of cells using Imaris software (Bitplane, Imaris x64 9.0.2) [24]. Using the Filament algorithm, the software was able to calculate the length of each cell dendrite and number of branch points. This was done by first selecting the appropriate region of interest for the desired cell. If there were nearby cells, the somas of those cells were included in the region of interest. Since the software builds the dendrites based on extension from the largest set diameter, doing this can prevent unwanted filaments bridging the dendrites of two or more cells. The dendrite diameter parameters were set with the largest diameter being the soma size and the smallest diameter at 0.5 μm. To further prevent unwanted connections, the option to ‘Remove disconnected segments’ was used in the dendrite building menu with the distance threshold set at 5 μm. After the algorithm finished detecting the dendrites, non-specifically detected filaments were manually trimmed. Dendrite length and branch points were then calculated by the software.
Plaque-associated microglia density was analyzed by using NIS element software. Microglia were counted under the following rule: if more than 50% of the microglia soma was within a plaque, it was counted as one plaque-associated microglia. Plaque volume was analyzed as described in the previous section (amyloid plaque deposition analysis).
Mouse behavioral test
The Morris water maze test was performed as previously described in order to test changes in mouse spatial learning and memory [25, 26]. Mice were randomly grouped by gender, genotypes, and treatments (which the experimenter was blind to) and then transferred to the testing environment at least 0.5 h before tests. During the test, mice were trained to find a hidden platform (20 cm diameter) in an open swimming pool (130 cm diameter) filled with 21°C water (nontoxic white dye was used to hide the platform). Four trials were performed each day for 12 days. Each trial started at a different position (NW, N, E, SE) while the platform was kept in the same location (SW). Each trial lasted 60 s followed by 30 s during which the mice were left on the platform to memorize its location. After 12 days of training, mice were subjected to a 60-s probe test, which started from a new position (NE) and the platform was removed. The frequency with which mice swam across the previous platform location as well as swimming speed and latency was recorded and analyzed with HVS Image software (HVS Image 2015). Probe results were analyzed using the platform location crossed frequency value.
Statistics
Statistical analyses were performed using Graph-Pad Software (Prism 7). For mouse lifespan, body weight, neurogenesis, synaptic density, hippocampal microglia density, Morris water maze learning curve, probe, and swimming speed, two-way ANOVA followed by Bonferroni post hoc analysis were performed. For hippocampal and cortical total plaque-occupied percentage, individual plaque volume, plaque-associated microglia density, microglia dendrite length, and microglia branch point number, unpaired Student’s t-test were performed. Results are presented as mean ± s.e.m. T-values and p-values were used to represent the variation and the significance level, respectively. p < 0.05 was considered as statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant.
RESULTS
MK0677 did not mitigate hippocampal Aβ loading in 5xFAD mice
To determine whether MK0677 mitigates hippocampal lesions in 5xFAD mice, we first sought to determine the optimal dose of MK0677. nonTg and 5xFAD mice at 3 months old received either saline or MK0677 at 1.5 or 3 mg/kg via daily i.p. injection for 30 days (Fig. 1A). 5xFAD mice at 3 months old have little to no hippocampal lesions and indiscernible behavioral change, both of which become prominent at 4 months of age (the endpoint of treatment) [20, 23, 25, 27, 28]. The doses of MK0677 were chosen based on previous studies [15, 19, 29]. Although 1.5 mg/kg MK0677 showed little effect on nonTg or 5xFAD mouse survival as compared with saline treatment, MK0677 at 3 mg/kg significantly increased the mortality of 5xFAD mice (Fig. 1B). Of note, 1.5 mg/kg MK0677 did not affect mouse body weight (Fig. 1C). We therefore selected 5xFAD mice that received the 1.5 mg/kg dose of MK0677 for biological response assays. Next, we examined the impact of MK0677 treatment on hippocampal Aβ deposition in 5xFAD mice. To this end, immunofluorescent staining using the specific antibody against Aβ was performed to visualize Aβ plaques. 5xFAD mice exhibited similar levels of total Aβ plaque-occupied volume regardless of MK0677 treatment (Fig. 1D1 and D3, in percentage values, 5xFAD saline 1.792 ± 0.169 versus 5xFAD MK0677 1.889 ± 0.206, p > 0.05). In addition, the measurement of the size of each single Aβ plaque showed that MK0677 administration did not significantly alter the pattern of Aβ accumulation in the hippocampus (Fig. 1D2 and D3, 5xFAD saline 11649 ± 1044.82 μm3 versus 5xFAD MK0677 12508 ± 929.80 μm3, p > 0.05). These results indicate that MK0677 at 1.5 mg/kg is not protective against hippocampal Aβ deposition, which is supported by previous studies showing minimal effects of ghrelin and its mimetic on Aβ deposition in transgenic AD mouse models [14, 16].
Fig. 1.
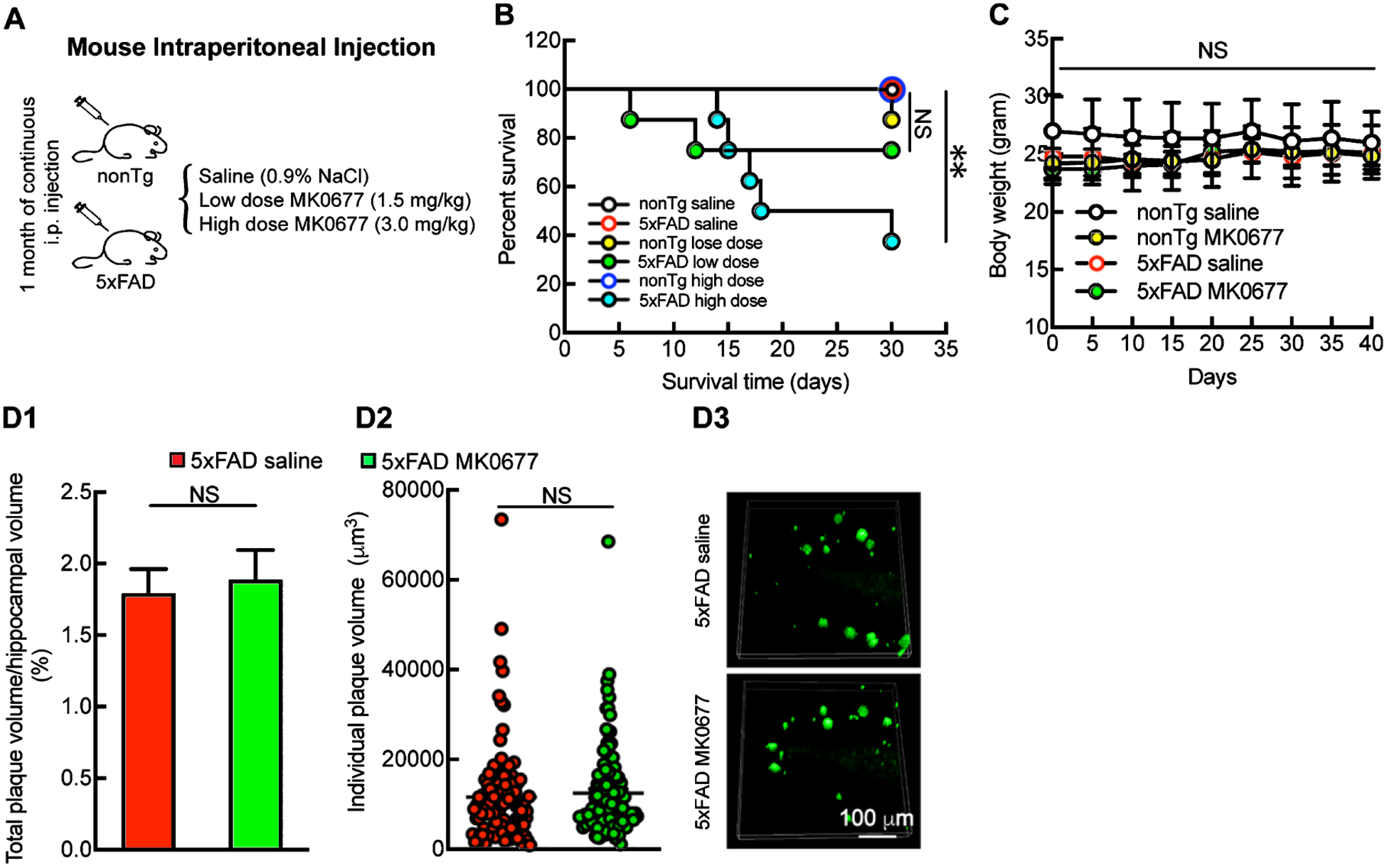
Unchanged hippocampal Aβ deposition in MK0677-treated 5xFAD mice. A) Daily MK0677 intraperitoneal Injection on mice. B) 3.0 mg/kg MK0677 treatment had significant impact on 5xFAD lifespan. Two-way ANOVA followed by Bonferroni post hoc analysis. **p < 0.01 between nonTg saline and 5xFAD 3.0 mg/kg MK0677groups; NS, no significant difference between nonTg saline and 5xFAD 1.5 mg/kg MK0677 groups, n = 4–8 mice. C) Mouse body weight remained the same during 1.5 mg/kg MK0677 treatment. NS, no significant difference among all the groups, n = 4–8 mice. D1, D2) 1.5 mg/kg MK0677 treatment had no influence on amyloid plaque-occupied volume on 5xFAD mice. Unpaired Student’s t-test. t = 0.3613, NS, not significant, n = 4 mice per group. D2) 1.5 mg/kg MK0677 treatment did not change the single amyloid plaque volume in mice hippocampi. Unpaired Student’s t-test. t = 0.6155, NS, not significant, n = 4 mice each group. D3) Representative images for amyloid plaque staining. Scale bar = 100 μm.
MK0677 promoted hippocampal neurogenesis in 5xFAD mice
In view of the neurogenic effect of GHSR1α activation [10] and defective neurogenesis in AD-relevant pathological settings [30], we examined neurogenesis in the DG by immunostaining of DCX, a specific marker of immature granule cells [31]. Compared with their nonTg littermates, 5xFAD mice exhibited a substantial decrease in DCX-positive cells in the dentate gyrus (Fig. 2A and B, nonTg saline 2.183 ± 0.454 number/10,000 μm2 versus 5xFAD saline 0.789 ± 0.147 number/10,000 μm2, p < 0.05), suggesting impaired neurogenesis in Aβ-rich milieus. Although nonTg MK0677-treated mice at 1.5 mg/kg only induced a marginal increase in DCX-positive cells (Fig. 2A and B, nonTg saline 2.183 ± 0.454 number/10,000 μm2 versus nonTg MK0677 2.412 ± 0.319 number/10,000 μm2, p > 0.05), 5xFAD mice with 1.5 mg/kg MK0677 treatment showed a remarkably augmented number of DCX-positive cells in their dentate gyri (Fig. 2A and B, 5xFAD saline 0.789 ± 0.147 number/10,000 μm2 versus 5xFAD MK0677 2.038 ± 0.303 number/10,000 μm2, p < 0.05). Importantly, the age- and gender-matched Ghsr null mice displayed a substantially reduced number of DCX-positive cells and showed no response to MK0677’s neurogenic effect (Fig. 2A and B, Ghsr null saline 0.914 ± 0.161 number/10,000 μm2 versus Ghsr null MK0677 0.934 ± 0.132 number/10,000 μm2, p > 0.05), indicating the close relationship of GHSR1α signaling and neurogenesis. Our findings are in agreement with previous reports that GHSR1α activation promotes hippocampal neurogenesis in AD mouse models [14].
Fig. 2.
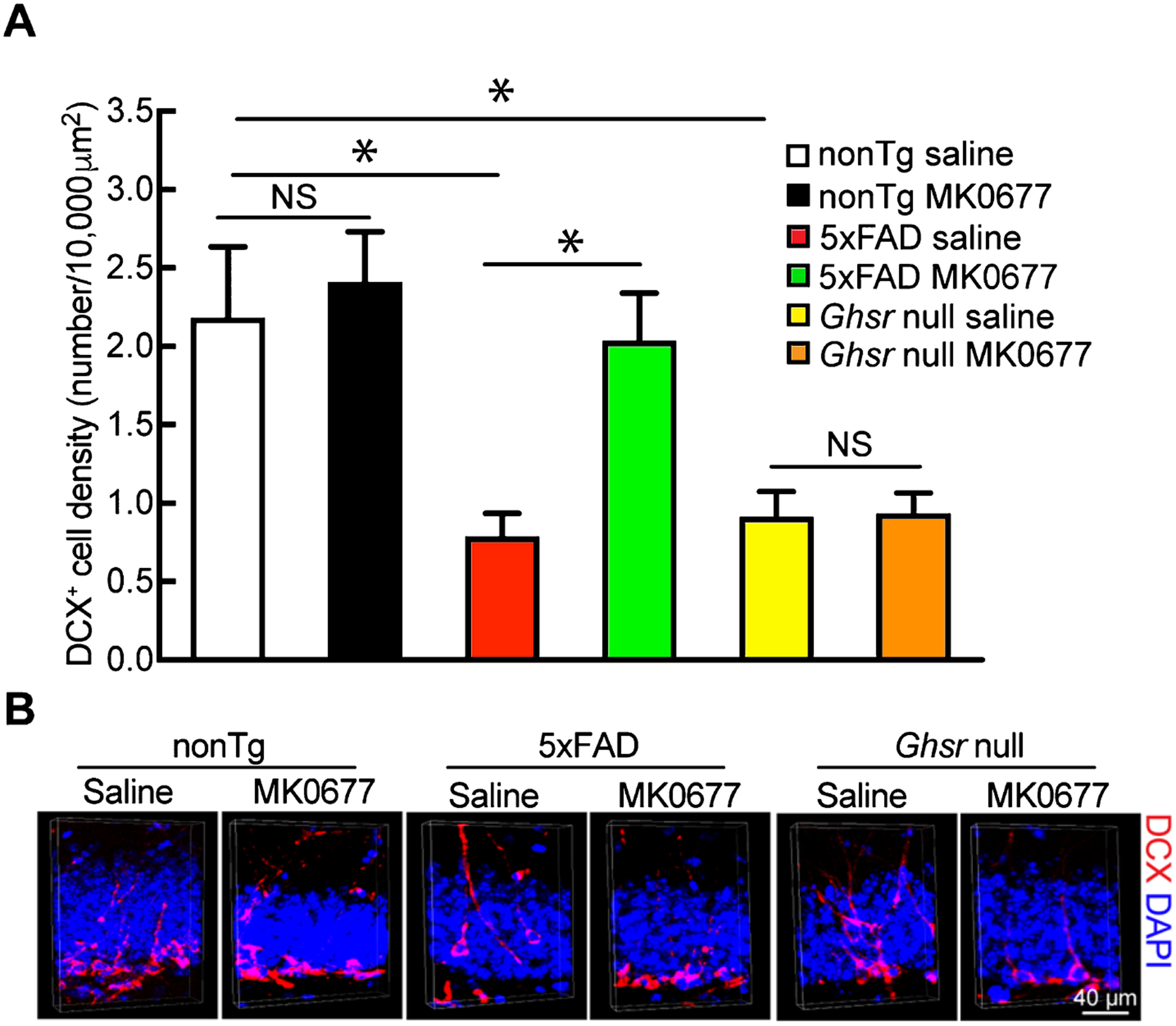
MK0677 treatment significantly increased neurogenesis in 5xFAD mice. A) 5xFAD mice presented increased DCX+ cells after MK0677 treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. t = 3.541, *p < 0.05 nonTg saline versus 5xFAD saline; t = 0.5837, NS, no significant difference between nonTg saline and nonTg MK0677 treatment groups; t = 3.172; *p < 0.05 5xFAD saline versus 5xFAD MK0677; t = 3.223, *p < 0.05 nonTg saline versus Ghsr null saline; t = 0.04955; NS, no difference between Ghsr null saline and MK0677 treatment groups. n = 4 mice per group. B) Representative images for neurogenesis, DCX+ cells represent newly generated neurons (red), nuclei were labeled with DAPI (blue). Scale bar = 40 μm.
MK0677 did not alleviate hippocampal synaptic loss in 5xFAD mice
Although MK0677 at the tested dose did not affect hippocampal Aβ deposition, it was unclear whether or not elevated neurogenesis would mitigate hippocampal synaptic loss by replenishing degenerated neurons. In an effort to elucidate this, we measured synaptic density in the CA1 region, which is an AD-sensitive hippocampal area [32] with pronounced synaptic loss and abundant GHSR1α expression [5–7]. Synapses were identified by colocalized staining of postsynaptic density 95 (PSD95, postsynaptic marker) and vesicular glutamate transporter 1 (vGlut1, presynaptic marker) [23]. In comparison with their nonTg counterparts, saline-treated 5xFAD mice demonstrated significantly decreased synaptic density in the CA1 region (Fig. 3A and B, in comparison with nonTg saline, nonTg saline 1.000 ± 0.0137 versus 5xFAD saline 0.573 ± 0.0361, p < 0.001). NonTg mice showed a mild response to MK0677 treatment, displayed by a slight increase in synaptic density in the CA1 region (Fig. 3A and B, in comparison with nonTg saline, nonTg saline 1.000 ± 0.0137 versus nonTg MK 0677 1.150 ± 0.0657, p > 0.05). However, such 5xFAD genotypic synaptic loss was not significantly attenuated by MK0677 administration (Fig. 3A and B, in comparison with nonTg saline, 5xFAD saline 0.573 ± 0.0361 versus 5xFAD MK0677 0.610 ± 0.0178, p > 0.05). In addition, we also examined synaptic density in the DG region, where principal neurons are born [33]. Similarly, MK0677 treatment on 5xFAD mice demonstrated little beneficial effect on synaptic loss in the DG (Supplementary Figure 1A and B, in comparison with nonTg saline, 5xFAD saline 0.720 ± 0.0375 versus 5xFAD MK0677 0.681 ± 0.0554, p > 0.05).
Fig. 3.
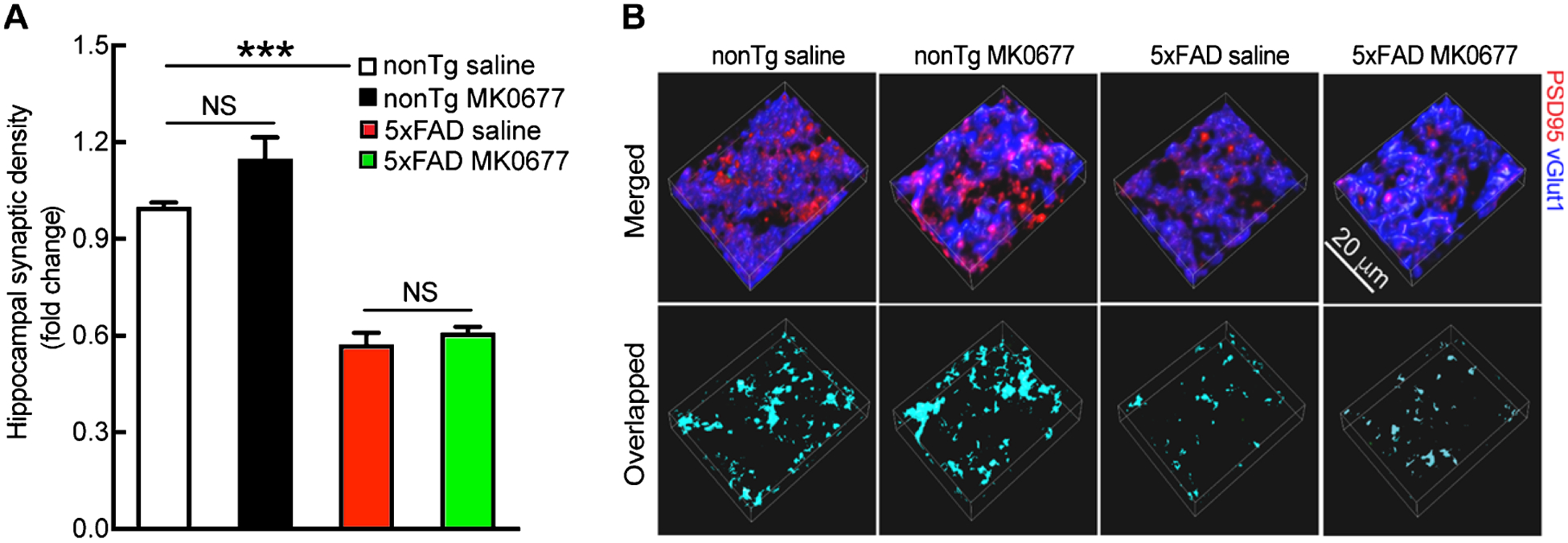
Synaptic density remained unaltered in 5xFAD regardless of MK0677 treatment. A) Saline and MK0677-treated 5xFAD mice showed similar synaptic density. Two-way ANOVA followed by Bonferroni post hoc analysis. t = 7.725, ***p < 0.001 between nonTg saline and 5xFAD saline groups; t = 2.704, NS, no significant difference for nonTg saline and MK0677 treatment groups; t = 0.6671, NS, no significant difference for 5xFAD saline and MK0677 treatment groups. n = 4 mice per group. B) Representative images of synapse staining. vGlut1 (blue) and PSD95 (red) were used as pre- and post-synaptic markers, respectively. The overlaid staining of vGlut1 and PSD95 indicates synapses. Scale bar = 20 μm.
MK0677 did not attenuate microglial response to Aβ
Microglial activation in response to Aβ toxicity and neuronal death is a pronounced AD brain pathology, which is proposed to be a key indicator of the neuroinflammation that is involved in AD pathogenesis [34–36]. To determine whether MK0677 affects hippocampal microglial activation, we stained microglia using an antibody against a microglial marker, ionized calcium-binding adaptor molecule 1 (Iba-1) [37]. As opposed to their nonTg littermates, saline-treated 5xFAD mice demonstrated substantially increased microglial density in the hippocampus (Fig. 4A1 and A2, nonTg saline 0.483 ± 0.0308 density/10,000 μm3 versus 5xFAD saline 1.139 ± 0.114 density/10,000 μm3, p < 0.001). Such genotypic change was not prevented by MK0677 treatment (Fig. 4A1 and A2, 5xFAD saline 1.139 ± 0.114 density/10,000 μm3 versus 5xFAD MK0677 1.069 ± 0.0095 density/10,000 μm3, p > 0.05). Moreover, saline- and MK0677-treated 5xFAD mice showed no significant difference in the density of plaque-associated microglia (Fig. 4B1 and B2, 5xFAD saline 5.538 ± 0.261 number/10,000 μm3 versus 5xFAD MK0677 4.682 ± 0.164 number/10,000 μm3, p > 0.05), length of total process (Fig. 4C1 and C3, 5xFAD saline 560.48 ± 64.706 μm versus 5xFAD MK0677 599.19 ± 34.908 μm, p > 0.05), or the number of branch points (Fig. 4C2 and C3, 5xFAD saline 36.528 ± 6.576 versus 5xFAD MK0677 40.521 ± 3.661, p > 0.05). These results do not demonstrate any effect of MK0677 on hippocampal microglial response to Aβ.
Fig. 4.
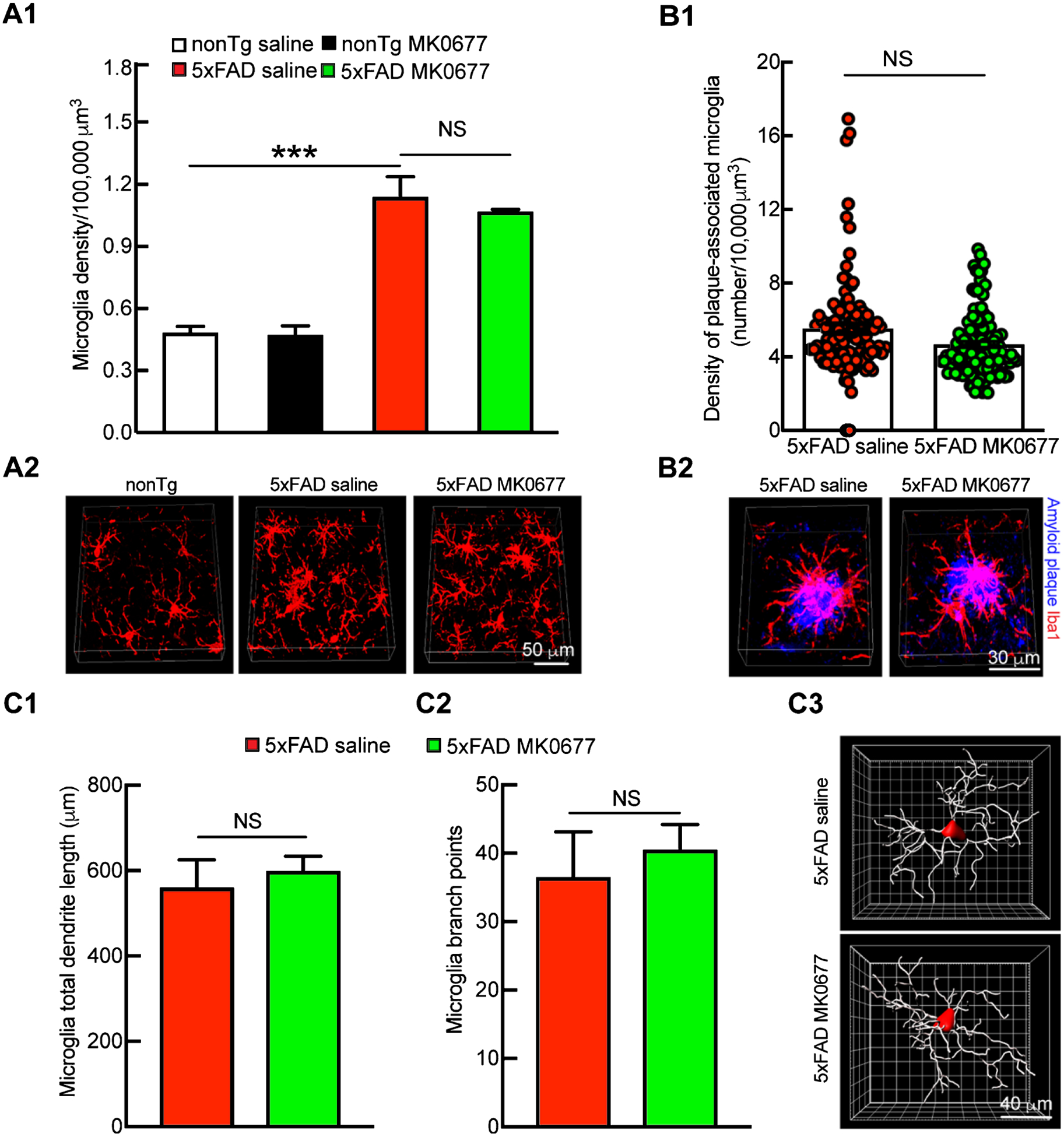
MK0677 did not cause microglia-related changes in 5xFAD mice. A1) Microglia density remained the same in 5xFAD mice after MK0677 treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. t = 8.244, ***p < 0.001 between nonTg saline and 5xFAD saline groups; t = 0.1297, NS, no significant difference for nonTg saline and MK0677 treatment groups; t = 0.8793, NS, no significant difference for 5xFAD saline and MK0677 treatment groups. n = 4 mice per group. A2) 3D representative images for hippocampal microglia density. Iba1 (red) antibody were used to mark microglia. Scale bar = 50 μm. B1) Plaque-associated microglia density remained unchanged after MK0677 treatment in 5xFAD mice. Unpaired Student’s t-test. t = 2.814, NS, not significant, n = 4 mice per group. B2) 3D representative images generated using NIS element software. Scale bar = 30 μm. C1, C2) Microglia total dendrite length and branch point number did not change in 5xFAD mice with MK0677 treatment. Unpaired Student’s t-test. t = 0.5612 for microglia total dendrite length comparison between 5xFAD saline and MK0677 treatment groups, t = 0.6058 for microglia branch points comparison between 5xFAD saline and MK0677 treatment groups. NS, not significant. n = 3 mice per group. C3) 3D representative images generated using Imaris software. Scale bar = 40 μm.
MK0677 did not improve spatial learning and memory in 5xFAD mice
To determine whether MK0677 preserves 5xFAD mouse cognition, especially AD-sensitive hippocampus-associated cognition, we examined mouse spatial reference learning and memory using the Morris water maze. Saline-treated 5xFAD mice exhibited pronounced damage to spatial reference learning (Fig. 5A) and memory (Fig. 5B, nonTg saline 7 ± 1.225 versus 5xFAD saline 3.25 ± 0.25, p < 0.05), which were in sharp contrast with their nonTg littermates (Fig. 5A, B). Despite its little effect on nonTg mouse spatial navigation (Fig. 5A) or memory (Fig. 5B, nonTg saline 7 ± 1.225 versus nonTg MK0677 8.143 ± 0.800, p > 0.05), MK0677 treatment failed to show any protective effect with regards to genotypic cognitive impairment in 5xFAD mice (Fig. 5A, B). Of note, the administration of MK0677 did not affect the swimming speed of the experimental mice (Fig. 5C, nonTg saline 0.36 ± 0.0204 m/s, nonTg MK0677 0.347 ± 0.0163 m/s, 5xFAD saline 0.338 ± 0.0222 m/s, 5xFAD MK0677 0.324 ± 0.0243 m/s, p > 0.05). Such findings echo the ineffectual function of MK0677 treatment on the development of hippocampal synaptic density in 5xFAD mice.
Fig. 5.
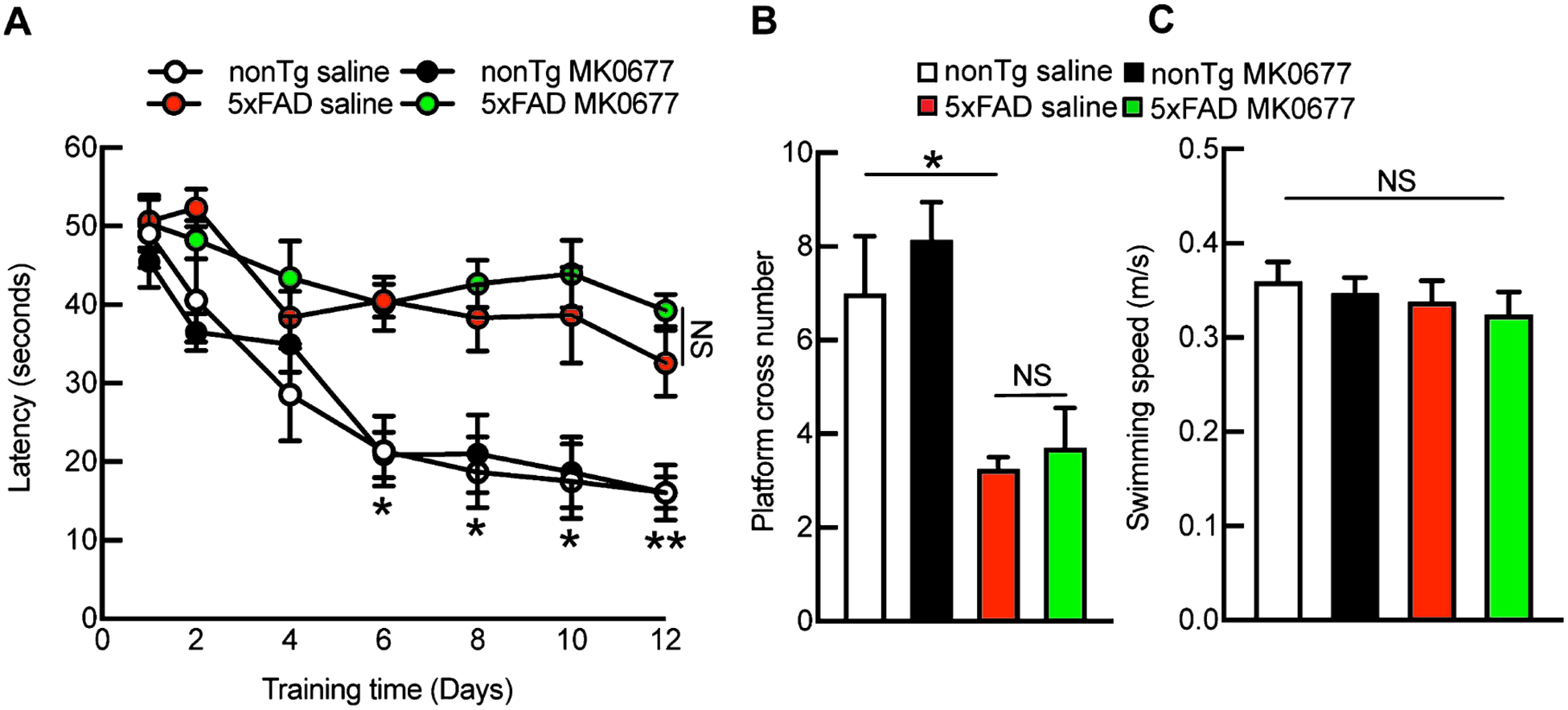
MK0677 treatment did not improve 5xFAD mice spatial learning and memory ability. A) Morris water maze learning curves had no difference between saline (0.9% NaCl) and MK0677-treated 5xFAD mice. *p < 0.05, **p < 0.01 for nonTg saline and 5xFAD with and without MK0677 treatment. NS, no significant difference between 5xFAD with and without MK0677 treatment. B) 5xFAD saline and MK0677 mice showed similar spatial reference memory. t = 2.597, *p < 0.05 between nonTg saline and 5xFAD saline treatment; t = 0.8931, NS, no significant difference between nonTg w/, w/o MK0677 treatment; t = 0.3628, NS, no significant difference between 5xFAD w/, w/o MK0677 treatment. C) Swimming speed remained the same among four experimental groups. Two-way ANOVA followed by Bonferroni post hoc analysis. t = 0.3967 between nonTg saline and MK0677 treatment, t = 0.6343 between nonTg and 5xFAD saline, t = 0.4530 between 5xFAD saline and MK0677 treatment, NS, no significant difference among all the groups, n = 5–7 mice per group.
DISCUSSION
Although the detailed mechanisms underlying the ghrelin/GHSR1α system’s regulation of hippocampal synaptic physiology remain to be elucidated, three pathways have been proposed including direct modulation, metabolic regulation, and neurogenic effect. The direct modulation of hippocampal synaptic function through GHSR1α is evidenced by the observation that GHSR1α activation promotes α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) trafficking, resulting in enhanced AMPAR incorporation into hippocampal synapses [13]. Such an effect is associated with the activation of protein kinase A (PKA), which is a key in GHSR1α downstream signaling [13, 38]. Moreover, Kern and colleagues have shown that GHSR1α heteromerizes with hippocampal dopamine receptor D1 (DRD1), instigating DRD1 Gαq-Ca2+ signaling, and culminating in the initiation of hippocampal synaptic plasticity [39]. In addition to direct action, the ghrelin/GHSR1α system may modulate hippocampal function through its regulation of brain metabolism [12]. Indeed, the pituitary, the hypothalamus and the hippocampus are GHSR1α-expressing regions in central nervous system [5, 40]. GHSR1α in the pituitary and the hypothalamic arcuate nucleus as well as ventrome-dial nuclei regulates the production and secretion of multiple critical hormones such as growth hormone, adrenocorticotropin, and thyroid-stimulating hormone, as well as others that affect food intake, metabolism, and stress responses [41–44]. These metabolic effects of the ghrelin/GHSR1α system may promote hippocampal synaptic function and facilitate hippocampus-associated cognition [45, 46]. Lastly, recent studies have also highlighted the link between GHSR1α activation and enhanced hippocampal neurogenesis and its potential contribution to hippocampal synaptic regulation in health and disease [47]. Since AMPAR deregulation [48–50], brain dysmetabolism [51, 52], and neurogenesis defects [30, 53, 54] are featured pathological changes accompanying AD, GHSR1α activation has its potential to remedy hippocampal synaptic failure in AD.
In previous studies, different groups have validated the beneficial effects of GHSR1α ligands, including ghrelin and acyl ghrelin, as well as GHSR1α agonists, including MK0677 and LY444711, on hippocampal neurogenesis, long-term potentiation, and cognition against Aβ toxicity in rodent models [14, 16, 18, 55]. Although we have observed impaired neurogenesis in the dentate gyrus in 5xFAD mice and the neurogenic effect of MK0677 treatment, which is in agreement with a previous study [14], our results do not support the protective effect of GHSR1α activation against the development of Aβ-induced synaptic deficits or cognitive impairment. The ineffectiveness of MK0677 determined in our study is in line with a previous clinical trial using MK0677 for the treatment of AD [19]. Such discrepancies between previous studies and ours may arise from the use of different types of experimental systems. Two of the aforementioned studies employed Aβ40- or Aβ42-injected mouse or rat models [18, 55]. The acute Aβ toxicity model may not represent the complexity of neuronal changes in response to chronic Aβ production and accumulation. Another group challenged a transgenic AD mouse model on a high glycemic index diet [16]; therefore, the protective effect of GHSR1α activation determined in this study may, at least in part, result from the influence of a GHSR1α agonist against high glucose-induced stress in Aβ-rich settings. It has also been proposed that hippocampal neurogenesis could be a potential avenue for AD treatment [56, 57]. Despite this theory, the enhanced hippocampal neurogenesis by the administration of MK0677 failed to alleviate synaptic loss in the hippocampal CA1 region or mouse cognitive impairment in our study. It cannot be excluded that MK0677-enhanced neurogenesis is not potent enough to replenish a sufficient number of neurons capable of attenuating hippocampal lesions in 5xFAD mice. In fact, whether or not neurogenesis can rescue hippocampal function in diseases has yet to reach a consensus. It also remains to be seen whether (and if so, how many) hippocampal progenitor cells can differentiate into granule neurons and subsequently form functional synaptic neural networks [57, 58]. Moreover, it is unclear whether MK0677-promoted neurogenesis implements any yet-undetermined influence(s) on brain function, especially in 5xFAD mice. Additionally, nonTg and 5xFAD mice displayed different responses to MK0677-mediated neurogenesis in the DG, which seems to implicate enhanced sensitivity to neurogenic stimulations in AD-relevant conditions. Further studies on this issue could serve to bring light to these questions.
Lastly, Jeong and colleagues showed that MK0677 decreases Aβ deposition and microglial activation in 5xFAD mouse neocortices [15]. In current study, we did not detect any effect of MK0677 on hippocampal Aβ loading or microglial activation. In fact, our quantitative analysis failed to show any influence of MK0677 on total plaque-occupied volume (Supplementary Figure 2A and C, in percentage values, 5xFAD saline 1.289 ± 0.323 versus 5xFAD MK0677 1.737 ± 0.274, p > 0.05), single plaque size (Supplementary Figure 2B and C, 5xFAD saline 9591 ± 1174 μm3 versus 5xFAD MK0677 12735 ± 1069.5 μm3, p > 0.05), the density of total microglia (Supplementary Figure 3A1 and A2, 5xFAD saline 1.738 ± 0.0591 /10,000 μm3 versus 5xFAD MK0677 1.628 ± 0.0718 /10,000 μm3, p > 0.05), or the density of plaque-associated microglia (Supplementary Figure 3B1 and B2, 5xFAD saline 3.963 ± 0.362 number/10,000 μm3 versus 5xFAD MK0677 3.558 ± 0.388 number/10,000 μm3, p > 0.05) in the neocortex of MK0677-treated 5xFAD mice. Our observations of the inconsequential impact of ghrelin or other GHSR1α agonist on brain Aβ loading is in agreement with the results from several previous studies [14, 16]. We cannot exclude the possibility that MK0677 at high doses, such as 5 mg/kg used in Jeong’s study [15], would affect Aβ production and/or deposition. However, we found that MK0677 at 3 mg/kg significantly increased mortality of 5xFAD mice, which prevented our use of higher doses of MK0677. Therefore, it is suggested to err on the side of caution when using high doses of MK0677 as a therapeutic strategy for AD treatment even if it is effective in the reduction of brain amyloidosis.
In summary, we have shown that GHSR1α activation by its agonist MK0677 promotes hippocampal neurogenesis but fails to mitigate hippocampal synaptic lesions and cognitive impairment in 5xFAD mice. Our findings correlate with those from a failed clinical trial [19] regardless of the argument that said trial treated the patients at a critically advanced stage. To counter this, we administrated MK0677 to mice at the asymptomatic stage when there is little to mild brain Aβ deposition and indiscernible synaptic injury and failed to detect the preventive effect of GHSR1α activation. Therefore, GHSR1α activation through its agonists does not seem to be an effective strategy for AD prevention. Of note, in a recent study we found increased serum levels of acyl ghrelin in patients with mild cognitive impairment due to AD, which negatively correlates with the patients’ cognitive performance [59]. This observation, together with the ineffectiveness of MK0677 as seen in our study and a previous clinical trial [19], seems to suggest a suppressed GHSR1α response to its ligand- or agonist-induced activation in AD-relevant pathophysiological settings. Indeed, we have determined that complexation with Aβ inhibits ligand-induced activation of GHSR1α and thus suppresses GHSR1α’s regulation of dopamine receptor D1 (DRD1), leading to hippocampal synaptic deficits [60]. We therefore form a “ghrelin resistance” hypothesis of AD hippocampal synaptic failure. In this case, the detailed mechanisms of GHSR1α deregulation in AD-related conditions need further investigation. Nevertheless, the simplest interpretation of our results is that GHSR1α activation by MK0677 alone has limited therapeutic potential for AD prevention and treatment.
Supplementary Material
ACKNOWLEDGMENTS
Supported by research funding from NIH (R00AG 037716, R01AG053588, and R01AG059753) and Alzheimer’s Association (AARG-16-442863) to H.D.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0779r1).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-190779.
REFERENCES
- [1].Perl DP (2010) Neuropathology of Alzheimer’s disease. Mt Sinai J Med 77, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Adler DH, Wisse LEM, Ittyerah R, Pluta JB, Ding SL, Xie L, Wang J, Kadivar S, Robinson JL, Schuck T, Trojanowski JQ, Grossman M, Detre JA, Elliott MA, Toledo JB, Liu W, Pickup S, Miller MI, Das SR, Wolk DA, Yushkevich PA (2018) Characterizing the human hippocampus in aging and Alzheimer’s disease using a computational atlas derived from ex vivo MRI and histology. Proc Natl Acad Sci U S A 115, 4252–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cui R, Liu M (2019) Hippocampus analysis by combination of 3D DenseNet and Shapes for Alzheimer’s disease diagnosis. IEEE J Biomed Health Inform 23, 2099–2107. [DOI] [PubMed] [Google Scholar]
- [4].Broglio F, Papotti M, Muccioli G, Ghigo E (2007) Brain-gut communication: Cortistatin, somatostatin and ghrelin. Trends Endocrinol Metab 18, 246–251. [DOI] [PubMed] [Google Scholar]
- [5].Mani BK, Walker AK, Lopez Soto EJ, Raingo J, Lee CE, Perelló M, Andrews ZB, Zigman JM (2014) Neuroanatomical characterization of a growth hormone secretagogue receptor-green fluorescent protein reporter mouse. J Comp Neurol 522, 3644–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hsu TM, Hahn JD, Konanur VR, Noble EE, Suarez AN, Thai J, Nakamoto EM, Kanoski SE (2015) Hippocampus ghrelin signaling mediates appetite through lateral hypothalamic orexin pathways. Elife 4, e11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mani BK, Osborne-Lawrence S, Mequinion M, Lawrence S, Gautron L, Andrews ZB, Zigman JM (2017) The role of ghrelin-responsive mediobasal hypothalamic neurons in mediating feeding responses to fasting. Mol Metab 6, 882–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cabral A, De Francesco PN, Perello M (2015) Brain circuits mediating the orexigenic action of peripheral ghrelin: Narrow gates for a vast kingdom. Front Endocrinol (Lausanne) 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hauberg K, Kohlmeier KA (2015) The appetite-inducing peptide, ghrelin, induces intracellular store-mediated rises in calcium in addiction and arousal-related laterodorsal tegmental neurons in mouse brain slices. Peptides 65, 34–45. [DOI] [PubMed] [Google Scholar]
- [10].Kim C, Kim S, Park S (2017) Neurogenic effects of ghrelin on the hippocampus. Int J Mol Sci 18, E588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hsu TM, Noble EE, Reiner DJ, Liu CM, Suarez AN, Konanur VR, Hayes MR, Kanoski SE (2018) Hippocampus ghrelin receptor signaling promotes socially-mediated learned food preference. Neuropharmacology 131, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA, Morley JE, Pinto S, Sherwin RS, Xu L, Yamada KA, Sleeman MW, Tschop MH, Horvath TL (2006) Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci 9, 381–388. [DOI] [PubMed] [Google Scholar]
- [13].Ribeiro LF, Catarino T, Santos SD, Benoist M, van Leeuwen JF, Esteban JA, Carvalho AL (2014) Ghrelin triggers the synaptic incorporation of AMPA receptors in the hippocampus. Proc Natl Acad Sci U S A 111, E149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Moon M, Cha MY, Mook-Jung I (2014) Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J Alzheimers Dis 41, 233–241. [DOI] [PubMed] [Google Scholar]
- [15].Jeong YO, Shin SJ, Park JY, Ku BK, Song JS, Kim JJ, Jeon SG, Lee SM, Moon M (2018) MK-0677, a ghrelin agonist, alleviates amyloid beta-related pathology in 5XFAD mice, an animal model of Alzheimer’s disease. Int J Mol Sci 19, E1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kunath N, van Groen T, Allison DB, Kumar A, Dozier-Sharpe M, Kadish I (2015) Ghrelin agonist does not foster insulin resistance but improves cognition in an Alzheimer’s disease mouse model. Sci Rep 5, 11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Eslami Z, Ghassempour A, Aboul-Enein HY (2017) Recent developments in liquid chromatography-mass spectrometry analyses of ghrelin and related peptides. Biomed Chromatogr 31, doi: 10.1002/bmc.3796 [DOI] [PubMed] [Google Scholar]
- [18].Santos VV, Stark R, Rial D, Silva HB, Bayliss JA, Lemus MB, Davies JS, Cunha RA, Prediger RD, Andrews ZB (2017) Acyl ghrelin improves cognition, synaptic plasticity deficits and neuroinflammation following amyloid beta (Abeta1–40) administration in mice. J Neuroendocrinol 29, doi: 10.1111/jne.12476 [DOI] [PubMed] [Google Scholar]
- [19].Sevigny JJ, Ryan JM, van Dyck CH, Peng Y, Lines CR, Nessly ML, Group MKPS (2008) Growth hormone secretagogue MK-677: no clinical effect on AD progression in a randomized trial. Neurology 71, 1702–1708. [DOI] [PubMed] [Google Scholar]
- [20].Eimer WA, Vassar R (2013) Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, Jones JE, Deysher AE, Waxman AR, White RD, Williams TD, Lachey JL, Seeley RJ, Lowell BB, Elmquist JK (2005) Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest 115, 3564–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qi Wang JT, Hao C, Heng D, Lan G (2019) Amyloid beta-mediated KIF5A deficiency disrupts anterograde axonal mitochondrial movement. Neurobiol Dis 127, 410–418. [DOI] [PubMed] [Google Scholar]
- [23].Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, Gauba E, Lu L, Pascual JM, Kroener S, Du H (2016) Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun 7, 11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cengiz P, Zafer D, Chandrashekhar JH, Chanana V, Bogost J, Waldman A, Novak B, Kintner DB, Ferrazzano PA (2018) Developmental differences in microglia morphology and gene expression during normal brain development and in response to hypoxia-ischemia. Neurochem Int 127, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lu L, Guo L, Gauba E, Tian J, Wang L, Tandon N, Shankar M, Beck SJ, Du Y, Du H (2015) Transient cerebral ischemia promotes brain mitochondrial dysfunction and exacerbates cognitive impairments in young 5xFAD mice. PLoS One 10, e0144068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Morris RGM (2008) Morris water maze. Scholarpedia 3, 6315. [Google Scholar]
- [27].Wang L, Guo L, Lu L, Sun H, Shao M, Beck SJ, Li L, Ramachandran J, Du Y, Du H (2016) Synaptosomal mitochondrial dysfunction in 5xFAD mouse model of Alzheimer’s disease. PLoS One 11, e0150441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gauba E, Chen H, Guo L, Du H (2019) Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol Dis 121, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zheng H, Bailey A, Jiang MH, Honda K, Chen HY, Trum-bauer ME, Van der Ploeg LH, Schaeffer JM, Leng G, Smith RG (1997) Somatostatin receptor subtype 2 knockout mice are refractory to growth hormone-negative feedback on arcuate neurons. Mol Endocrinol 11, 1709–1717. [DOI] [PubMed] [Google Scholar]
- [30].Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N, Avila J, Llorens-Martin M (2019) Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25, 554–560. [DOI] [PubMed] [Google Scholar]
- [31].Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L (2005) Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21, 1–14. [DOI] [PubMed] [Google Scholar]
- [32].Padurariu M, Ciobica A, Mavroudis I, Fotiou D, Baloyannis S (2012) Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr Danub 24, 152–158. [PubMed] [Google Scholar]
- [33].Drew LJ1, Fusi S, Hen R(2013) Adult neurogenesis in the mammalian hippocampus: Why the dentate gyrus? Learn Mem 26, 710–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- [35].Wes PD, Sayed FA, Bard F, Gan L (2016) Targeting microglia for the treatment of Alzheimer’s disease. Glia 64, 1710–1732. [DOI] [PubMed] [Google Scholar]
- [36].Visan I (2017) Alzheimer’s disease microglia. Nat Immunol 18, 876. [DOI] [PubMed] [Google Scholar]
- [37].Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57, 1–9. [DOI] [PubMed] [Google Scholar]
- [38].Cuellar JN, Isokawa M (2011) Ghrelin-induced activation of cAMP signal transduction and its negative regulation by endocannabinoids in the hippocampus. Neuropharmacology 60, 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kern A, Mavrikaki M, Ullrich C, Albarran-Zeckler R, Brantley AF, Smith RG (2015) Hippocampal dopamine/DRD1 signaling dependent on the ghrelin receptor. Cell 163, 1176–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, Smith RG, Van der Ploeg LH, Howard AD (1997) Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res 48, 23–29. [DOI] [PubMed] [Google Scholar]
- [41].Grey CL, Grayfer L, Belosevic M, Chang JP (2010) Ghrelin stimulation of gonadotropin (LH) release from goldfish pituitary cells: Presence of the growth hormone secretagogue receptor (GHS-R1a) and involvement of voltage-sensitive Ca2+ channels. Mol Cell Endocrinol 317, 64–77. [DOI] [PubMed] [Google Scholar]
- [42].Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660. [DOI] [PubMed] [Google Scholar]
- [43].Gianotti L, Ramunni J, Lanfranco F, Maccagno B, Giordano R, Broglio F, Maccario M, Muller EE, Ghigo E, Arvat E (2001) Recombinant human IGF-I does not modify the ACTH and cortisol responses to hCRH and hexarelin, a peptidyl GH secretagogue, in humans. J Endocrinol Invest 24, 67–71. [DOI] [PubMed] [Google Scholar]
- [44].Takaya K, Ariyasu H, Kanamoto N, Iwakura H, Yoshimoto A, Harada M, Mori K, Komatsu Y, Usui T, Shimatsu A, Ogawa Y, Hosoda K, Akamizu T, Kojima M, Kangawa K, Nakao K (2000) Ghrelin strongly stimulates growth hormone release in humans. J Clin Endocrinol Metab 85, 4908–4911. [DOI] [PubMed] [Google Scholar]
- [45].McGregor G, Harvey J (2018) Regulation of hippocampal synaptic function by the metabolic hormone, leptin: Implications for health and neurodegenerative disease. Front Cell Neurosci 12, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].McGregor G, Malekizadeh Y, Harvey J (2015) Minire-view: Food for thought: Regulation of synaptic function by metabolic hormones. Mol Endocrinol 29, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Huang HJ, Chen XR, Han QQ, Wang J, Pilot A, Yu R, Liu Q, Li B, Wu GC, Wang YQ, Yu J (2019) The protective effects of Ghrelin/GHSR on hippocampal neurogenesis in CUMS mice. Neuropharmacology 155, 31–43. [DOI] [PubMed] [Google Scholar]
- [48].Jurado S (2017) AMPA receptor trafficking in natural and pathological aging. Front Mol Neurosci 10, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang Y, Guo O, Huo Y, Wang G, Man HY (2018) Amyloid-beta induces AMPA receptor ubiquitination and degradation in primary neurons and human brains of Alzheimer’s disease. J Alzheimers Dis 62, 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Guntupalli S, Widagdo J, Anggono V (2016) Amyloid-beta-induced dysregulation of AMPA receptor trafficking. Neural Plast 2016, 3204519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Thomas SC, Alhasawi A, Appanna VP, Auger C, Appanna VD (2015) Brain metabolism and Alzheimer’s disease: The prospect of a metabolite-based therapy. J Nutr Health Aging 19, 58–63. [DOI] [PubMed] [Google Scholar]
- [52].de la Monte SM, Tong M (2014) Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol 88, 548–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Maruszak A, Pilarski A, Murphy T, Branch N, Thuret S (2014) Hippocampal neurogenesis in Alzheimer’s disease: Is there a role for dietary modulation? J Alzheimers Dis 38, 11–38. [DOI] [PubMed] [Google Scholar]
- [54].Mu Y, Gage FH (2011) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Eslami M, Sadeghi B, Goshadrou F (2018) Chronic ghrelin administration restores hippocampal long-term potentiation and ameliorates memory impairment in rat model of Alzheimer’s disease. Hippocampus 28, 724–734. [DOI] [PubMed] [Google Scholar]
- [56].Sailor KA, Ming GL, Song H (2006) Neurogenesis as a potential therapeutic strategy for neurodegenerative diseases. Expert Opin Biol Ther 6, 879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].DeCarolis NA, Eisch AJ (2010) Hippocampal neurogenesis as a target for the treatment of mental illness: A critical evaluation. Neuropharmacology 58, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Baptista P, Andrade JP (2018) Adult hippocampal neuro-genesis: Regulation and possible functional and clinical correlates. Front Neuroanat 12, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cao X, Zhu M, He Y, Chu W, Du Y, Du H (2018) Increased serum acylated ghrelin levels in patients with mild cognitive impairment. J Alzheimers Dis 61, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tian J, Guo L, Sui S, Driskill C, Phensy A, Wang Q, Gauba E, Zigman JM, Swerdlow RH, Kroener S, Du H (2019) Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a role in Alzheimer s disease. Sci Transl Med 11, eaav6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.