

Summary
Microtubules are non-covalent dynamic polymers essential for the life of all eukaryotic cells. Their dynamic behavior is regulated by a large array of cellular effectors. In vitro microtubule assays have been instrumental in dissecting the mechanism of microtubule associated proteins. In this chapter we focus on microtubule severing enzymes katanin and spastin. They are AAA ATPases that generate internal breaks in microtubules by extracting tubulin dimers out of the microtubule lattice. We present detailed protocols for TIRF microscopy-based assays that were instrumental in proving that these enzymes not only sever microtubules but also remodel the microtubule lattice by promoting the exchange of lattice GDP-tubulin with GTP-tubulin from the soluble pool. This activity modulates microtubule dynamics and supports microtubule-dependent microtubule amplification in the absence of a nucleating factor.
Keywords: cytoskeleton, microtubule, microtubule severing, microtubule repair, spastin, katanin, microtubule dynamics, GTP-tubulin, rescue, AAA ATPase
1. Introduction
Microtubules are dynamic cytoskeletal polymers that are essential for basic cellular functions such as cell division, intracellular transport and differentiation. Microtubules stochastically grow and depolymerize from their ends, a process known as dynamic instability (1). Microtubules grow through the addition of GTP-tubulin at their ends. Lattice incorporation triggers the hydrolysis of GTP to GDP. The GDP-tubulin lattice is labile and is protected from depolymerization by the GTP-tubulin cap which results from the lag between the rate of GTP-tubulin incorporation and GTP hydrolysis (1, 2, 3). Microtubule end dynamics are regulated in cells by a vast array of microtubule associated proteins (4, 5).
Microtubule severing enzymes are ATPases associated with various cellular activities (AAA ATPases) that use the energy of ATP hydrolysis to break microtubules in the middle (6). Microtubule severing activity was initially observed in metaphase Xenopus laevis extracts (7). The protein responsible for this activity was later isolated and identified through biochemical purification and named katanin after the Japanese word for sword “katana” (8). Since this original discovery, several other microtubule severing enzymes have been identified: spastin (9, 10), fidgetin (11) and katanin-like proteins (12, 13). Microtubule severing enzymes are essential for cilia biogenesis, cell division, and morphogenesis, and their mutation is associated with debilitating neurodevelopmental and neurodegenerative diseases (6).
Light microscopy based in vitro microtubule assays were instrumental in establishing the activity of microtubule severing enzymes and proving that they were breaking microtubules along their lengths and not depolymerizing them from their ends (7, 9, 10). However, these assays used stabilized microtubules (taxol or GMPCPP) immobilized to glass using antibodies, neutravidin-biotin linkages or a rigor kinesin and thus missed two important factors that characterize in vivo microtubules (1) their dynamic behavior and (2) the presence of μM concentrations of soluble GTP-tubulin. Using a combination of negative stain electron microscopy (EM) and total internal reflection fluorescence microscopy (TIRFM), we recently showed that microtubule severing enzymes, katanin and spastin sever microtubules by removing tubulin dimers from the microtubule lattice and that these nanodamage sites are repaired by the spontaneous incorporation of GTP-tubulin from the soluble tubulin pool (14), thus creating GTP-islands along the microtubule. These transient GTP-tubulin islands can protect the microtubule from depolymerization and promote microtubule rescue (14). When a microtubule is severed, the severed plus-end emerges with a higher density of GTP-tubulin stabilizing it against instantaneous depolymerization. The synergy between the increased rescue frequency and increased stability of the newly severed plus ends results in an amplification of microtubule number and mass (14). Severing-mediated microtubule amplification explains the paradoxical in vivo findings that loss of severing enzymes leads to a reduction in microtubule mass in C. elegans meiotic spindles, plant microtubule cortical arrays and Drosophila neurons (15, 16, 17, 18, 19).
In this chapter, we describe protocols to observe severing enzyme mediated repair of stabilized and non-stabilized microtubules and severing-enzyme mediated microtubule amplification. These assays can be used to decipher the biophysical mechanism of microtubule severing enzymes and their effects on microtubule dynamics as well as for the identification and characterization of cellular factors that regulate their activity.
2. Materials
2.1. Equipment
Multi-color TIRF motorized inverted microscope (e.g., Nikon Ti-E 2000 with perfect focus).
A high NA TIRF objective (e.g., 1.49 NA Nikon CFI Apo TIRF 100x).
488 and 640 nm solid-state lasers (Sapphire, Coherent).
Appropriate filters and dichroic mirrors.
Cooled electron-multiplying charged-coupled device (EMCCD) camera (e.g., iXON 897, Andor).
Controller software (e.g., MicroManager (20)).
Objective heater (Bioptechs).
2.2. Materials and Solutions
Plasma cleaned/piranha cleaned and silanized slides and coverslips (see Note 1).
Neutravidin (Thermo Fisher Cat# 31000).
BRB80: 80 mM PIPES, 1 mM MgCl2, and 1 mM EGTA, pH 6.8 (adjusted with KOH).
Severing enzyme buffer: 20 mM HEPES, 300 mM KCl, 10 mM MgCl2, 15% glycerol, 5 mM DTT or 1 mM TCEP, pH 7.0; pass through a 0.1 μm filter and keep on ice before use; use to dilute severing enzyme.
BB: BRB80 with 0.1% 2-mercaptoethanol.
BBC: BB with 2 mg/ml casein.
1% methylcellulose 4000 cP.
Oxygen scavengers (catalase, glucose oxidase and 2 M glucose) prepared as described previously (21) (see Note 2).
Purified spastin or katanin (see Section 2.3).
Porcine brain tubulin (see Section 2.4).
GMPCPP-stabilized microtubules (see Section 2.5) (see Note 3).
GMPCPP-stabilized unmodified microtubule seeds (see Section 2.5).
Pre-perfusion mixture: BBC supplemented with 50 mM KCl, 1 mM ATP, 1% Pluronic F127, 20 mM glucose, glucose oxidase and catalase (see Note 4).
Tubulin solution: BRB80, 0.5 mM GTP, 1% Pluronic F127, 2.5 mg/ml casein, 1 μM HiLyte488-labeled tubulin.
Wash solution: BRB80 with 1.5 mg/ml casein, 10 mM 2-mercaptoethanol, 1% Pluronic F127, 20 mM glucose, glucose oxidase and catalase.
Enzyme solution: severing enzyme at desired concentration in pre-perfusion mixture.
Severing assay buffer: 50 mM KCl, 0.5% Pluronic F127, 0.2 mg/ml casein, 1.5% glycerol, 0.1% methylcellulose 4000 cP, 1 mM GTP, 1 mM ATP, 20 mM glucose, glucose oxidase and catalase in BB.
2.3. Purified severing enzymes
Purify spastin or katanin according to (21, 22) and store in small aliquots at −80°C.
Thaw an aliquot of enzyme and spin for 10 minutes at 4°C and 279,000g to remove any aggregates (see Note 5).
Remove the supernatant and measure the concentration using absorbance at 280 nm or Bradford assay.
Enzymes are stable on ice for several hours at 20 μM.
2.4. Tubulin (see Note 6)
Porcine brain unlabeled tubulin, HiLyte488 tubulin, HiLyte647 tubulin, and biotin-tubulin (Cytoskeleton, Inc.). Tubulin labeled with other fluorophores can also be used.
Quickly thaw an aliquot of unlabeled porcine brain tubulin and place on ice.
Resuspend 20 μg of lyophilized, labeled tubulin in ice-cold BRB80.
Spin the unlabeled and labeled tubulin in separate tubes for 10 minutes at 4°C and 279,000g to remove any aggregates (see Note 5).
Carefully remove the top portion of the supernatant without touching the bottom of the tube that may contain tubulin aggregates. Measure the concentration by Bradford assay.
Keep the tubulin on ice. Use as quickly as possible as tubulin aggregates over time.
2.5. Double-cycled, GMPCPP-stabilized microtubules and unmodified microtubule seeds [adapted from (23)]
For dynamic and GMPCPP-capped GDP microtubules, we used GMPCPP microtubule seeds polymerized from unmodified tubulin purified from tsa201 cells as described previously (24). Unmodified microtubules are severed at a lower rate compared to brain microtubules (25). Therefore, by using unmodified microtubule seeds, severing enzymes preferentially sever the dynamic or GDP-brain microtubule extensions and not the unmodified GMPCPP-microtubule seeds at the spastin or katanin concentrations used in these assays. Alternatively, sea urchin axonemes can also be used (26) to grow dynamic brain microtubule extensions.
For fluorescently labeled, GMPCPP-stabilized brain microtubules, mix 1 μl of 1 mg/ml biotin tubulin, 10 μls of 2 mg/ml HiLyte 647 tubulin, and 39.5 μls of 2 mg/ml unlabeled brain tubulin. For unmodified microtubule seeds, mix 2 μls of 2 mg/ml biotin tubulin and 98 μls of 2 mg/ml unmodified tubulin. Incubate on ice for 5 minutes.
Ultracentrifuge the tubulin mixture to remove any tubulin aggregates at 279,000g for 10 minutes at 4°C (see Note 5).
Add GMPCPP to a final concentration of 0.5 mM and incubate at 37°C in a water bath (or heat block) for 1 hour.
Pellet the microtubules using an ultracentrifuge at 126,000g for 15 minutes at 37°C.
Remove the supernatant. Gently wash the pellet with warm BRB80.
Re-suspend the microtubule pellet in cold BRB80 such that the final tubulin concentration is ~ 2 mg/ml.
Incubate on ice for 30 minutes, mix by pipetting gently every 10 minutes.
Add GMPCPP to a final concentration of 0.5 mM. For GMPCPP-stabilized brain microtubules, incubate in 37°C water bath (or heat block) between 4 to 16 hours. For GMPCPP-stabilized unmodified seeds, incubate in 37°C water bath (or heat block) for 30 minutes to obtain short microtubules.
Spin microtubules at 126,000g for 15 minutes at 37°C.
Remove the supernatant without touching the pellet. Gently wash the pellet with warm BRB80.
Gently re-suspend the microtubule seeds in warm BRB80 with a cut tip to prevent microtubule shearing.
GMPCPP-stabilized microtubules are stored at 30°C and should be used within 2–3 days. For GMPCPP-stabilized seeds, flash-freeze in small aliquots. On the day of performing the assays, dilute the seed aliquot 1:10 in warm BB and place at 37°C for 1 hour before use and then keep at room temperature.
3. Methods
All steps are performed at room temperature unless stated otherwise.
3.1. Generation and detection of microtubule nanoscale damage on GMPCPP-stabilized microtubules using spastin or katanin (Fig. 1)
Fig 1.
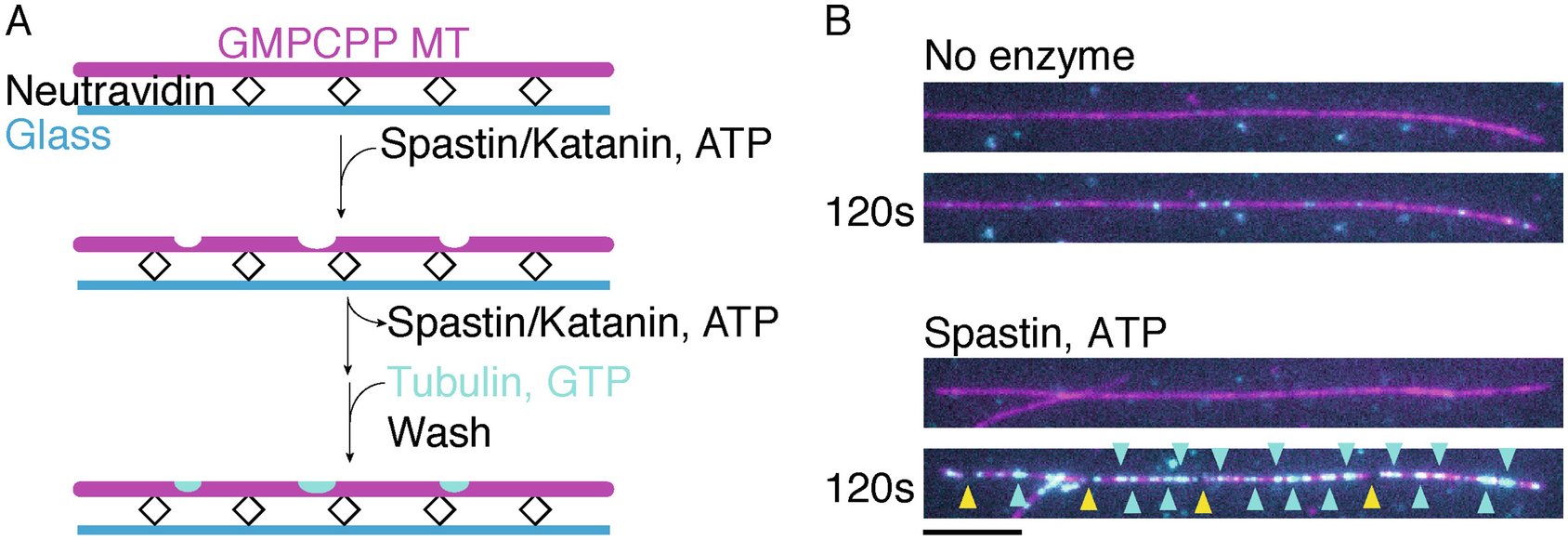
A) Schematic of assay for visualizing severing enzyme mediated nanodamage and healing of GMPCPP-stabilized microtubules. B) Top panels, GMPCPP stabilized microtubule (magenta) not treated with severing enzymes shows no tubulin incorporation (cyan) along the microtubule lattice. Bottom panels, GMPCPP stabilized microtubule incubated 120 s with 2 nM spastin and ATP shows tubulin incorporation (cyan) along the microtubule lattice. Cyan arrows indicate sites of tubulin incorporation, yellow arrows indicate severing events. Scale bar, 5 μm.
Assemble chamber as described previously (21).
Perfuse room temperature BRB80 into the chamber.
Perfuse cold 0.1 mg/ml Neutravidin and incubate for 5–10 minutes.
Wash the chamber with BRB80 and BBC.
Dilute the microtubules with warm BB and mix with a cut tip to avoid shearing. Perfuse the microtubules into the chamber and incubate for 10 minutes (see Note 7).
Wash chamber three times with BBC (see Note 8).
Acquire images of multiple areas and save the coordinates for these areas. To avoid photodamage, use short exposures and low laser power. The purpose of this step is to quickly visualize the microtubules before starting the assay with the severing enzymes and to count the number of severing events.
-
Perfuse enzyme solution into the chamber and incubate for 30–120 sec. The nanodamage extent varies steeply with incubation time, so the time has to be well-controlled. We typically use enzyme concentrations between 2 and 20 nM.
Stop here if you want to obtain nanodamaged microtubules for subsequent assays (for example with motor proteins to investigate the effects of the nanodamage on motility) and do not want to visualize repair.
Perfuse tubulin solution and incubate for 5 minutes. Tubulin labeled with a different dye than the microtubules will allow visualization of the incorporation of tubulin at sites of nanodamage introduced by the enzyme in Step 9 (see Note 11).
Wash chamber three times with the wash solution and visualize microtubules and incorporated tubulin.
3.2. Two-color severing assays with GMPCPP-capped GDP microtubules to monitor lattice nanodamage and repair (Fig. 2).
Fig 2.
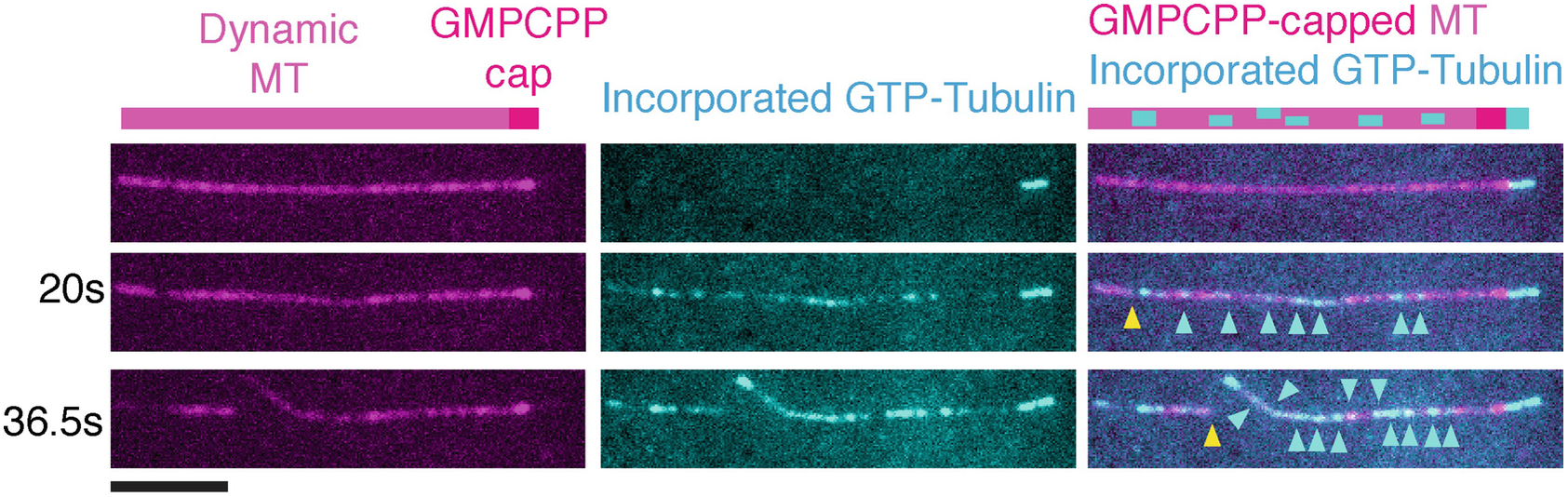
Time course of GMPCPP capped dynamic microtubule extensions (magenta) exposed to 10 nM spastin in the presence of 7 μM GTP tubulin (cyan). Cyan arrows indicate sites of tubulin incorporation, yellow arrows indicate severing events. Scale bar, 5 μm.
The following protocol describes the generation of GMPCPP-capped microtubule extensions (adapted from (27, 28)). GMPCPP-capped microtubules are needed in order to be able to exchange the tubulin concentration in the chamber without microtubule depolymerization.
Perfuse BRB80 into the chamber.
Incubate the chamber with 0.1 mg/ml Neutravidin for 5–10 minutes.
Wash the chamber with BBC.
Incubate the chamber with unmodified microtubule seeds or axonemes for 10 minutes (see Notes 7, 8).
Wash the chamber with BBC.
Polymerize 10% or 20% Hilyte 647-labeled dynamic microtubules at 16 μM tubulin in severing assay buffer for 10–12 minutes on the microscope (see Notes 7 – 12).
Perfuse in 6 μM of 10% Hilyte 647-labeled tubulin in severing assay buffer supplemented with 0.5 mM GMPCPP instead of GTP (see Note 13).
Wash the chamber with severing assay buffer.
Start image acquisition as 10 nM spastin and 7 μM tubulin (or any tubulin concentration that is investigated) in severing assay buffer is perfused into the chamber. A frame rate of 2 Hz or higher is sufficient with 100 ms exposure in each channel.
Seal the chamber to prevent evaporation.
3.3. Severing assays with dynamic microtubules (Fig. 3)
Fig 3.
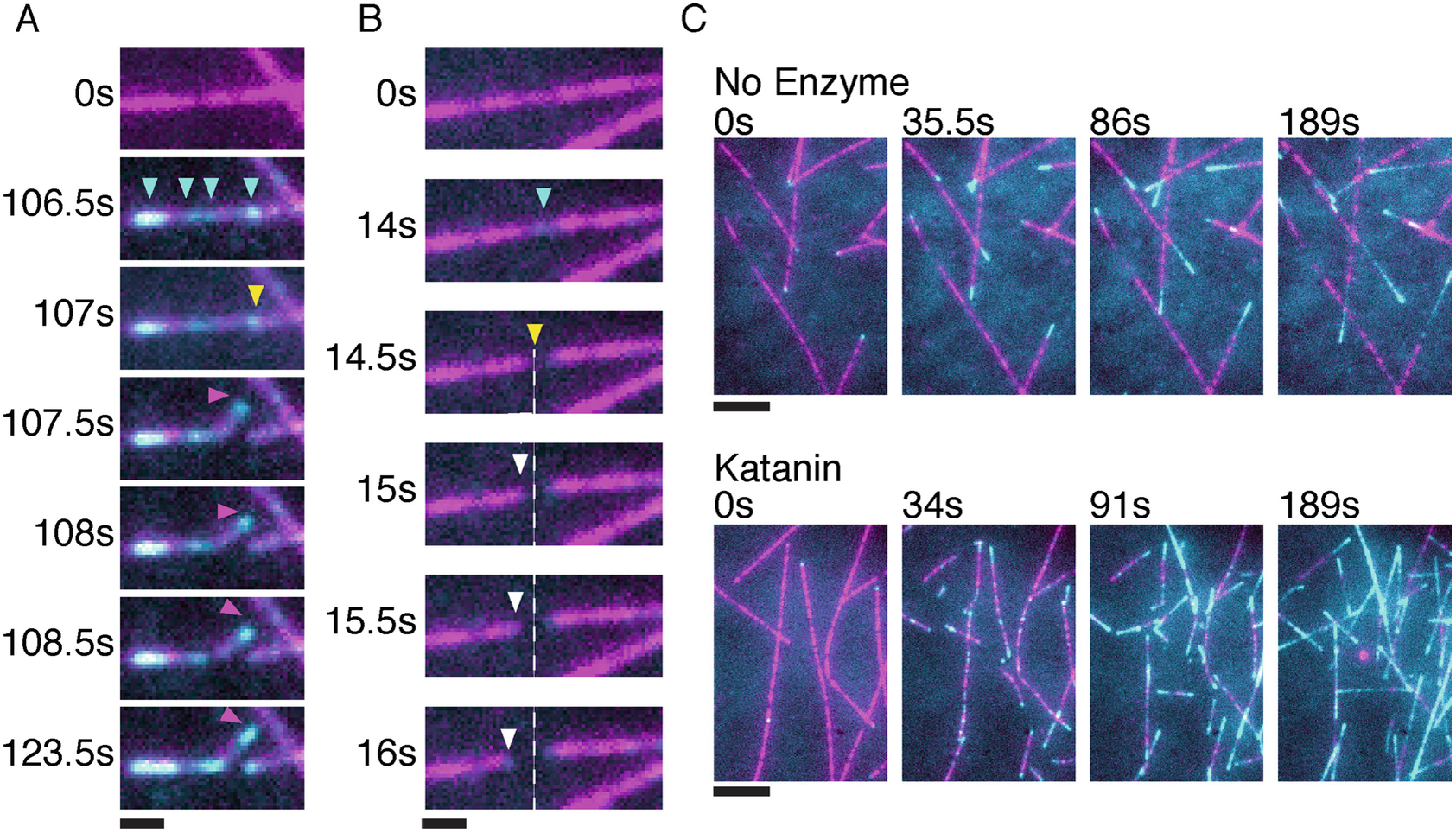
Incorporation of soluble tubulin (cyan) along the lattice of dynamic microtubules (magenta) in the presence of 25 nM katanin and 12 μM GTP tubulin. A) Time course of a severing enzyme generated plus-end that is stable and grows. Cyan arrows indicate sites of tubulin incorporation, yellow arrow shows severing event and magenta arrow follows the stable new plus end. B) Time course of a severing enzyme generated plus-end that is unstable and depolymerizes. Cyan arrow indicates site of tubulin incorporation, yellow arrow indicates a severing event and white arrow follows the depolymerizing new plus end. The dotted line marks the severing enzyme generated new plus-end. Scale bar, 1 μm. C) Top panels, time lapse images showing microtubule dynamics at 12 μM tubulin in the absence of severing enzymes. The last frame is bleach corrected in the microtubule (magenta) channel. Bottom panels, time lapse images showing microtubule number and mass amplification through katanin severing. Cyan shows newly incorporated tubulin only at the growing ends (top panel) or at the growing ends and along the microtubule shaft in the presence of enzyme and ATP (bottom panel). Scale bar, 5 μm.
Perfuse BRB80 to the chamber.
Incubate the chamber with 0.1 mg/ml Neutravidin for 10 minutes.
Wash the chamber with BBC.
Incubate the chamber with seeds for 10 minutes (see Notes 7, 8).
Wash the chamber with BBC.
Polymerize 10% Hilyte 647-labeled dynamic microtubules at 12 μM tubulin in severing assay buffer (see Notes 7 – 12).
Perfuse 10% Hilyte 488-labeled tubulin at 12 μM and spastin or katanin (5 – 25 nM) in severing assay buffer into the chamber. Start image acquisition during perfusion. A range of tubulin and enzyme concentrations should be tested to investigate the effects of tubulin lattice incorporation.
Seal the chamber to prevent evaporation.
3.4. Quantification
Quantification can be performed using FIJI (29).
3.4.1. Quantification of the intensity of incorporated tubulin and microtubule severing rates
Manually select microtubules with a line selection.
Measure background-subtracted average intensity of the fluorescent tubulin incorporated (MatLab scripts can be used to generate background-subtracted line scans and detect the peaks on line scans, measure their full-width-at-half-maximum and intensity).
To measure severing rate, count the number of mesoscale severing events per microtubule length as a function of time (25).
3.4.2. Scoring the stability of the newly severed plus ends in dynamic microtubule assays
Determine microtubule polarity. This can be done by (1) measuring the growth rate of the two ends or (2) by imaging during the polymerization phase since the plus-end grows faster. After severing, keep track of the polarity of the severed microtubules that are no longer connected to the coverslip through the seed.
Once a severing event occurs, follow the newly severed end frame by frame. If there is no depolymerization (in the 647 microtubule channel) immediately following severing, tubulin incorporation (in the 488 channel) is seen at the microtubule end and growth persists, the newly severed end is considered stable (Fig. 3A). If the newly severed end depolymerizes, loss of fluorescent microtubule intensity (in the 647 microtubule channel) at the new plus end is observed (Fig. 3B).
4. Conclusion
In this chapter, we describe protocols to monitor severing enzyme-catalyzed tubulin exchange along the microtubule lattice as well as severing enzyme-mediated microtubule amplification. These assays lay down the foundation for understanding the effects of microtubule severing enzymes on microtubule dynamics and organization and how their activities synergize with other microtubule associated proteins, such as CLASP (30) or depolymerizing factors like MCAK (31) that can interact with the newly severed microtubule ends or the severing enzyme generated GTP-tubulin islands.
5. Notes
Proteins adsorb non-specifically to untreated glass. Therefore, it is essential for all glass surfaces to be clean and passivated for these assays. Otherwise, severing enzymes and tubulin will non-specifically adsorb to the glass. This is especially important for severing enzymes as they are used at low concentrations in these assays. A detailed protocol for piranha cleaned glass can be found in (21). Alternatively, glass can be plasma cleaned with Argon for 3 minutes using the Expanded Plasma Cleaner (Harrick Plasma, Ithaca, NY). Rinse the glass three times with MilliQ water, incubate for 15 minutes in 0.1 M KOH and continue from the KOH step in (21).
If chambers have fluorescent aggregates, filter the catalase/glucose oxidase through a 0.1 μm filter.
For healing assays using stabilized microtubules, taxol-stabilized microtubules can be used instead of GMPCPP-stabilized microtubules. To polymerize taxol microtubules see (21). If taxol microtubules are used, make sure to add taxol to all buffers.
Solutions 13–17 should be prepared just before use.
Make sure the ultracentrifuge tubes and Eppendorf tubes are pre-chilled prior to placing protein into them.
Severing enzyme activity is very sensitive to aggregated tubulin because the enzymes will bind to the tubulin aggregates instead of microtubules. Therefore, it is essential to remove all aggregates and work with high-quality, pure samples. To remove tubulin aggregates, ultra-centrifuge tubulin at 279,000g for 10 minutes at 4°C. If tubulin aggregates persist, cycle the tubulin through one cycle of polymerization and depolymerization (32, 33) or purify a new batch of tubulin.
The extent of nanoscale damage and/or severing depends on the density of microtubules in the chamber and how strongly they are attached to the coverslip with the neutravidin-biotin linkage. The same applies if a rigor kinesin mutant is used to immobilize the microtubules on the glass (9, 34). Therefore, if quantitation is attempted, it is important to use similar microtubule densities in all chambers and the same immobilization protocol.
Do not add any cold solutions to the chamber once microtubules are added as this will depolymerize the microtubules.
The perfusion volumes used in the protocol will vary depending on the width of the chambers. Typically, our chambers hold ~7 μls. We usually perfuse volumes 2–3 times the chamber volume.
To prevent evaporation from the edges of the chamber, leave a few μls of perfusion solution at the ends of the channel and place a humidity cover over the chamber.
Add tubulin after the addition of all other components.
From this step onwards, assays are performed on the microscope at 30°C.
To avoid de novo microtubule nucleation in the chamber, add the 0.5 mM GMPCPP just before perfusing the tubulin mixture into the chamber. The incubation time with the GMPCPP tubulin will vary. Therefore, image the chamber intermittently to see if GMPCPP extensions are growing.
Acknowledgement
A.R.M. is supported by the intramural programs of the National Institute of Neurological Disorders and Stroke (NINDS) and the National, Heart, Lung and Blood Institute (NHLBI).
References
- 1.Mitchison T, Kirschner M (1984) Dynamic instability of microtubule growth. Nature 312:237–242 [DOI] [PubMed] [Google Scholar]
- 2.Carlier M-F (1982) Guanosine-5′-triphosphate hydrolysis and tubulin polymerization. Mol. Cell. Biochem 47:97–113 [DOI] [PubMed] [Google Scholar]
- 3.Carlier MF, Pantaloni D (1981) Kinetic analysis of guanosine 5’-triphosphate hydrolysis associated with tubulin polymerization. Biochemistry 20:1918–1924 [DOI] [PubMed] [Google Scholar]
- 4.Alfaro-Aco R, Petry S (2015) Building the Microtubule Cytoskeleton Piece by Piece. J. Biol. Chem 290:17154–17162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akhmanova A, Steinmetz MO (2015) Control of microtubule organization and dynamics: two ends in the limelight. Nat. Rev. Mol. Cell Biol 16:711–726 [DOI] [PubMed] [Google Scholar]
- 6.Mcnally FJ, Roll-Mecak A (2018) Microtubule-severing enzymes: From cellular functions to molecular mechanism. J. Cell Biol 217:4057–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vale RD (1991) Severing of stable microtubules by a mitotically activated protein in Xenopus egg extracts. Cell 64:827–839 [DOI] [PubMed] [Google Scholar]
- 8.Mcnally FJ, Vale RD (1993) Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell 75:419–429 [DOI] [PubMed] [Google Scholar]
- 9.Roll-Mecak A, Vale RD (2005) The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr. Biol 15:650–655 [DOI] [PubMed] [Google Scholar]
- 10.Evans KJ, Gomes ER, Reisenweber SM et al. (2005) Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J. Cell Biol 168:599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukherjee S, Diaz Valencia JD, Stewman S et al. (2012) Human Fidgetin is a microtubule severing the enzyme and minus-end depolymerase that regulates mitosis. Cell Cycle 11:2359–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang D, Rogers GC, Buster DW et al. (2007) Three microtubule severing enzymes contribute to the “Pacman-flux” machinery that moves chromosomes. J. Cell Biol 177:231–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonbuchner TM, Rath U, Sharp DJ (2010) KL1 is a novel microtubule severing enzyme that regulates mitotic spindle architecture. Cell Cycle 9:2403–2411 [DOI] [PubMed] [Google Scholar]
- 14.Vemu A, Szczesna E, Zehr EA et al. (2018) Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science 361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roll-Mecak A, Vale RD (2006) Making more microtubules by severing: a common theme of noncentrosomal microtubule arrays? J. Cell Biol 175:849–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srayko M, O’toole ET, Hyman AA et al. (2006) Katanin disrupts the microtubule lattice and increases polymer number in C. elegans meiosis. Curr. Biol 16:1944–1949 [DOI] [PubMed] [Google Scholar]
- 17.Sherwood NT, Sun Q, Xue M et al. (2004) Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2:e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindeboom JJ, Nakamura M, Hibbel A et al. (2013) A mechanism for reorientation of cortical microtubule arrays driven by microtubule severing. Science 342:1245533. [DOI] [PubMed] [Google Scholar]
- 19.Burk DH, Ye ZH (2002) Alteration of oriented deposition of cellulose microfibrils by mutation of a katanin-like microtubule-severing protein. Plant Cell 14:2145–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edelstein AD, Tsuchida MA, Amodaj N et al. (2014) Advanced methods of microscope control using muManager software. Journal of biological methods 1(2):e10 10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziółkowska NE, Roll-Mecak A, In vitro microtubule severing assays. Methods Mol. Biol 1046, 323–334 (2013). doi: 10.1007/978-1-62703-538-5_19; [DOI] [PubMed] [Google Scholar]
- 22.Zehr E, Szyk A, Piszczek G et al. (2017) Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat. Struct. Mol. Biol 24:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gell C, Bormuth V, Brouhard GJ, Cohen DN, Diez S, Friel CT, Helenius J, Nitzsche B, Petzold H, Ribbe J, Schaffer E, Stear JH, Trushko A, Varga V, Widlund PO, Zanic M & Howard J (2010) Microtubule dynamics reconstituted in vitro and image by single-molecule fluorescence microscopy. Methods Cell Biol. 95:221–245 [DOI] [PubMed] [Google Scholar]
- 24.Vemu A, Garnham CP, Lee D-Y et al. (2014) Generation of differentially modified microtubules using in vitro enzymatic approaches. Methods Enzymol. 540:149–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valenstein ML, Roll-Mecak A (2016) Graded control of microtubule severing by tubulin glutamylation. Cell 164:911–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbons IR, Fronk E (1979) A latent adenosine triphosphatase form of dynein 1 from sea urchin sperm flagella. J. Biol. Chem 254:187–196 [PubMed] [Google Scholar]
- 27.Severin FF, Sorger PK, Hyman AA (1997) Kinetochores distinguish GTP from GDP forms of the microtubule lattice. Nature 388:888–891 [DOI] [PubMed] [Google Scholar]
- 28.Aumeier C, Schaedel L, Gaillard J et al. (2016) Self-repair promotes microtubule rescue. Nat. Cell Biol 18:1054–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindelin J, Arganda-Carreras I, Frise E et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9:676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Bassam J, Kim H, Brouhard G et al. (2010) CLASP promotes microtubule rescue by recruiting tubulin dimers to the microtubule. Dev. Cell 19:245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walczak CE, Mitchison TJ, Desai A (1996) XKCM1: a Xenopus kinesin-related protein that regulates microtubule dynamics during mitotic spindle assembly. Cell 84:37–47 [DOI] [PubMed] [Google Scholar]
- 32.Weisenberg RC, Borisy GG, Taylor EW (1968) The colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 7:4466–4479 [DOI] [PubMed] [Google Scholar]
- 33.Borisy GG, Marcum JM, Olmsted JB et al. (1975) Purification of tubulin and associated high molecular weight proteins from porcine brain and characterization of microtubule assembly in vitro. Ann. N. Y. Acad. Sci 253:107–132 [DOI] [PubMed] [Google Scholar]
- 34.Hartman JJ, Mahr J, Mcnally K et al. (1998) Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell 93:277–287 [DOI] [PubMed] [Google Scholar]