

Abstract
Structure–activity relationship studies of a reported menthol-based transient receptor potential cation channel subfamily M member 8 channel (TRPM8) antagonist, guided by computational simulations and structure-based design, uncovers a novel series of TRPM8 antagonists with >10-fold selectivity versus related TRP subtypes. Spiro[4.5]decan-8-yl analogue 14 inhibits icilin-evoked Ca2+ entry in HEK-293 cells stably expressing human TRPM8 (hTRPM8) with an IC50 of 2.4 ± 1.0 nM, while in whole-cell patch-clamp recordings this analogue inhibits menthol-evoked currents with a hTRPM8 IC50 of 64 ± 2 nM. Molecular dynamics (MD) simulations of compound 14 in our homology model of hTRPM8 suggest that this antagonist forms extensive hydrophobic contacts within the orthosteric site. In the wet dog shakes (WDS) assay, compound 14 dose-dependently blocks icilin-triggered shaking behaviors in mice. Upon local administration, compound 14 dose dependently inhibits cold allodynia evoked by the chemotherapy oxaliplatin in a murine model of peripheral neuropathy at microgram doses. Our findings suggest that 14 and other biphenyl amide analogues within our series can find utility as potent antagonist chemical probes derived from (−)-menthol as well as small molecule therapeutic scaffolds for chemotherapy-induced peripheral neuropathy (CIPN) and other sensory neuropathies.
Keywords: Menthol, molecular dynamics simulations, chemical probe, TRPM8, ory neuropathy, TRP channel
Graphical Abstract
INTRODUCTION
TRPM8 (transient receptor potential cation channel subfamily M member 8 channel), often referred to as the “cold receptor”, is a polymodal nociceptor of the somatosensory nervous system and a major sensor of cold nociception in humans.1 TRPM8 is expressed, albeit differentially compared to other temperature sensitive TRP channels TRPA1 and TRPV1, in primary sensory neurons (Aδ and C-fibers) of the dorsal root ganglia (DRG) and trigeminal ganglia (TG)2 as well as in some other non-neuronal tissues. Within primary sensory neurons, TRPM8 is present on both the peripheral and central nerve terminals, supporting a role in the somatosensory and central nervous systems.3 This cold-sensing nociceptor initiates sensory nerve impulses after activation by mildly cool temperatures (15–25 °C), chemical agents such as (−)-menthol ((1R,2S,5R)-5-methyl-2-(1-methylethyl)-cyclohexanol), the “super-agonist” icilin (3-(2-hydroxyphenyl)-6-(3-nitrophenyl)-3,4-dihydropyrimidin-2(1H)-one, also known as AG-3–5), and the endogenous lipid PIP2.4 Maximal stimulation of TRPM8 by menthol occurs via a ligand stoichiometry of four menthol molecules each occupying a monomer of one functional TRPM8 tetramer.5
TRPM8 appears to be a potential target for multiple therapeutic interventions. TRPM8 agonists have been explored for benign prostatic hyperplasia and prostate cancer, while antagonists have spurred more interest, for cold hypersensitivity in neuropathic pain, and migraine, among others.6
A number of proprietary TRPM8 antagonists, discovered largely through high throughput screening of in-house chemical libraries, are known.6 However, the precise molecular details of how these antagonists bind to human TRPM8 and subsequently influence its gating mechanism, as well as their underlying pharmacophores, remains to be elucidated, especially in the wake of recent success with TRPM8 and TRP channel structural biology revealed through cryo-electron microscopy (cryo-EM).7,8
In the TRPM8-mediated Ca2+ flux assay, well-known TRPM8 antagonists appear to manifest either modest or variable potencies. For example, quinoline-based PF-05105679 (Pfizer, Phase 1, NCT01393652) inhibits the effects of voltage- and WS-12-mediated activation at human TRPM8 with IC50 values of 103 ± 29.4 nM and 181 ± 7.21 nM,1,9 while 5,6,7,8-tetrahydro-1,7-naphthyridine analogue AMG2850 (Amgen) exhibits IC50 values ranging from 7 to 156 nM, against menthol, icilin, and cold activation at rat and human TRPM8.10,11 2-Benzyloxy-benzoic acid amide derivative AMTB (Bayer) affords weaker inhibition against icilin-mediated responses at human TRPM8, with a pIC50 of 6.23 ± 0.02.12 To date, no TRPM8 antagonists have advanced to clinical use; for example, PF-05105679 does not possess a measurable therapeutic index at high doses (600 and 900 mg) required to achieve unbound plasma concentrations greater than its IC50 value.1 Likewise, Phase 1 studies of the nicotinic acid analogue AMG 33313 (Amgen, NCT02132429) were terminated for undisclosed reasons. There remains a need for introducing more TRPM8 antagonists that are potent, reasonably specific and thereby can sustain the possibility of successfully passing through clinical trials after successful preclinical development.
Moreover, a variety of both agonist and antagonist scaffolds with wide chemical diversity have been used to decipher the role of TRPM8 in various disease states, such as orofacial and neuropathic pain,14 overactive bladder (OAB) and painful bladder syndrome,12 diseases involving thermoregulation,15 oral squamous cell carcinoma,16 nicotine addictive behaviors,17 and chronic obstructive pulmonary disease.18 In some cases, use of these ligands as tool molecules to probe the pharmacology of TRPM8 have led to conflicting data, particularly in determining its role in chronic neuropathic pain.3,14,19 Highly potent chemical probes based on the cognate ligand (−)-menthol with an antagonist profile are needed to revisit the TRPM8 field from an early drug discovery perspective, which currently lacks such small molecules of this chemotype. De novo design of these scaffolds is enabled by contemporary knowledge of the TRPM8 small molecule binding site.
Recent cryo-EM structures of both apo- and agonist-bound avian TRPM8 (TRPM8FA, PDB codes 6BPQ (4.1 Å), 6NR2 (4 Å), and 6NR3 (3.4 Å)),7,8 coupled with supporting in vitro and in vivo mutagenesis studies at mouse, rat, squirrel, and human orthologues, point to a definitive binding site for the prototypical ligands menthol and icilin that is likely decoupled from cold sensing. Mutagenesis studies have suggested a conserved Tyr745, located in the middle of the transmembrane S1 helix of the voltage-sensor-like domain (VSLD),7 is crucial for both menthol binding, as determined by radioligand displacement studies using [3H]-menthol,20 as well as efficacy.21 These studies are consistent with the structure of avian TRPM8 complexed with menthol analogue WS-12 (PDB 6NR2).8 Constructs of the isolated S1–S4 sensing domain also suggest this region to be crucial for menthol recognition, as determined by microscale thermophoresis and NMR.22 Other menthol-sensitive residues include Tyr1005 and Leu1009, both of which are conserved21 and located in the TRP helix transmembrane domain, near the cytosolic domain interface. The positional equivalent of these two residues can be seen in the WS-12 bound structure (Tyr1004 and Leu1008 in TRPM8FA). This pocket also binds icilin with similar residues, including Tyr7458,21 and Leu1009,21 as well as Gly805, Asn799, and Asp802 in the S3 helix.8,23 The cryo-EM structure of icilin-bound avian TRPM8 reinforces the notion that Tyr745, a residue conserved across all TRPM subtypes,24 is a common TRPM8 residue for both menthol and icilin recognition, despite their different chemotypes, while Asn799 and Asp802 are necessary for Ca2+-dependent icilin binding, along with Gln782 and Glu785. Additional icilin-binding residues line the orthosteric site within the VSLD and TRP helix, including Phe838, Arg841, His844, and Tyr1004 (Phe839, Arg842, His845, and Tyr1005 at hTRPM8, respectively).8 This ligand-binding site also overlaps with a crucial voltage- and cold-sensitive residue: a conserved Arg841 in the S4 helix (Arg842 at hTRPM8),20 indicating that the ability of TRPM8 to function as a polysensor of ligand and voltage stimuli is derived in part from this common site, composed of S1–S4 helices within the VSLD. Transgenic models of hibernating ground squirrels and rats implicate residues responsible for cold vs icilin activation, scattered throughout the VSLD in the loops linking the TM helices, at positions 726 (pre-S1 domain), 762 (S1–S2 loop), 819 (S3–S4 loop), and 927, 946, and 947 in the loop connecting the pore helix (PH) and S6 helix.25 From the sequence alignments of rat, squirrel, and human TRPM8 orthologues,7,25 four of the six cold-conferring residues in rats are conserved in the human orthologue (Tyr726, Ser762, Ser819 and Asn947), raising the possibility that ligand vs cold activation in humans occurs at topographically distinct regions. On the other hand, the remaining two mismatched residues are conserved in humans and cold-tolerant hibernating ground squirrels (Ala927, His946), suggesting either that these residues do not support a cold phenotype, or contribute to a cold response via allosteric regulation.
In light of these recent developments in the TRPM8 field, we hypothesize that reported small molecule TRPM8 antagonists with both structural similarities and conformational rigidity can reveal similar molecular determinants for ligand recognition, via their poses from molecular dynamics (MD) simulations in our human TRPM8 homology model based on the avian cryo-EM structure TRPM8FA (PDB 6BPQ, ~4.1 Å).7 In turn, these insights can allow for the design of novel TRPM8 chemical probes and therapeutic scaffolds for TRPM8-mediated sensory neuropathies. We employed this structure-based approach using reported, selective TRPM8 antagonists such as (−)-menthyl 126 (rTRPM8 IC50: 20 ± 2 nM; 50 ± 10 nM) and 5,6,7,8-tetrahydro-1,7-naphthyridine AMG285010,11 (Figure 1), containing similar hydrophobic moieties such as (−)-menthyl and trifluoropropanyl groups, as well as amides/ureas, and cycloalkyl and aromatic 6-membered rings. These moieties are also present in other well-known TRPM8 antagonists such as 2-benzyloxy-benzoic acid amide derivative AMTB as well as TRPV1 antagonists with bifunctional TRPM8 antagonist activity, such as pyridylpiperazine carboxamide BCTC and urea SB-452533 (Figure 1). Using this approach, we herein report the discovery of a novel biphenyl amide series with potent TRPM8 antagonist activity at hTRPM8 and tractable structure–activity relationships (SARs) to inform our drug discovery efforts.
Figure 1.
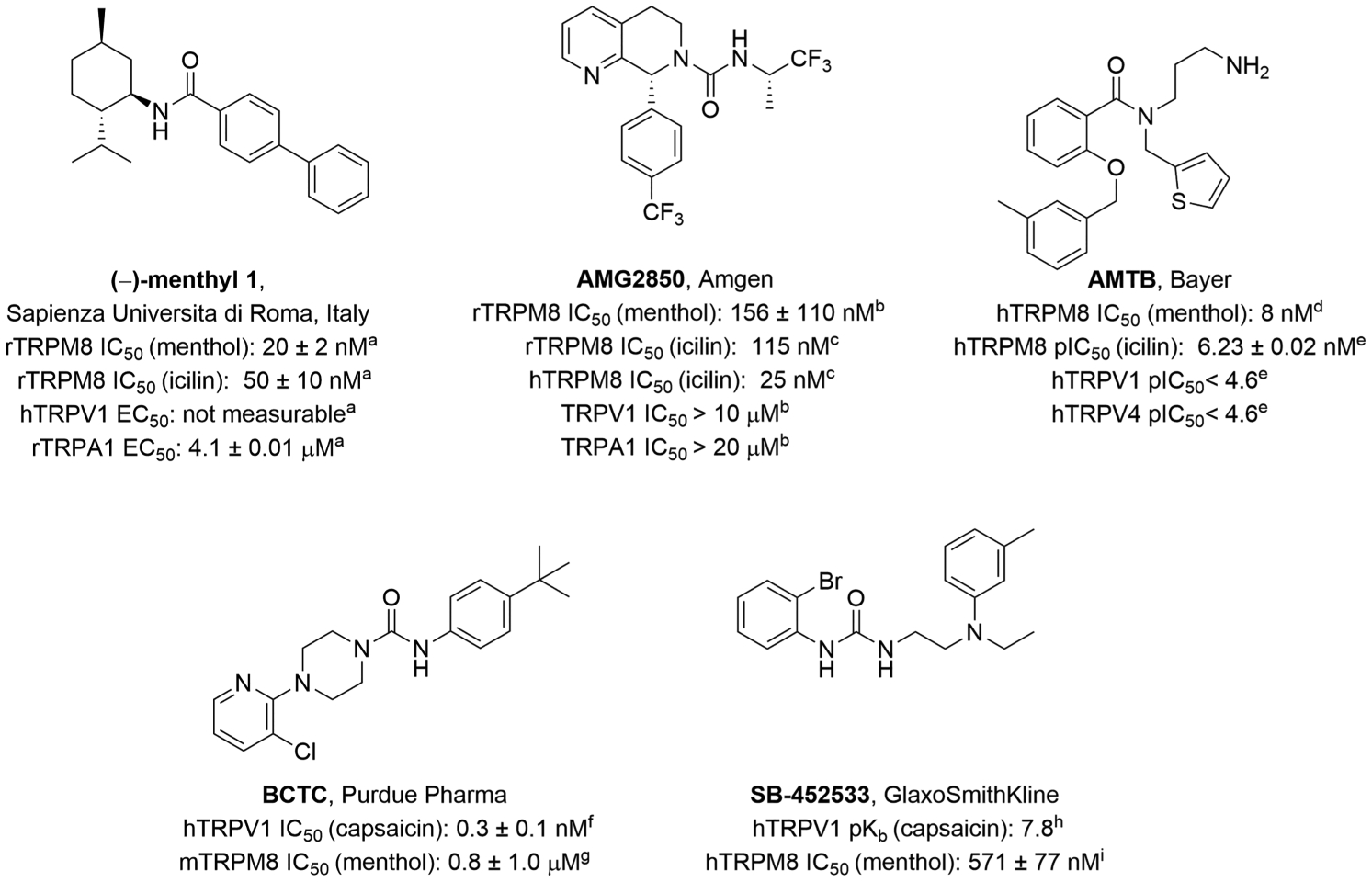
Reported TRPM8 antagonists. IC50 values (antagonists) determined in the Ca2+ flux assay against the effect of menthol and/or icilin: (a) ref 26, (b) ref 11, (c) ref 10, (d) ref 27, (e) ref 12, (f) ref 28, (g) ref 29, (h) ref 30, and (i) ref 31. Icilin: 3-(2-hydroxyphenyl)-6-(3-nitrophenyl)-3,4-dihydropyrimidin-2(1H)-one.
PHARMACOLOGICAL CHARACTERIZATION
Evaluation of TRPM8 Agonist and Antagonist Activity Using Ca2+ Imaging.
Analogues 2–15 were evaluated for their ability to modulate TRPM8 functions with Fura-2 based Ca2+ imaging in HEK-293 cells stably expressing hTRPM8. The latter were a kind gift from Thomas Voets (Laboratory of Ion Channels Research, KU Leuven, Belgium). The concentration of intracellular free Ca2+ ([Ca2+]i) was monitored as a Fura-2 fluorescence ratio (F355/F380).32–34
The analogues were assessed against Ca2+ influx ([Ca2+]i) evoked by icilin (AG-3–5).33 The latter was used at 500 nM, as maximum Ca2+ entry was found to occur at this concentration in our dose–response study. We chose icilin to stimulate TRPM8 in our assay, since it was markedly more potent (EC50: ~70 nM vs 6 μM) than (−)-menthol in our initial concentration–response studies (Figure S2) and this broadly agrees with previous reports.33,34
TRPV1 and TRPA1 Agonist and Antagonist Ca2+ Flux Assay (Selectivity Profiling).
Compounds with hTRPM8 IC50 values ≤ 100 nM were evaluated for off-target effects at two related temperature-sensitive subtypes hTRPA1 and hTRPV1, using high-throughput FLIPR-based Ca2+ flux assays at Eurofins Pharma Discovery Services (St. Charles, MO). Compounds were evaluated at single concentrations corresponding to approximately 90% target coverage at TRPM8, followed by single point assays at 10-fold higher concentrations to demonstrate >10-fold selectivity. Agonist (intrinsic) activity was evaluated in recombinant HEK293 cell lines stably expressing either human TRPA1 or TRPV1 and normalized to the maximum effect (Emax) of the agonists allyl isothiocyanate or capsaicin, respectively. Antagonist activity was evaluated by measuring the ability of the analogues to inhibit AITC- or capsaicin-induced increases in cytosolic [Ca2+] by inhibiting Ca2+ influx in these cell lines, and compared to the effect of the ruthenium red (TRPA1) and capsazepine (TRPV1) used as reference antagonists.
Whole-Cell Patch Clamp Electrophysiology of 14.
The functional activity of 14 in calcium flux assays was confirmed with whole-cell patch-clamp electrophysiology. Human TRPM8 whole-cell patch-clamp electrophysiology was done at 23 ± 1 °C on human embryonic kidney (HEK) 293 cells (ATCC CRL-1573) authenticated by polymorphic genetic marker testing (DNA Diagnostics Center Medical) as described previously.22,35 A series of 500 μM menthol and variable concentration (from 0 to 1 μM) 14 solutions were used. Cellular current response was measured at each concentration of 14. To determine the IC50, normalized currents from six cells were averaged at each concentration of antagonist, and the standard error of the mean was calculated at each point. The data were fit to a single site competition model to obtain IC50. The IC50 whole-cell patch-clamp experiments were deliberately performed against saturating (500 μM) menthol concentrations, because menthol currents are not Ca2+ dependent and give reproducible and robust currents. Similarly, studying the inhibitory effects of 14 against saturating menthol allows direct comparison to other published TRPM8 antagonism studies that used these conditions in whole-cell patch-clamp measurements.32,36–38
In Vitro Metabolic Stability.
Compound 14 was evaluated for metabolic stability in mouse liver microsomes by Absorption Systems (Exton, PA), by determining the percent remaining at 0, 10, 20, 30, and 60 min when incubated with mouse liver microsomes in the presence of NADPH. The half-life (t1/2) and in vitro intrinsic clearance (CLint) of 14 was calculated, in addition to the control (testosterone).
RESULTS AND DISCUSSION
The available functional data on known TRPM8 antagonists including (−)-menthyl 1 and AMG2850 are from rat and human TRPM8, for which we are yet to have solved structures, while the structure of the avian orthologue has recently been elucidated using cryo-EM.7 To first evaluate the degree of sequence conservation across these species, we carried out sequence alignment of the transmembrane S1–S4 helices (VSLD), S5–S6 helices (pore domain), pore helix (PH) and TRP helix of avian, human, and rat TRPM8 (Figure S1). As detailed above, residues within the VSLD and TRP helix are reported to be crucial for agonist (menthol) binding, agonist (menthol, icilin) response or efficacy and voltage sensitivity. Interestingly, the high degree of homology between full-length avian and human TRPM8 (83%)7 is retained within this transmembrane region containing the ligand binding site (86%); additionally there is a comparatively high sequence identity (87%) between the avian and rat orthologues in this same region (Figure S1). We therefore considered the avian TRPM8 structure to be a reliable template for building a homology model of hTRPM8 with a view to first predicting plausible binding modes of known TRPM8 antagonists (−)-menthyl 1 and AMG2850 and establishing a relevant structure–function correlation. The latter, in turn, could inform our structure-based strategy for designing novel TRPM8 antagonist probes.
MD Simulations.
To date, no structure of hTRPM8 is available. We first built the homology model of hTRPM8 based on the cryo-EM structure of the collared flycatcher Ficedula albicollis (TRPM8FA, PDB 6BPQ),7 with which hTRPM8 shares 83% sequence identity to full length TRPM8FA and 86% in region containing the ligand binding site. We mainly focused on the transmembrane region of TRPM8, composed of the VSLD (S1–S4 helices, also termed the sensing domain, SD) and pore domains, as well as the TRP helix, and built our homology model of hTRPM8 with a sequence from Gln671 to Asn1010 (340 residues). The VSLD and TRP helix binds small molecule agonists,8 as discussed above, while the isolated VSLD (including the pre-S1 domain) is reported to recapitulate binding phenotypes that agree with functional activation of full-length hTRPM8 (1104 residues).22
A Ca2+ ion was placed in the VLSD, coordinating to Glu782, Gln785, Asn799, and Asp802, to allow our human model to more closely resemble the cryo-EM structures of related human melastatin subtypes M2 (PDB 6MJ2)39 and M4 (PDB 6BQV),24 and provide structural context for our Ca2+ flux studies using the TRPM8 agonist icilin as an activator (Table 1). These structures capture a bound Ca2+ ion coordinating to four side chains oriented similarly in the VLSD, three of which are conserved among TRPM8, TRPM2, and TRPM4: Glu782 (TRPM8, S2 helix)/Glu843 (TRPM2, S2)/Glu828 (TRPM4, S2); Gln785 (TRPM8, S2)/Gln846 (TRPM2, S2)/Gln831 (TRPM4, S2); and Asn799 (TRPM8, S3)/Asn869 (TRPM2, S3)/Asn865 (TRPM4, S3). Taking into consideration the remaining nonconserved Ca2+ binding residue, we chose Asp802 (S3) as the final chelating amino acid to satisfy the tetrahedral complex, corresponding to Asp868 in the M4 subtype (S3 helix). Notably, both Asn799 and Asp802 are well-known icilin sensitive residues.23 The recently reported icilin-bound structure of TRPM8 (PDB 6NR3, 3.4 Å) reveals a Ca2+ ion bound to these four residues within the now well-defined Ca2+ binding site, in agreement with our homology model.
Table 1.
IC50 Values of 1 Analogues in Nanomolar, Determined by Cellular Ca2+ Flux Assays in HEK-293 Cells Stably Expressing hTRPM8 and Represented as Mean ± SEM from Three Independent Experimentsa
 |
Functional Activity | |
|---|---|---|
| Cmpd | R1 | hTRPM8 potcncy IC50 ± SEM |
| 1 | 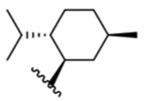 |
16 ± 1 μM |
| 2 | 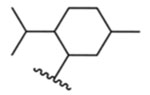 |
468 ± 1 nM |
| 3 | 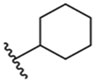 |
49 ± 1 μM |
| 4 | 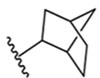 |
7.1 ±0.9 μM |
| 5 | 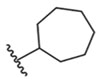 |
52 ± 1 nM |
| 6 | 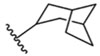 |
692 ± 1 nM |
| 7 | 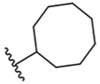 |
62 ± 1 nM |
| 8 | 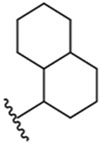 |
21 ± 1 μM |
| 9 | 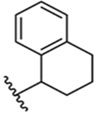 |
55 + 1 μM |
| 10 (diastereomeric mixture) | 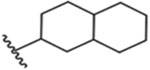 |
6± 1 nM |
| 11 | 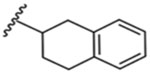 |
52 ± 1 nM |
| 12 (isolated diastereomer) | 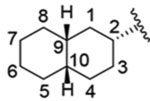 |
1.4 ± 1.0 nM |
| 13 | 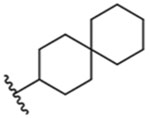 |
40 ± 1 nM |
| 14 | 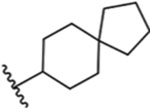 |
2.4 ± 1.0 nM |
| 15 | 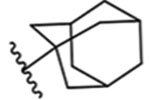 |
16 ± 1 nM |
| RQ-00203078 | 3± 1 nM | |
| icilin | EC50 | 74 ± 3 nM |
| (−)-menthol | EC50 | 5.7 ± 0.72 μM |
IC50 is defined by concentration of the compounds that inhibit 50% of icilin-evoked Ca2+ entry.
Sequentially, we docked (−)-menthyl 1, AMG2850, and 14 into the ligand binding site of hTRPM8, carried out 100 ns molecular dynamics (MD) simulations for each compound in hTRPM8, and analyzed the results as detailed below. (−)-Menthyl 1, described as a potent and selective TRPM8 antagonist26 at rat TRPM8 in Ca2+ flux assays (IC50 vs 20 μM menthol, 20 ± 2 nM; IC50 vs 0.25 μM icilin, 50 ± 10 nM), is currently used in the literature as a TRPM8 tool molecule (also described as OMDM233)40 as well as AMG2850.41
(−)-Menthyl 1 and hTRPM8.
As shown in Figure 2c, our results show that both the root-mean-square deviation (RMSD) of hTRPM8 (about 3.9 Å) and (−)-menthyl 1 (1.8 Å) kept stable after 50 ns during the simulation, indicating that the time scale of 100 ns is reasonable.
Figure 2.
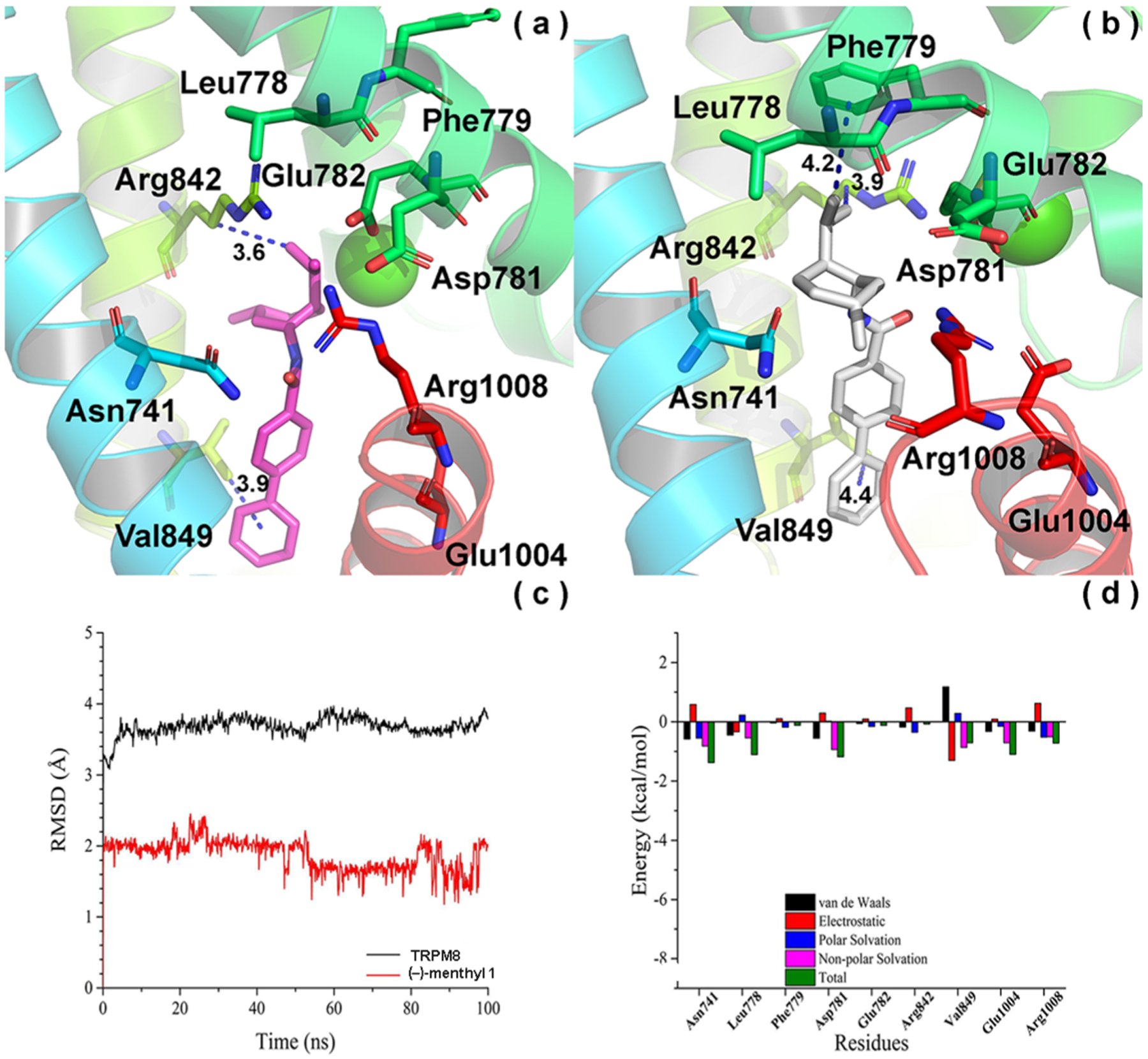
Convergence parameters of (−)-menthyl 1 and hTRPM8 during MD simulation. (a) Docking pose (pre-MD) of 1 in hTRPM8. (−)-menthyl 1 is shown as magenta sticks. Important residues are shown as sticks. Individual helices are colored as follows: S1 (blue), S2 (faded green), S4 (yellow green), TRP helix (red). The Ca2+ ion (green) is shown in CPK representation. (b) Binding mode (post-MD) of 1 in hTRPM8 after 100 ns MD. (−)-Menthyl 1 is shown as gray sticks. (c) RMSD of 1 and hTRPM8 during MD simulation. (d) Energy decomposition of key residues in hTRPM8 that contributed to the binding of 1. Our hTRPM8 homology model was constructed using the cryo-EM structure of TRPM8FA (PDB 6BPQ) as a template.
The biphenyl of 1 kept stable during the MD simulation, projecting into the base of the binding cavity in an extended conformation, toward the TRP helix, which forms strong hydrophobic interactions with Asn741 (S1, ~4.0 Å, not shown) and a conserved Val849 (S4, 4.4 Å), as shown in Figure 2b, occupying a similar space as the corresponding methoxyphenyl of WS-12.8 This pose is consistent with SAR studies on this scaffold, in which a biphenyl substitution affords the highest TRPM8 potency among all analogues studied.26
In contrast to the biphenyl moiety, we found that the flexible part of 1, the (−)-menthyl moiety, endures a change in conformation during the MD simulation, when comparing its binding mode between pre-MD (Figure 2a) and post-MD (Figure 2b). However, interactions between 1 and some residues can be observed during the MD. For example, the (−)-menthyl of 1 interacts with a conserved Arg1008 (TRP helix, 3.5 Å, not shown) (Figure 2b), which assumes a folded conformation similar to that seen in the WS-12 bound structure, and may represent a common TRPM8 binding residue for small molecule ligands. To explore the contribution of these residues to the binding of 1, we decomposed the free energy using the MM/GBSA method, as shown in Figure 2d. From the energy decomposition, we found that Arg1008 contributed greatly to the binding of 1. Although the distance between 1 and Arg842 (S4) was about 3.9 Å, the (−)-menthyl moiety approaches a conserved Leu778 (S2, ~3.8 Å, not shown) during the MD simulation (Figure 2b), overlapping with the (−)-menthyl and nitrophenyl binding regions of WS-12 and icilin, respectively,8 making Arg842 contribute less to the binding free energy than Leu778. The results are consistent with that of the free energy decomposition using the MM/GBSA method (Figure 2d). Interestingly, the (−)-menthyl group does not contact Tyr745 (S1), as suggested for (−)-menthol by radioligand displacement studies, as well as the WS-12-bound structure, likely due to the anchoring nature of the biphenyl, which forms extensive hydrophobic interactions with the base of the orthosteric site. Our studies here provide the first forays into investigating menthol-based antagonist mediated molecular events using the TRPM8 structural biology described to date.
AMG2850 and hTRPM8.
We also performed 100 ns MD simulation for AMG2850 with hTRPM8. We found that the RMSD of hTRPM8 equilibrates (about 4.4 Å) after 60 ns, as shown in Figure 3c. The RMSD of AMG2850 kept stable at 1.6 Å (Figure 3c). Due to the stability of AMG2850, the time scale of 100 ns is reasonable to explore its potential binding mode. Comparing the binding mode of AMG2850 between pre-MD (Figure 3a) and post-MD (Figure 3b), we found that several important interactions between AMG2850 and hTRPM8 kept stable. For example, a conserved Arg842 (S4) forms a strong hydrogen bond with the urea carbonyl (3.1/3.0 Å) of AMG2850, similar to the WS-12 amide carbonyl and icilin dihydropyrimidinone carbonyl,8 suggesting a common H-bond acceptor feature for TRPM8 ligand recognition via this common residue. Consistent with this predicted interaction, SAR studies of the AMG2850 precursor series, containing a tetrahydrothienopyridine core, highlight the importance of the urea moiety, where deletion of this group negates TRPM8 activity (derivatives 24–26 reported by Tamayo et al.42). Our MD results also show that a conserved Trp798 (S3) forms a hydrogen bond with the trifluorophenyl CF3 group of AMG2850, with a distance of 3.2 Å, in agreement with SAR studies of the precursor tetrahydrothienopyridine series, wherein a 4-CF3 affords optimal functional and pharmacokinetic properties.42 Interestingly, the inactive S-enantiomer of a related tetrahydroisoquinoline analogue of AMG2850 positions this trifluorophenyl in the opposite direction, away from Trp798, while the corresponding R-enantiomer has a reported TRPM8 IC50: 56 ± 24 nM (derivatives 87–88 reported by Tamayo et al.42). Moreover, Leu841 (S4) (distance: 4.2 Å, not shown) and the backbone of Arg842 (distance: 3.3 Å, not shown) interact with the trifluorophenyl CF3 group of AMG2850 via a hydrophobic interaction. Tyr1005 (conserved residue, TRP helix) forms strong hydrophobic interactions with the 5,6,7,8-tetrahydro-1,7-naphthyridine, with a distance of 4.4 Å. This residue (Tyr1004 in TRPM8FA) hydrogen bonds to the icilin nitro group and WS-12 amide nitrogen, suggesting another common TRPM8 binding residue, in agreement with functional studies of menthol.21 Again, this interaction is consistent with the SAR data for this series, suggesting the importance of hydrophobic interactions between the ion channel and the azatetrahydroquinoline. Modifications including removal of the aromatic ring, expansion of the piperidine by one carbon (to an azepine), and conformational restriction via methyl substitution at the 8-position, results in significant loss or negates TRPM8 activity (derivatives 32, 72, and 86 reported by Horne et al.10 and Tamayo et al.42), which may disturb this hydrophobic interaction. Alternatively, these analogues may disrupt the hydrogen bond between Arg842 and the urea carbonyl of AMG2850, resulting in a dramatic loss of TRPM8 activity.
Figure 3.
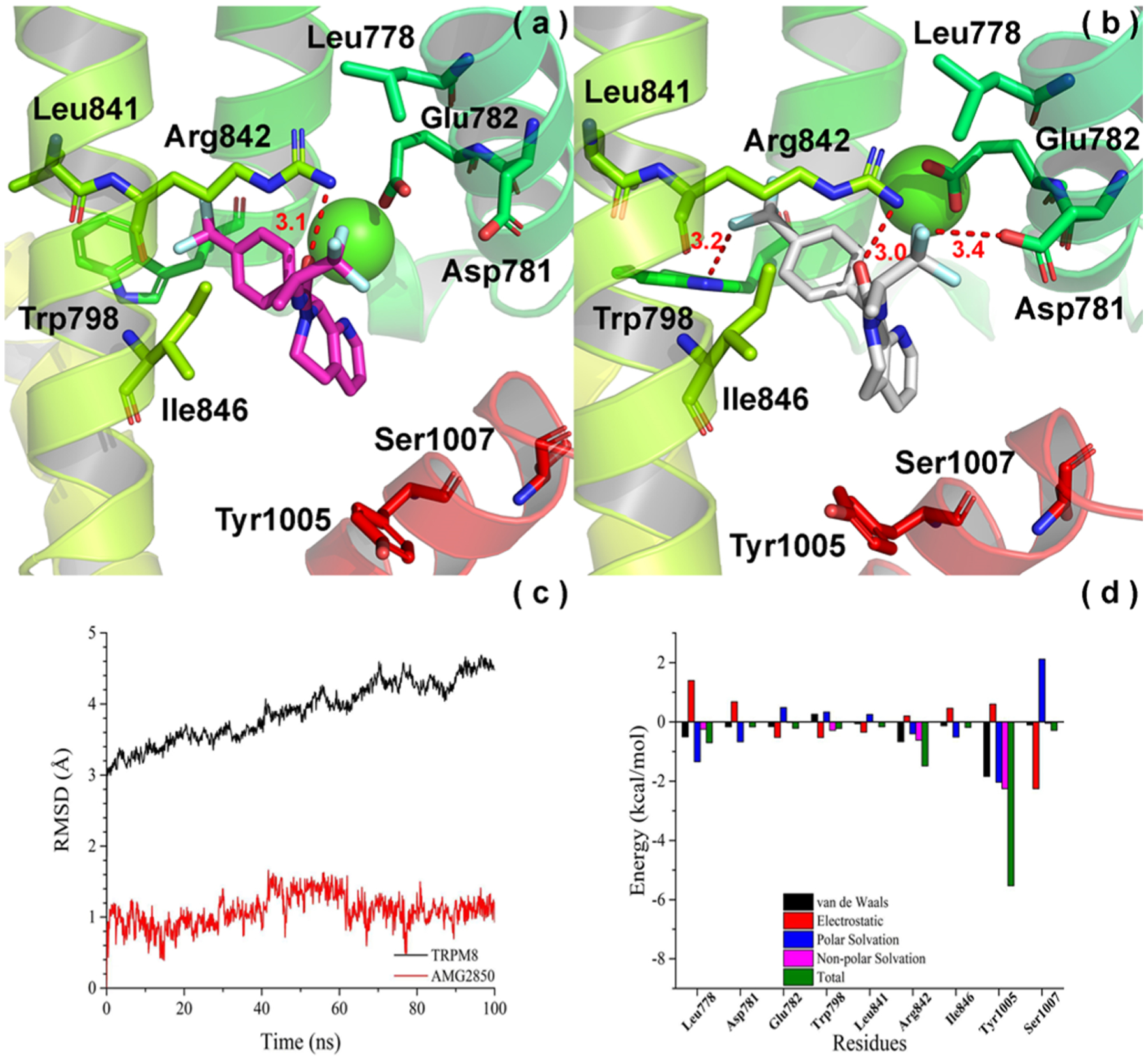
Convergence parameters of AMG2850 and hTRPM8 during MD simulation. (a) Docking pose (pre-MD) of AMG2850 in hTRPM8. AMG2850 is shown as magenta sticks. Important residues are shown as sticks. Individual helices are colored as follows: S2 (faded green), S3 (green), S4 (yellow-green), TRP helix (red). The Ca2+ ion (green) is shown in CPK representation. (b) Binding mode (post-MD) of AMG2850 in hTRPM8 after 100 ns MD. AMG2850 is shown as gray sticks. (c) RMSD of AMG2850 and hTRPM8 during MD simulation. (d) Energy decomposition of key residues in hTRPM8 that contributed to the binding of AMG2850. Our hTRPM8 homology model was constructed using the cryo-EM structure of TRPM8FA (PDB 6BPQ) as a template.
On the opposite side of the molecule, we observe that Asp781 (S2) interacts with the trifluoropropanyl CF3 via hydrogen bonding with a distance of 3.4 Å. Similar to the previously discussed interactions, this interaction is consistent with SAR studies of this region on the azatetrahydroquinolinone and tetrahydroisoquinoline scaffolds, where the trifluoropropanyl group affords high TRPM8 potency.10 Together with hydrogen bonding interactions with Arg842 and Trp798, these orient AMG2850 in a T-shape conformation, with either ends of the molecule spanning the upper portion of the VSLD pocket, vs the extended conformation seen with (−)-menthyl 1. On the other hand, the azatetrahydroquinoline ring of AMG2850 penetrates deeply into the cavity, toward the TRP helix, similar to the biphenyl of 1. Moreover, we observe that Leu778 forms strong hydrophobic interactions with trifluoropropanyl CF3 with a distance of 3.6 Å (not shown), similar to the (−)-menthyl of 1. These results are consistent with that of the free energy decomposition using the MM/GBSA method (Figure 3d).
Notably, from our MD studies, we did not find any direct interactions between the two antagonists studied and the Ca2+ binding residues, which could point to a possible structure-based explanation of their ability to function as antagonists. We did observe interactions between AMG2850 and Arg842, which could in theory reinforce the tetrahedral Ca2+ complex in human TRPM8 via Asp802, similar to that seen in the human TRPM4 structure (corresponding TRPM4 residues are Arg905 and Asp868).24 Such interactions with Arg842 could work to destabilize the bound Ca2+, resulting in channel inhibition. On the other hand, the TRPM8FA (avian) structure does not depict the corresponding residue Arg841 as interacting with Ca2+ binding residues, but instead makes a direct interaction with the icilin carbonyl.8
Structure-Based Design Strategy.
By superimposing the predicted binding modes of AMG2850 and (−)-menthyl 1 from MD simulations, we observe several important residues that form a hydrophobic pocket within the VSLD, in the S2 and TRP helix (Figure 4). Leu778 (S2) interacts with both the (−)-menthyl and trifluoropropanyl groups of 1 and AMG2850. In the same vicinity, Arg1008 (TRP helix) form a key hydrophobic interaction with the (−)-menthyl group. These interactions allow the aforementioned groups to superimpose in the VSLD, and suggests that the 1 scaffold can be substituted with other lipophilic groups such as ring-expanded analogues of the core cyclohexyl ring, bi- and tricyclic rings, aromatic-containing groups and spiro-substituted bicyclics.
Figure 4.
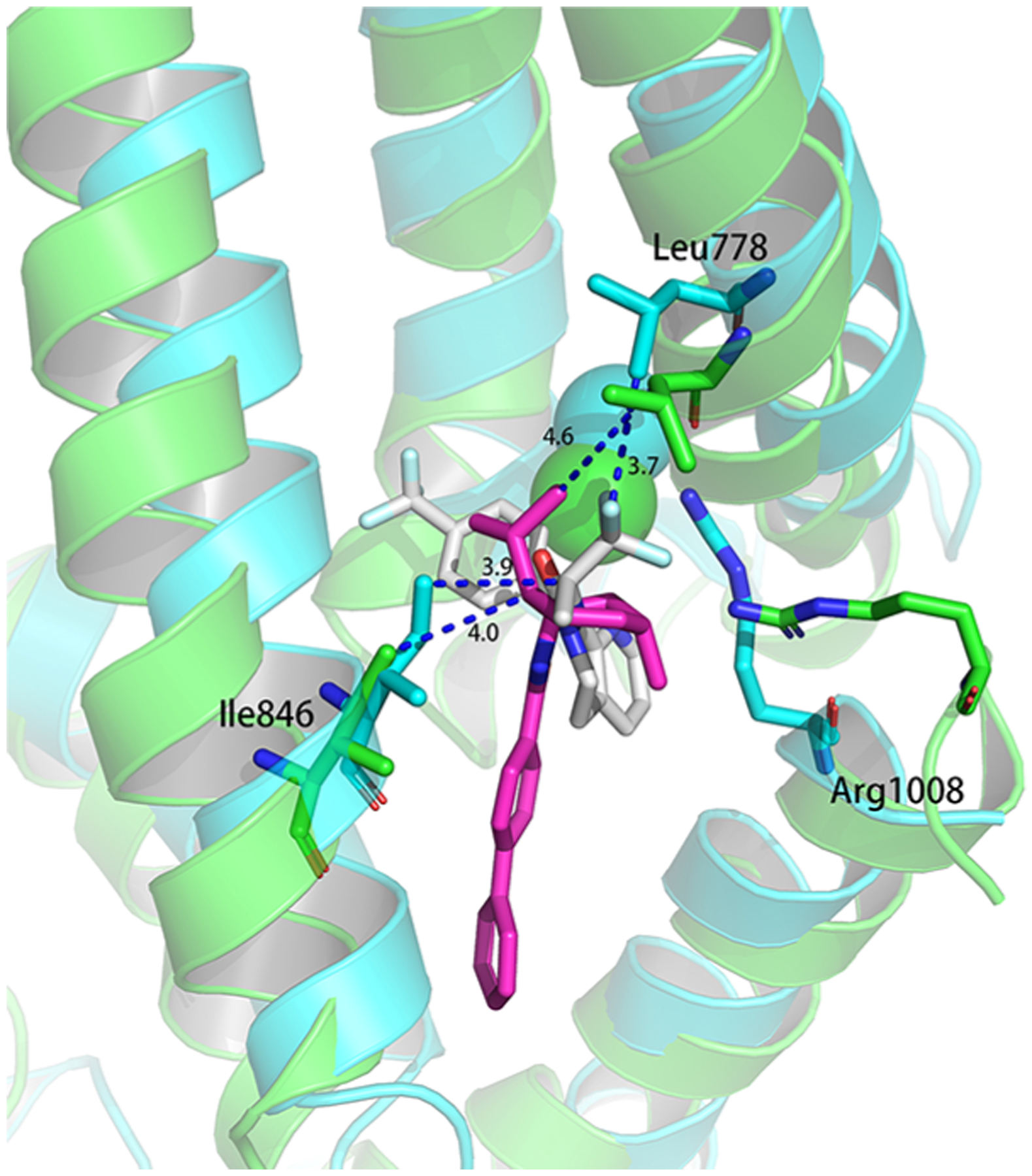
Superimposition of AMG2850 and (−)-menthyl 1 from their MD poses. (Green and magenta, MD pose of TRPM8 and (−)-menthyl 1; blue and gray, MD pose of TRPM8 and AMG2850).
We explored these substitutions on the 1 scaffold at human TRPM8 via ratiometric Ca2+ imaging (representative experiments are shown in Figure 5, corresponding histograms are shown in Figure S3), while retaining the biphenyl, as this was the optimal substitution for potent TRPM8 antagonist activity, from previous SAR studies.26 To this end, various lipophilic substitutions of the (−)-menthyl moiety of 1 were prepared as shown in Scheme 1, via standard EDCI/HOBt-mediated amidations of commercially available substituted amines and [1,1′-biphenyl]-4-carboxylic acid I-1.
Figure 5.
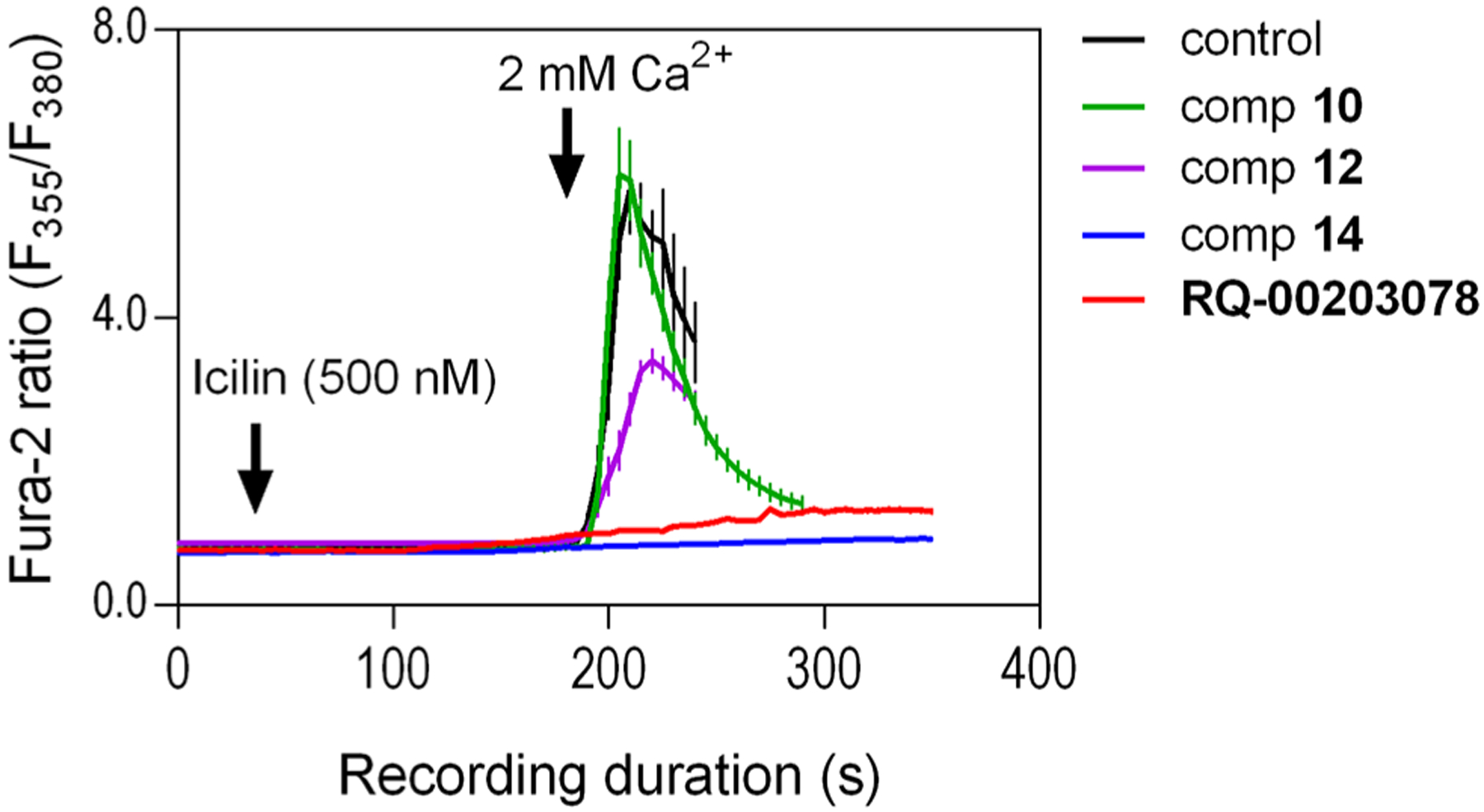
Evaluation of test compounds for any effect on icilin-evoked Ca2+ entry signals using fura-2 based Ca2+ imaging of HEK-293 cells stably expressing human TRPM8. The fura-2 loaded cells bathed in Ca2+-free extracellular solution were exposed to icilin (500 nM) with or without the synthesized compounds or the known TRPM8 antagonist RQ-00203078 at various concentrations (For this figure, all were at 3 nM. The corresponding histogram is provided in Figure S3). After ~4 min of compound addition, Ca2+ free bath solution was replaced by the one containing 2 mM Ca2+. Ca2+ influx was monitored as fura-2 fluorescence ratio (F355/F380). Each data point represents mean ± SEM (n ≥ 35 cells from three independent experiments done in three different days).
Scheme 1. Synthesis of 2–15.
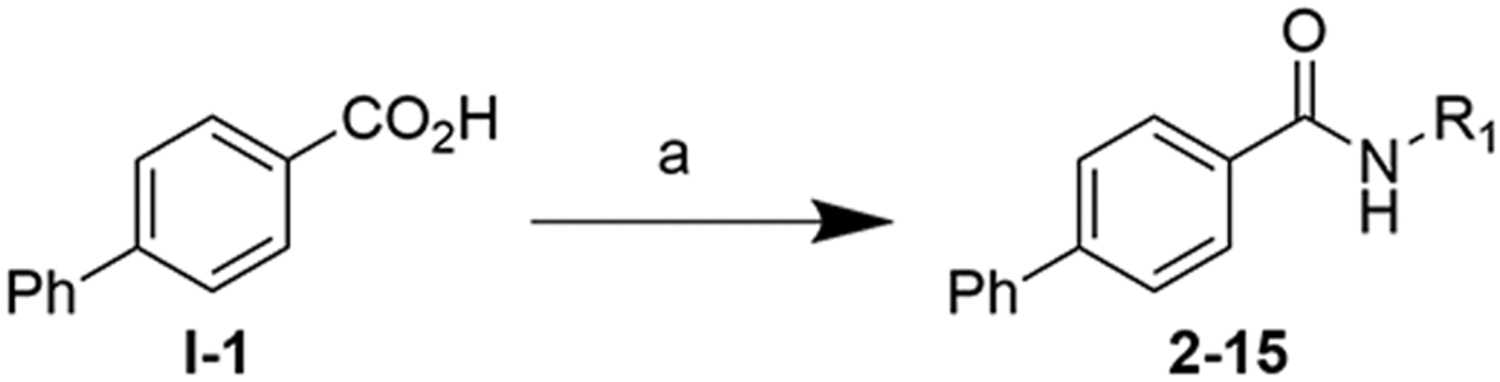
a aReagents and conditions: (a) R1-NH2, EDCI, HOBt, Et3N.
Structure–Activity Relationship (SAR) Analysis of (−)-Menthyl 1.
Dose–response studies of the prototypical icilin in this assay induces Ca2+ influx at an EC50 of 74 ± 3 nM (Figure S2a) which appears to be more potent than the previously reported values lying within the 120–200 nM range,29,33 but this could very well be due difference in cell type (CHO vs HEK cells)33 or species-dependence variation in icilin sensitivity (human versus mouse).29 In our hands, icilin-evoked Ca2+ signals began to plateau at 500 nM. We used RQ-00203078 as a positive control as well as a reference standard for our SAR studies, due to its potent TRPM8 antagonist activity.43 In our assays, RQ-00203078 inhibits icilin-evoked Ca2+ signals at an IC50 of 2.96 ± 0.99 nM (Figure S4), which is lower than the value (~8 nM) previously reported against 30 μM menthol-evoked Ca2+ influx in HEK cells expressing hTRPM8.43
For our synthesized analogues, IC50 values are shown in Table 1, and expressed as the concentration required to produce half-maximal inhibition of icilin-stimulated Ca2+ influx. We also resynthesized (−)-menthyl 1 using the methodology reported by Ortar and colleagues26 to serve as an additional standard for our biochemical assays at hTRPM8, due to its potent antagonist activity at rTRPM8 in calcium flux assays (IC50 vs 20 μM menthol, 20 ± 2 nM; IC50 vs 0.25 μM menthol, 50 ± 10 nM) and weak intrinsic (agonist) activity at hTRPV1 and rTRPA1. Interestingly, 1 shows dramatically reduced potency (IC50 of 16 ± 1 μM) in our assays in HEK-293 cells transfected with human TRPM8 (Table 1). Differences in observed IC50 values in the calcium flux assay at human vs rat TRPM8 suggests species differences among TRPM8 orthologues for this compound. Species differences are known to exist between different TRPM8 orthologues despite high sequence conservation.35,44 Additional studies at rTRPM8 could be performed on all analogues of this series to determine whether similar potency shifts are observed. Although human TRPM8 is highly homologous to the rat orthologue, with 96% sequence identity in the VSLD domain, pore domain, and TRP helix (residues 734–1009, calculated by Clustal Omega, data not shown), there are a small number of different amino acid residues, possibly accounting for the discrepancy seen between these orthologues in the calcium flux assay. One such residue at position 1007, in the TRP helix, is situated in the binding pocket (serine at hTRPM8, asparagine at rTRPM8). Studies are ongoing to further characterize 1 and will be reported in due course.
Removing the chirality of lead 1 to give isomeric mixture 2 increases hTRPM8 activity by 30-fold (Table 1) in our assays (hTRPM8 IC50: 468 ± 1 nM) relative to 1, indicating the mixture contains isomers with a more optimal stereochemical arrangement at the C1, C2 and C5 positions than 1. Relative to menthol isomeric mixture 2, removal of the C5 methyl and C2 isopropyl groups (as in 3) and conformational restriction of the core cyclohexyl ring (as in 4) affords reduced TRPM8 potency, in the micromolar range, highlighting the importance of the entire (−)-menthyl scaffold as well as the conformational flexibility of the cyclohexyl ring, and possibly displacing hydrophobic interactions with Leu778 and Arg1008, as suggested by our MD studies.
On the other hand, ring expanded analogues (cycloheptyl 5 and cyclooctyl 7) improve hTRPM8 potency by 8–9-fold relative to 2, with IC50 values of 52 ± 1 and 62 ± 1 nM, respectively, suggesting the ability to capture additional interactions in the pocket. In order to rationalize these results, we docked analogues 5 and 7 into our hTRPM8 homology model (Figure 6). For both analogues, the biphenyl ring is positioned between the S1 and S4 helices and TRP helix, similar to 1. The biphenyl ring of both analogues forms strong hydrophobic interactions with Val849 (conserved, S4, 3.8/3.8 Å), the same interaction seen for the 1 biphenyl, in addition to Phe738 (conserved, S1, 3.5/3.7 Å), Leu1001 (conserved, TRP helix, 3.7/3.7 Å), and menthol- and icilin-sensitive Tyr1005 (conserved, TRP helix, 4.0/4.0 Å). The 1-phenyl group forms edge-to-face π–π stacking with both Phe738 and Tyr1005. These additional interactions may contribute to the enhanced potency of these analogues in the Ca2+ flux assay, versus 1. Interestingly, the 5,6,7,8-tetrahydro-1,7-naphthyridine of AMG2850 forms hydrophobic contacts with Tyr1005 (Figure 3), in the same vicinity as the 1-phenyl of 1 (Figure 4). This reinforces the notion of Tyr1005 as a common small molecule binding residue. Both the amide NH and carbonyl O of 5 and 7 interact with Arg1008 (conserved, TRP helix) with strong hydrogen-bond interactions, possibly driving its potent functional activity vs that seen in 1 where this residue only forms hydrophobic contacts with the (−)-menthyl moiety. These results suggest that Arg1008 is a key residue for the recognition of both 1 and its analogues. In addition, the cycloheptyl and cyclooctyl moieties of both ligands approached three residues with similar hydrophobic interactions, including voltage- and icilin-sensitive Arg842 (conserved, S4, 4.2/4.0 Å), icilin-sensitive His845 (conserved, S4, 3.7/4.0 Å), and Ile846 (conserved, S4, 3.6/3.5 Å), located on the opposite side of the VSLD as Leu778 and Arg1008. Together, these results suggest that the orthosteric site is able to accommodate larger hydrophobic substitutions than that seen in 1, with a number of other lipophilic residues as mentioned above, while the biphenyl provides an anchoring interaction toward the floor of the cavity.
Figure 6.
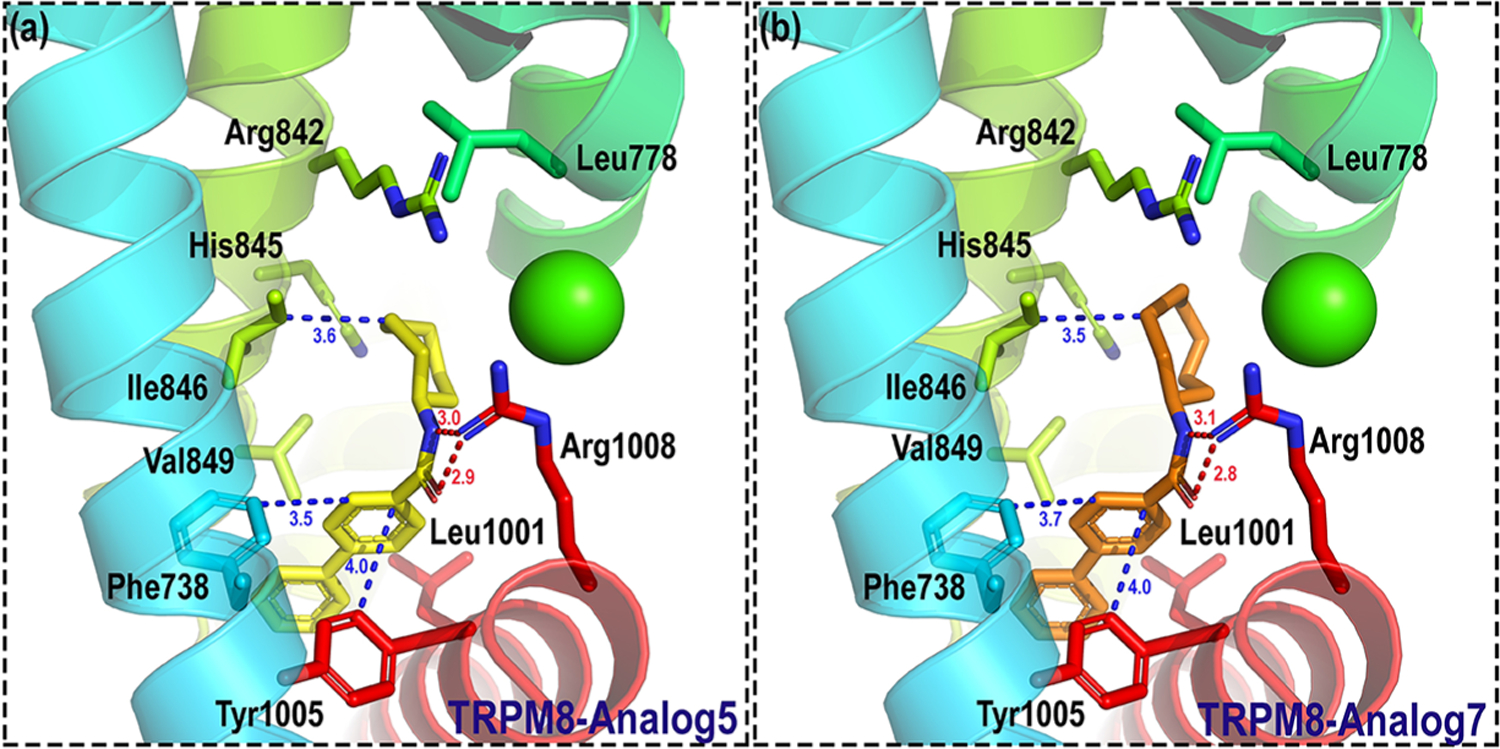
Binding mode of 5 and 7 in hTRPM8. Analogues 5 and 7 are shown as yellow and orange sticks, respectively. Important residues are shown as sticks. Individual helices are colored as follows: S1 (blue), S2 (faded green), S4 (yellow green), and TRP helix (red). The Ca2+ ion (green) is shown in CPK representation. Our hTRPM8 homology model was constructed using the cryo-electron microscopy (cryo-EM) structure of TRPM8FA (PDB 6BPQ) as a template.
Similar to 3 and 4, conformational restriction of 5 (to bicyclo[3.2.1]octan-3-yl 6) results in a sharp decrease in potency, possibly due to the loss of multiple H-bond contacts to Arg1008. Exploration with bicyclic and tricyclic ring replacements were carried out to further probe the lipophilic space around our series.
Decahydronapthalen-1-yl 8 results in a significant drop in hTRPM8 potency, relative to 5 and 7, in the micromolar range, indicating that the pocket is of a discriminate volume; similarly, tetrahydronaphthalen-1-yl 9 suffers a 3-fold loss of potency vs 8, again suggesting the need for flexibility at this region of the scaffold. In striking contrast, decahydronaphthalen-2-yl analogue 10, a mixture of two isomers, enhances TRPM8 potency by 3500-fold relative to 8 (IC50: 6 ± 1 nM) and has 10-fold higher potency than both the cycloheptyl 5 and cyclooctyl 7 analogues, suggesting the decahydronapthalen-2-yl ring occupies the lipophilic pocket to maximize available hydrophobic interactions. Consistent with the SAR trends seen with 8 and 9, substitution with a tetrahydronaphthalen-2-yl group (analogue 11) decreases hTRPM8 potency by 9-fold, though it retains nanomolar potency at hTRPM8 with an IC50 of 52 ± 1 nM. Separating out one of the isomers of analogue 10 via chromatography to give 12 gratifyingly affords potency in the single digit nanomolar range, with an IC50 of 1.4 ± 1.0 nM, 4-fold higher than the diastereomeric mixture. It is possible that 10 forms multiple contacts with residues within the VSLD to give high hTRPM8 potency, similar to the case of the single isomer 12. LC-MS analysis of 10 and 12 (Figures S20 and S22, respectively) indicates that 10, a mixture of two diastereomers, is composed of >85% of the 12 isomer, thus accounting for its potent IC50 value.
The relative stereochemical configuration of 12 was established by 1H, 13C, gCOSY, NOESY, gHSQCAD, gHMBCAD and gH2BC (Table S6, Figures S24–S30). Analysis of NMR data including comparison of decalin ring proton and carbon chemical shifts to literature values indicates that the compound is a cis-decalin with the biphenyl amide moiety in the 2-position. Analysis of NOESY data indicates that the amide group is trans to the decalin bridge protons (see structure in Table S6; absolute stereochemistry cannot be established).
The structure was determined to be cis-decalin because the carbon and proton chemical shifts are more consistent with that configuration (see Dodziuk et al.45). cis-Decalin bridge carbons (C9,10) resonate at 36.1 ppm, whereas those in trans-decalin resonate at 42.5 ppm. Furthermore, the bridgehead protons resonate at lower ppm in the trans-decalin than that observed for this structure (compare 1.91, 1.68 vs 0.87 ppm for trans-decalin). Also see Browne et al.46
The relative stereochemistry of the amide moiety is a bit more challenging to ascertain. NOESY data is consistent with the proposed structure; however, the severe chemical shift overlap, particularly with H10 and H1, renders unambiguous assignment impossible. NOESY data does establish that H9 (1.91 ppm) and H2 (4.01 ppm) are on the same face. This result coupled with the chemical shift data is consistent with a cis-decalin substituted in the trans-2-position with a biphenyl carboxamide.
We then probed spiro-substituted bicyclic replacement of the (−)-menthyl group of lead 1. Spiro [5.5]-undecan-3-yl analogue 13 retains a similar potency as 5 and 7 with an IC50 of 40 ± 1 nM, though not in the high nanomolar range as in the case of decahydronapthalen-1-yl analogues 10 and 12, suggesting the complementary region of the receptor cavity is of a fixed volume and cannot tolerate a spiro-arrangement of the two cyclohexyl rings.
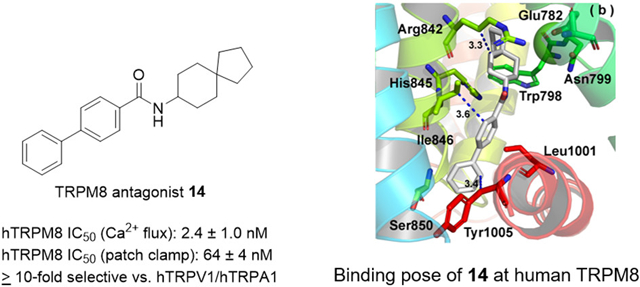
Ring contraction of 13 by one carbon (to spiro[4.5]decan-8-yl analogue 14) recapitulates the potent activity of decahydronapthen-1-yl isomer 12 (IC50: 2.4 ± 1.0 nM), with comparable activity as the highly potent TRPM8 antagonist RQ-00203078 in our assays.43 Spiro[4.5]decan-8-yl 14 is of similar potency as 12, further suggesting the constrained or discerning nature of the pocket. The binding site for this series could be determined unequivocally using structural biology techniques such as cryo-EM or X-ray crystallography to produce a ligand bound structure of human TRPM8, coupled with mutagenesis studies. Currently, the structure of human TRPM8 is not known. Notably, analogue 14 shares several common structural features with other potent TRPM8 antagonists reported in the scientific and patent literature. Both a spiro group (spiro-isoxazoline) and biphenyl aromatic arrangement are also seen in highly potent benzimidazole-based TRPM8 antagonist JNJ41876666/TC-I 201447 (cTRPM8 IC50 (icilin): 0.8 nM). Another TRPM8 antagonist reported by Glenmark Pharmaceuticals, (R)-(−)-10e (hTRPM8 IC50 (45Ca2+ uptake): 8.9 nM), contains a 3,4-dihydrospiro[chromene-2,4′-piperi-dine.48 Similarly, Raqualia Pharma reported a series of azaspiro TRPM8 antagonists containing ring systems such as 1-oxa-3-azaspiro[4.5]decan-2-ones, 1,3-diazaspiro[4.5]decane-2,4-diones and 1-oxa-3-azaspiro[4.5]decane-2,4-diones, linked via a ketone, to phenyl rings para substituted with aromatic and heterocyclic rings.49
We then constructed a dose–response curve of 14 from the data shown in Figure S3. We selected 14 for further characterization, given its lack of chiral centers unlike 12. As shown in Figure S5, compound 14 dose-dependently inhibits icilin-induced Ca2+ influx with an IC50 = 2.4 ± 1.0 nM and, at higher concentrations, nearly abolishes the Ca2+ influx (Figures S3 and S5).
To determine the ability of 14 to inhibit menthol in the Ca2+ flux assay, we first evaluated the menthol response in our HEK-293 cells stably expressing hTRPM8 and found an EC50 ~ 6 μM (Figure S2b). The observed menthol potency was comparable to the values previously reported for hTRPM8 in similar calcium imaging of HEK293 cells.50,51 The lowest concentration at which compound 14 was still able to cause significant reduction in icilin-evoked TRPM8-mediated Ca2+ entry was 3 nM (Figure S5). We tested compound 14 at the same lowest (i.e., 3 nM) concentration against the maximal (100 μM) menthol-evoked TRPM8-mediated Ca2+ entry (Figure S6a).51 As can be seen in Figure S6b, compound 14 at 3 nM was also able to significantly (P < 0.01) reduce TRPM8-mediated Ca2+ entry triggered by menthol. These studies provide additional support for exploring the effects of 14 in this assay using menthol as an agonist, in addition to our SAR studies carried out with icilin.
Adamantyl 15 affords slightly lower potency than 14, with an IC50 of 16 ± 1 nM, indicating that tricyclic rings are tolerated, as well as bicyclics 10–14. Altogether, the antagonist activity of 10, 12, 14, and 15 in the Ca2+ flux assay, with IC50 values in the ≤10 nM range in human TRPM8-transfected cells, exceeds or is comparable to the Ca2+ flux activity of reported TRPM8 antagonists with in vivo activity, including PF-05105679 (Pfizer, Phase 1, hTRPM8 IC50 = 181 nM),9 AMG 333 (Amgen, Phase 1, hTRPM8 IC50 = 13 ± 4 nM),13 AMG2850 (Amgen),10,11 RQ-00203078 (RaQualia Pharma),43 AMTB (Bayer),12 arylglycine derivative 12 and vinylcycloalkyl-substituted benzimidazole 25 (Janssen),52,53 DFL23448 and DFL23693,14 M8-B,15 M8-An,54 tryptophan derivative 14,38 and others. None of the analogues with hTRPM8 IC50 values ≤ 100 nM in the Ca2+ flux assay (5, 7, 10–15), were able to stimulate the channel alone at a single high concentration of 10 μM (Figure S7), in agreement with results obtained in our antagonist assays.
In Vitro Selectivity Profiling at TRPA1 and TRPV1.
We next determined the selectivity profile of our novel hits 5, 7, and 10–15 vs two temperature-related TRP subtypes, hTRPA1 and hTRPV1. Many TRP ligands have inverse behavior, for example, the TRPM8 agonist menthol blocks TRPV155 and TRPA1,56,57 while activating TRPA1 at lower concentrations.57 Similarly, the TRPV1 agonist capsaicin is able to inhibit TRPM8-mediated responses.55 On the other hand, TRPV1 antagonists capsaze-pine, BCTC, and SB-452533 and others exhibit dose-dependent blockade of TRPM8.29,31 These studies suggest ligand recognition overlap between TRPM8, TRPA1, and TRPV1. Similar to TRPM8, TRPA1 is activated by cold temperatures in vitro and in vivo, though at slightly lower temperatures.58,59 Icilin also activates TRPA1 at μM concentrations in the Ca2+ flux assay.58
In our TRPA1 Ca2+ flux assays, dose–response studies of the TRPA1 agonist AITC affords an EC50 of 7.62 ± 0.89 μM (Figure S8), in agreement with previous studies.60 The TRPA1 antagonist ruthenium red, used as a standard and positive control in our assays, dose-dependently blocks AITC (10 μM)-activated [Ca2+]i release at an IC50 of 162 ± 33 nM (dose–response curve shown in Figure S9), in the same range as that of Klionsky and colleagues,61 who report an IC50 of 29 ± 6 nM vs the effect of 80 μM AITC. At TRPV1, the EC50 and IC50 values for the TRPV1 agonist capsaicin (EC50 19 ± 2.5 nM, Figure S10) and antagonist capsazepine (IC50 451 ± 48 nM, Figure S11) are similar to values reported by El Kouhen et al.62 Curves included in the supplemental figures are listed at ±95% Confidence interval.
Among the compounds with significant TRPM8 antagonist activity, all analogues with the exception of 5, 7, and 11 had negligible intrinsic (agonist) and antagonist activity in Ca2+ flux assays at both hTRPV1 and hTRPA1, at concentrations corresponding to approximately 2× their hTRPM8 IC50 values and at a 10-fold greater concentration than the former value. Specifically, none of the compounds showed greater than 25% activation at hTRPA1 and hTRPV1 when normalized to the Emax concentration of the reference agonists AITC (300 μM) and capsaicin (3 μM) at both concentrations tested. Similarly, none of these analogues were able to inhibit either subtype at greater than 25% when challenged with the EC80 concentrations of AITC and capsaicin (10 and 0.1 μM, respectively). For example, even at a 17-fold higher concentration than its hTRPM8 IC50 value of 6 ± 1 nM, analogue 10 does not activate or inhibit hTRPA1 or hTRPV1. Similarly, derivatives 12–15 do not affect hTRPA1- and hTRPV1-mediated responses at concentrations 21-, 25-, 13-, and 19-fold higher than their hTRPM8 IC50 values of 1.4 ± 1, 40 ± 1, 2.4 ± 1.0, and 16 ± 1 nM, respectively. Thus, many of the derivatives in our series are at least 10-fold selective for TRPM8 vs related thermoTRPs TRPA1 and TRPV1. Future work will focus on dose–response studies.
Whole-Cell Patch Clamp Electrophysiology of 14.
Whole-cell patch clamp electrophysiology was used to further investigate and validate the antagonist activity of 14 at hTRPM8, using the cognate ligand menthol to stimulate the channel (Figure 7). In patch clamp experiments, activation of TRPM8 by icilin in transfected HEK-293 cells or TRPM8-expressing Xenopus ooctyes is dependent on the presence of extracellular Ca2+,4 additionally, icilin activates the channel with highly variable latencies and marked desensitization,23 an effect not seen with menthol. Considering these things, we further explored the ability of 14 to inhibit menthol-mediated responses at hTRPM8, as detailed in previous publications.32,36,37 In contrast to a Ca2+ and pH dependent icilin-evoked response in whole-cell recordings, menthol induces robust activation of TRPM8 in the voltage-clamp assay,33 allowing for better comparison studies. Considering these things, we evaluated the ability of 14 to affect currents induced by the agonist menthol.
Figure 7.
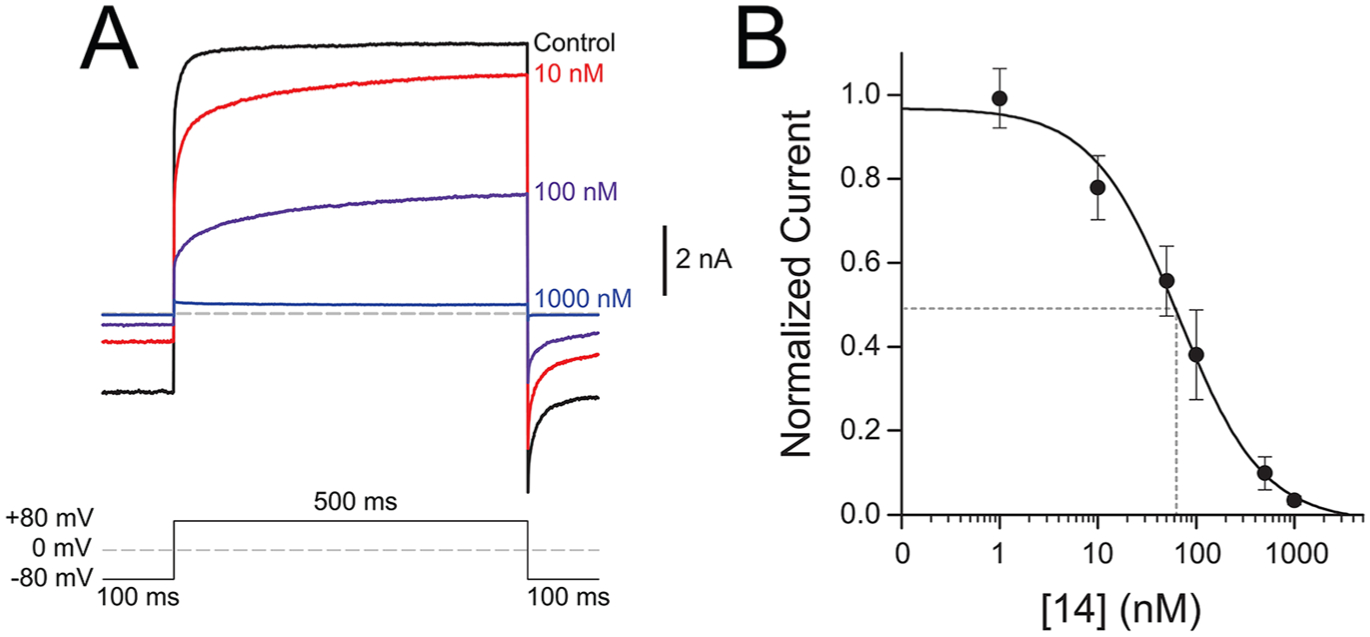
Inhibition of menthol-evoked TRPM8 currents by 14. Currents were measured by whole-cell patch-clamp electrophysiology of HEK-293 cells transiently transfected with human TRPM8. (A) Average current traces (n = 4) measured by a + 80 mV voltage pulse upon exposure to a saturating concentration of 500 μM menthol and varying concentrations of 14. (B) Dose response of 14 measured at concentrations from 1 nM to 1 μM at +80 mV in the presence of 500 μM menthol. Current response was normalized to the maximum current magnitude measured without antagonist for each cell. Data was fit with a single binding site Hill equation, and the IC50 was calculated to be 64 ± 2 nM (n = 6 cells). Error bars represent standard error of the mean.
In agreement with Ca2+ flux studies using icilin, this analogue inhibits menthol-induced TRPM8 responses in a concentration-dependent manner, with an IC50 of 64 ± 4 nM, demonstrating that 14 is a bona fide and functionally robust TRPM8 antagonist, with the ability to inhibit TRPM8 responses at nanomolar concentrations, elicited by different chemical stimuli. The antagonist activity of 14 surpasses a tryptamine-based antagonist by 6-fold in the same assay,36 with similar potency as β-lactam antagonists containing three labile ester groups.37 Although the IC50 value of 14 does not approach that of the well-known TRPM8 antagonist PBMC in this assay (IC50: 0.4–0.6 nM),32 this benzyloxy-phenylmethylcarbamate analogue of AMTB, containing labile ester and carbamate groups, appears to have limited utility as a chemical probe. PBMC gives variable responses in different behavioral models of neuropathic pain and is unable to block cold sensitization in the CIPN model.32 Altogether, 14 can overcome assay-specific nuances related to different chemical activators, demonstrating robust, highly potent antagonist activity in both the Ca2+ flux and whole cell patch clamp assay when challenged with both icilin and menthol. 14 is well characterized pharmacologically and has TRPM8 activity in a number of assays vs other small molecule TRPM8 ligands, demonstrating its viability as a chemical tool to probe TRPM8-mediated pharmacology. As discussed below, 14 interacts with Arg842 and Tyr1005, also shown to bind to icilin and menthol analogue WS-12. Binding to these residues may suggest why 14 is able to inhibit both icilin and menthol-mediated responses in Ca2+ flux and electrophysiology assays, respectively; however site specific mutagenesis studies could shed further insight.
We next sought to rationalize the potent antagonist activity of 14 in the context of our human TRPM8 homology model, by carrying out 100 ns MD simulations.
Compound 14 and hTRPM8.
We carried out 100 ns MD simulation for our highly potent antagonist, spiro[4.5]decan-8-yl analogue 14, in hTRPM8. We found that both the RMSD of hTRPM8 (about 3.9 Å) and 14 (1.9 Å) kept stable after 30 ns as shown in Figure 8c, indicating the time scale of 100 ns is reasonable.
Figure 8.
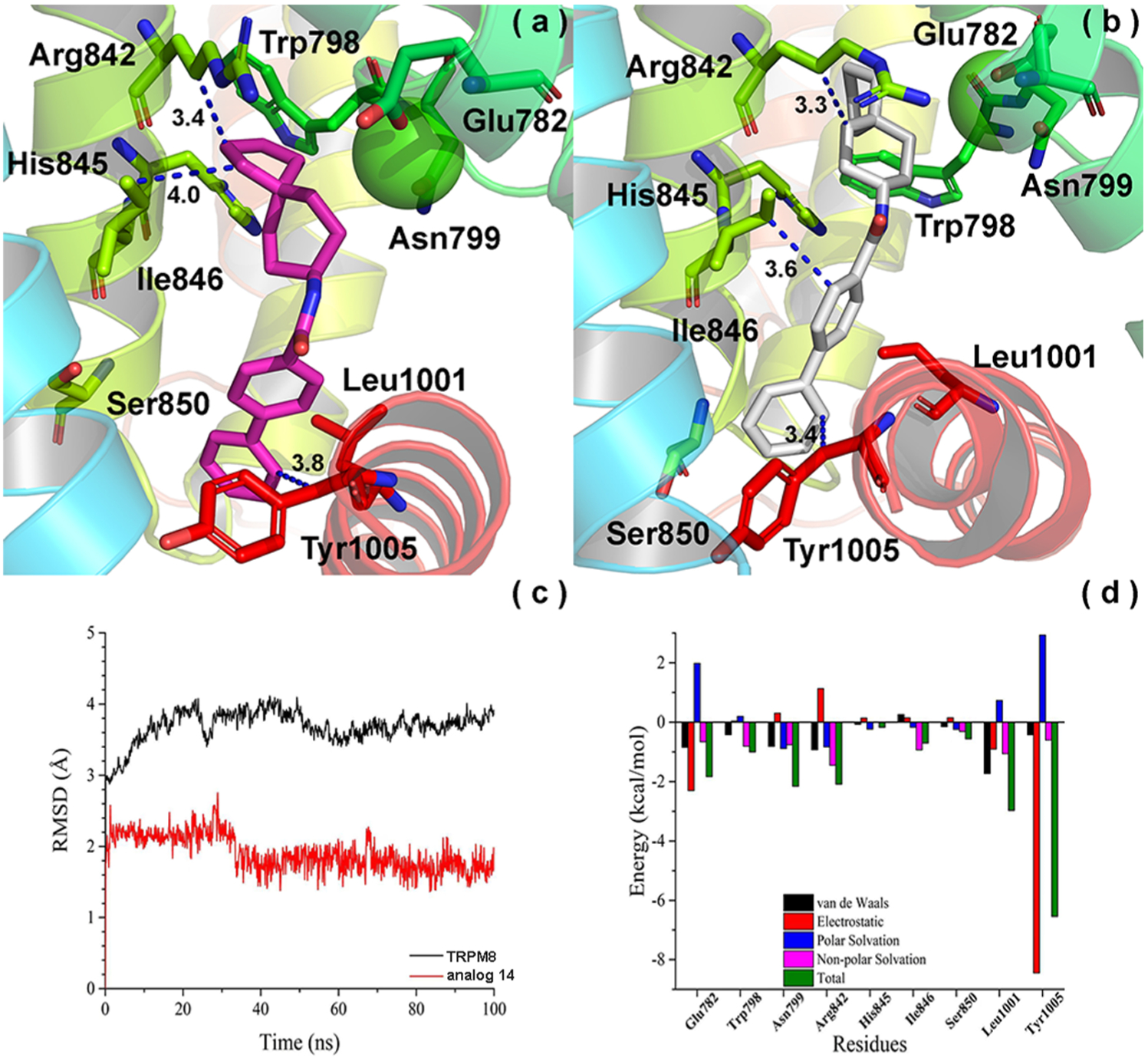
Convergence parameters of 14 and hTRPM8 during MD simulation. (a) Binding mode (pre-MD) of 14 in hTRPM8. 14 is shown as magenta sticks. Important residues are shown as sticks. Individual helices are colored as follows: S1 (blue), S2 (faded green), S3 (green), S4 (yellow-green), TRP helix (red). The Ca2+ ion (green) is shown in CPK representation. (b) Binding mode (post-MD) of 14 in hTRPM8. (c) RMSD of 14 and hTRPM8. (d) Energy decomposition of key residues in hTRPM8 that contributed to the binding of 14. Our hTRPM8 homology model was constructed using the cryo-EM structure of TRPM8FA (PDB 6BPQ) as a template.
As shown in Figure 8a, the cyclopentyl of 14 forms strong hydrophobic interactions with conserved Arg842 (S4, side chain, 3.4 Å) and Ile846 (S4, 4.0 Å) residues within the lower S4 helix, similar to the cycloheptyl and cyclooctyl of analogues 5 and 7 (Figure 6). The biphenyl contacts conserved Leu1001 (~3.1 Å, not shown) and Tyr1005 (menthol-sensitive, 3.8 Å) residues in the TRP helix before MD simulation. Figure 8b shows the binding pose of 14 within TRPM8 after MD simulation. We observe that the interactions between the biphenyl of 14 and Ile846 (3.6 Å)/Leu1001 (3.3 A, not shown)/Tyr1005 (3.4 Å) kept stable during the MD, as shown in Figure 8b. Ile846 shifts instead to make hydrophobic contacts with the biphenyl group, similar to Leu1001 and Tyr1005. Interactions with Leu1001 and Tyr1005 are again consistent with the docked poses of 5 and 7, as discussed previously.
Ile846, Leu1001, and Tyr1005 anchor the biphenyl into the base of the VSLD, where it is more deeply buried than the biphenyl of 1, again suggesting this as a plausible rationale for agonist- vs antagonist-mediated behavior for these chemotypes. Importantly, the hydrophobic interaction observed between Tyr1005 and the biphenyl is also seen in the AMG2850 post-MD pose. This residue also contacts WS-12 and icilin via polar interactions as discussed previously, reinforcing the notion of Tyr1005 as a common TRPM8 residue for ligand recognition. Unlike 1 and WS-12, the aromatic-containing groups do not overlap in the VSLD.
To our surprise, the spiro[4.5]decan-8-yl group maintains hydrophobic contacts with Arg842, though with the cyclohexyl ring. Together with Ile846, Leu1001, and Tyr1005, the energy decomposition of the key residues further supports the notion that these four residues may contribute greater than others to the recognition of 14 in TRPM8, as shown in Figure 8d. Arg842 represents another common residue for small molecule binding suggested from previous cryo-EM studies8 and our MD studies on AMG2850, on the opposite side of the VSLD vs the menthol-binding site, approaching the bound Ca2+ ion and coordinating residues in the S2–S3 helices. This forces the molecule into an extended confirmation. This region of the VSLD orthosteric site (lower S2 helix, and S3–S4 helices) contains a number of hydrophobic as well as polar residues, and it may preferentially accommodate larger hydrophobic substitutions on our biphenyl amide series such as those seen in our potent nanomolar antagonists 5, 7, and 10–15 vs the menthol-binding region in the S1 and upper S2 helix, providing structural context for our experimentally observed SAR trends. Residues within this vicinity (Glu782, Arg842 (Arg841 in TRPM8FA) and His845 (His844 in TRPM8FA), among others) are reported to adopt multiple conformations, allowing TRPM8 to bind both WS-12 and icilin.8
The presence of bulky residues such as Trp798 (S3) and His845 (S4), 3.6 and 3.8 Å away from the spiro cyclohexyl group, may serve to sterically repulse the decahydronapthalen-1-yl and tetrahydronaphthalen-1-yl rings of 8 and 9, effectively decreasing their TRPM8 potencies to the micromolar range, vs other analogues in our series with a greater degree of conformational flexibility at this region (as in 5 and 7) or a more optimal bicyclic arrangement (10–14).
Interestingly, 14 does not interact with Arg1008 (as is the case for 1, 5, and 7) or form hydrogen bonds, but instead it forms extensive hydrophobic contacts within the binding cavity, similar to that seen in the recently reported structures of antagonists AMTB and JNJ41876666/TC-I 2014 bound to Parus major (parrot) TRPM8 (pmTRPM8) (PDB 6O6R and 6O72, respectively).63 Comparison of the post-MD pose of 14 with the JNJ41876666/TC-I 2014-complexed structure reveals the biphenyls of both antagonists superimpose within the VSLD, supporting our MD results, while the spiro-containing moieties are oriented oppositely in the pocket. Opposite orientations seen for the spiro[4.5]decan-8-yl of 14 vs the spiro-isoxazoline of JNJ41876666/TC-I 2014 are attributed to projection of either group off of an amide (trigonal planar geometry, 120°) vs a conformationally rigid benzimidazole, respectively.
In Silico Selectivity Profiling at TRPA1 and TRPV1.
In order to rationalize the selectivity profile of 14, we docked this analogue into the cryo-EM structures of rTRPV1 (PDB 3J5R)64 and hTRPA1 (PDB 3J9P)65 and compared these poses to the pre-MD (docked) pose in our hTRPM8 homology model. Interestingly, the binding pockets of TRPM8, TRPV1, and TRPA1 occupy different regions of the VLSD (S1–S4), the pore domain formed by S5–S6, and the pore helix (Figure 9). Vanilloid ligands resiniferatoxin (RTX), a capsaicin-like homovanillyl ester antagonist, and the competitive antagonist capsazepine bind to TRPV1 residues in the S3 and S4 helices, and the S4–S5 linker at 2.9 and 3.4 Å.64 Taken together with the liganded cryo-EM structure of avian TRPM8, whose binding pocket is defined within the S1–S4 helices and TRP helix, these structures suggest the absence of a central binding site for structurally disparate natural product ligands for both TRP subtypes, namely, menthol and capsaicin, respectively. Rather, these molecules bind to different areas within the VSLD of each channel. While the structure of TRPA1 is not liganded (~4 Å), cryo-EM along with mutagenesis studies of a key Phe909 residue suggests that the natural product agonist allyl isothiocyanate (AITC) and antagonist A-967079 bind in a site defined by residues in the S5, S6, and pore helix,65 again suggesting that prototypical TRP channel agonists from nature bind to different pockets on their respective subtypes.
Figure 9.
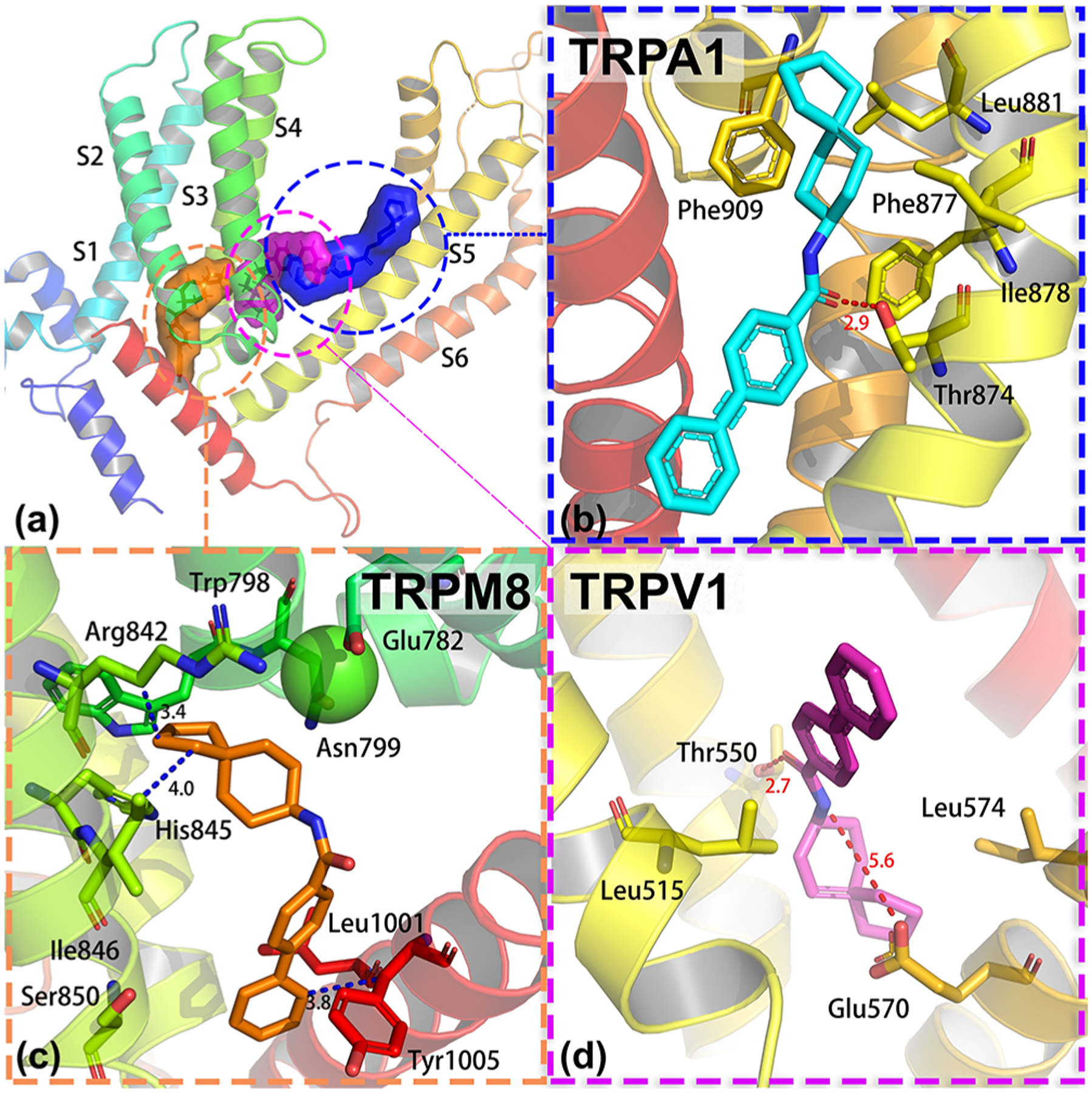
Binding pocket of 14 in TRPM8, TRPA1, and TRPV1 and their binding mode. (a) Position of the binding pocket of 14 in TRPM8, TRPA1, and TRPV1. (b) Binding mode of 14 in hTRPA1 (PDB 3J9P). (c) Binding mode of 14 in hTRPM8. Our hTRPM8 homology model was constructed using the cryo-electron microscopy (cryo-EM) structure of TRPM8FA (PDB 6BPQ) as a template. (d) Binding mode of 14 in hTRPV1 (PDB 3J5R).
In order to rationalize the selectivity of our compound, we docked 14 to TRPA1 and TRPV1 and compared these results to the docked pose (pre-MD) of 14 at TRPM8 (docking energy of −9.9 kcal/mol at TRPM8). As shown in Figure 9c, the docked pose shows 14 forms extensive interactions with TRPM8. Several important residues contributed to the binding of 14, including Trp798, Arg842, His845, Ile846, Leu1001, and Tyr1005. Figure 9b shows the interactions between 14 and TRPA1. The binding pocket of TRPA1 revealed a unique density within a pocket formed by S5, S6, and the first pore helix, including Phe909 (pore helix 1), Leu881 (S5), Ile878 (S5), Phe877 (S5), and Thr874 (S5), which is consistent with the reported binding site.65 By analyzing the interactions, we found that Thr874 forms a hydrogen bond with 14, while Phe909, Leu881, Ile878, and Leu871 contributed to the hydrophobic interactions. Figure 9d shows the binding pose of 14 in TRPV1. The pocket in TRPV1 was formed by five trans-membrane domains from two adjacent monomers, including S3, S4, the S4–S5 linker, S5, and S6. Several important residues within the binding pocket are shown in Figure 9d, including Tyr511 (S3), Leu515 (S3), Leu574 (S4), Thr550 (S4), Arg557 (S4–S5 linker), Glu570 (S5 of the first monomer), and Leu672 (S6 of the second monomer). We found that Thr550 forms a strong hydrogen bond with 14, while Leu515 and Leu574 form hydrophobic contacts with 14. The docking energies of 14 at TRPM8, TRPA1, and TRPV1 are −9.9, −7.1, and −8.3 kcal/mol, indicating that 14 should preferentially bind to TRPM8. From our high throughput FLIPR assays in Table 2, 14 is modestly selective (13-fold) for TRPM8 vs TRPA1 and TRPV1. Future studies will focus on testing higher concentrations of 14 at TRPA1 and TRPV1 to determine any dose–response effects and a more comprehensive selectivity profile.
Table 2.
Intrinsic and Antagonist Functional Activities of Selected Biphenyl Amides at TRPA1 and TRPV1 Receptors, Determined in Ca2+ Flux Assays
| compd | concn tested (×TRPM8 IC50) | hTRPA1% activation (n = 3–5)b | hTRPA1% inhibition (n = 3)c | hTRPV1% activation (n = 3)b | hTRPV1% inhibition (n = 3)c |
|---|---|---|---|---|---|
| 5, | 100 nM | 0.1 ± 0.7 | −2 ± 5 | 0.0 ± 1.0 | −3 ± 4 |
| hTRPM8 IC50: | 1 ,μM | 0.8 ± 0.5 | 4 ± 5 | 0.6 ± 1.5 | 24 ± 1 |
| 52 ± 1 nM | |||||
| 7, | 100 nM | −1.0 ± 0.1 | −13 ± 4 | −1.9 ± 0.7 | −4 ± 4 |
| hTRPM8 IC50: | 1 ,μM | −1.5 ± 0.9 | 0.2 ± 6 | −3.2 ± 0.6 | 14 ± 4 |
| 62 ± 1 nM | |||||
| 10, | 10 nM | 0.2 ± 0.4 | −8 ± 3 | 2.4 ± 0.8 | −13 ± 4 |
| hTRPM8 IC50: | 100 nM | −1.1 ± 0.2 | −1 ± 10 | 2.3 ± 0.9 | 2 ± 3 |
| 6 ± 1 nM | |||||
| 11, | 100 nM | −0.1 ± 0.5 | 3 ± 6 | −1.8 ± 0.9 | −3.1 ± 0.6 |
| hTRPM8 IC50: | 1 ,μM | −0.8 ± 0.6 | 4 ± 1 | −0.7 ± 1.5 | 13 ± 3 |
| 52 ± 1 nM | |||||
| 12, | 3 nM | −2.3 ± 1.0 | −0.3 ± 3 | 2 ± 1 | −12 ± 3 |
| hTRPM8 IC50: | 30 nM | −2.1 ± 0.5 | 5 ± 9 | 3 ± 1 | 1.0 ± 2 |
| 1.4 ± 1.0 nM | |||||
| 13, | 100 nM | 0.2 ± 0.5 | −1 ± 5 | −1.0 ± 0.4 | −3 ± 4 |
| hTRPM8 IC50: | 1 ,μM | −0.2 ± 1.1 | −7 ± 9 | 0.7 ± 0.2 | 5±2 |
| 40 ± 1 nM | |||||
| 14, | 3 nM | −0.8 ± 0.4 | −6 ± 5 | 1 ± 1 | −9 ± 2 |
| hTRPM8 IC50: | 30 nM | 0.1 ± 1.2 | −3 ± 2 | 1 ± 2 | −0.6 ± 5 |
| 2.4 ± 1.0 nM | |||||
| 15, | 30 nM | −2.6 ± 0.5 | −7 ± 5 | 1.0 ± 0.2 | −6 ± 5 |
| hTRPM8 IC50: | 300 nM | −1.5 ± 0.4 | −7 ± 7 | 1 ± 2 | 3 ± 4 |
| 16 ± 1 nM |
Data are normalized to the maximum response triggered by 500 nM icilin.
Data are normalized to the concentration corresponding to the Emax value of TRPA1 and TRPV1 agonists AITC (300 μM) and capsaicin (3 μM), respectively.
Data are normalized to the EC80 of AITC (10 μM) and capsaicin (0.1 μM).
Metabolic stability testing of 14 was carried out in mouse liver microsomes to determine its suitability as a TRPM8 chemical probe in vivo. 14 has a half-life (t1/2) of 30 min in mouse liver microsomes and an intrinsic clearance (CLint) of 0.0455 mL/min/mg; indicating sufficient metabolic stability to conduct the behavioral assays (Table S5). 14 may undergo CYP450 hydroxylation of the spiro or aromatic rings, leading to t1/2 = 30 min.
In Vivo Characterization of 14.
Wet Dog Shakes Assay.
To determine whether the observed ability of compound 14 to potently inhibit TRPM8 activity in vitro also translates in TRPM8-dependent antinociceptive activity in vivo, a “wet dog shakes” (WDS) assay was performed.66,67 In this assay, icilin (10 mg/kg intraperitoneally i.p.) was injected in mice treated with 14 or vehicle. Mice TRPM8 is known to share high homology with rat TRPM8 that has been used for in vitro assays.36 Icilin injections in mice produce vivid and quantifiable TRPM8-mediated shaking behaviors.68 Analogue 14 (1, 30, and 50 mg/kg) or its vehicle were administered subcutaneously (s.c.) 30 min before icilin injection.
Cold Allodynia.
Increasing evidence suggests that TRPM8 plays a critical role in mouse models of chemotherapy-induced neuropathic pain evoked by oxaliplatin (OXP), a condition mimicking cold hypersensitivity provoked by chemotherapy-induced peripheral neuropathy (CIPN).69 Both acute and chronic OXP-induced cold hypersensitivity have been reproduced in rats and correlated with TRPM8 expression and function.70 Mizoguchi et al.71 reported that, in a rodent model, acute cold allodynia after OXP injection was alleviated by the TRPM8 blockers N-(2-aminoethyl)-N-[4-(benzyloxy)-3-methoxybenzyl]-N′-(1S)-1-(phenyl) ethyl] urea and TC-I 2014. According to these findings, we investigated the effect of our antagonist 14 in an OXP-induced cold allodynia model, using acetone for cooling stimulation. Considering that the cold pain threshold is increased from ~12 to ~26 °C in OXP-treated patients, acetone stimulation is considered to evoke pain in OXP-treated mice.72
As shown in Figure 10, compound 14 produces marked, dose-dependent antinociceptive behavior. At the highest dose of 50 mg/kg, 14 inhibits WDS like cold hypersensitivity in mice with an efficacy comparable to that observed with a standard analgesic dose of gabapentin (25 mg/kg, s.c.)
Figure 10.
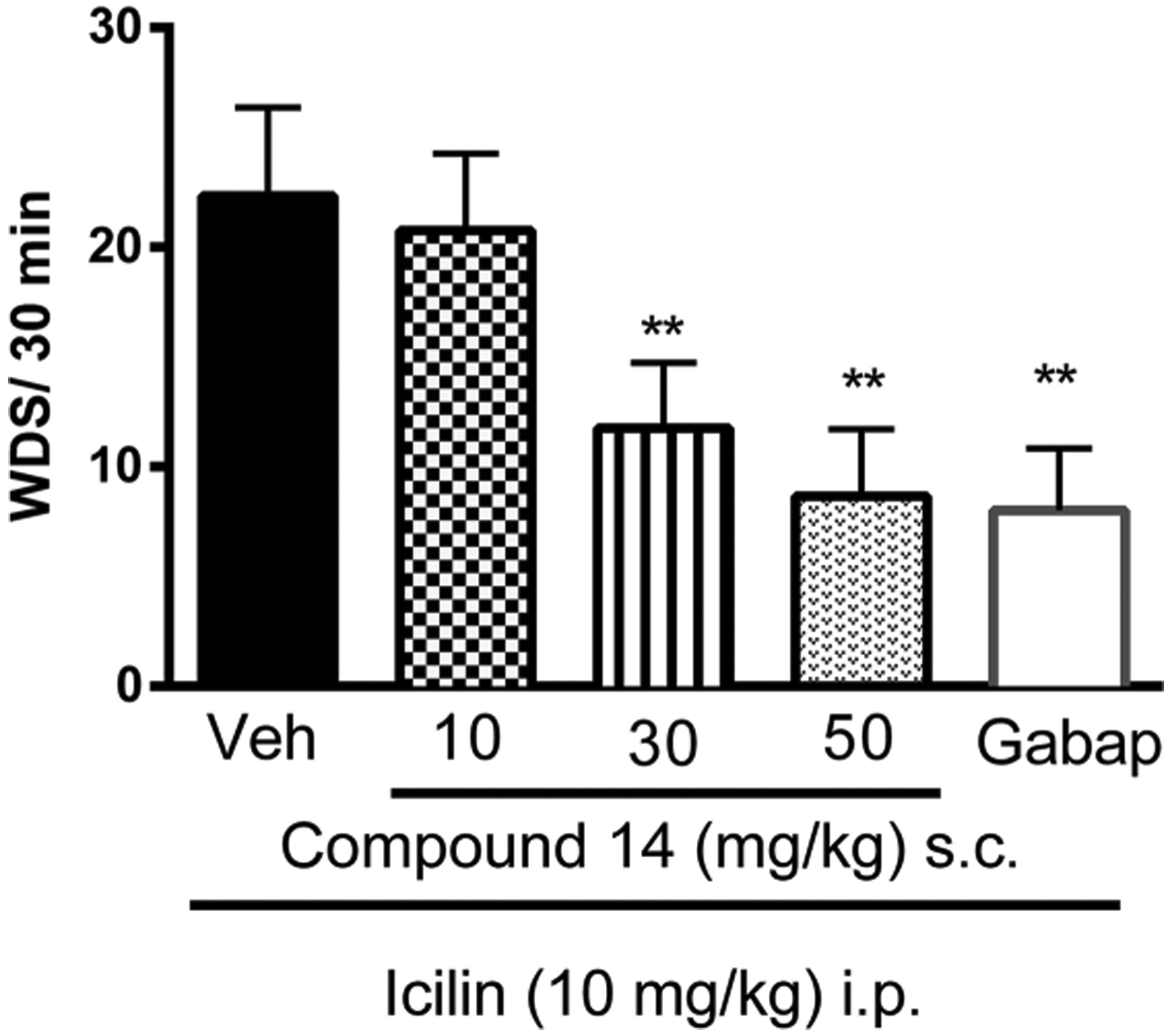
Effect of compound 14 on icilin-induced WDS in ICR (CD1) mice. Compound or vehicle was administered 30 min before icilin injection. Gabapentin (25 mg/kg) was administered 1 h before icilin injection. After i.p. injection of icilin (10 mg/kg), the number of wet dog shakes (WDS) was counted over 30 min. Data are given as mean ± SEM (n = 6). Two-way ANOVA with Bonferroni post hoc test. **P < 0.01.
These results link the antagonist activity of 14 in both the Ca2+ flux and patch-clamp assay to TRPM8 mechanistically, in a whole animal model, since icilin-induced WDS behaviors are not reproducible in TRPM8 null mice.68 This data demonstrates a key attribute for a chemical or pharmacological probe molecule.
ThermoTRPs, including TRPM8, TRPA1, TRPV1, TRPV2, and TRPV4, are described in the literature as potential new targets for treating chemotherapy-induced peripheral neuropathy (CIPN).73 Defects in sensory perception to cold are manifested as cold allodynia, defined as pain experienced from a normally nonpainful cold stimulus, and cold hyperalgesia, a heightened pain response from a painful cold stimulus,74 among others. Such cold deficiencies are seen clinically in chemo-therapy-induced allodynia75,76 and other pain states. Abnormal acute and sensory responses to cold exposure occurs following the first rounds of treatment with the chemotherapeutic agent oxaliplatin,77 the most commonly used antitumor treatment for colorectal cancers and others;78 additionally, oxaliplatin precipitates increased sensitivity to menthol, perhaps due to overexpression or activity of TRPM8.69,75
Preclinically, oxaliplatin-induced hypersensitivity to innocuous and noxious cold stimuli proceeds through a TRPM8-dependent mechanism, as confirmed by knockout studies.79 TRPM8 blockade with the bifunctional TRPM8/TRPV1 antagonist capsazepine dose-dependently inhibits oxaliplatin-induced cold allodynia in vivo, an effect not seen with TRPV1 antagonist 5′-iodoresiniferatoxin.80 Similarly, TRPM8 mRNA expression in the DRG significantly increases following cold allodynia induced by single injection of oxaliplatin, an effect functionally linked to an increase of response to TRPM8 stimulation in vivo.80 These studies are in agreement with previous literature reporting similar increases in TRPM8 mRNA expression in L4-L6 DRG following induction of cold hyperalgesia by oxaliplatin treatment,81 as well as treatment with the oxaliplatin metabolite oxalate. In vitro, TRPM8 mRNA levels were also significantly increased in oxaliplatin-treated DRG neurons.82 Analogue 14 was evaluated for its ability to block sensory responses to cold in a model of peripheral neuropathic pain induced by oxaliplatin, as cold hypersensitivity is the hallmark of oxaliplatin-induced neuropathy.79 Currently, no pharmacotherapies are available for CIPN, despite advances in cancer treatment with new and more targeted chemo-therapies.83 Duloxetine, used for diabetic neuropathy, has only a modest analgesic effect in patients with CIPN induced by oxaliplatin.84
The activity of compound 14 was evaluated 7 days after three intraperitoneal injections of OXP (6 mg/kg) in C57/BL6 mice, when cold allodynia had developed. After single subcutaneous administration of 14 (0.1 and 1 μg), attenuated cold allodynia is evident at 15 min and reaches maximum inhibition 30 min after administration (Figure 11) in a dose dependent manner. This data suggests that 14 may be a viable therapeutic scaffold for the treatment of CIPN.
Figure 11.
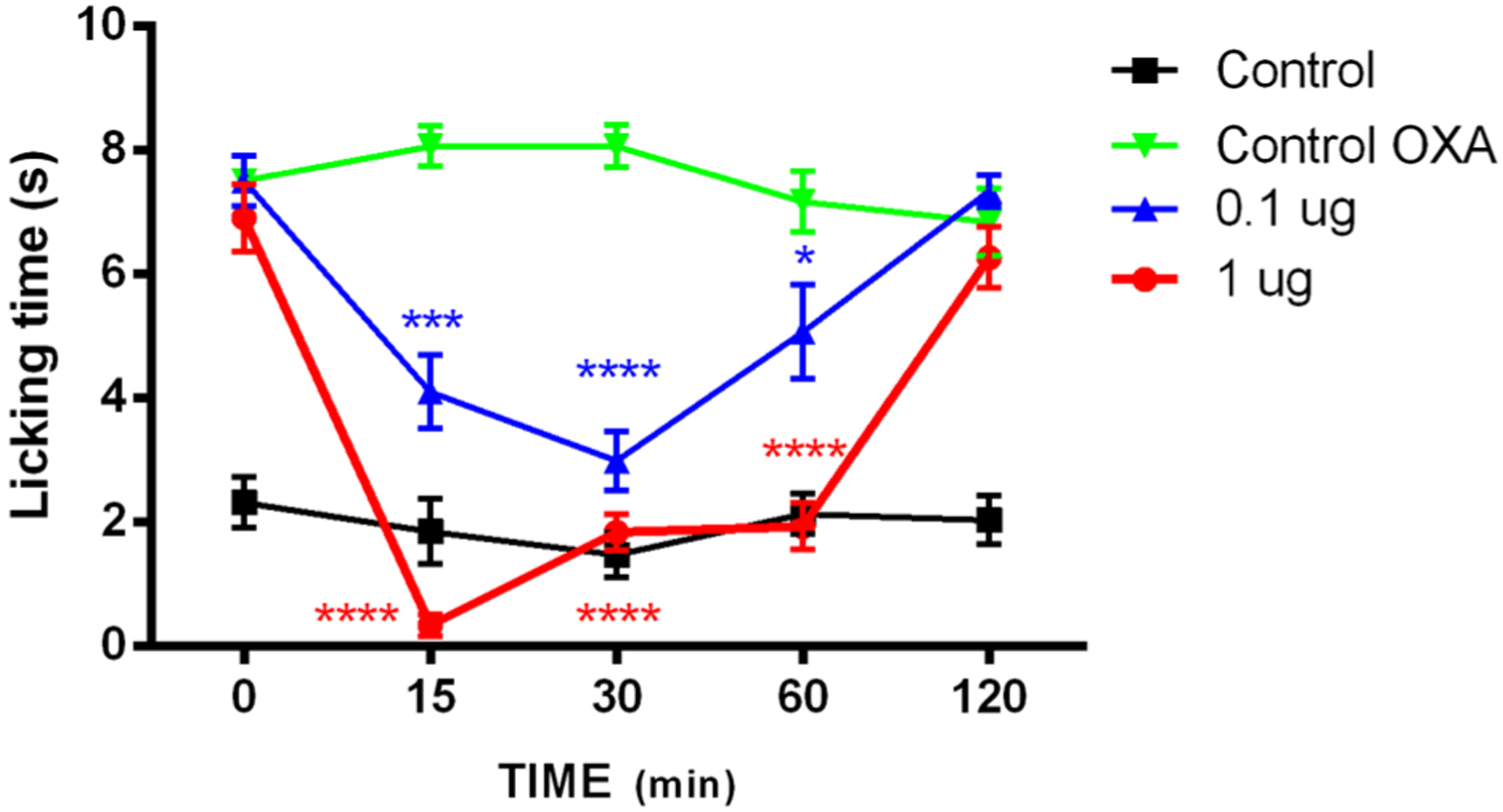
Effect of compound 14 on Oxaliplatin (OXP)-induced cold allodynia in male C57 mice. Mice were given three i.p. injection of OXP (6 mg/kg) or the vehicle (saline and a 5% mannitol solution, black line (control)) on alternate days. At day 7 after administration, cold allodynia was evaluated by the acetone test. Time-course of cold allodynia without compound injection (green line) and injection of compound 14 at 0.1 μg (blue line) or at 1.0 μg (red line). Data are given as mean ± SEM n = 6. One-way ANOVA with Dunnett’s post hoc test. *P < 0.05; ***P < 0.001; ****P < 0.0001.
The antinociceptive activity of 14 surpasses that of another reported, potent TRPM8 antagonist (R)-(−)-10e in the same assay.48 (R)-(−)-10e inhibits oxaliplatin-induced nocifensive paw licking, though at >10 000-fold higher doses when administered p.o. Similarly, a tryptophan-based TRPM8 antagonist reverses oxaliplatin-induced cold allodynia in C57/BL6 mice at the same doses as 14;38 however, its effects are short lasting, perhaps due to a labile ester group. Our 14 significantly suppresses OXP effects at 60 min, equal to two t1/2 values, suggesting that additional phenomena are at hand. For example, 14 may function in vivo via a drug depot effect, imparted by its lipophilicity. Of equal importance, 14 and other menthol-like antagonists reported herein may be useful chemical tools to explore the role of TRPM8 in other pain states such as chronic neuropathic pain in vivo, for which the role of this ion channel is controversial.85 Such small molecule scaffolds can then provide pharmacotherapies for sensory neuropathies.
In conclusion, probing replacements of the (−)-menthyl ring of 1 with lipophilic moieties, as suggested by computational studies of well-known TRPM8 antagonist scaffolds carried out in our human TRPM8 homology model, allows for the discovery of a novel series of menthol-like small molecule ligands with highly potent hTRPM8 antagonist activity in the Ca2+ flux assay and >10-fold selectivity vs TRPV1 and TRPA1. These menthol-optimized analogues may find use as chemical probes to investigate TRPM8 pharmacology, structure–function studies, and structural biology studies, along with menthol-based agonists reported by Dendreon (D-3263 and others) and Proctor & Gamble, with nanomolar agonist activity at human TRPM8.86–88 This series may also provide leads for pharmacotherapies for CIPN induced by oxaliplatin and other TRPM8-related sensory neuropathies, as 14 is able to attenuate oxaliplatin-induced cold allodynia in vivo. Additionally, these compounds may also aid in the identification of a TRPM8 pharmacophore for the rational design of small molecule ligands and therapeutic scaffolds. One of the most potent analogues identified (14), has superior antagonist activity in both Ca2+ flux and patch-clamp assays, and can dose-dependently inhibit the effects of multiple TRPM8 activators (icilin, menthol and cold) across a battery of in vitro and in vivo assays. 14 is readily accessible synthetically from a simple amidation reaction, and possesses no chiral centers, thus enhancing its attractiveness. To our knowledge, 14 is one of the most potent TRPM8 antagonists described to date. These SAR studies suggests its spiro[4.5]-decan-8-yl moiety is a preferred scaffold for TRPM8 recognition vs the cognate ligand (−)-menthol and represents an optimal menthol surrogate. The field awaits a TRPM8 structure complexed with a menthol-derived antagonist to address subtleties surrounding channel activation vs inactivation, particularly in the context of natural product-based scaffolds. Our antagonist 14 represents a viable candidate for these studies, to parse antagonist- vs agonist-mediated molecular events at TRPM8, along with menthol analogue WS-12.
METHODS
Molecular Modeling.
Homology Modeling of hTRPM8.
We built our human TRPM8 model based on the cryo-EM structure of the collared flycatcher Ficedula albicollis (TRPM8FA, PDB 6BPQ, resolution ~ 4.1 Å),7 with which hTRPM8 shares 83% sequence identity to TRPM8FA. The cryo-EM structure was downloaded from the Protein Data Bank (http://www.pdb.org/pdb/). SYBYL-X 1.389 was applied to repair all the residues and minimize the energy. The full sequence of hTRPM8 (Q7Z2W7, 1104 residues) was retrieved from the UniProtKB/Swiss-Prot (http://www.uniprot.org/uniprot/). The sequence alignments and homology modeling used our reported protocol.90
Modeller 9.1891 was used to construct the hTRPM8 model. The detailed parameters can be found in our previous publications.90,92–95 Once the 3D models were generated, SYBYL-X 1.3 was used to conduct the energy minimizations. Briefly, the parameters defined in the SYBYL were as follows: Gradient was set to 0.5 kcal/mol, max iterations were set to 5000, force field was set to MMFF94s, and the charges were set to MMFF94. All these settings are the same as described in our recent publication.90,92–95 Then proSA-web Z-scores96 and PROCHECK Ramachandran plots97 were used to check the overall quality of our 3D models.
Molecular Docking Ligands into hTRPM8.
The docking program Surflex-Dock GeomX (SFXC) implemented in SYBYL-X 1.3 was used to build TRPM8-ligand complexes, in which the total score was expressed in −log10(Kd).98 Then MOLCAD module in SYBYL-X 1.3 was applied to explore the potential binding pocket of hTRPM8, which included the residues Phe738 (S1), Tyr745 (S1), Glu782 (S2), Glu785 (S2), Trp798 (S3), Asn799 (S3), Asp802 (S3), Ala805 (S3), Arg842 (S4), His845 (S4), Ile846 (S4), Val849 (S4), Glu1004 (TRP helix), Tyr1005 (TRP helix), Arg1008 (TRP helix), and Leu1009 (TRP helix). All the parameters and the protocol of molecular docking can be found in our previous work.90,92–95
Molecular Dynamics (MD) Simulations.
In order to accelerate the MD simulation, we selected the transmembrane domain of hTRPM8 (residues from Gln671 to Asn1010) to carry out the simulations of TRPM8 with different ligands. Each monomer of the TRPM8 receptor was complexed with one ligand, so there were four (same) ligands for each simulation. Then the system was solvated into a 0.15 mol/L NaCl solution, including 514 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipids, 51 470 water molecules, and 156 Cl− and 140 Na+ ions. The initial conformation of TRPM8 and ligands were taken from molecular docking studies. The sizes of the initial simulation boxes were set to ~151 Å × 151 Å × 119 Å. The AMBER ff14SB force field99 was applied to TRPM8 and the AMBER Lipid14 force field100 was applied to lipids. Water molecules were treated with the TIP3P water model.101 Then all the MD simulations were conducted using the PMEMD.mpi and PMEMD.cuda modules in the AMBER16102–104 package. In order to avoid possible steric clashes, we performed several minimization steps for each system. Then, each system was gradually heated from 0 to 300 K during the heating stage, followed by equilibrium and production stages (300 K) with a time step of 2 fs. A periodic boundary condition was employed to maintain constant temperature and pressure (NPT) ensembles. The pressure was set to 1 atm and controlled by the anisotropic (x−, y−, z−) pressure scaling protocol with a pressure relaxation time of 1 ps. The temperature was kept constrained by Langevin dynamics with a collision frequency of 2 ps−1.105,106 Long-range electrostatics were handled by the particle mesh Ewald (PME) method,107,108 and the cutoff value of real-space interactions was set to 10 Å. All covalent bonds involving hydrogen atoms were constrained by the SHAKE algorithm.109 Each system was carried out with 100 ns MD simulation and the trajectory of each simulated system was saved every 100 ps.
Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) Calculation.
For the saved trajectories of each MD simulation, the MM/GBSA110–112 method was applied to calculate the binding energies of TRPM8 under the treatment of different ligands. In order to calculate the mean binding energy, 50 snapshots were extracted from each trajectory every 200 ps from 90 to 100 ns using the following formula:
where ΔEbind is the binding energy and ΔEMM denotes the sum of molecular mechanical energies in vacuo and can be further divided into the contributions from electrostatic, van der Waals, and internal energies. This term could be computed through a molecular mechanics method. ΔESOL is the solvation energy which includes the polar solvation energy (ΔEGB) calculated with the generalized Born (GB) approximation model113,114 and the nonpolar part (ΔESA) obtained by fitting solvent accessible surface area (SASA)115 with the linear combinations of pairwise overlaps (LCPO) model.116,117 Additionally, the energies of each residue were decomposed into the backbone and side-chain atoms. Through energy decomposition, we can analyze the contributions of the key residues to the biological activity.118
Chemistry.
Thin layer chromatography was performed on Analtech silica gel GF 250 um TLC plates. The plates were visualized with a 254 nM UV light and staining with iodine. Flash chromatography was carried out on F60 silica gel, 230–400 mesh, 60 Å (Silicycle SiliaFlash). Automated flash chromatography was carried out on a Biotage Isolera Prime automated flash purification system with a multivariable wavelength (200–400 nm) detector (model ISO-PSV), using Biotage SNAP Ultra cartridges (10 or 25 g) packed with Biotage HP-Sphere 25 micron spherical silica, using a binary gradient of solvent A (dichloromethane)/solvent B (diethyl ether) (for 2: 99/1 → 94/6, for 4: 100/0 → 96/4, for 5, 6, 9, 10, 12, 14, 15: 100/0 → 98/2, for 7: 96/4 → 94/6; for 13: 100/0 → 94/6) at a flow rate of 12 or 25 mL/min. Eluted peaks were monitored at 254 and 280 nm. NMR was recorded on a Bruker Ascent 400 MHz NMR using CDCl3. Mass spectra were obtained on an Agilent 1290 Infinity and 6490 Triple Quad LC/MS using electrospray ionization (ESI) mode. LC-UV-MS of 2 and its isomers (2A, 2B, 2C) and 10 and 12 was carried out using an Accela UHPLC system (Thermo Scientific, San Jose, CA) consisting of an Accela 1250 pump, open autosampler, and PDA detector followed by connection to a Thermo Q Exactive Orbitrap mass spectrometer. Chromatographic separation was performed on an Agilent Eclipse XDB C18 column (1.8 μm particles and dimensions of 2.1 mm × 100 mm, with a 2.1 mm × 5 mm guard column) using mobile phases consisting of A: 0.1% formic acid in LC-MS grade water (v/v) and B: 0.1% formic acid in acetonitrile (v/v) and two different elution methods. A gradient elution method was used for 2 and its isomers (2A, 2B, 2C), which consisted of 0% B to 95% B over 8 min followed by column cleaning and equilibration time for a total of 20 min. An isocratic elution method was used for 10 and 12, which was 58% B held constant for 20 min. For both chromatographic methods, the flow rate was 300 μL/min, and 5 μL of sample were injected. UV detection was achieved using the PDA detector over the range of 200–600 nm and specific channel wavelengths were monitored (280, 254, and 214 nm). The mass spectrometer parameters consisted of electrospray ionization in positive ion mode, scanning in full MS mode at 3.7 kV electrospray voltage, resolution setting of 70,000, AGC target 1e6, maximum IT 100 ms, and scan range m/z 150–1000. Data processing was performed using Qual Browser in Thermo Xcalibur version 4.1.31.9. Elemental analyses were performed by Atlantic Microlabs, Norcross, GA. The purity of all final compounds was greater than 99.6%.
General Amidation Procedure.
To a stirred solution of [1,1′-biphenyl]-4-carboxylic acid (1.00–1.5 equiv) in THF (0.1 M) was added EDCI (1.04–1.56 equiv), HOBt (1.04–1.56 equiv), and Et3N (1.2–3.3 equiv), and the reaction was stirred at room temperature for 1 h. To this mixture was added a solution of the substituted amine (1.0–1.2 equiv) in THF, and the reaction was stirred at 40 °C for 2–3 h or room temperature for 1–3 days. The reaction was diluted with EtOAc and H2O. The combined organic layers were washed with saturated NaCl(aq), dried over Na2SO4, filtered, and concentrated.
N-(2-Isopropyl-5-methylcyclohexyl)-[1,1′-biphenyl]-4-carboxamide (2, a Mixture of Three Isomers Present as a 30:21:49 ratio by LCMS).
Prepared according to the general procedure using 5-methyl-2-(propan-2-yl)cyclohexan-1-amine (62 mg, 0.40 mmol). The crude residue was purified via flash chromatography using hexane/diethyl ether/NH4OH (100/0/0 → 95/5/0.5), followed by automated flash chromatography to afford 83 mg of the title material in 62% yield. 1H NMR (mixture) (400 MHz, CDCl3) δ 7.87–7.78 (m, 6H), 7.69–7.63 (m, 7H), 7.63–7.58 (m, 6H), 7.51–7.35 (m, 8H), 6.22 (d, J = 9.0 Hz, 2H), 6.16 (d, J = 8.4 Hz, 1H), 5.82 (d, J = 9.5 Hz, 1H), 4.63–4.55 (m, 2H), 4.39 (tt, J = 7.8, 3.9 Hz, 1H), 4.03 (qd, J = 10.9, 3.9 Hz, 1H), 2.15–2.07 (m, 1H), 2.06–1.91 (m, 4H), 1.90–1.60 (m, 4H), 1.56–1.23 (m, 3H), 1.22–1.11 (m, 6H), 1.09–0.82 (m, 27H). MS (ESI) (mixture) m/z 336.30 (M + H)+. LCMS (mixture), RT (Peak 1) = 8.76 min, m/z (M + H)+ = 336.2; LCMS RT (Peak 2) = 8.84 min, m/z (M+H)+ = 336.2; LCMS RT (Peak 3) = 9.05 min, m/z (M+H)+ = 336.2. Anal. Calcd For C23H29NO (mixture): C, 82.34; H, 8.71; N, 4.18. Found: C, 82.07; H, 8.58; N, 4.29.
Isomeric separation of 2 (50 mg) was performed by preparative thin layer chromatography (TLC) (2000 um, 20 × 20 cm, Analtech) using hexane/EtOAc/NH4OH (79.6/20.0/0.4). The plate was exposed twice to the aforementioned solvent system to afford isomer 2A (15 mg) and a mixture of isomers 2B and 2C (33 mg). Additional preparative TLC of the isomer 2B and 2C mixture was carried out using a gradient of hexane/EtOAc/NH4OH (89.6/9.9/0.5 → 84.6/14.9/0.5) to afford isomer 2B (2.3 mg) and isomers 2B and 2C (16.6 mg).
Isomer 2A, Isolated.
1H NMR (400 MHz, CDCl3) δ 7.86–7.84 (m, 2H), 7.70–7.67 (m, 2H), 7.65–7.62 (m, 2H), 7.51–7.48 (m, 2H), 7.44–7.39 (m, 1H), 6.24 (d, J = 8 Hz, 1H), 4.64–4.59 (m, 1H), 2.08–2.03 (m, 1H), 1.99–1.95 (m, 1H), 1.85–1.81 (m, 1H), 1.54 (br s, 1H), 1.48–1.41 (m, 1H), 1.28 (s, 1H), 1.22–1.04 (m, 3H), 1.00–0.92 (m, 9H). LCMS RT = 9.26 min, m/z (M + H)+ = 336.2. TLC (SiO2) Rf 0.37 (8:2 hexane/diethyl ether + 2 drops NH4OH).
Isomer 2B, Isolated.
1H NMR (400 MHz, CDCl3) δ 7.85–7.82 (m, 2H), 7.67–7.60 (m, 4H), 7.49–7.36 (m, 3H), 5.79 (d, J = 8 Hz, 1H), 4.03 (qd, J = 10.9, 3.8 Hz, 1H), 2.16–2.12 (m, 1H), 2.05–1.85 (m, 1H), 1.79–1.70 (m, 1H), 1.25–1.15 (m, 5H), 1.04–1.00 (m, 1H), 0.95–0.85 (m, 9H). LCMS RT = 8.85 min, m/z (M + H)+ = 336.2. TLC (SiO2) Rf 0.31 (8:2 hexane/diethyl ether + 2 drops NH4OH).
Isomer 2C (Obtained as a 70:30 Ratio of 2B:2C).
1H NMR (400 MHz, CDCl3) δ 7.87–7.83 (m, 2H), 7.69–7.67 (m, 2H), 7.64–7.62 (m, 2H), 7.51–7.47 (m, 2H), 7.43–7.39 (m, 1H), 6.18 (2C, d, J = 8 Hz, 0.5H), 5.84 (2B, J = 12 Hz, 1H), 4.39 (2C, tt, J = 7.8, 3.9 Hz, 1H), 4.05 (2B, qd, J = 10.9, 3.8 Hz, 1H), 2.16–2.12 (m, 1H), 2.05–1.99 (m, 1H), 1.89 (br s, 1H), 1.81–1.61 (m, 4H), 1.57–1.54 (m, 1H), 1.48–1.15 (m, 4H), 1.06–0.98 (2C, m, 4H), 0.96–0.88 (2B, m, 9H). LCMS RT (Isomer B) = 8.93 min, m/z (M + H)+ = 336.2. LCMS RT (Isomer C) = 9.00 min, m/z (M+H)+ = 336.2. TLC (Isomer C) (SiO2) Rf 0.28 (8:2 hexane/diethyl ether +2 drops NH4OH).
N-Cyclohexyl-[1,1′-biphenyl]-4-carboxamide (3).
Prepared according to the general procedure using cyclohexylamine·HCl (342 mg, 2.52 mmol). The crude residue was purified via trituration using 30 mL hexane/ether (90/10), followed by trituration with 15 mL MeOH to afford 536 mg of the title material in 76% yield. 1H NMR (400 MHz, CDCl3) δ 7.86–7.79 (m, 2H), 7.68–7.57 (m, 4H), 7.51–7.42 (m, 2H), 7.42–7.34 (m, 1H), 6.00 (d, J = 8.1 Hz, 1H), 4.01 (dddd, J = 14.7, 10.7, 8.0, 4.0 Hz, 1H), 2.07–2.04 (m, 2H), 1.77 (dt, J = 13.7, 3.7 Hz, 2H), 1.67 (dt, J = 12.7, 3.7 Hz, 1H), 1.52–1.37 (m, 2H), 1.33–1.14 (m, 3H). MS (ESI) m/z 280.3 (M + H)+. Anal. Calcd For C19H21NO·0.3H2O: C, 80.13; H, 7.65; N, 4.92. Found: C, 80.15; H, 7.37; N, 4.99.
N-(Bicyclo[2.2.1]heptan-2-yl)-[1,1′-biphenyl]-4-carboxamide (4, Single Isomer).
Prepared according to the general procedure using bicyclo[2.2.1]heptan-2-amine·HCl (106 mg, 0.72 mmol). The crude residue was purified via automated flash chromatography to afford 80 mg of the title material in 38% yield. 1H NMR (400 MHz, CDCl3) δ 7.88–7.77 (m, 2H), 7.69–7.58 (m, 4H), 7.51–7.42 (m, 2H), 7.47–7.34 (m, 1H), 6.17 (d, J = 7.2 Hz, 1H), 4.35 (dddd, J = 11.3, 6.8, 4.4, 1.6 Hz, 1H), 2.59 (br s, 1H), 2.39–2.14 (m, 2H), 1.71–1.44 (m, 2H), 1.39 (ddt, J = 9.9, 3.2, 1.7 Hz, 1H), 1.34–1.23 (m, 1H), 0.86 (ddd, J = 12.9, 4.7, 3.0 Hz, 1H). MS (ESI) m/z 292.4 (M+H)+. Anal. Calcd For C20H21NO: C, 82.44; H, 7.26; N, 4.81. Found: C, 82.05; H, 7.10; N, 4.84.
N-Cycloheptyl-[1,1′-biphenyl]-4-Carboxamide (5).
Prepared according to the general procedure using cycloheptylamine (100 mg, 0.88 mmol). The crude residue was purified via automated flash chromatography to afford 200 mg of the title material in 77% yield. 1H NMR (400 MHz, CDCl3) δ 7.86–7.78 (m, 2H), 7.69–7.57 (m, 4H), 7.50–7.43 (m, 2H), 7.42–7.35 (m, 1H), 6.07 (d, J = 8.1 Hz, 1H), 4.18 (ddt, J = 12.8, 8.4, 3.6 Hz, 1H), 2.17–1.98 (m, 3H), 1.73–1.60 (m, 4H), 1.58–1.49 (m, 5H). MS (ESI) m/z 294.3 (M + H)+. Anal. Calcd For C20H23NO: C, 81.87; H, 7.90; N, 4.77. Found: C, 81.84; H, 7.76; N, 4.67.
N-(Bicyclo[3.2.1]octan-3-yl)-[1,1′-biphenyl]-4-carboxamide (6).
Prepared according to the general procedure using bicyclo[3.2.1]-octan-3-amine·HCl (90 mg, 0.55 mmol). The crude residue was purified via automated flash chromatography to afford 54 mg of the title material in 32% yield. 1H NMR (400 MHz, CDCl3) δ 7.84–7.77 (m, 2H), 7.70–7.56 (m, 4H), 7.51–7.42 (m, 2H), 7.42–7.34 (m, 1H), 5.86 (d, J = 8.4 Hz, 1H), 4.33 (tdt, J = 11.5, 8.4, 5.8 Hz, 1H), 2.31 (br s, 2H), 2.06–1.97 (m, 2H), 1.69 (br s, 4H), 1.54–1.44 (m, 1H), 1.40 (d, J = 11.2 Hz, 1H), 1.33–1.23 (m, 2H). MS (ESI) m/z 306.3 (M+H)+. Anal. Calcd For C21H23NO: C, 82.58; H, 7.59; N, 4.59. Found: C, 82.35; H, 7.68; N, 4.58.
N-Cyclooctyl-[1,1′-biphenyl]-4-carboxamide (7).
Prepared according to the general procedure using cyclooctylamine (500 mg, 3.93 mmol). The crude residue was purified via flash chromatography using hexane/EtOAc (95/5→ 30/70), followed by automated flash chromatography to afford 605 mg of the title material in 50% yield. 1H NMR (400 MHz, CDCl3) δ 7.86–7.78 (m, 2H), 7.68–7.56 (m, 4H), 7.51–7.42 (m, 2H), 7.42–7.34 (m, 1H), 6.08 (d, J = 8.1 Hz, 1H), 4.23 (ddq, J = 12.0, 8.0, 3.7 Hz, 1H), 2.01–1.89 (m, 2H), 1.78–1.60 (m, 12H). MS (ESI) m/z 308.3 (M+H)+. Anal. Calcd For C21H25NO: C, 82.04; H, 8.20; N, 4.56. Found: C, 81.93; H, 8.10; N, 4.58.
N-(Decahydronaphthalen-1-yl)-[1,1′-biphenyl]-4-carboxamide (8, Mixture of Three Isomers).
Prepared according to the general procedure using decahydronaphthalen-1-amine HCl (91 mg, 0.483 mmol). The crude residue was purified via trituration using 15 mL MeOH to afford 136 mg of the title material in 84% yield. 1H NMR (400 MHz, CDCl3) δ 7.87–7.79 (m, 6H), 7.72–7.57 (m, 12H), 7.51–7.42 (m, 6H), 7.42–7.35 (m, 3H), 6.05 (d, J = 8.5 Hz, 1H), 5.82 (d, J = 9.3 Hz, 2H), 4.15 (ddt, J = 11.8, 7.9, 4.0 Hz, 1H), 3.89–3.75 (m, 2H), 2.16–2.06 (m, 3H), 1.97–0.81 (m, 45H). MS (ESI) m/z 334.30 (M + H)+. Anal. Calcd For C23H27NO·0.3 H2O: C, 81.52; H, 8.21; N, 4.13. Found: C, 81.44; H, 7.94; N, 4.16.
N-(1,2,3,4-Tetrahydronaphthalen-1-yl)-[1,1′-biphenyl]-4-carbox-amide (9).
Prepared according to the general procedure using 1,2,3,4-tetrahydro-1-naphthylamine (250 mg, 1.70 mmol). The crude residue was purified via automated flash chromatography to afford 384 mg of the title material in 69% yield. 1H NMR (400 MHz, CDCl3) δ 7.90–7.82 (m, 2H), 7.69–7.57 (m, 4H), 7.51–7.43 (m, 2H), 7.42–7.34 (m, 2H), 7.24–7.12 (m, 3H), 6.37 (d, J = 8.4 Hz, 1H), 5.48–5.38 (m, 1H), 2.94–2.75 (m, 2H), 2.24–2.12 (m, 1H), 2.03–1.86 (m, 3H). MS (ESI) m/z 328.20 (M + H)+. Anal. Calcd For C23H21NO: C, 84.37; H, 6.46; N, 4.28. Found: C, 84.14; H, 6.37; N, 4.17.
N-(Decahydronaphthalen-2-yl)-[1,1′-biphenyl]-4-carboxamide (10, a Mixture of Two Isomers Present as a 86:13 ratio by LCMS) and N-((2SR,9RS,10SR)-Decahydronaphthalen-2-yl)-[1,1′-biphenyl]-4-carboxamide (12, Single Isomer).
Prepared according to the general procedure using decahydronaphthalen-2-amine (63 mg, 0.41 mmol). The crude residue was purified via automated flash chromatography to afford 10 (111 mg, 80%) and 12 (28 mg, 20%). 1H NMR (mixture) (400 MHz, CDCl3, 10) δ 7.87–7.78 (m, 3H), 7.69–7.57 (m, 6H), 7.50–7.42 (m, 3H), 7.42–7.35 (m, 1H), 6.00 (d, J = 8.1 Hz, 1H), 5.94 (d, J = 8.4 Hz, 0.31H), 4.20 (s, 0.26H), 4.00 (tdt, J = 11.9, 8.2, 4.2 Hz, 1H), 2.18–1.59 (m, 9H), 1.57–0.84 (m, 8H). MS (ESI) (mixture) m/z 334.3 (M + H)+. LCMS (mixture) RT (Peak 1 (major isomer) corresponds to 12) = 7.91 min, m/z (M + H)+ = 334.2; LCMS RT (Peak 2 (minor isomer)) = 8.70 min, m/z (M+H)+ = 334.2. Anal. Calcd For C23H27NO (mixture): C, 82.84; H, 8.16; N, 4.20. Found: C, 82.57; H, 7.97; N, 4.20. TLC (major isomer, corresponds to 12) (SiO2) Rf 0.54 (8:2 hexane/EtOAc). TLC (minor isomer) (SiO2) Rf 0.59 (8:2 hexane/EtOAc).
1H NMR (400 MHz, CDCl3, 12, single isomer) δ 7.87–7.79 (m, 2H), 7.70–7.57 (m, 4H), 7.51–7.43 (m, 2H), 7.42–7.35 (m, 1H), 6.00 (d, J = 8.1 Hz, 1H), 4.01 (tdt, J = 12.0, 8.4, 4.3 Hz, 1H), 1.96–1.58 (m, 8H), 1.56–1.20 (m, 8H). MS (ESI) (single isomer) m/z 334.3 (M + H)+. LCMS (single isomer) RT = 8.04 min, m/z (M + H)+ = 334.2. Anal. Calcd For C23H27NO (single isomer): C, 82.84; H, 8.16; N, 4.20. Found: C, 82.73; H, 8.13; N, 4.14. TLC (SiO2) Rf 0.54 (8:2 hexane/EtOAc).
1H, 13C, gCOSY, NOESY, gHSQCAD, gHMBCAD, gH2BC of 12.
All experiments were run on an Agilent DDR2 500 MHz spectrometer equipped with an OneNMR probe running at 499.90 and 125.71 MHz for 1H and 13C resonance frequencies, respectively. Experiments were run at ambient temperature using CDCl3 as the solvent. 1H spectra were referenced against residual protonated solvent (7.26 ppm) and 13C to 77 ppm. Spectra were processed using MestReNova (12.0.3) NMR processing program.
1H NMR.
Ten second relaxation delay, 8013 Hz spectral window, 2.04 s acquisition time, 8 scans. Baseline correction applied using Whittaker smoother (automatic detection).
13C NMR.
Broadband 1H-decoupled, 1 s relaxation delay, 512 scans, 31 250 Hz spectral window, 1.05 s acquisition time. Baseline correction applied using Whittaker smoother (automatic detection) and 0.5 Hz exponential multiplication was applied.
gCOSY.
One second relaxation delay, 4 scans per increment, 200 increments, 4882 Hz spectral window, 0.15 s acquisition time. F1 and F2 were multiplied by Sine Square II function (50%), as implemented by MestReNova, prior to Fourier transform. Linear prediction (Zhu-Bax) to 1024 points in F1 was applied. Baseline correction was applied to both dimensions using Whittaker smoother (automatic detection).
gH2BC.
One second relaxation delay, 8 scans per increment, 200 increments, 8013, 25 141 Hz spectral window (1H, and 13C, respectively), 0.15 s acquisition time. F1 was multiplied by a Gaussian and F2 by Sine Square II function (50%), as implemented by MestReNova, prior to Fourier transform. Baseline correction was applied to both dimensions using Whittaker smoother (automatic detection). Linear prediction (Zhu-Bax) to 2048 points in F1 was applied.
gHMBCAD.
One second relaxation delay, 8 scans per increment, 320 increments, 8013, 30 166 Hz spectral window (1H, and 13C, respectively), 0.15 s acquisition time. F2 was multiplied by a Gaussian and F1 by Sine Square II function (50%), as implemented by MestReNova, prior to Fourier transform. Baseline correction was applied to both dimensions using Whittaker smoother (automatic detection). Linear prediction (Zhu-Bax) to 2048 points in F1 was applied.
gHSQCAD.
One second relaxation delay, 4 scans per increment, 200 increments; 8013, 25141 Hz spectral window (1H, and 13C, respectively), 0.15 s acquisition time. F1 and F2 were multiplied by a Gaussian prior to Fourier transform. Baseline correction was applied to both dimensions using Whittaker smoother (automatic detection). Linear prediction (Zhu-Bax) to 2048 points in F1 was applied.
N-(1,2,3,4-Tetrahydronaphthalen-2-yl)-[1,1′-biphenyl]-4-carbox-amide (11).
Prepared according to the general procedure using 1,2,3,4-tetrahydronaphthalen-2-amine·HCl (197 mg, 1.34 mmol). The crude residue was purified via trituration with 15 mL MeOH to afford 328 mg of the title material in 75% yield. 1H NMR (400 MHz, CDCl3) δ 7.95–7.93 (m, 2H), 7.77–7.71 (m, 4H), 7.57–7.50 (m, 3H), 7.37 (s, 1H), 7.23–7.26 (m, 3H), 6.29 (d, J = 4 Hz, 1H), 4.65 (br s, 1H), 3.39 (d, J = 20 Hz, 1H), 3.07 (br s, 1H), 2.88–2.94 (m, 1H), 2.33–2.30 (m, 1H), 2.09–2.00 (m, 1H), 1.70 (s, 1H). MS (ESI) m/z 328.2 (M + H)+. Anal. Calcd For C23H21NO: C, 84.37; H, 6.46; N, 4.28. Found: C, 84.10; H, 6.42; N, 4.25.
N-(Spiro[5.5]undecan-3-yl)-[1,1′-biphenyl]-4-carboxamide (13).
Prepared according to the general procedure using spiro[5.5]-undecan-3-amine·HCl (90 mg, 0.44 mmol). The crude residue was purified via automated flash chromatography to afford 130 mg of the title material in 84% yield. 1H NMR (400 MHz, CDCl3) δ 7.86–7.79 (m, 2H), 7.69–7.57 (m, 4H), 7.51–7.43 (m, 2H), 7.42–7.35 (m, 1H), 6.02 (d, J = 8.1 Hz, 1H), 3.97 (dddd, J = 14.9, 10.7, 8.3, 4.2 Hz, 1H), 1.88 (ddd, J = 13.1, 6.2, 3.8 Hz, 3H), 1.67 (d, J = 13.6 Hz, 3H), 1.42 (br s, 8H), 1.25 (td, J = 12.7, 4.4 Hz, 4H). MS (ESI) m/z 348.4 (M + H)+. Anal. Calcd For C24H29NO: C, 82.95; H, 8.41; N, 4.03. Found: C, 82.99; H, 8.42; N, 3.99.
N-(Spiro[4.5]decan-8-yl)-[1,1′-biphenyl]-4-carboxamide (14).
Prepared according to the general procedure using spiro[4.5]decan-8-amine·HCl (91 mg, 0.48 mmol). The crude residue was purified via automated flash chromatography to afford 110 mg of the title material in 68% yield. 1H NMR (400 MHz, CDCl3) δ 7.87–7.79 (m, 2H), 7.69–7.57 (m, 4H), 7.51–7.43 (m, 2H), 7.42–7.35 (m, 1H), 6.01 (d, J = 8.1 Hz, 1H), 4.05–3.93 (m, 1H), 2.00–1.91 (m, 2H), 1.67–1.60 (m, 3H), 1.58–1.31 (m, 11H). MS (ESI) m/z 334.3 (M + H)+. Anal. Calcd For C23H27NO: C, 82.84; H, 8.16; N, 4.20. Found: C, 82.57; H, 8.16; N, 4.22.
N-(Adamantan-1-yl)-[1,1′-biphenyl]-4-carboxamide (15).
Prepared according to the general procedure using 1-adamantanamine· HCl (100 mg, 0.54 mmol). The crude residue was purified via automated flash chromatography to afford 110 mg of the title material in 62% yield. 1H NMR (400 MHz, CDCl3) δ 7.83–7.75 (m, 2H), 7.67–7.57 (m, 4H), 7.51–7.42 (m, 2H), 7.42–7.34 (m, 1H), 5.84 (s, 1H), 2.15 (br s, 9H), 1.73 (br s, 6H). MS (ESI) m/z 332.3 (M + H)+. Anal. Calcd For C23H25NO·0.1H2O: C, 82.89; H, 7.62; N, 4.20. Found: C, 82.87; H, 7.47; N, 4.18.
In Vitro Pharmacology.
Evaluation of TRPM8 Agonist and Antagonist Activity Using Ca2+ Imaging.
Fura-2 based Ca2+-imaging was used to evaluate the effects of the synthesized compounds on icilin-evoked Ca2+ signals. HEK-293 cells stably expressing human TRPM8 (a kind gift from Professor Thomas Voets, Laboratory of Ion Channels Research, KU Leuven, Belgium) were first loaded with Fura-2 AM for 45 min and then another 40 min was allowed for de-esterification of the dye. The cells were then bathed in Ca2+-free HBSS buffer and incubated with a test compound for 5 min, and then icilin (500 nM, from Tocris), the well-known TRPM8 agonist, was added to the bath solution. At 1 min after icilin addition, 2 mM Ca2+ was added to the bath solution.
Ratio fluorescence images were captured using a QIClick digital CCD camera (QImaging, BC, Canada) mounted on a Nikon Eclipse Ti–S Microscope. Consecutive excitation was provided by a Dual OptoLED Power Supply (Cairn), alternating between both 355 nm (F355) and 380 nm (F380) wavelength LEDs. Emission fluorescence was collected at 510 nm (470 nm–550 nm). 12-bit images were acquired in every 5 seconds with MetaFluor (Molecular Devices, USA). The fluorescence at each time-point was extracted for both 355 and 380 nm wavelengths, corrected for autofluorescence and the 355 nm/380 nm ratios (F355/F380) were then calculated to represent Ca2+ influx. All Ca2+ imaging experiments were done in room (22 °C) temperature. For comparison, peak Ca2+ response for each compound was normalized to that of icilin (500 nM). RQ-00203078 was purchased from Tocris.
For obtaining the concentration–response profile of icilin, Fura-2 loaded HEK-293 cells stably expressing human TRPM8 in different Petri dishes were bathed in Ca2+ free HBSS and were first incubated with icilin in graded concentrations (10−9 to 10−6 M). One min after icilin addition, 2 mM Ca2+ was added to the bath solution and the resultant Ca2+ influx (as Fura-2 fluorescence ratios) were recorded. EC50 was obtained from the corresponding concentration–response curve.
To determine the antagonist activity of our designed compounds, cells bathed in Ca2+-free Hank’s balanced salt solution (Ca2+ free HBSS) were first incubated for 3 min with different concentrations (1 nM, 3 nM, 10 nM, 30 nM, 100 nM, 300 nM, 1 μM, 3 μM, 10 μM, 30 μM, and 100 μM) of analogues 2–15. Then 500 nM icilin was added and after another 1 min, Ca2+-free HBSS was replaced by HBSS containing 2 mM free Ca2+ and the resultant Ca2+ signal was measured. The TRPM8 antagonist RQ-00203078 was used as a positive control and for comparison purposes,43 due to its reported high nanomolar potency (<10 nM) at hTRPM8 vs other antagonists in the literature. Analogues with IC50 values ≤ 100 nM were tested alone at a single concentration of 10 μM for their intrinsic (agonist) ability to induce Ca2+ influx, and compared to the effect of 500 nM icilin.
TRPA1 and TRPV1 Selectivity Assays.
The Ca2+ flux assay utilized Fluo-8 Ca2+ dye and fluorescence were measured at excitation wavelength of 470–495 nm and emission wavelength of 515–575 nm using FLIPRTETRA plate reader (Molecular Devices). A total of eight compounds were tested for both agonist (intrinsic) and antagonist activity at concentrations corresponding to approximately IC90 against TRPM8 and 10× IC90 to demonstrate a 10-fold selectivity. Recombinant HEK293 cell lines stably expressing human TRPA1 (Eurofins cat #CYL3066) and human TRPV1 (Eurofins cat #CYL3063) were used for the study. Cells were cultured and maintained in DMEM-F12 medium supplemented with 10% FBS, 1% nonessential amino acids, and 400 μg/mL geneticin. Allyl isothiocyanate (AITC, Millipore Sigma) and capsaicin (Tocris) were used as reference agonists, whereas ruthenium red (Tocris) and capsazepine (Millipore Sigma) were reference antagonists against TRPA1 and TRPV1, respectively. For the experiments, cells were plated in flat-bottom collagen-coated 384 well plates and incubated at 37 °C 5% CO2. After 24 h of incubation, the media was aspirated and 40 μL of dye loading buffer (modified Hanks Balanced Salt Solution (HBSS) where HBSS was supplemented to contain 20 mM HEPES and 2.5 mM Probenecid at pH 7.4) containing Fluo-8 Ca2+ dye at 5 μg/mL was added to the cells in each well and incubated at 30 °C (5% CO2) in a humidified chamber for 80 min prior to starting the FLIPR Tetra protocol. Test compounds were initially dissolved in DMSO to make 300× the final assay concentrations, and then diluted in assay buffer (modified Hanks Balanced Salt Solution (HBSS) where HBSS was supplemented to contain 20 mM HEPES at pH 7.4) to the final dilutions. For the agonist assay, test compound(s), vehicle controls, and reference agonist (at Emax = 300 μM (AITC) and 3 μM (capsaicin)) were added to the assay plate for 180 s after a fluorescence baseline was established. For the antagonist assay, cells were preincubated with the test compounds or positive controls (known antagonists) for 5 min at room temperature and then challenged with EC80 concentration of reference agonists (AITC EC80 = 10 μM, capsaicin EC80 = 0.1 μM) for 180 s after establishment of a fluorescence baseline.
Agonist: 25% activation when normalized to Emax.
Antagonist: 25% inhibition when normalized to EC80.
Whole Cell Patch Clamp Electrophysiology.
HEK-293 Cell Culture.
Human embryonic kidney (HEK) 293 cells (ATCC CRL-1573) were cultured in growth medium comprising 90% Dulbecco’s modified Eagle’s medium, 10% fetal bovine serum, 100 U·mL−1 penicillin-streptomycin, and 2 mM l-glutamine (Gibco). Cells were cultured in 35 mm polystyrene dishes (Falcon) at 37 °C in the presence of 5% CO2. Cells were transiently transfected with human TRPM8 in a pIRES2 plasmid containing EGFP as a reporter. This plasmid expresses bicistronic mRNA with an internal ribosome entry site (IRES) positioned between the TRPM8 gene and the fluorescent protein reporter gene such that the reporter is not covalently fused to TRPM8. Transient transfection was performed using Fugene 6 transfection reagent (Promega) and 0.5 μg of plasmid in a 35 mm dish (Falcon) at a ratio of 3 μL transfection reagent per μg of plasmid according to manufacturer protocol. HEK-293 cells were authenticated by polymorphic genetic marker testing (DNA Diagnostics Center Medical).
Whole-Cell Patch-Clamp Electrophysiology.
At 48 h following transfection, cells were released from culture dishes by brief exposure to 0.25% trypsin/EDTA (Gibco), resuspended in supplemented DulDMEM, plated on glass coverslips and allowed to recover for 1–2 h at 37 °C in 5% CO2. Cells that exhibited green fluorescence, indicating successful transfection, were selected for electrophysiology measurements. Cells were placed in a chamber with extracellular solution containing (in mM) NaCl 132, KCl 4.8, MgCl2 1.2, CaCl2 2, HEPES 10, and glucose 5, with the pH adjusted to 7.4 using NaOH and the osmolality adjusted to 310 mOsm using sucrose. Pipets were filled with a solution containing (in mM) K+ gluconate 135, KCl 5, MgCl2 1, EGTA 5, and HEPES 10; pH was adjusted to 7.2 with KOH, and osmolality was adjusted to 300 mOsm using sucrose. Chemicals were obtained from Sigma-Aldrich. Osmolality was measured using a Vapro 5600 vapor pressure osmometer (Wescor). Whole-cell voltage-clamp current measurements were performed using an Axopatch 200B amplifier (Axon Instruments) and pClamp 10.3 software (Axon Instruments). Data was acquired at 2 kHz and filtered at 1 kHz. Patch pipettes were pulled using a P-2000 laser puller (Sutter Instruments) from borosilicate glass capillaries (World Precision Instruments) and heat-polished using a MF-830 microforge (Narishige). Pipettes had resistances of 2–5 MΩ as measured in the extracellular solution. A reference electrode was placed in a 2% agar bridge made with a composition similar to the extracellular solution, without glucose added. Experiments were performed at 23 ± 1 °C. Menthol and 14 stock solutions were prepared in DMSO at 1 M and 3.6 mM, respectively. A series of perfusion solutions were prepared by diluting menthol to 500 μM menthol and 14 to varying concentrations (from 1 to 1000 nM) in extracellular solution. A perfusion capillary was placed close to the cell and solution was perfused throughout the recording. Current response from each cell was measured under each concentration of 14. Only one cell was measured per coverslip to prevent confounding effects of previous exposure to menthol and 14.
Data Analysis.
For each cell, current magnitude at each concentration of 14 was normalized by dividing by the maximum current measured at 500 μM menthol and without the antagonist. Normalized currents from six cells were averaged at each concentration of antagonist, and the standard error of the mean was calculated at each point. The data were plotted in SigmaPlot and fit to a single site competition model using the equation =, where I is the current intensity and [antagonist] is the concentration of 14 at a particular I value.
Hepatic Metabolic Stability.
Stability in Liver Microsomes.
Male CD-1 mouse liver microsomes (lot#1710069) were purchased from XenoTech. The reaction mixture, minus NADPH, was prepared as described below. Analogue 14 was added into the reaction mixture at a final concentration of 1 μM. The control compound, testosterone, was run simultaneously with 14 in a separate reaction. An aliquot of the reaction mixture (without cofactor) was equilibrated in a shaking water bath at 37 °C for 3 min. The reaction was initiated by the addition of the cofactor, and the mixture was incubated in a shaking water bath at 37 °C. Aliquots (100 μL) were withdrawn at 0, 10, 20, 30, and 60 min. 14 and testosterone samples were immediately combined with 400 μL of ice-cold 50/50 acetonitrile (ACN)/H2O containing 0.1% formic acid and internal standard to terminate the reaction. The samples were then mixed and centrifuged to precipitate proteins. All samples were assayed by LC-MS/MS using electrospray ionization. Analytical conditions are compared to the PARR at time 0 to determine the percent remaining at each time point. Half-lives and intrinsic clearance were calculated using GraphPad software, fitting to a single-phase exponential decay equation (Table 3).
Table 3.
Hepatic Metabolic Stability Parameters
| reaction composition | |
|---|---|
| liver microsomes | 0.5 mg/mL |
| NADPH (cofactor) | 1 mM |
| potassium phosphate, pH 7.4 | 100 mM |
| magnesium chloride | 5 mM |
| analogue 14 | 1 μM |
| testosterone control | |||
|---|---|---|---|
| control compd | half-life (min) | CLint (mL/min/mg protein) | acceptable range (t1/2, min) |
| testosterone | 3.7 | 0.372 | ≤15 |
| analytical method | ||||
|---|---|---|---|---|
| liquid chromatography | ||||
| column | Thermo BDS Hypersil C18 30 × 2.1 mm, 3 μm, with guard column | |||
| M.P. buffer | 25 mM ammonium formate buffer, pH 3.5 | |||
| aqueous reservoir (A) | 90% water, 10% buffer | |||
| organic reservoir (B) | 90% acetonitrile, 10% buffer | |||
| flow rate | 350 μL/min | |||
| gradient program | time (min) | %A | %B | |
| 0.0 | 100 | 0 | ||
| 1.0 | 0 | 100 | ||
| 1.5 | 0 | 100 | ||
| 1.6 | 100 | 0 | ||
| 2.5 | 100 | 0 | ||
| total run time | 2.5 min | |||
| autosampler | 10 μL injection volume | |||
| autosampler wash | water/methanol/2-propanol: 1/1/1; with 0.2% formic acid | |||
| mass spectrometer | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| instrument | PE SCIEX API 4000 | ||||||||
| interface | turbo ionspray | ||||||||
| mode | multiple reaction monitoring | ||||||||
| method | 2.5 min duration | ||||||||
| settings | test article | ± | Q1 | Q3 | DP | EP | CE | CXP | IS |
| analogue 14 | + | 334.3 | 198.1 | 83 | 10 | 30 | 11 | 5500 | |
| TEM: 500 | CAD: 7 | CUR: 20 | GS1: 20 | GS2: 3 | |||||
In Vivo Assays.
C57-mice (≈30g) (Harlam, Holland) were used for the study. All experiments were approved by the Institutional Animal and Ethical Committee of the Universidad Miguel Hernandez where experiments were conducted and they were in accordance with the guidelines of the Economic European Community and the Committee for Research and Ethical Issues of the International Association for the Study of Pain. All parts of the study concerning animal care were performed under the control of veterinarians.
Wet Dog Shakes Assay.
Icilin, a TRPM8 agonist, was dissolved in 20% DMSO and 1% Tween 80 in distilled water and injected intraperitoneally (i.p.) in a volume of 10 mg/kg. Each animal was acclimatized for 30 min for two consecutive days before icilin administration. The compound 14 stock was prepared in DMSO and diluted in saline for injections. Gabapentin was dissolved in saline and administered s.c. at the dose of 25 mg/kg 60 min prior to icilin injection. Control animals received the vehicle injection.
Cold Allodynia.
Oxaliplatin (Tocris) was dissolved in water with gentle warming and was subcutaneously (s.c.) injected on days 1, 3, and 5 at a 6 mg/kg dose. The day 7 after administration, experiments were performed. Together with Oxaliplatin injection, saline and a 5% mannitol solution were intraperitoneally injected to prevent kidney damage and dehydration. The compounds 14 stock was prepared in DMSO (Sigma-Aldrich) and diluted in saline for injections. Compounds at different doses (0.1–1 μg) was injected into the plantar surface (25 μL) of the right hind paw of mice. Cold chemical thermal sensitivity was assessed using acetone drop method. Mice were placed in a metal mesh cage and allowed to habituate for approximately 30 min in order to acclimatize them. A freshly dispensed acetone drop (10 μL) was applied gently on to the midplantar surface of the hind paw. Cold chemical sensitive reaction with respect to paw licking was recorded as a positive response (nociceptive pain response). The responses were measured for 20 s with a digital stopwatch. For each measurement, the paw was sampled twice and the mean was calculated. The interval between each application of acetone was approximately 5 min.
Supplementary Material
Funding
Research reported in this publication was supported by the National Institutes of Health NIDA P30DA035778 (X.-Q.X.) and NIGMS R01GM112077 (W.D.V.H.); Marshall University School of Pharmacy intramural grant (V.B.J.); Cambridge Trust (S.R.); (T.R.); and Spain Ministry of Economy and Competitiveness SAF2015-66275-C02-01 (A.F.-M.).
ABBREVIATIONS USED
- gCOSY
gradient correlation spectroscopy
- NOESY
nuclear Overhauser effect spectroscopy
- gHSQCAD
gradient-enhanced heteronuclear single quantum coherence with adiabatic pulses
- gHMBCAD
gradient-enhanced heteronuclear multiple bond correlation with adiabatic pulses
- gH2BC
heteronuclear 2-bond correlation
- Q1
quadrupole 1
- Q3
quadrupole 3
- DP
declustering potential
- EP
entrance potential
- CE
collision energy
- CXP
collision cell exit potential
- IS
ionspray voltage
- TEM
temperature
- CAD
collisionally activated dissociation
- CUR
curtain gas
- GS1
gas 1
- GS2
gas 2
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.9b00404.
Sequence alignment; concentration–response curves for icilin, (−)-menthol, RQ-00203078, analogue 14, AITC, ruthenium red, capsaicin, capsazepine; Ca2+ imaging data for analogs at human TRPM8; liver microsome data; LCMS and 1H NMR spectra of analogues 2, 2A, 2B, 2C, 10, and 12; 2D NMR spectra of analogue 12; human TRPM8 model coordinates; Human TRPM8 Homology Model PDB-formatted coordinates for our computational model (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Winchester WJ, Gore K, Glatt S, Petit W, Gardiner JC, Conlon K, Postlethwaite M, Saintot PP, Roberts S, Gosset JR, Matsuura T, Andrews MD, Glossop PA, Palmer MJ, Clear N, Collins S, Beaumont K, and Reynolds DS (2014) Inhibition of TRPM8 channels reduces pain in the cold pressor test in humans. J. Pharmacol. Exp. Ther 351, 259–269. [DOI] [PubMed] [Google Scholar]
- (2).Kobayashi K, Fukuoka T, Obata K, Yamanaka H, Dai Y, Tokunaga A, and Noguchi K (2005) Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J. Comp. Neurol 493, 596–606. [DOI] [PubMed] [Google Scholar]
- (3).Proudfoot CJ, Garry EM, Cottrell DF, Rosie R, Anderson H, Robertson DC, Fleetwood-Walker SM, and Mitchell R (2006) Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain. Curr. Biol 16, 1591–1605. [DOI] [PubMed] [Google Scholar]
- (4).McKemy DD, Neuhausser WM, and Julius D (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58. [DOI] [PubMed] [Google Scholar]
- (5).Janssens A, and Voets T (2011) Ligand stoichiometry of the cold- and menthol-activated channel TRPM8. J. Physiol 589, 4827–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Perez de Vega MJ, Gomez-Monterrey I, Ferrer-Montiel A, and Gonzalez-Muniz R (2016) Transient Receptor Potential Melastatin 8 Channel (TRPM8) Modulation: Cool Entryway for Treating Pain and Cancer. J. Med. Chem 59, 10006–10029. [DOI] [PubMed] [Google Scholar]
- (7).Yin Y, Wu M, Zubcevic L, Borschel WF, Lander GC, and Lee SY (2018) Structure of the cold- and menthol-sensing ion channel TRPM8. Science 359, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yin Y, Le SC, Hsu AL, Borgnia MJ, Yang H, and Lee SY (2019) Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 363, No. eaav9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Andrews MD, Af Forselles K, Beaumont K, Galan SR, Glossop PA, Grenie M, Jessiman A, Kenyon AS, Lunn G, Maw G, Owen RM, Pryde DC, Roberts D, and Tran TD (2015) Discovery of a Selective TRPM8 Antagonist with Clinical Efficacy in Cold-Related Pain. ACS Med. Chem. Lett 6, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Horne DB, Tamayo NA, Bartberger MD, Bo Y, Clarine J, Davis CD, Gore VK, Kaller MR, Lehto SG, Ma VV, Nishimura N, Nguyen TT, Tang P, Wang W, Youngblood BD, Zhang M, Gavva NR, Monenschein H, and Norman MH (2014) Optimization of potency and pharmacokinetic properties of tetrahydroisoquinoline transient receptor potential melastatin 8 (TRPM8) antagonists. J. Med. Chem 57, 2989–3004. [DOI] [PubMed] [Google Scholar]
- (11).Gavva NR, Davis C, Lehto SG, Rao S, Wang W, and Zhu DX (2012) Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol. Pain 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lashinger ES, Steiginga MS, Hieble JP, Leon LA, Gardner SD, Nagilla R, Davenport EA, Hoffman BE, Laping NJ, and Su X (2008) AMTB, a TRPM8 channel blocker: evidence in rats for activity in overactive bladder and painful bladder syndrome. Am. J. Physiol. Renal Physiol 295, F803–810. [DOI] [PubMed] [Google Scholar]
- (13).Horne DB, Biswas K, Brown J, Bartberger MD, Clarine J, Davis CD, Gore VK, Harried S, Horner M, Kaller MR, Lehto SG, Liu Q, Ma VV, Monenschein H, Nguyen TT, Yuan C, Youngblood BD, Zhang M, Zhong W, Allen JR, Chen JJ, and Gavva N (2018) Discovery of TRPM8 Antagonist (S)-6-(((3-fluoro-4-(trifluoromethoxy)phenyl)(3-fluoropyridin-2-yl)methyl)carbamo yl)nicotinic acid (AMG 333), a Clinical Candidate for the Treatment of Migraine. J. Med. Chem 61, 8186–8201. [DOI] [PubMed] [Google Scholar]
- (14).De Caro C, Russo R, Avagliano C, Cristiano C, Calignano A, Aramini A, Bianchini G, Allegretti M, and Brandolini L (2018) Antinociceptive effect of two novel transient receptor potential melastatin 8 antagonists in acute and chronic pain models in rat. Br. J. Pharmacol 175, 1691–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Almeida MC, Hew-Butler T, Soriano RN, Rao S, Wang W, Wang J, Tamayo N, Oliveira DL, Nucci TB, Aryal P, Garami A, Bautista D, Gavva NR, and Romanovsky AA (2012) Pharmacological blockade of the cold receptor TRPM8 attenuates autonomic and behavioral cold defenses and decreases deep body temperature. J. Neurosci 32, 2086–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Okamoto Y, Ohkubo T, Ikebe T, and Yamazaki J (2012) Blockade of TRPM8 activity reduces the invasion potential of oral squamous carcinoma cell lines. Int. J. Oncol 40, 1431–1440. [DOI] [PubMed] [Google Scholar]
- (17).Willis DN, Liu B, Ha MA, Jordt SE, and Morris JB (2011) Menthol attenuates respiratory irritation responses to multiple cigarette smoke irritants. FASEB J 25, 4434–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lin AH, Liu MH, Ko HB, Perng DW, Lee TS, and Kou YR (2017) Inflammatory Effects of Menthol vs. Non-menthol Cigarette Smoke Extract on Human Lung Epithelial Cells: A Double-Hit on TRPM8 by Reactive Oxygen Species and Menthol. Front. Physiol 8, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Patel R, Goncalves L, Leveridge M, Mack SR, Hendrick A, Brice NL, and Dickenson AH (2014) Anti-hyperalgesic effects of a novel TRPM8 agonist in neuropathic rats: a comparison with topical menthol. Pain 155, 2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Voets T, Owsianik G, Janssens A, Talavera K, and Nilius B (2007) TRPM8 voltage sensor mutants reveal a mechanism for integrating thermal and chemical stimuli. Nat. Chem. Biol 3, 174–182. [DOI] [PubMed] [Google Scholar]
- (21).Bandell M, Dubin AE, Petrus MJ, Orth A, Mathur J, Hwang SW, and Patapoutian A (2006) High-throughput random mutagenesis screen reveals TRPM8 residues specifically required for activation by menthol. Nat. Neurosci 9, 493–500. [DOI] [PubMed] [Google Scholar]
- (22).Rath P, Hilton JK, Sisco NJ, and Van Horn WD (2016) Implications of Human Transient Receptor Potential Melastatin 8 (TRPM8) Channel Gating from Menthol Binding Studies of the Sensing Domain. Biochemistry 55, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chuang HH, Neuhausser WM, and Julius D (2004) The super-cooling agent icilin reveals a mechanism of coincidence detection by a temperature-sensitive TRP channel. Neuron 43, 859–869. [DOI] [PubMed] [Google Scholar]
- (24).Autzen HE, Myasnikov AG, Campbell MG, Asarnow D, Julius D, and Cheng Y (2018) Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 359, 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Matos-Cruz V, Schneider ER, Mastrotto M, Merriman DK, Bagriantsev SN, and Gracheva EO (2017) Molecular Prerequisites for Diminished Cold Sensitivity in Ground Squirrels and Hamsters. Cell Rep 21, 3329–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ortar G, De Petrocellis L, Morera L, Moriello AS, Orlando P, Morera E, Nalli M, and Di Marzo V (2010) (−)-Menthylamine derivatives as potent and selective antagonists of transient receptor potential melastatin type-8 (TRPM8) channels. Bioorg. Med. Chem. Lett 20, 2729–2732. [DOI] [PubMed] [Google Scholar]
- (27).Lampe T, Alonso-Alija C, Beck H, Rosentreter U, Sandner P, Stahl E, and Stelte-Ludwig B (2007) Preparation of substituted 2-benzyloxy-benzoic acid amide derivatives as Cold Menthol Receptor 1 (CMR-1) modulators for treating and preventing urological diseases or disorders. Patent WO 2007017093 A1. [Google Scholar]
- (28).Gavva NR, Klionsky L, Qu Y, Shi L, Tamir R, Edenson S, Zhang TJ, Viswanadhan VN, Toth A, Pearce LV, Vanderah TW, Porreca F, Blumberg PM, Lile J, Sun Y, Wild K, Louis JC, and Treanor JJ (2004) Molecular determinants of vanilloid sensitivity in TRPV1. J. Biol. Chem 279, 20283–20295. [DOI] [PubMed] [Google Scholar]
- (29).Behrendt HJ, Germann T, Gillen C, Hatt H, and Jostock R (2004) Characterization of the mouse cold-menthol receptor TRPM8 and vanilloid receptor type-1 VR1 using a fluorometric imaging plate reader (FLIPR) assay. Br. J. Pharmacol 141, 737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rami HK, Thompson M, Wyman P, Jerman JC, Egerton J, Brough S, Stevens AJ, Randall AD, Smart D, Gunthorpe MJ, and Davis JB (2004) Discovery of small molecule antagonists of TRPV1. Bioorg. Med. Chem. Lett 14, 3631–3634. [DOI] [PubMed] [Google Scholar]
- (31).Weil A, Moore SE, Waite NJ, Randall A, and Gunthorpe MJ (2005) Conservation of functional and pharmacological properties in the distantly related temperature sensors TRVP1 and TRPM8. Mol. Pharmacol 68, 518–527. [DOI] [PubMed] [Google Scholar]
- (32).Knowlton WM, Daniels RL, Palkar R, McCoy DD, and McKemy DD (2011) Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS One 6, No. e25894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Andersson DA, Chase HW, and Bevan S (2004) TRPM8 activation by menthol, icilin, and cold is differentially modulated by intracellular pH. J. Neurosci 24, 5364–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Andersson DA, Nash M, and Bevan S (2007) Modulation of the cold-activated channel TRPM8 by lysophospholipids and polyunsaturated fatty acids. J. Neurosci 27, 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hilton JK, Salehpour T, Sisco NJ, Rath P, and Van Horn WD (2018) Phosphoinositide-interacting regulator of TRP (PIRT) has opposing effects on human and mouse TRPM8 ion channels. J. Biol. Chem 293, 9423–9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bertamino A, Ostacolo C, Ambrosino P, Musella S, Di Sarno V, Ciaglia T, Soldovieri MV, Iraci N, Fernandez Carvajal A, de la Torre-Martinez R, Ferrer-Montiel A, Gonzalez Muniz R, Novellino E, Taglialatela M, Campiglia P, and Gomez-Monterrey I (2016) Tryptamine-Based Derivatives as Transient Receptor Potential Melastatin Type 8 (TRPM8) Channel Modulators. J. Med. Chem 59, 2179–2191. [DOI] [PubMed] [Google Scholar]
- (37).de la Torre-Martinez R, Bonache MA, Llabres-Campaner PJ, Balsera B, Fernandez-Carvajal A, Fernandez-Ballester G, Ferrer-Montiel A, Perez de Vega MJ, and Gonzalez-Muniz R (2017) Synthesis, high-throughput screening and pharmacological characterization of beta-lactam derivatives as TRPM8 antagonists. Sci. Rep 7, 10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bertamino A, Iraci N, Ostacolo C, Ambrosino P, Musella S, Di Sarno V, Ciaglia T, Pepe G, Sala M, Soldovieri MV, Mosca I, Gonzalez-Rodriguez S, Fernandez-Carvajal A, Ferrer-Montiel A, Novellino E, Taglialatela M, Campiglia P, and Gomez-Monterrey I (2018) Identification of a Potent Tryptophan-Based TRPM8 Antagonist With in Vivo Analgesic Activity. J. Med. Chem 61, 6140–6152. [DOI] [PubMed] [Google Scholar]
- (39).Wang L, Fu TM, Zhou Y, Xia S, Greka A, and Wu H (2018) Structures and gating mechanism of human TRPM2. Science 362, No. eaav4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).De Petrocellis L, Ligresti A, Schiano Moriello A, Iappelli M, Verde R, Stott CG, Cristino L, Orlando P, and Di Marzo V (2013) Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol 168, 79–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ha MA, Smith GJ, Cichocki JA, Fan L, Liu YS, Caceres AI, Jordt SE, and Morris JB (2015) Menthol attenuates respiratory irritation and elevates blood cotinine in cigarette smoke exposed mice. PLoS One 10, No. e0117128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tamayo NA, Bo Y, Gore V, Ma V, Nishimura N, Tang P, Deng H, Klionsky L, Lehto SG, Wang W, Youngblood B, Chen J, Correll TL, Bartberger MD, Gavva NR, and Norman MH (2012) Fused piperidines as a novel class of potent and orally available transient receptor potential melastatin type 8 (TRPM8) antagonists. J. Med. Chem 55, 1593–1611. [DOI] [PubMed] [Google Scholar]
- (43).Ohmi M, Shishido Y, Inoue T, Ando K, Fujiuchi A, Yamada A, Watanabe S, and Kawamura K (2014) Identification of a novel 2-pyridyl-benzensulfonamide derivative, RQ-00203078, as a selective and orally active TRPM8 antagonist. Bioorg. Med. Chem. Lett 24, 5364–5368. [DOI] [PubMed] [Google Scholar]
- (44).Caceres AI, Liu B, Jabba SV, Achanta S, Morris JB, and Jordt SE (2017) Transient Receptor Potential Cation Channel Subfamily M Member 8 channels mediate the anti-inflammatory effects of eucalyptol. Br. J. Pharmacol 174, 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dodziuk H, Jaszunski M, and Schilf W (2005) 1H and 13C NMR chemical shifts and spin-spin coupling constants in trans- and cisdecalins. Magn. Reson. Chem 43, 639–646. [DOI] [PubMed] [Google Scholar]
- (46).Browne LM, Klinck RE, and Stothers JB (1979) Carbon-13 NMR Studies: 82.† Carbon-13 spectra of several 10-methyl-cisdecalins. Conformational preferences of some cis-decalin derivatives. Org. Magn. Reson 12, 561–568. [Google Scholar]
- (47).Player MR, Parks DJ, Parsons W, Meegalla SK, Illig CR, and Ballentine SK (2010) Heterocyclic benzimidazoles as TRPM8 modulators. Patent US 20100093788 A1. [Google Scholar]
- (48).Chaudhari SS, Kadam AB, Khairatkar-Joshi N, Mukhopadhyay I, Karnik PV, Raghuram A, Rao SS, Vaiyapuri TS, Wale DP, Bhosale VM, Gudi GS, Sangana RR, and Thomas A (2013) Synthesis and pharmacological evaluation of novel N-aryl-3,4-dihydro-1’H-spiro[chromene-2,4’-piperidine]-1’-carboxamides as TRPM8 antagonists. Bioorg. Med. Chem 21, 6542–6553. [DOI] [PubMed] [Google Scholar]
- (49).Shishido Y, Ohmi M, and Ando K (2015) Preparation of azaspiro derivatives as TRPM8 antagonists. Patent WO 2015136947 A1. [Google Scholar]
- (50).Klein AH, Iodi Carstens M, McCluskey TS, Blancher G, Simons CT, Slack JP, Furrer S, and Carstens E (2011) Novel menthol-derived cooling compounds activate primary and second-order trigeminal sensory neurons and modulate lingual thermosensitivity. Chem. Senses 36, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bodding M, Wissenbach U, and Flockerzi V (2007) Characterisation of TRPM8 as a pharmacophore receptor. Cell Calcium 42, 618–628. [DOI] [PubMed] [Google Scholar]
- (52).Zhu B, Xia M, Xu X, Ludovici DW, Tennakoon M, Youngman MA, Matthews JM, Dax SL, Colburn RW, Qin N, Hutchinson TL, Lubin ML, Brandt MR, Stone DJ, Flores CM, and Macielag MJ (2013) Arylglycine derivatives as potent transient receptor potential melastatin 8 (TRPM8) antagonists. Bioorg. Med. Chem. Lett 23, 2234–2237. [DOI] [PubMed] [Google Scholar]
- (53).Calvo RR, Meegalla SK, Parks DJ, Parsons WH, Ballentine SK, Lubin ML, Schneider C, Colburn RW, Flores CM, and Player MR (2012) Discovery of vinylcycloalkyl-substituted benzimidazole TRPM8 antagonists effective in the treatment of cold allodynia. Bioorg. Med. Chem. Lett 22, 1903–1907. [DOI] [PubMed] [Google Scholar]
- (54).Patel R, Goncalves L, Newman R, Jiang FL, Goldby A, Reeve J, Hendrick A, Teall M, Hannah D, Almond S, Brice N, and Dickenson AH (2014) Novel TRPM8 antagonist attenuates cold hypersensitivity after peripheral nerve injury in rats. J. Pharmacol. Exp. Ther 349, 47–55. [DOI] [PubMed] [Google Scholar]
- (55).Takaishi M, Uchida K, Suzuki Y, Matsui H, Shimada T, Fujita F, and Tominaga M (2016) Reciprocal effects of capsaicin and menthol on thermosensation through regulated activities of TRPV1 and TRPM8. J. Physiol. Sci 66, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Macpherson LJ, Hwang SW, Miyamoto T, Dubin AE, Patapoutian A, and Story GM (2006) More than cool: promiscuous relationships of menthol and other sensory compounds. Mol. Cell. Neurosci 32, 335–343. [DOI] [PubMed] [Google Scholar]
- (57).Karashima Y, Damann N, Prenen J, Talavera K, Segal A, Voets T, and Nilius B (2007) Bimodal action of menthol on the transient receptor potential channel TRPA1. J. Neurosci 27, 9874–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, and Patapoutian A (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829. [DOI] [PubMed] [Google Scholar]
- (59).Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, Nilius B, and Voets T (2009) TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 106, 1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, and Fanger CM (2007) TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. U. S. A 104, 13525–13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Klionsky L, Tamir R, Gao B, Wang W, Immke DC, Nishimura N, and Gavva NR (2007) Species-specific pharmacology of Trichloro(sulfanyl)ethyl benzamides as transient receptor potential ankyrin 1 (TRPA1) antagonists. Mol. Pain 3, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).El Kouhen R, Surowy CS, Bianchi BR, Neelands TR, McDonald HA, Niforatos W, Gomtsyan A, Lee CH, Honore P, Sullivan JP, Jarvis MF, and Faltynek CR (2005) A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel and selective transient receptor potential type V1 receptor antagonist, blocks channel activation by vanilloids, heat, and acid. J. Pharmacol. Exp. Ther 314, 400–409. [DOI] [PubMed] [Google Scholar]
- (63).Diver MM, Cheng Y, and Julius D (2019) Structural insights into TRPM8 inhibition and desensitization. Science 365, 1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gao Y, Cao E, Julius D, and Cheng Y (2016) TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Paulsen CE, Armache JP, Gao Y, Cheng Y, and Julius D (2015) Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 525, 552. [DOI] [PubMed] [Google Scholar]
- (66).Wei ET (1983) Inhibition of shaking movements in rats by central administration of cholinergic and adrenergic agents. Psychopharmacology (Berl) 81, 111–114. [DOI] [PubMed] [Google Scholar]
- (67).Wei ET, and Seid DA (1983) AG-3–5: a chemical producing sensations of cold. J. Pharm. Pharmacol 35, 110–112. [DOI] [PubMed] [Google Scholar]
- (68).Colburn RW, Lubin ML, Stone DJ Jr., Wang Y, Lawrence D, D’Andrea MR, Brandt MR, Liu Y, Flores CM, and Qin N (2007) Attenuated cold sensitivity in TRPM8 null mice. Neuron 54, 379–386. [DOI] [PubMed] [Google Scholar]
- (69).Kono T, Satomi M, Suno M, Kimura N, Yamazaki H, Furukawa H, and Matsubara K (2012) Oxaliplatin-induced neurotoxicity involves TRPM8 in the mechanism of acute hypersensitivity to cold sensation. Brain Behav 2, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ling B, Authier N, Balayssac D, Eschalier A, and Coudore F (2007) Behavioral and pharmacological description of oxaliplatin-induced painful neuropathy in rat. Pain 128, 225–234. [DOI] [PubMed] [Google Scholar]
- (71).Mizoguchi S, Andoh T, Yakura T, and Kuraishi Y (2016) Involvement of c-Myc-mediated transient receptor potential melastatin 8 expression in oxaliplatin-induced cold allodynia in mice. Pharmacol. Rep 68, 645–648. [DOI] [PubMed] [Google Scholar]
- (72).Deuis JR, Dvorakova LS, and Vetter I (2017) Methods Used to Evaluate Pain Behaviors in Rodents. Front. Mol. Neurosci 10, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Naziroglu M, and Braidy N (2017) Thermo-Sensitive TRP Channels: Novel Targets for Treating Chemotherapy-Induced Peripheral Pain. Front. Physiol 8, 1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Jensen TS, and Finnerup NB (2014) Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol 13, 924–935. [DOI] [PubMed] [Google Scholar]
- (75).Binder A, Stengel M, Maag R, Wasner G, Schoch R, Moosig F, Schommer B, and Baron R (2007) Pain in oxaliplatin-induced neuropathy–sensitisation in the peripheral and central nociceptive system. Eur. J. Cancer 43, 2658–2663. [DOI] [PubMed] [Google Scholar]
- (76).Attal N, Bouhassira D, Gautron M, Vaillant JN, Mitry E, Lepere C, Rougier P, and Guirimand F (2009) Thermal hyperalgesia as a marker of oxaliplatin neurotoxicity: a prospective quantified sensory assessment study. Pain 144, 245–252. [DOI] [PubMed] [Google Scholar]
- (77).Grothey A (2003) Oxaliplatin-safety profile: neurotoxicity. Semin. Oncol 30, 5–13. [DOI] [PubMed] [Google Scholar]
- (78).Kelland L (2007) The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 7, 573–584. [DOI] [PubMed] [Google Scholar]
- (79).Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V, Couette B, Busserolles J, Courteix C, Noel J, Lazdunski M, Eschalier A, Authier N, and Bourinet E (2011) Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med 3, 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Gauchan P, Andoh T, Kato A, and Kuraishi Y (2009) Involvement of increased expression of transient receptor potential melastatin 8 in oxaliplatin-induced cold allodynia in mice. Neurosci. Lett 458, 93–95. [DOI] [PubMed] [Google Scholar]
- (81).Kawashiri T, Egashira N, Kurobe K, Tsutsumi K, Yamashita Y, Ushio S, Yano T, and Oishi R (2012) L type Ca(2)+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol. Pain 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Ta LE, Bieber AJ, Carlton SM, Loprinzi CL, Low PA, and Windebank AJ (2010) Transient Receptor Potential Vanilloid 1 is essential for cisplatin-induced heat hyperalgesia in mice. Mol. Pain 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Staff NP, Grisold A, Grisold W, and Windebank AJ (2017) Chemotherapy-induced peripheral neuropathy: A current review. Ann. Neurol 81, 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Shapiro CL (2016) Highlights of Recent Findings on Quality-of-Life Management for Patients With Cancer and Their Survivors. JAMA Oncol 2, 1401–1402. [DOI] [PubMed] [Google Scholar]
- (85).Moran MM, and Szallasi A (2018) Targeting nociceptive transient receptor potential channels to treat chronic pain: current state of the field. Br. J. Pharmacol 175, 2185–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Yelm KE, Wos JA, Bunke GM, Frederick H, Haught JC, Hoke SH, Sreekrishna KT, and Lin Y (2017) Synthesis of aryl cyclohexane carboxamide derivatives useful as sensates in consumer products. Patent US 20170057911 A1. [Google Scholar]
- (87).Wos JA, Yelm KE, Bunke GM, Frederick HA, Reilly M, Haught JC, Sreekrishna KT, and Lin Y (2017) Synthesis of cyclohexane ester derivatives useful as cooling sensates in consumer products. Patent WO 2017106279 A1 [Google Scholar]
- (88).Moreno O, Natarajan S, and Duncan DF (2008) Small-molecule modulators of Trp-p8 activity. Patent US 20080255185 A1 [Google Scholar]
- (89).SYBYL-X, ver. 1.3, Tripos International, St. Louis, MO, 2010. [Google Scholar]
- (90).Xie XQ, Chen JZ, and Billings EM (2003) 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins: Struct., Funct., Genet 53, 307–319. [DOI] [PubMed] [Google Scholar]
- (91).Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, and Sali A (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct 29, 291–325. [DOI] [PubMed] [Google Scholar]
- (92).Chen JZ, Wang J, and Xie XQ (2007) GPCR structure-based virtual screening approach for CB2 antagonist search. J. Chem. Inf. Model 47, 1626–1637. [DOI] [PubMed] [Google Scholar]
- (93).Feng Z, Alqarni MH, Yang P, Tong Q, Chowdhury A, Wang L, and Xie XQ (2014) Modeling, molecular dynamics simulation, and mutation validation for structure of cannabinoid receptor 2 based on known crystal structures of GPCRs. J. Chem. Inf. Model 54, 2483–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Teramachi J, Silbermann R, Yang P, Zhao W, Mohammad KS, Guo J, Anderson JL, Zhou D, Feng R, Myint KZ, Maertz N, Beumer JH, Eiseman JL, Windle JJ, Xie XQ, Roodman GD, and Kurihara N (2016) Blocking the ZZ domain of sequestosome1/p62 suppresses myeloma growth and osteoclast formation in vitro and induces dramatic bone formation in myeloma-bearing bones in vivo. Leukemia 30, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Feng Z, Pearce LV, Xu X, Yang X, Yang P, Blumberg PM, and Xie XQ (2015) Structural insight into tetrameric hTRPV1 from homology modeling, molecular docking, molecular dynamics simulation, virtual screening, and bioassay validations. J. Chem. Inf. Model 55, 572–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Wiederstein M, and Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35, W407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Laskowski RA, MacArthur MW, Moss DS, and Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr 26, 283–291. [Google Scholar]
- (98).Jain AN (1996) Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J. Comput.-Aided Mol. Des 10, 427–440. [DOI] [PubMed] [Google Scholar]
- (99).Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Dickson CJ, Madej BD, Skjevik AA, Betz RM, Teigen K, Gould IR, and Walker RC (2014) Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput 10, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- (102).Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, and Walker RC (2012) Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013) Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- (104).Case D, Cerutti D, Cheatham T, Darden T, Duke R, Giese T, Gohlke H, Goetz A, Greene D, and Homeyer N (2016) AMBER 16; University of California: San Francisco. [Google Scholar]
- (105).Loncharich RJ, Brooks BR, and Pastor RW (1992) Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers 32, 523–535. [DOI] [PubMed] [Google Scholar]
- (106).Izaguirre JA, Catarello DP, Wozniak JM, and Skeel RD (2001) Langevin stabilization of molecular dynamics. J. Chem. Phys 114, 2090–2098. [Google Scholar]
- (107).Darden T, York D, and Pedersen L (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092. [Google Scholar]
- (108).Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995) A smooth particle mesh Ewald method. J. Chem. Phys 103, 8577–8593. [Google Scholar]
- (109).Ryckaert JP, Ciccotti G, and Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 23, 327–341. [Google Scholar]
- (110).Yang P, Wang L, and Xie XQ (2012) Latest advances in novel cannabinoid CB(2) ligands for drug abuse and their therapeutic potential. Future Med. Chem 4, 187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Hawkins GD, Cramer CJ, and Truhlar DG (1996) Parametrized Models of Aqueous Free Energies of Solvation Based on Pairwise Descreening of Solute Atomic Charges from a Dielectric Medium. J. Phys. Chem 100, 19824–19839. [Google Scholar]
- (112).Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, and Cheatham TE 3rd (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res 33, 889–897. [DOI] [PubMed] [Google Scholar]
- (113).Tsui V, and Case DA (2000) Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 56, 275–291. [DOI] [PubMed] [Google Scholar]
- (114).Bashford D, and Case DA (2000) Generalized born models of macromolecular solvation effects. Annu. Rev. Phys. Chem 51, 129–152. [DOI] [PubMed] [Google Scholar]
- (115).Sitkoff D, Sharp KA, and Honig B (1994) Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem 98, 1978–1988. [Google Scholar]
- (116).Still WC, Tempczyk A, Hawley RC, and Hendrickson T (1990) Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc 112, 6127–6129. [Google Scholar]
- (117).Weiser J, Shenkin PS, and Still WC (1999) Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem 20, 217–230. [Google Scholar]
- (118).Hu J, Feng Z, Ma S, Zhang Y, Tong Q, Alqarni MH, Gou X, and Xie XQ (2016) Difference and Influence of Inactive and Active States of Cannabinoid Receptor Subtype CB2: From Conformation to Drug Discovery. J. Chem. Inf. Model 56, 1152–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.