

SUMMARY
The Cullin-RING E3 ligase (CRL) family is commonly hijacked by pathogens to redirect the host ubiquitin proteasome machinery to specific targets. During HIV infection, CRL5 is hijacked by HIV Vif to target viral restriction factors of the APOBEC3 family for ubiquitination and degradation. Here, using a quantitative proteomics approach we identify the E3 ligase ARIH2 as a regulator of CRL5-mediated APOBEC3 degradation. The CUL5Vif/CBFß complex recruits ARIH2 where it acts to transfer ubiquitin directly to the APOBEC3 targets. ARIH2 is essential for CRL5-dependent HIV infectivity in primary CD4+ T-cells. Furthermore, we show that ARIH2 cooperates with CRL5 to prime other cellular substrates for polyubiquitination, suggesting this may represent a general mechanism beyond HIV infection and APOBEC3 degradation. Taken together, these data identify ARIH2 as a co-factor in the Vif-hijacked CRL5 complex that contributes to HIV infectivity and demonstrate the operation of the E1-E2-E3/E3-stubstrate ubiquitination mechanism in a viral infection context.
Graphical Abstract
eTOC Blurb
Degradation of APOBEC3 proteins by HIV Vif hijacking Cullin RING E3 ligase 5 is essential for HIV proliferation. Hüttenhain et al. discovered the host factor ARIH2 which works with CUL5Vif/CBFß via a tag-team mechanism to target APOBEC3 proteins for ubiquitination and is essential for HIV proliferation in primary T cells.
INTRODUCTION
Viruses depend on cellular machinery for replication. This requires interactions of viral and host proteins, resulting in hijack or suppression of normal host functions (Goff, 2007; Hsu and Spindler, 2012). Accordingly, discovery of host-virus interactions is important for understanding mechanisms of viral infection including replication, antiviral host responses, and viral subversion of host defenses (Shah et al., 2015). Moreover, these interactions can be leveraged to develop agents that target the interface between virus and host proteins for use as antiviral therapeutics or vaccines (Watanabe et al., 2014). Proteomic approaches based on purification of affinity tagged viral proteins followed by mass spectrometry (AP-MS) analyses have emerged as effective tools to discover host-pathogen interactions (Batra et al., 2018; Davis et al., 2015; Eckhardt et al., 2018; Jager et al., 2011a; 2011b; Jean Beltran et al., 2017; Y. Luo et al., 2016; Mirrashidi et al., 2015; Penn et al., 2018; Ramage et al., 2015; Shah et al., 2018; Watanabe et al., 2014). These studies showed that members of the Ubiquitin Proteasome System (UPS), especially of Cullin RING E3 ligase (CRL) family, are commonly hijacked by pathogens to redirect host cell E3 ligases to ubiquitinate host proteins for degradation (Huh et al., 2007; Jager et al., 2011a; Mahon et al., 2014; Sato et al., 2009).
In recent years, a flurry of reports revealed many different potential routes by which CRLs can regulate and mediate ubiquitination in vitro. This in part stems from a single Cullin-RING catalytic core serving as a subunit of numerous different E3s, each with different substrate receptors. One important principle to emerge from these studies is that CRLs assemble on-demand – substrate binding to a substrate receptor initiates a cascade of reactions that transiently stabilize the assembly of CRL via Cullin modification with the ubiquitin-like protein NEDD8 (Emberley et al., 2012; Enchev et al., 2012; Fischer et al., 2011; Pierce et al., 2013; Reitsma et al., 2017; S. Wu et al., 2013; Zemla et al., 2013). Neddylated CRLs, like other RING E3s, do not themselves directly transfer ubiquitin, but rather employ other enzymes that carry ubiquitin on an active site cysteine from which ubiquitin is transferred to the receptor-bound substrate. It was thought that neddylated CRLs utilize only E2 enzymes as ubiquitin carriers (Deshaies and Joazeiro, 2009), although recently a new mechanism was discovered for CRL1-4 involving the RING-Between-RING (RBR) E3 ARIH1 which is recruited to transfer ubiquitin to some CRL substrates via an E1-E2-E3/E3-substrate or “tag-team” cascade (Dove et al., 2017; Duda et al., 2013; Kelsall et al., 2013; Scott et al., 2016). Yet, despite the hundreds of endogenous biological processes – including cell division and differentiation – regulated by human CRLs, and even more hijacked by pathogens, their underlying ubiquitin transfer mechanism(s) have remained elusive for decades.
To understand how CRLs hijacked by viruses mediate ubiquitination of their substrates, we focus on the Cullin-RING E3 ligase 5 (CRL5) complex, which plays a central role in HIV infection. CRL5 is hijacked by the primate lentiviral protein Vif, together with the transcriptional cofactor core-binding factor beta (CBFß), to target viral restriction factors of the APOBEC3 family for ubiquitination and degradation (Jager et al., 2011b; Mehle et al., 2004; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003; Zhang et al., 2011) (Figure 1A). In the absence of Vif, APOBEC3 proteins are potent mediators of the antiviral response. Through incorporation into virions and introduction into subsequent infected cells, the APOBEC3 enzymes catalyze deamination of specific Cytosine bases, resulting in their transformation to Uracils during synthesis of the minus strand viral DNA, which in turn results in non-functional virions (Wissing et al., 2010). Recruitment of CBFß to CRL5 is required for this activity, through assembling a properly-folded CRL5-Vif-CBFß complex, in part by inhibiting Vif oligomerization and also by directly activating CRL5-Vif via the interaction (Jager et al., 2011b; Kim et al., 2013). The interaction with CRL5-Vif precludes CBFß’s association with the RUNX family of transcription factors, thereby also perturbing the expression of RUNX target genes (Kim et al., 2013). This conserved dual hijacking of both host ubiquitination and transcriptional regulation is beneficial to HIV infectivity (Kane et al., 2015; Kim et al., 2013).
Figure 1. Approach for discovery and functional validation of host-pathogen interactions.

(A) Schematic of Vif recruitment of CBFß to the CUL5 E3 ligase complex for ubiquitination and degradation of APOBEC3G.
(B) Schematic of the approach for discovering host-pathogen interactions using quantitative proteomics and functional validating the interactions by CRISPR-Cas9 based KO in primary cells.
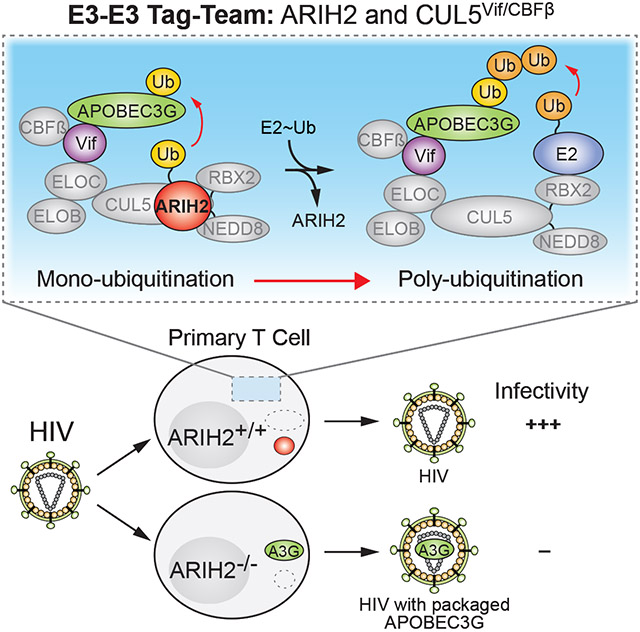
Notably, NEDD8 and the specific neddylation pathway for CUL5 were found to be crucial for HIV infectivity (Stanley et al., 2012), raising the question of which other cellular regulators of CRL5 could play roles in HIV infection. Since efforts have been made to design inhibitors targeting the interaction between Vif and APOBEC3 proteins (Pery et al., 2015), identifying cellular regulators of APOBEC3 degradation via CRL5 could provide an enzymatic route for pharmaceutical targeting. Here, we address this gap using a quantitative, differential proteomics approach revealing HIV-dependent remodeling of CRL5 E3 ligase composition under infection conditions, coupled with CRISPR Cas9-based gene knockout validation of targets, and biochemical studies of ubiquitination (Figure 1B). By showing that one of the Vif-dependent interactors identified, the RBR E3 ubiquitin ligase ARIH2, is essential for CRL5-dependent HIV infectivity in primary CD4+ T-cells, we define a discrete ubiquitination mechanism utilized in an essential human biological process and hijacked by HIV as well as demonstrate the E1-E2-E3/E3-stubstrate ubiquitination mechanism in cells. Furthermore, we show that ARIH2 also cooperates with CRL5 to prime other cellular substrates for polyubiquitination, suggesting this may represent a general mechanism beyond HIV infection and APOBEC3 degradation.
The combined proteomic and genetic approach presented here, which allows for the study of differential host interactions networks in response to virus infection followed by functional validation of host-virus interactions, will be broadly applicable to other viral systems and facilitate the discovery of anti-viral targets (Figure 1B).
RESULTS
The CRL5 protein interaction network is modulated by HIV infection
To study how CRL5 is remodeled during HIV infection and to identify cellular regulators mediating infection, we used a quantitative AP-MS approach to build a differential protein interaction network of CRL5 in the presence and absence of HIV infection (Figure 2A). We generated three stable Jurkat T cell lines, each expressing a tetracycline-inducible affinity-tagged version of a central component of the Vif-hijacked CRL5 complex: CUL5, the scaffold of CRL5; ELOB, which forms together with ELOC the adaptor of CRL5; and CBFß, a transcriptional regulator that is redirected by HIV Vif to CRL5, and that is required for structural reasons to assemble an activated, properly-folded CRL5-Vif complex that also contains CBFß (Jager et al., 2011b; Kim et al., 2013) (Figure 1A). We selected cell lines with affinity tagged proteins expressed at similar levels compared to endogenous protein levels to preserve endogenous protein complex stoichiometry of CRLs and to avoid artifacts that are inherent with overexpression of scaffold proteins (Gibson et al., 2013). Both affinity tagged CUL5 and CBFß were expressed in equivalent amounts when compared to the endogenous proteins, while only levels of affinity tagged ELOB exceeded endogenous levels (Figure S1A).
Figure 2. CUL5-ELOB-CBFß protein interaction network in the context of HIV infection.

(A) Affinity purification coupled to mass spectrometry (AP-MS) workflow for discovering Vif-dependent protein interactors of the CUL5 E3 ligase complex.
(B) Combined network of specific interactors for CUL5, CBFß and ELOB obtained from infection with HIV wt and mock. Different colors and shapes of the nodes denote gene ontology enriched biological processes for interacting proteins, different edge colors denote condition in which the interaction has been observed (see legend).
Cells expressing the tagged proteins as well as parental Jurkat T cells were subjected to infection with replication-deficient, Vesicular Stomatitis Virus Glycoprotein (VSVg) pseudotyped HIV-1 NL4-3 virus, a Vif-deficient version of the same virus strain (HIV ΔVif), or a mock serving as a non-infection condition (mock) (Figure 2A) (Cronin et al., 2005). Spinoculation of the cultures at a high multiplicity of infection (MOI) resulted in reproducible infection rates ranging from 75 to 90% across the lines expressing different affinity tagged constructs and viruses (Figure S1B). Following 24h of infection, anti-FLAG immune complexes were purified and subjected to global, quantitative analysis by MS (Figure 2A). Specific interactors of the three bait proteins were determined using SAINT high confidence interactor scoring by comparing affinity purifications from bait-expressing versus parental Jurkat T cells (H. Choi et al., 2011) (Figure 2A, Table S1). Proteins with a false discovery rate (FDR) below 0.15 were subsequently validated using a targeted proteomics approach, which allows for accurate, reproducible and consistent quantification across samples and conditions (Huttenhain et al., 2012). We developed targeted proteomics assays specific for all potential interactors with a SAINT FDR below 0.15 based on the global MS analysis. The assays were then used to quantify potential interactors in a targeted fashion across all infection conditions and controls, followed by statistical analysis using the MSstats framework (M. Choi et al., 2014). Proteins significantly enriched in anti-FLAG immune complexes compared to controls were considered validated interactors and integrated into the CUL5-CBFß-ELOB network consisting of 138 proteins involved in 192 interactions (Figure 2B, Table S1).
The interaction network was enriched for proteins involved in molecular processes associated with CRL function, such as protein ubiquitination, signal transduction and activation of CRLs by conjugation with the ubiquitin like protein NEDD8 (Figure 2B, Figure S1C). Six of the 192 interactions were observed only in the absence of HIV infection, and 26 were observed only in the presence of HIV infection (Figure 2B, Table S1). CBFß served as an internal control, showing HIV-dependent interactions as expected, since in the absence of infection it is only known to function as a transcriptional regulator through interaction with RUNX family members, while in the presence of HIV infection it associates with the CRL5 complex (Jager et al., 2011b; Kim et al., 2013; Wong et al., 2011; Zhang et al., 2011). Notably, besides functioning as adaptor protein together with ELOC in CRL5 and CRL2 complexes, ELOB forms the heterotrimeric Elongin (SIII) complex with ELOA and ELOC, which activates elongation by RNA polymerase II, and thus a large proportion of observed ELOB interactors are involved in transcriptional regulation (Figure 2B) (Aso et al., 1995; Kim et al., 2013). We did not recover APOBEC3 proteins, the known substrates of CUL5Vif/CBFß, in our AP-MS experiments, which can be explained by the transient nature of ligase-substrate interactions, which makes them difficult to capture (Harper and Tan, 2012) and by low level, if any, expression of APOBEC3 protein in Jurkat T cells. While previous studies have shown that CRL5 plays a central functional role during HIV infection, this global, unbiased CRL5 interaction network in the context of infection forms the starting point to determine additional HIV-mediated cellular regulators of the complex.
We next aimed to determine how Vif modulates the CRL5 interaction network, which could point to cellular regulators required for Vif-mediated degradation of APOBEC3 proteins. Accordingly, the abundances of all validated CUL5, ELOB and CBFß interactors were compared between wt to Vif-deficient HIV infection. Vif-dependent interactors were defined by significant enrichment with at least one bait during wt infection (Figure 3A, 3B, Table S2). The 14 Vif-dependent interactors include the expected Vif and CBFß (Figure 3B). Consistent with previous results, we found all core complex components of CRL5 and CRL2 physically interacting with CBFß only in the presence of Vif (Jager et al., 2011b; 2011a).
Figure 3. Vif-dependent changes on the CUL5-ELOB-CBFß interaction network.

(A) Relative abundance comparison of all specific interactors for CUL5, CBFß, and ELOB, respectively, under HIV wt and HIV ΔVif infection conditions shown as volcano plots. Volcano plots describe the log2 fold change and −log10 p-value of each interactor comparing HIV wt to ΔVif infection as determined by MSstats (M. Choi et al., 2014). The dashed line represents a p-value cutoff of 0.05. Proteins indicated in red are significantly upregulated enriched under HIV wt infection (p-value cutoff 0.05, n = 4), protein indicated in black represent known bait interactors, while grey dots are all other interactors. Data has been acquired by a targeted proteomics strategy.
(B) Hierarchical clustering for relative abundance comparison of interactors for CUL5, CBFß, and ELOB, that showed a significant fold change comparing HIV wt and HIV ΔVif infection (p-value cutoff 0.05) for at least one of the bait proteins. Besides known CUL5 and CUL2 complex components as well as Vif and CBFß, two known Vif-dependent factors recruited to CUL5 complex, the analysis reveals five so far functionally uncharacterized Vif-dependent interactors, namely ARIH1, ARIH2, AMBRA1, DCAF11, and ALIX.
(C) Protein abundances of selected Vif-dependent interactors across all baits and infection conditions. Data from four independent experiments are presented as mean ± SEM.
See also Table S2.
In addition to the known interactions, our approach identified five interactions enriched in wt over Vif-deficient infections: ARIH1, ARIH2, AMBRA1, ALIX, and DCAF11 (Figure 3B). Notably, AMBRA1 and DCAF11, both substrate receptors of the CRL4 complex, have been previously shown to physically interact with Vif (Jager et al., 2011a; Y. Luo et al., 2016). A recent study from our lab discovered that AMBRA1 regulates the activity of CRL5 and CRL2 complexes by ubiquitination and degradation of ELOC (Chen et al., 2018). The four other factors, ARIH1, ARIH2, ALIX, and DCAF11, have either not been connected to Vif previously or their function with Vif is unknown. ALIX is a member of the ESCRT machinery known to regulate HIV budding (Fujii et al., 2009; Morita et al., 2011; Votteler and Sundquist, 2013) and appears to be recruited to CBFß and ELOB in a Vif-dependent manner (Figure 3C). DCAF11, also known as WDR23, interacts with all three bait proteins in a Vif-dependent manner (Figure 3C) and has been shown to be a component of the CUL4ADCAF11 E3 ligase, which ubiquitinates the stem-loop binding protein (SLBP) to regulate the life cycle of histone transcripts (Brodersen et al., 2016; Djakbarova et al., 2016).
ARIH1 and ARIH2, both members of the RBR E3 ligase family, were recently shown to interact with neddylated CRL complexes (Kelsall et al., 2013). NEDD8 and its covalently-linked Cullin are both required to relieve autoinhibition and expose the catalytic cysteines of the ARIH-family E3s (Duda et al., 2013; Kelsall et al., 2013). Additionally, an in vitro study recently identified a “tag-team” mechanism by which ARIH1 collaborates with neddylated CRL1, CRL2, and CRL3 E3s to transfer the first ubiquitin directly onto their substrate receptor-bound substrates (Scott et al., 2016). Interestingly, both ARIH1 and ARIH2 interact with CBFß in a Vif-dependent manner (Figure 3C), suggesting that Vif may use the tag-team mechanism together with ARIH1 and ARIH2 for substrates of the CUL2Vif/CBFß and CUL5Vif/CBFß E3 ligases, respectively. To investigate the potential impact on HIV infection of the four Vif-dependent host factors identified here, ARIH1, ARIH2, ALIX or DCAF11, we selected them for further functional validation.
ARIH2 is functionally relevant for HIV infection
To functionally validate the potential importance of Vif-dependent host factors, we used a recently developed platform for CRISPR-Cas9 genome engineering in primary CD4+ T cells (Hultquist et al., 2019; 2016; Schumann et al., 2015) (Figure 4A). Briefly, for each gene of interest, four different guide RNAs were designed, complexed with Cas9 and electroporated into primary CD4+ T cells derived from six different donors (Hultquist et al., 2019) (Figure 4A). The expanded polyclonal knockout (KO) T cells for each guide were infected with replication competent HIV-1 NL4-3 Nef-IRES Gfp reporter virus at a low MOI. Cells were collected three, five and seven days after infection, fixed and analyzed by flow cytometry to determine infection rates (Figure 4A). Besides the Vif-dependent factors identified here, we included genes with known HIV infection phenotypes upon KO in our validation screen. Amongst them were CXCR4, the co-receptor for HIV virus entry (Hultquist et al., 2016), and other CUL5vif/CBFß complex members, such as ELOB, ELOC, and CBFß (Jager et al., 2011b). KO efficiencies for all genes and all guides in polyclonal primary CD4+ T cells were evaluated by western blot analysis or tracking of indels by decomposition (TIDE) using PCR (Brinkman et al., 2014) (Figure S2A, S2B).
Figure 4. ARIH2 knockout in primary T cells leads to a reduced HIV infection rate.

(A) Workflow for functional validation of CUL5 factors using CRISPR knockouts followed by HIV infection in primary T cells, adapted from Hultquist et al (Hultquist et al., 2016).
(B) Relative HIV infection rate in primary T cells comparing CRISPR knockouts of selected genes to non-targeting control (day 5 of spread infection, NT = non-targeting control). Each gene was knocked out using 2-4 guide RNAs in 2-6 donors, infections for each donor and knockout were performed in triplicates.
(C) Western blots confirming ARIH2 knockout for all four guide RNAs in primary T cells from two donors and the corresponding HIV infection rate at day 5 of spread infection. For each donor spread HIV infections rate over seven days is shown for each ARIH2 guide RNA compared to controls, non-targeting guide RNA and CXCR4 knockout (n = 3, data from three replicate infections are presented as mean ± SEM, NT = non-targeting control).
See also Figure S2.
Out of the four factors, ARIH2 KO showed a strong and reproducible reduction in HIV infectivity after 5 days of spreading infection when compared to non-targeting (NT) controls (Figure 4B). Despite successful KO based on western blot (Figure S2A) or TIDE results (Figure S2B), no significant change in HIV infectivity was observed for ARIH1, ALIX and DCAF11 KO when compared to non-targeting (NT) controls (Figure 4B). The strength of ARIH2’s phenotype on HIV infectivity was comparable or even stronger than the one observed for other components of the CUL5vif/CBFß ligase, namely ELOB, ELOC and CBFß (Figure 4B). All four guide RNAs targeting ARIH2 resulted in a significant decrease in infection rate, which is reproducible across donors (Figure 4C,Figure S2C). The guide that was least effective in ablating ARIH2 protein levels also showed the smallest impact on HIV replication (Figure 4B, 4C, Figure S2C). While the spread infection data showed a moderate effect of ARIH2 KO on HIV infection rate at an early time point after HIV infection (day 3), the phenotype was most clear at day 5 of spreading infection (Figure 4C, Figure S2C). In contrast to CXCR4 KO, which affects initial infection, the primary T cell results for ARIH2 point to a function in later stages of the HIV life cycle, either by reducing the formation of newly formed virus particles or by decreasing their infection rate. Taken together, the results derived from the initial AP-MS experiments and the functional validation in primary CD4+ T cells suggest a central role for ARIH2 in CRL5 function during HIV infection.
ARIH2 tag-teams with CRL5Vif/CBFß to accelerate polyubiquitination of APOBEC3 proteins in vitro
To test the potential role of ARIH2 in CRL5Vif/CBFß-mediated APOBEC3 ubiquitination, we performed a series of in vitro ubiquitination assays considering factors required both for HIV restriction in host cells and potentially for the tag-team mechanism involving ARIH RBR E3s (Figure 5A). First, modification of CRL5 with the ubiquitin like protein NEDD8 is required for APOBEC3 ubiquitination and viral infectivity (Stanley et al., 2012). Second, ubiquitination of APOBEC3 proteins requires Vif, CBFß as well as a neddylated Cullin-RING core. From the perspective of ARIH RBR E3s, in vitro ARIH2’s catalytic cysteine is sequestered without encountering neddylated CUL5-RBX2, although mechanisms and function of catalytic cysteine release remain unknown (Kelsall et al., 2013). Accordingly, neddylated CRL5Vif/CBFß complex was reconstituted using purified recombinant proteins. A3G and A3F ubiquitination was assayed to test the activity of the reconstituted complex using the well characterized ubiquitin conjugating enzyme UBE2R1 (hCDC34), which is capable of forming specific K48-linked ubiquitin chains on A3G (Jager et al., 2011b) (Figure 5A). To test if ARIH2 functions together with neddylated CRL5Vif/CBFß to ubiquitinate A3G, we first performed an in vitro ubiquitination time course in the presence and absence of ARIH2 and the ubiquitin conjugating enzyme UBE2L3 (hUBCH7), which was previously shown to transfer ubiquitin to the active site of ARIH2 in the presence of neddylated CUL5-RBX2 (Kelsall et al., 2013) (Figure 5B). While A3G polyubiquitination was observed in both cases to a certain extent, polyubiquitination in the presence of ARIH2 and UBE2L3 was not only significantly enhanced but also substantially accelerated (Figure 5B). The rapid Ubiquitin (Ub) chain formation on A3G was specific for ARIH2 working together with CRL5Vif/CBFß, since replacing with the related ARIH1 in the same reaction did not show enhanced polyubiquitination of A3G (Figure 5C), and dependent on UBE2L3, which transfers Ub to ARIH2’s RING2 catalytic cysteine (Figure 5D).
Figure 5. ARIH2 monoubiquitinates APOBEC3G to prime it for accelerated polyubiquitination via CUL5Vif/CBFß in vitro.

(A) Schematic describing the in vitro ubiquitination reaction.
(B) Immunoblot showing time course for APOBEC3G ubiquitination reactions in the presence and absence of ARIH2/UBE2L3. In the presence of ARIH2 and UBE2L3, formation of extensive ubiquitination chains on APOBEC3G via neddylated CUL5vif/CBFß (N8~CRL5Vif/CBFß) is accelerated and enhanced compared to treatment with neddylated CUL5vif/CBFß and UBE2R1 alone.
(C) Acceleration of APOBEC3G ubiquitination is specific for ARIH2 and cannot be phenocopied by replacing ARIH2 with ARIH1 (Reaction 3).
(D) Acceleration of APOBEC3G ubiquitination is dependent on UBE2L3.
(E) ARIH2 monoubiquitinates A3G at multiple sites to prime for CRL5Vif/CBFß dependent Ub chain extension. Immunoblot showing APOBEC3G ubiquitination reactions in the presence and absence of UBE2R1, ARIH2 and CUL5vif/CBFß, Ub K48R, Methyl-Ub or wild-type Ub. Methyl-Ub (Reaction 4) and K48R-Ub (Reaction 5) recapitulate the pattern observed for the reaction that focuses on ARIH2 activity on A3G ubiquitination (Reaction 3, absence of UBE2R1).
(F) Ubiquitination sites on APOBEC3G derived from the in vitro assay in the presence of ARIH2 and UBE2L3 overlap with previously described APOBEC3G sites in cells (Albin et al., 2013).
See also Figure S3.
Since ARIH1 was described to prime CRL substrates for polyubiquitination through initial monoubiquitination (Scott et al., 2016), we next sought to examine if ARIH2 has the same effect on A3G. To this end, we performed the in vitro ubiquitination assay in the presence and absence of UBE2R1, which restricts the reaction only to ARIH2 activity. Additionally, we replaced wt Ub with Methyl-Ub or a K48R Ub mutant in the reaction, both of which are not able to form any Ub chain or K48-linked Ub chains, respectively, and therefore function as chain terminators. In the absence of UBE2R1, A3G polyubiquitination disappears and is instead replaced by enhanced multi-monoubiquitination (Figure 5E). Similar products were observed by replacing wt Ub with both Ub chain terminators (Figure 5E), suggesting that ARIH2 monoubiquitinated A3G in the presence of neddylated CUL5Vif/CBFß on multiple lysine residues. Finally, we mapped the A3G Ub sites obtained in our in vitro ubiquitination assay by MS and identified sites on K63, K249, K297, K303, and K334, which recapitulated Ub sites that have been previously detected on A3G in vivo (Figure 5F) (Albin et al., 2013). All in vitro ubiquitination reactions described for A3G were also performed for A3F, another potent HIV restriction factor of the APOBEC3 family which is degraded by CUL5Vif/CBFß during HIV infection (Hultquist et al., 2011), and the results mirror those observed for A3G (Figure S3).
Ablation of ARIH2 restores restriction activity of APOBEC3G
We have observed that ARIH2 is responsible for priming ubiquitination of A3G through CRL5 and that ablation of ARIH2 in primary CD4+ T cells results in a late block to infection. Next, we sought to determine if the infection phenotype observed in primary T cells upon ablation of ARIH2 is a direct result of decreased A3G ubiquitination and degradation. Based on our observations, we predicted that absence of ARIH2 would lead to higher cellular A3G levels and subsequent packaging into newly formed HIV-1 particles, leading to less infectious virions. To test this hypothesis, we then performed an assay to monitor packaging of A3G into virus particles in the presence and absence of ARIH2. We first stably depleted ARIH2 in HEK293T cells using a CRISPR-Cas9 approach based on guide RNA sequences validated in primary CD4+ T cells (Sanjana et al., 2014; Shalem et al., 2014). Next ARIH2 ablated cell lines as well as HEK293T parental cells were used to produce replication competent wt or ΔVif HIV-1 virus particles in the presence of A3G (Figure 6A). Increasing amounts of ARIH2 were co-transfected into the cells in order to rescue the phenotype. After 48h, viral supernatants were purified and used to infect CEM-Gfp reporter cells to assess virus titers (Figure 6A). Additionally, lysates of parental and ARIH2 ablated HEK293T cells as well as virus-like particles (VLPs) were collected concurrently to assay A3G expression and packaging.
Figure 6. ARIH2 collaborates with CUL5vif/CBFß to degrade APOBEC3G and enable HIV-1 infectivity.

(A) Schematic for APOBEC3G packaging assay. HEK293T parental and ARIH2 ko cells are transfected either with Vif-proficient or ΔVif containing HIV-1 NL4-3 proviral expression constructs, A3G, and ARIH2. After 48h, virus like particles (VLPs) are harvested and their titers are determined on CEM-Gfp reporter cells. Cells and VLPs are harvested and prepared for immunoblotting. HIV-infected CEM-Gfp cells are harvested after 48h and analyzed by flow cytometry to determine Gfp positive cells.
(B) Infectivity of Vif-proficient and ΔVif HIV-1 produced using two different HEK293T-ARIH2 KO cell lines as well as HEK293T parental cells transfected with proviral construct, A3G, and ARIH2 as indicated (n = 3; data from three replicate infections are presented as mean ± SEM; * < 0.05, ** < 0.01, *** < 0.001). Immunoblots are shown for the indicated proteins in virus-producing cells and VLPs.
In contrast to parental HEK293T cells, ARIH2 depleted cell lines showed increased packaging of A3G into VLPs generated from wt HIV-1, which led to 40% reduced infectivity of the VLPs (Figure 6B, condition 4). However, the amount of A3G packaged into wt HIV-1 virions produced in ARIH2 KO cell lines did not completely phenocopy the ΔVif HIV-1 virions in terms of A3G packaging and infectivity (Figure 6B, condition 1-3), indicating that A3G is still ubiquitinated and degraded in the absence of ARIH2 but to a much lesser degree. Cotransfection of increasing amounts of ARIH2 in KO cells fully rescued the A3G packaging and infectivity phenotype in both KO cell lines (Figure 6B, condition 5 and 6). For one ARIH2 KO cell line, this decrease in cellular A3G did not appear dose-dependent, likely due to the higher transfection efficiency of this clone and the high levels of exogenous expression relative to endogenous levels. For the highest amount of ARIH2 transfected, we observed a reduction in cellular levels of A3G even in ΔVif HIV-1 infected cells (Figure 6B, condition 3). Since ARIH2 modulates the activity of CRL5 broadly (see below), overexpression of ARIH2 could enhance endogenous regulatory mechanisms resulting in less A3G even in the absence of Vif. Notably, this was not reflected in the virus particles, potentially due to saturating levels of A3G. Alternately, it may be that A3G is only being depleted from a specific cellular compartment under these conditions and that the pool of A3G capable of packaging is unaffected. Indeed, recent studies have shown that part of the function of Vif is to sequester A3G away from sites of virus assembly (Binning et al., 2018), and that A3G must be in the right place at the right time to enter the virus particle. Regardless, the results of the A3G degradation and packaging assay in combination with the proteomic, in vitro, and primary cell data, strongly support a model wherein ARIH2 enhances vif-mediated degradation of A3G.
ARIH2 broadly regulates cellular CRL5-based ubiquitination
Our findings suggested that ARIH2 might use the same tagging cascade with CRL5Vif/CBFß for ubiquitinating and degrading APOBEC3 proteins during HIV infection as described for ARIH1 and CRL1-3 (Dove et al., 2017; Kelsall et al., 2013; Scott et al., 2016). Therefore, we hypothesized that ARIH2 might regulate ubiquitination of other CRL5-substrates. To investigate this hypothesis, we performed AP-MS experiments of ARIH2 wt and ARIH2 (C310S) mutant purified from HEK293T cells (Figure S3A). These experiments revealed that ARIH2 interacts with multiple components of CRL5 complexes, including CUL5 itself as well as multiple CUL5 substrate receptors such as ASB9 (Figure S3A, Table S3). Using in vitro ubiquitination assays, we showed that ARIH2 is required for the in vitro ubiquitination of the previously identified CUL5ASB9 E3 ligase substrate CKB (Debrincat et al., 2007) (Figure S3B, Figure S3C). We propose a model in which ARIH2 interacts with neddylated CRL5 to transfer the first Ub to multiple sites on a substrate, which is followed by polyubiquitination through the CRL5 E2 conjugating enzyme (Figure 7).
Figure 7. Model of how ARIH2 tag-teams with neddylated CRL5 complexes to ubiquitinate CRL substrates.

(SR = substrate receptor)
DISCUSSION
Accumulating evidence has pointed to a central role of the UPS in viral pathogenesis (H. Luo, 2016; Tang et al., 2018). Viral proteins subvert or manipulate the UPS by redirecting host cell E3 ligases in order to induce ubiquitination and degradation of antiviral restriction factors or to derail the immune response. Many viruses utilize Cullin RING E3 ligases (Huh et al., 2007; Jager et al., 2011a; Mahon et al., 2014; Sato et al., 2009), the largest family of E3 ligases, which form multi-subunit complexes subjected to various sources of regulation. While the core components of the CRL complexes hijacked by viral proteins and a number of substrates targeted for ubiquitination during viral infection are known (Mahon et al., 2014), the mechanisms by which Ub is actually transferred to targets has remained a major unanswered question.
Characterizing the mechanism of Ub attachment in virus-host systems is critically important for rational drug design strategies that target these processes. However, this is not a straightforward question as several mechanisms have been described including, but not limited to, the recruitment and use of a single E2 enzyme (Deshaies and Joazeiro, 2009). Sequential recruitment of two functionally distinct E2 enzymes, one for priming substrates by directly marking them with Ub and another for extending polyubiquitin chains, has been suggested for both CRL1 and CRL4 E3s (Lu et al., 2018; K. Wu et al., 2010). Furthermore, CRL1-3 were shown to utilize an E1-E2-E3/E3 tagging cascade where the RBR E3 ligase ARIH1 catalyzes addition of the first monoubiquitin moieties onto the substrate protein followed by polyubiquitin chain extension by the respective CRL E3 (Dove et al., 2017; Scott et al., 2016). Yet, which mechanism(s) work on endogenous substrates of CRLs remains largely unknown. As a result, the use of unbiased approaches to identify and characterize proteins mediating ubiquitination are often critical to uncovering their complex mechanisms.
While much is already known about the assembly of the CUL5Vif/CBFß complex during HIV infection to target and degrade the APOBEC3 proteins (Jager et al., 2011b; Mehle et al., 2004; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003; Zhang et al., 2011), the mechanics of ubiquitination and the identities of the enzymes employed to directly ligate Ub have remained unknown. To understand how CRL5 hijacked by HIV Vif mediates ubiquitination of APOBEC3 family members, we employed a combination of quantitative AP-MS, CRISPR-Cas9 gene ablation in primary CD4+ T cells, and in vitro ubiquitination assays. Using quantitative AP-MS of the CUL5Vif/CBFß complex in the context of HIV infection, we discovered that the RBR E3 ligase ARIH2 interacts with CUL5vif/CBFß in an HIV Vif-dependent manner (Figure 2, Figure 3). While ARIH2 has been previously shown to bind CRL5 in vitro, this study demonstrates the function of this interaction in a biological process in cells.
Using in vitro ubiquitination assays we could then show that in the presence of ARIH2 and UBE2L3 (hUBCH7), the physiological E2 for RBR-type E3 ligases (Kelsall et al., 2013), there is a rapid increase in the amount of polyubiquitinated A3G and A3F as well as acceleration of the reaction compared to neddylated CUL5Vif/CBFß and UBE2R1 (hCDC34) alone (Figure 5B, S5A). Our in vitro data suggest that this effect is driven by a tag-team mechanism in which ARIH2 together with UBE2L3 act as initiators, priming A3G and A3F bound to neddylated CUL5Vif/CBFß with monoubiquitination, followed by chain elongation through UBE2R1 (Figure 5D, 5E, S5C, S5D), a mechanism that is analogous to what was described for ARIH1 (Dove et al., 2017; Scott et al., 2016). Combined with the mapping of Ub sites on in vitro ubiquitinated A3G which recapitulate previous sites detected in cells (Figure 5F) (Albin et al., 2013) our results strongly suggest that this mechanism also leads to polyubiquitination of A3G in vivo. Beyond targeting APOBEC3 proteins during HIV infection, we further provided evidence that ARIH2 also tag-teams with other CRL5s to accelerate polyubiquitination of substrates, including CKB (Figure 7, Figure S3).
CRISPR-based knockouts of ARIH2 in primary CD4+ T cells resulted in about 50% reduced infectivity across all donors and guide RNAs, a phenotype that was at least as strong and consistent as that for ELOB, ELOC and CBFß in this same assay (Figure 4B). While other selected Vif-dependent factors -ARIH1, ALIX, and DCAF11 - did not show a significant effect on HIV infection rate in our assay (Figure 4B), these results do not imply that they are not bona fide Vif-dependent interactors. Rather, these results indicate that these factors are not essential for the replication of this virus in primary CD4+ T cells and exploration of these factors in other cell types or with other virus strains is warranted. While CD4+ T cells are the primary target of HIV infection, HIV infects other cells types, such as macrophages and dendritic cells, and in fact these cell types may comprise important reservoirs of latently infected cells that allow the virus to persist.
Based on the increased ubiquitination of APOBEC3 proteins in the presence of ARIH2 in vitro and the decreased replicative capacity of the virus in ARIH2 knock-out primary T cells, we hypothesized a mechanism wherein ARIH2 depletion results in reduced A3G ubiquitination, enhanced A3G packaging, and less infectious virus. Indeed, when we produced HIV virions in HEK293T cells lacking ARIH2, increased amounts of A3G were packaged and the infectivity of the virions was reduced (Figure 6B). HIV virions produced in ARIH2-ablated cells did not completely phenocopy virions produced from Vif-deficient HIV in terms of infectivity and A3G packaging, because APOBEC3 proteins can still be polyubiquitinated in the absence of ARIH2, but to a much lesser degree.
Taken together, we demonstrated that ARIH2 and CUL5Vif/CBFß work via a tag-team mechanism to target the APOBEC3 proteins for ubiquitination and that ARIH2 is therefore essential for HIV proliferation in primary CD4+ T cells (Figure 4 and 6). Importantly, this shows that an E1-E2-E3/E3-substrate cascade directly regulates a biological process, and demonstrates that the E1-E2-E3/E3-substrate cascade is hijacked by a virus to accelerate ubiquitination and degradation of antiviral restriction factors. While we believe that this mechanism plays a role for multiple CRL5 substrates, as suggested by association of ARIH2 with diverse CRL5 adaptors (Figure S3), we specifically demonstrate its importance for HIV, which hijacks the mechanism using Vif. A crucial element of the tag-teaming mechanism is a requirement for Cullin neddylation to relieve autoinhibition of the ARIH-family RBR (Scott et al., 2016). The employment of ARIH2 as a ubiquitin carrying enzyme provides a mechanistic rationale for the requirement of the distinctive neddylation pathway for CUL5 in HIV infectivity (Stanley et al., 2012).
To date, the studies of hijacking ubiquitination machinery have raised prospects of targeting, such as the interaction between APOBEC3 and Vif (Pery et al., 2015), but these demand the challenges of inhibiting protein-protein interactions, especially with broad shallow surfaces. However, enzyme activities represent more attractive targets. Our study raises the possibility that ARIH2 could be an attractive target for therapeutic intervention, because it is an enzyme, undergoes conformational activation, and selectively only targets CRL5. Furthermore, it is essential for the propagation of HIV infection, but our study as well as previous studies showed that ARIH2 is a non-essential gene in cells (Wang et al., 2015).
Finally, the approach that we used here combining quantitative AP-MS with functional testing through CRISPR-Cas9 genome engineering in primary cells can be adapted to other viruses or pathogens to help study the host complexes that they hijack. Insights from these types of studies promise not only to promote more comprehensive understanding of viral infections but also to provide therapeutic targets for intervention.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Nevan J. Krogan (nevan.krogan@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
VSVg-pseudotyped HIV-1 NL4-3 virus production.
ENV-deficient (ΔEnv) HIV-1 NL4-3 was generated using site-directed mutagenesis to delete the start codon and replace Env’s Tyr7 and Leu10 with stop codons. All Env mutations maintain identical coding sequence for overlapping Vpu open reading frame. Vif-deficient (ΔVif) HIV-1 NL4-3 was generated using site-directed mutagenesis to delete the start codon and replace Vif’s Lys22 and Arg23 with stop codons (Stanley et al., 2012). All Vif mutations maintain identical coding sequence for overlapping Pol open reading frame. A T175 flask HEK293T cells were transfected with 22.02ug of HIV-1 NL4-3 ΔEnv plasmid and 2.98ug pcDNA/VSVg using acidified PEI pH4 in lactate-buffered saline (LBS), which yielded a stoichiometric ratio of 3:1 provirus to envelope. Viral supernatant was collected 48 hours post transfection, and cleared by centrifugation and filtration through a 0.45 micron filter. Virus was precipitated by addition of sterile 50% PEG-6000 and 4M NaCl to final concentrations of 8.5% and 0.3M, respectively, followed by incubation at 4 degrees for two hours, centrifugation and resuspension in Phosphate buffered saline (PBS). Viral titer was quantified by titration on Jurkat T cells followed by fixation, staining with anti-HIV-1 Core Antigen Clone K57 FITC (Beckmann Coulter) and detection of HIV infected cells by flow cytometry.
HIV-1 NL4-3 Nef-IRES Gfp Virus production.
HIV NL4-3 Nef-IRES-GFP AS1 reporter virus (Schindler et al., 2003) was generated by transfection into 293T cells using Polyjet transfection reagent (SignaGen). Viral supernatant was collected 48 hours post transfection, and cleared by centrifugation and filtration through a 0.45 micron filter. Virus was precipitated by addition of sterile 50% PEG-6000 and 4M NaCl to final concentrations of 8.3% and 0.3M, respectively, followed by storage at 4 degrees for two hours, centrifugation and resuspension of 50X concentrated stocks. Viral titer was quantified by titration on stimulated primary CD4+ T cells followed by flow cytometry to detect GFP positive (HIV infected) cells.
Generation of stable Jurkat cells.
Jurkat TRex cells (Invitrogen) were cultured in RPMI/10%FBS plus Pen/Strep and 10 μg/ml Blasticidin (Gibco). Stable Jurkat cell clones were generated by electroporation with the linearized vector, limiting dilution followed by selection with 300μg/ml Zeocin (Invitrogen). Affinity tagged protein expression levels were tested by inducing expression with 1 μg/ml doxycycline for up to 16 h, collecting cell pellets, lysing them in 2xLaemmli Sample Buffer (Biorad) and boiling them for 20 min at 95 degrees. Cell lysates were analyzed by Western Blot analysis probing with antibodies against CUL5 (1:2000 dil., Bethyl), CBFß (1:500 dil., Santa Cruz), ELOB (1:500 dil., Abcam), and FLAG (1:5000 dil., Sigma-Aldrich). GAPDH (1:5000 dil., Sigma-Aldrich) was used as a loading control.
Generation of CRISPR knockout HEK293T cells.
Sequences for all guide RNAs targeting ARIH2 and validated in primary T cells were cloned into the LentiCRISPR2 vector containing two expression cassettes, for the Cas9 protein and the guide RNAs, as well as puromycin resistance (Sanjana et al., 2014; Shalem et al., 2014). Lentiviral constructs were transiently transfected into HEK293T cells using PolyJet transfection reagent (3ul PolyJet:1ug DNA) (SignaGen) and placed under 1 μg/ml puromycin (Sigma-Aldrich) selection to select successfully transfected cells. After 48h of selection, single ARIH2 depleted clones were generated for each guideRNA by limiting dilutions. Each clone was characterized by WB to assess reduction of ARIH2 expression and by sequencing to confirm gene editing on ARIH2.
Isolation of primary CD4+ T cells.
Human T Cell Isolation and Culture Leukoreduction chambers from healthy, anonymous donors were purchased from Blood Centers of the Pacific and processed within 12 hours. Primary CD4+ T-cells were harvested by positive selection using a FABian automated enrichment system and CD4 isolation kit (IBA Lifesciences, Goettingen, Germany). Isolated CD4+ T cells were suspended in complete Roswell Park Memorial Institute (RPMI) media, consisting of RPMI-1640 [UCSF Cell Culture Facility (CCF)] supplemented with 5mM 4-(2- hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, UCSF CCF), 2mM Glutamine (UCSF CCF), 50μg/mL penicillin/streptomycin (P/S, UCSF CCF), 5mM nonessential amino acids (UCSF CCF), 5mM sodium pyruvate (UCSF CCF), and 10% fetal bovine serum (FBS, Atlanta Biologicals). These cells were immediately stimulated on anti-CD3 coated plates [coated overnight with 10μg/mL αCD3 (UCHT1, Tonbo Biosciences)] in the presence of 5μg/mL soluble anti-CD28 (CD28.2, Tonbo Biosciences). Cells were stimulated for 48 hours prior to electroporation.
METHOD DETAILS
Cloning of affinity tagged constructs.
Open reading frames for CBFß, ELOB, and CUL5 were PCR amplified from templates available in house and ligated into the vector pcDNA4/TO (Invitrogen) carrying either a 5’ 3xFlag2xStrep or a 3’ 2xStrep3xFlag tag.
Infection of Jurkat cells and affinity purification.
For AP, 5x107 cells were induced with 1 μg/ml doxycycline for 16 h. Infections were performed by spinoculation with high MOI VSVg pseudotyped HIV-1 NL4-3 ΔEnv, HIV-1 NL4-3 ΔEnv ΔVif and mock in the presence of 1μg/ml Polyethylenimine (PEI) (Polysciences) and 16μg/ml Polybrene (Sigma-Aldrich). After 24h of infection, cells were treated 4h before harvesting and AP with 10μM MG132 to block proteasome activity and stabilize ligase-substrate interactions.
Following infection, cells were lysed in 500ul cold lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet P40, protease inhibitor (Roche) and phosphatase inhibitor (Roche), 5% Glycerol, 2mM COPS5 inhibitor 1,10-Phenathroline (Sigma-Aldrich)). The lysate was incubated with 30 μl anti-FLAG M2 Affinity Gel (Sigma-Aldrich) for 2h. The beads were washed 2x with lysis buffer containing 0.05% Nonidet P40 followed by three washes with lysis buffer without detergent and glycerol. Proteins were eluted with 30μl 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA containing either 100 μg/ml 3xFLAG peptide (ELIM) and 0.05% RapiGest (Waters). For MS analysis the AP eluates were reduced with 1 mM DTT at 37°C for 30 minutes then alkylated with 3 mM iodoacetamide for 45 minutes at room temperature, followed with quenching by addition of 3 mM DTT for 15 min. The samples were digested with sequencing grade modified trypsin (Promega) overnight at 37°C. The resulting peptides were cleaned up for MS analysis using Ultra Micro Spin C18 columns (The Nest Group). The final peptide sample was resuspended in 0.1% formic acid.
Shotgun mass spectrometric data acquisition.
Digested peptide mixtures were analyzed by LC-MS/MS on a Thermo Scientific Orbitrap Fusion mass spectrometry system equipped with a Thermo Scientific Easy nLC 1200 ultra high-pressure liquid chromatography and autosampler system. Samples were injected onto a C18 column (25 cm x 75 um I.D. packed with ReproSil Pur C18 AQ 1.9 μm particles) in 0.1% formic acid and then separated with an 80min gradient from 5% to 30% Buffer B (90% ACN/10% water/0.1% formic acid) at a flow rate of 300nl/min. The mass spectrometer collected data in a data-dependent fashion, collecting one full scan in the Orbitrap followed by collision-induced dissociation MS/MS scans in the dual linear ion trap for the most intense peaks from the full scan with a set maximum cycle time of 3 seconds. Dynamic exclusion was enabled for 20 seconds with a repeat count of 1. Charge state screening was employed to reject analysis of singly charged species or species for which a charge could not be assigned. The resulting data was analyzed using MaxQuant (version 1.5.2.8) for identification and quantification (Cox and Mann, 2008). SAINTexpress was applied to score protein networks components of CUL5, CBFß, and ELOB (H. Choi et al., 2011) (see QUANTIFICATION AND STATISTICAL ANALYSIS).
Targeted mass spectrometric data acquisition.
SRM assays were generated for selected interactors of CUL5, CBFß and ELOB. SRM assay generation was performed using Skyline (Maclean et al., 2010). For all targeted proteins, proteotypic peptides and optimal transitions for identification and quantification were selected based on the Skyline spectral library generated from the shotgun MS experiments. For each protein 2-5 peptides were selected based on intensity, peptide length as well as chromatographic performance. For each peptide the 4 best SRM transitions were selected based on intensity and peak shape.
Digested peptide mixtures were analyzed by LC-SRM on a Thermo Scientific TSQ Quantiva MS system equipped with a Proxeon Easy nLC 1200 ultra high-pressure liquid chromatography and autosampler system. Samples were injected onto a C18 column (25 cm x 75 μm I.D. packed with ReproSil Pur C18 AQ 1.9 μm particles) in 0.1% formic acid and then separated with an 80 min gradient from 5% to 40% Buffer B (90% ACN/10% water/0.1% formic acid) at a flow rate of 300 nl/min. SRM acquisition was performed operating Q1 and Q3 at 0.7 unit mass resolution. For each peptide the best 4 transitions were monitored in a scheduled fashion with a retention time window of 4 min and a cycle time fixed to 2 sec. Argon was used as the collision gas at a nominal pressure of 1.5 mTorr. Collision energies were calculated by, CE = 0.0348 * (m/z) + 0.4551 and CE = 0.0271 * (m/z) + 1.5910 (CE, collision energy and m/z, mass to charge ratio) for doubly and triply charged precursor ions, respectively. RF lens voltages were calculated by, RF = 0.1088 * (m/z) + 21.029 and RF = 0.1157 * (m/z) + 0.1157 (RF, RF lens voltage and m/z, mass to charge ratio) for doubly and triply charged precursor ions, respectively. The resulting data was analyzed with Skyline for identification and quantification of peptides (Maclean et al., 2010). MSstats was used for statistical analysis (M. Choi et al., 2014). The resulting protein interaction network was visualized using Cytoscape (Shannon et al., 2003) (see QUANTIFICATION AND STATISTICAL ANALYSIS).
Protein expression and purification for in vitro ubiquitination assays.
For experiments monitoring ubiquitination of A3G, all proteins except A3G were expressed in E. coli BL21 (DE3) cells, grown at 37°C to optical density ~0.6, and induced overnight at 18°C with 1mM IPTG. ARIH2 and UBE2L3 were expressed in a pGEX6p1 vector in E. coli BL21 (DE3) as GST-3C fusion proteins, purified by GST affinity chromatography, ion exchange chromatography, cleaved overnight at 4°C with 3C protease, purified by size exclusion chromatography, and stored in 25mM Hepes pH8, 200mM NaCl, 1mM DTT. The following proteins were purified as described previously: ARIH1 (Scott et al., 2016), UBE2R1 (Jager et al., 2011b), S. pombe Uba1 (Olsen and Lima, 2013) and NEDD8ylated Vif E3 (Jager et al., 2011b). C-terminal myc-tagged APOBEC3G was expressed in baculovirus infected Sf9 cells and purified as described previously (Binning et al., 2018).
For experiments monitoring ubiquitination of CKB, ARIH2, UBE2L3, UBE2F, NEDD8 E1 (APPBP1-UBA3), ubiquitin E1 (UBA1), NEDD8 and fluorescently labeled ubiquitin were prepared as previously described (Duda et al., 2013; D. T. Huang et al., 2009; Kelsall et al., 2013; Scott et al., 2014; Walden et al., 2003a; 2003b). CKB, ARIH2 were expressed in E.coli BL21 Gold (DE3) cells as either GST-Thrombin or GST-TEV fusion proteins. GST-RBX2-CUL5 was expressed in insect cells. These proteins were purified from cell lysates by glutathione affinity chromatography, and the GST was liberated by either TEV or thrombin cleavage overnight at 4°C. Cleavage products were further purified by ion exchange and ultimately by size exclusion chromatography on a Superdex SD200 column in 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT.
ASB9 with a TEV-cleavable C-terminal His-tag was coexpressed with ElonginB (residues 1-118) and ElonginC (residues 17-112) in E.coli BL21 Gold (DE3) cells. The complex was purified from cell lysates by Ni affinity chromatography. Following overnight treatment with TEV protease at 4°C, the complex was further purified by ion exchange and size exclusion chromagraphy using a Superdex SD200 column, in 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT.
The CUL5-RBX2 complex was neddylated by mixing 12 μM CUL5-RBX2, 1 μM UBE2F, 0.2 μM APPBP1-UBA3, 25 μM NEDD8 in 25 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2, and 1 mM ATP. NEDD8 was added and reactions proceeded for 8 min at room temperature before quenching by adding 10 mM DTT. After microcentrifugation at 13K rpm for 10 min, the NEDD8~CUL5-RBX2 was purified using a Superdex SD200 column, in 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT.
In vitro ubiquitination assay.
Ubiquitination assays of A3G were performed at room temperature (22°C) in 50mM Tris-HCl (pH 7.5), 50mM NaCl, and 2.5mM MgCl2. Ubiquitination reactions comprised the ubiquitin activating system, 1.2mM ATP, 45microM wild type, K48R mutant, or methyl-ubiquitin (Boston Biochem), 1.2microM Uba1, 4.8uM UBE2R1, 1.8uM UBE2L3, in addition to 300nM NEDD8ylated HIV-1 NL4-3 Vif E3, 300nM ARIH1 or ARIH2, and 1microM myc-tagged APOBEC3G, with a final reaction volume of 10uL. Unless otherwise stated, reactions were quenched after 1 hour with 2xSDS loading dye and boiling. Ubiquitinated A3G was detected using a monoclonal anti-c-myc antibody (Sigma-Aldrich). Ubiquitination of CKB was performed in pulse-chase format. Prior to the assay, E3 mixes were made by incubating NEDD8~CUL5-RBX2, ARIH2, and/or ASB9-ElonginB-ElonginC on ice for 30 min in 25 mM Tris pH 7.5, 50mM NaCl. The thioester-linked UBCH7~UB intermediate was generated by incubating 10 μM UBCH7, 15 μM fluorescently labeled UB, 400 nM UBA1 in 50mM HEPES, 100mM NaCl, 2.5mM MgCl2, 1mM ATP, pH 7 for 15 min at room temperature. This reaction was quenched by diluting the UBCH7~UB thioester conjugate to 0.6 μM in 50 mM Tris, 50 mM NaCl, 50 mM EDTA, 0.5 mg/ml BSA, pH 7.5 on ice. The chase reaction was performed by combining E3 mixes, UBCH7~UB, and/or CKB at final concentraions of 0.5 μM E3s, 0.3 μM, and 2 μM, respectively. Aliquots were terminated at indicated times with 2X SDS-PAGE sample buffer. Reaction products were separated on 4%-12% NuPAGE gels (Invitrogen) and visualized by scanning on a Typhoon imager (GE).
Detection of ubiquitination sites on A3G.
In vitro ubiquitination reaction for APOBEC3G was performed in the presence of ARIH2 and its E2 ligase UBE2L3 (see In vitro ubiquitination assays). For MS analysis the result of the reaction was reduced with 1 mM DTT at 37°C for 30 minutes followed by alkylation with 3 mM iodoacetamide for 45 minutes at room temperature, which was quenched by addition of 3 mM DTT for 15 min. The sample was digested with sequencing grade modified trypsin (Promega) overnight at 37°C. The resulting peptides were cleaned up for MS analysis using Ultra Micro Spin C18 columns (The Nest Group). The final peptide sample was suspended in 20 μl 0.1% formic acid. The sample was analyzed by MS as described in “Shotgun mass spectrometric data acquisition” and data analysis for identification of ubiquitination sites was performed using Maxquant as described in “Protein identification and quantification from shotgun proteomics” with addition of the di-Glycine variable modification on Lysine.
CRISPR-Cas9 mediated knockout in primary CD4+ T cells.
Electroporation was performed using the Amaxa P3 Primary Cell 96-well Nucleofector kit and 4D-Nucleofecter (Lonza). Recombinant S. pyogenes Cas9 protein used in this study contains two nuclear localization signal (NLS) peptides that facilitate transport across the nuclear membrane. The protein was expressed and purified as described (Anders and Jinek, 2014) and obtained from the QB3 Macrolab, University of California, Berkeley. Purified Cas9 protein was stored in 20 mM HEPES at pH 7.5 plus 150mM potassium chloride, 10% glycerol, and 1mM tris(2-carboxyethyl)phosphine (TCEP) at −80°C. crRNA for each gene were designed by Dharmacon. Each crRNA and the tracrRNA were chemically synthesized (Dharmacon) and suspended in 10mM Tris-HCl pH 7.4 to generate 160μM RNA stocks. Cas9 RNPs were prepared fresh for each experiment. crRNA and tracrRNA were first mixed 1:1 and incubated 30 minutes at 37°C to generate 80μM crRNA:tracrRNA duplexes. An equal volume of 40μM S. pyogenes Cas9-NLS was slowly added to the crRNA:tracrRNA and incubated for 15 minutes at 37°C to generate 20μM Cas9 RNPs. For each reaction, roughly 1x10^5 stimulated T cells were pelleted and suspended in 20μL P3 buffer. 4μl 20μM Cas9 RNP mix was added directly to these cells and the entire volume transferred to the 96-well reaction cuvette. Cells were electroporated using program EH-115 on the Amaxa 4D-Nucleofector (Lonza). 80μL pre-warmed complete RPMI was added to each well and the cells were allowed to recover for 30 minutes at 37°C. Cells were then re-stimulated using CD2/CD3/CD28 flow cytometry-compatible stimulation beads (Miltenyi Biotec, Auburn, CA, USA) in complete RPMI supplemented with 80 U/mL IL-2-IS (Miltenyi Biotec) and cultured in 96-well V-bottom dishes. All subsequent culturing of primary CD4+ T cells was performed in complete RPMI with 80 U/mL IL-2-IS. Two days post-electroporation media was changed on the cells, and four days post-electroporation cells were split by replica plating into three identical 96-well dishes. Seven days post-electroporation two plates were washed in PBS and banked for genomic DNA purification and western blot analysis respectively, and the third plate was used for low MOI HIV infection.
Low MOI HIV Infection of primary CD4+ T cells and FACS analysis.
Cells were replica plated in technical triplicate in U-bottom 96-well plates, and sufficient HIV NL4-3 Nef-IRES-GFP AS1 reporter virus (Schindler et al., 2003) was added to produce 0.5-2% infection in the first round of infection (according to titration data generated in previous donors) along with sufficient supplemented RPMI + 80 U/mL IL-2 to bring the total volume to 200μL per well. 3, 5, and 7 days post-infection, 100uL of cells were moved to a separate 96-well plate and fixed in 70uL of 4% formaldehyde. After collection of each time-point, fresh media was added to top-up media to 200uL. Flow cytometry was performed on a Becton Dickinson FACSCanto II (BD Biosciences) through the UCSF Laboratory for Cell Analysis. Analysis of flow cytometry data was performed using FlowJo flow cytometry analysis software (version 10.3.0). Quantification was done by first gating the live cell population, followed by gating on the GFP+ cells.
Packaging assay and infectivity measurements.
Vif-deficient (ΔVif) HIV-1 NL4-3 was generated using site-directed mutagenesis to delete the start codon and replace Vif’s Lys22 and Arg23 with stop codons (Stanley et al., 2012). All Vif mutations maintain identical coding sequence for overlapping Pol open reading frame. HEK293T cells were transfected with Vif-proficient and ΔVif containing HIV-1 NL4-3 proviral expression constructs (1000ng) (Mulder et al., 2010), HA-tagged A3G (100ng), and 2xStrep-3xFlag-tagged ARIH2 (3 and 9 ng) using TransIT-293 transfection reagent (Mirus). After 48h, viral particles were harvested and their titers were determined on CEM-Gfp reporter cells (Gervaix et al., 1997). Cell and viral-particle lysates were prepared for immunoblotting using the following antibodies: anti-vif, anti-GAPDH, anti-ARIH2 (detection of endogenous and co-transfected ARIH2) and anti-p24. HIV-infected CEM-Gfp cells were harvested after 72h and prepared for flow cytometry by fixation in 1% paraformaldehyde-1xPBS. Gfp fluorescence was measured on an Attune NxT flow cytometer (Thermo Fisher Scientific). All data were analyzed using FlowJo flow cytometry analysis software (version 10.3.0). Quantification was done by first gating the live cell population, followed by gating on the Gfp+ cells. P-values were calculated using t test statistics implemented in Prism 7.
CompPASS Interaction Proteomics for ARIH2.
Interaction proteomics using the CompPASS platform was performed as described (Sowa et al., 2009) with modifications. Briefly, 293T Flp-In or 293T cells containing N-terminally FLAG-HA tagged ARIH2 C310S were induced for 24 hours with 0.5 μg/mL DOX. Cells were lysed in 50mM Tris pH 7.5, 150 mM NaCl, 0.5% NP40 with protease inhibitors to generate whole cell lysates. Clarified lysates were filtered through 0.45 μm filters and immunoprecipitated with 30 μL anti-FLAG magnetic beads per replicate (Sigma-Aldrich). Complexes were washed 4x with lysis buffer and 2x with PBS and eluted with FLAG peptide at room temperature. Elutions were subjected to disulfide bond reduction using DTT and alkylation with iodoacetamide followed by TCA precipitation. TCA-precipitated proteins were trypsinzed, purified with Empore C18 extraction media (3M) and analyzed via LC-MS/MS with a LTQ-Velos linear ion trap mass spectrometer (Thermo) with an 18 cm3 125 μm (ID) C18 column and a 50 min 8%–26% acetonitrile gradient. Complexes were analyzed twice by LC-MS to generate technical duplicates. Spectra were searched with Sequest against a target-decoy human tryptic UNIPROT-based peptide database, and these results were loaded into the Comparative Proteomics Analysis Software Suite (CompPASS) (Sowa et al., 2009), to identify high confidence candidate interacting proteins (HCIPs). Individual experiments were analyzed using a stats table derived from analogous AP-MS data for 41 unrelated proteins to determine normalized weighted D-scores (NWD-score) and Z-scores based on spectral counts. To identify bait-associated proteins, proteins were filtered at a 2% false discovery rate for those with a NWD-score ≥ 1.0 and a Z-score ≥ 4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Protein identification and quantification from shotgun proteomics.
The raw data was analyzed using the MaxQuant algorithm (version 1.5.2.8) for the identification and quantification of peptides and proteins (Cox and Mann, 2008). Data were searched against a database containing SwissProt Human (downloaded 09/2017) sequences and HIV-1 NL4-3 sequences, concatenated to a decoy database where each sequence was randomized in order to estimate the false discovery rate (FDR). Variable modifications were allowed for methionine oxidation and protein N-terminus acetylation. A fixed modification was indicated for cysteine carbamidomethylation. Full trypsin specificity was required. The first search was performed with a mass accuracy of +/− 20 parts per million and the main search was performed with a mass accuracy of +/− 4.5 parts per million. A maximum of 5 modifications and 2 missed cleavages were allowed per peptide. The maximum charge was set to 7+. Individual peptide mass tolerances were allowed. For MS/MS matching, the mass tolerance was set to 0.8 Da and the top 8 peaks per 100 Da were analyzed. MS/MS matching was allowed for higher charge states, water and ammonia loss events. The data were filtered to obtain a peptide, protein, and site-level false discovery rate of 0.01. The minimum peptide length was 7 amino acids. Results were matched between runs with a time window of 2 minutes for biological replicates.
Scoring of specific interactors for CUL5, CBFß, and ELOB.
The following experimental design was used for the AP-MS experiments: For all baits and controls we prepared four biological replicates of each infection condition. Jurkat TRex parental cells (not expressing any affinity tagged bait) were used as control condition and were processed and analyzed in parallel with the bait protein expressing cell lines in order to avoid batch effects. Protein spectral counts were extracted from each AP sample and each biological replicate. Finally, we use SAINTexpress (Significance Analysis of INTeractome) to compare spectral counts for each bait AP sample and infection condition to the control AP sample and to assign confidence scores to observed PPIs (H. Choi et al., 2011; 2012). Protein spectral counts for each sample were calculated as the sum from the peptide spectral counts for one protein in a given sample. To discriminate bona fide protein interactors of CUL5, CBFß, and ELOB for each infection condition from the control, we set a FDR threshold of 0.15. To generate an overall list of candidate interactors for each bait, we combined the proteins with an FDR below 0.15 for each infection condition. The candidates were subsequently validated by targeted MS.
Protein quantification from targeted proteomics data.
SRM data was processed using Skyline (Maclean et al., 2010). Protein significance analysis was performed using MSstats (M. Choi et al., 2014). For defining specific interactors each protein was tested for abundance differences in bait protein APs under different infection conditions compared to control APs. Proteins with an adjusted P value < 0.05 and log2FC > 1 (in case of ELOB log2FC > 3) were considered specific interactors. To determine Vif-dependent interactors of CUL5, CBFß, and ELOB normalization across samples was conducted based on each bait protein as global standard protein. Each protein was tested for abundance differences comparing bait APs under HIV wt infection to HIV ΔVif infection. Proteins with an adjusted P value < 0.05 and log2FC > 1 were considered significant. Model-based sample quantification implemented in MSstats was used to calculate the intensity of each protein in each biological sample and replicate combining all SRM transition intensities.
Gene Ontology analysis.
Gene ontology enrichment for biological function of CUL5, CBFß, and ELOB interactors was performed using DAVID Bioinformatics Resources 6.8 (D. W. Huang et al., 2009a; 2009b).
DATA AND SOFTWARE AVAILABILITY
Shotgun proteomics data access.
RAW data and database search results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009012 (Jones et al., 2006; Vizcaíno et al., 2014). The data can be accessed using this link: https://www.ebi.ac.uk/pride/archive/projects/PXD009012.
Targeted proteomics data access.
Raw data and SRM transition files can be accessed, queried, and downloaded via Panorama (https://panoramaweb.org/) (Sharma et al., 2014) using the following link https://panoramaweb.org/project/UCSF%20-%20Krogan%20Lab/CRL5_HIV_interactome/begin.view?.
Supplementary Material
Table S1. Generation of CUL5-CBFß-ELOB Protein Interaction Network, Related to Figure 2
(A) SAINT protein interaction network scoring for CUL5, CBFß, and ELOB in the context of HIV wt infection.
(B) SAINT protein interaction network scoring for CUL5, CBFß, and ELOB in the context of Vif-deficient HIV infection.
(C) SAINT protein interaction network scoring for CUL5, CBFß, and ELOB in the context of mock infection.
(D) MSstats analysis for targeted proteomics validation of CUL5 interactors.
(E) MSstats analysis for targeted proteomics validation of ELOB interactors.
(F)MSstats analysis for targeted proteomics validation of CBFß interactors.
Table S2. Vif-dependent changes on CUL5-CBFß-ELOB Protein Interaction Network, Related to Figure 3
(A) MSstats results for Vif-dependent changes on CUL5 interactors.
(B) MSstats results for Vif-dependent changes on ELOB interactors.
(C) MSstats results for Vif-dependent changes on CBFß interactors.
Table S3. Protein interactor scoring for ARIH2 using CompPASS, Related to Figure 7.
Table S4. List of oligonucleotides used in this study, Related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE |
SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CUL5 (rabbit) | Bethyl | Cat# A302-173A |
| Anti-CBFß (rabbit) | Santa Cruz Biotechnology | Cat# sc-56751 |
| Anti-ELOB (rabbit) | Abcam | Cat# ab154854 |
| Anti-ARIH2 (rabbit) | Abcam | Cat# EPR7670 |
| Anti-ALIX (mouse) | Biolegend | Cat# 634502 |
| Anti-HA (mouse) | Covance | Cat# 16B12 |
| Anti-p24 (mouse) | NIH AIDS Reagent Program | Cat# 6521 |
| Anti-vif (rabbit) | NIH AIDS Reagent Program | Cat# 809 |
| Anti-HIV-1 Core Antigen Clone KC57 FITC (mouse) | Beckmann Coulter | Cat# 41116015 |
| Anti-GAPDH (mouse) | Sigma-Aldrich | Cat# G8795-100UL |
| Anti-Flag Hrp (mouse) | Sigma-Aldrich | Cat# A8592-1MG |
| Anti-APOBEC3G (mouse) | NIH AIDS Reagent Program | Cat# 10069 |
| Anti-Rabbit Hrp (goat) | Bio-RAD | Cat# 170-6515 |
| Anti-Mouse Hrp (goat) | Bio-RAD | Cat# 172-1011 |
| Anti-Goat Hrp (rabbit) | Bio-RAD | Cat# 172-1034 |
| Anti-c-myc | Sigma-Aldrich | Cat# M4439 |
| Anti-CD3 (UCHT1) | Tonbo Biosciences | Cat# 70-0038 |
| Anti-CD28 (CD28.2) | Tonbo Biosciences | Cat# 70-0289 |
| Bacterial and Virus Strains | ||
| HIV-1 NL4-3 ΔEnv | This study | N/A |
| HIV-1 NL4-3 ΔEnv ΔVif | This study | N/A |
| HIV-1 NL4-3 | Mulder et al., 2010 | N/A |
| HIV-1 NL4-3 ΔVif | Stanley et al., 2012 | N/A |
| HIV-1 NL4-3 Nef-IRES Gfp | Schindler et al., 2003; NIH AIDS Reagent Program | Cat# 11349 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Anti-FLAG® M2 Magnetic Beads | Sigma | Cat# M8823 |
| 3xFlag peptide | ELIM Biopharm | Custom order |
| RapiGest SF Surfactant | Waters | Cat# 186001861 |
| Sequencing grade trypsin | Promega | Cat# V5113 |
| MG132 | Calbiochem | Cat# 474790 |
| PolyJet transfection reagent | SigmaGen Laboratories | Cat# SL100688 |
| TransIT-293 Transfection Reagent | Mirus | Cat# MIR2700 |
| PhosStop™ phosphatase inhibitors | Roche | Cat# 04906837001 |
| cOmplete™ Protease Inhibitor Cocktail | Roche | Cat# 11836153001 |
| Cas9 protein | QB3 Macrolab, University of California, Berkeley | Custom order |
| Blasticidin | Gibco | Cat# R210-01 |
| Zeocin | Invitrogen | Cat# R250-01 |
| Doxycycline | Clontech | Cat# 631311 |
| Human IL-2 IS | Miltenyi Biotec | Cat# 130-097-745 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| 1,10-Phenanthroline | Sigma-Aldrich | Cat# 131377 |
| Polybrene (Hexadimethrine bromide) | Sigma-Aldrich | Cat# H9268 |
| Polyethylenimine (PEI) | Polysciences | Cat# 23966-1 |
| Deposited Data | ||
| Shotgun proteomics RAW and analyzed data | This study | https://www.ebi.ac.uk/pride/archive/projects/PXD009012 |
| Targeted proteomics RAW and analyzed data | This study | https://panoramaweb.org/project/UCSF%20-%20Krogan%20Lab/CRL5_HIV_interactome/begin.view? |
| Experimental Models: Cell Lines | ||
| T-REx Jurkat Cell Line | Jager et al., 2011a | N/A |
| 293T (ATCC CRL-3216) | ATCC | Cat# CRL-3216 |
| CUL5-2xStrep-3xFlag T-REx Jurkat Cell Line | This study | N/A |
| CBFß-2xStrep-3xFlag T-REx Jurkat Cell Line | This study | N/A |
| ELOV-2xStrep-3xFlag T-REx Jurkat Cell Line | This study | N/A |
| 293T ARIH2 KO cell line clone #1 | This study | N/A |
| 293T ARIH2 KO cell line clone #2 | This study | N/A |
| CEM Gfp cells | NIH AIDS Reagent Program, (Gervaix et al., 1997) | Cat# 3655 |
| Oligonucleotides | ||
| See Table S4 for all oligonucleotides | ||
| Recombinant DNA | ||
| pcDNA4: CUL5 | This study | N/A |
| pcDNA4: CBFß | This study | N/A |
| pcDNA4: ELOB | This study | N/A |
| CRISPR lenti: ARIH2 guide 1 | This study | N/A |
| CRISPR lenti: ARIH2 guide 2 | This study | N/A |
| pGEX-TEV-UBE2L3 (UBCH7) | Duda et al., 2013 | N/A |
| pGEX-TEV-ARIH2 | Kelsall et al., 2013 | N/A |
| pGEX2TK UB (C) | Scott et al., 2014 | N/A |
| pGEX NEDD8 | Walden et al., 2003a | N/A |
| pGEX APPBP1-UBA3 | Walden et al., 2003b | N/A |
| pGEX UBE2F | Huang et al., 2009 | N/A |
| pFastbac CUL5 | This study | N/A |
| pFastbac RBX2 | This study | N/A |
| pACYC Elongin C (17-112)-Elongin B (1-118) | This study | N/A |
| pET3a ASB9-TEV-His6 | This study | N/A |
| pGEX6p1 GST-3C-ARIH2 | This study | N/A |
| pGEX GST-TEV-ARIH1 | Duda et al., 2013 | N/A |
| pRSF Duet His6-GB1-TEV-CRL5/RBX2 | Jager et al., 2011b | N/A |
| pCDF Duet-1 Elongin B (1-118) Elongin C (17-112) | Jager et al., 2011b | N/A |
| pET Duet NL43 Vif/CBF-beta | Binning et al., 2018 | N/A |
| pET28b His6-thrombin-Sumo-Uba1 | Olsen and Lima 2013 | N/A |
| pHIS His6-TEV-UBE2R1 | Jager et al., 2011b | N/A |
| pGEX6p1 GST-3C-UBE2L3 | This study | N/A |
| pGEX27a His6-Ubiquitin | Binning et al., 2018 | N/A |
| pGEX-2TK GST-thrombin-PKA-NEDD8 | Huang et al., 2005 | N/A |
| pFastbac APOBEC3G-myc-TEV-GFPII | Binning et al., 2018 | N/A |
| pGEX-4T3 pGSTHsAPPBP1rbsUBA3 | Huang et al., 2005 | N/A |
| pHIS-TEV-UBE2F | Stanley et al., 2012 | N/A |
| Software and Algorithms | ||
| MaxQuant | Cox J et al., 2008 | http://www.coxdocs.org/doku.php?id=:maxquant:start |
| MSstats | M. Choi et al., 2014 | http://msstats.org |
| SAINTexpress | H. Choi et al., 2011 | http://saint-apms.sourceforge.net/ |
| Skyline | MacLean et al., 2010 | https://skyline.ms/project/home/software/Skyline/begin.view |
| Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Cytoscape | Shannon et al., 2003 | http://www.cytoscape.org/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| Other | ||
| Orbitrap Fusion Tribrid Mass Spectrometer | Thermo Fisher Scientific | Cat#IQLAAEGAAPFADBMBCX |
| TSQ Quantiva Triple Quadrupole Mass Spectrometer | Thermo Fisher Scientific | Cat#IQLAAEGAAXFAOUMZZZ |
| EASY-nLC 1200 System | Thermo Fisher Scientific | Cat#LC140 |
HIGHLIGHTS.
Quantitative proteomics identifies the E3 ligase ARIH2 as part of CUL5Vif/CBFß complex
CUL5Vif/CBFß complex recruits ARIH2 to transfer ubiquitin directly to APOBEC3
ARIH2 acts in an E1-E2-E3/E3 cascade to target degradation of restriction factor APOBEC3
ARIH2 is essential for efficient HIV infection in primary T cells
ACKNOWLEDGMENTS
R.H. is recipient of postdoctoral fellowships from the Swiss National Science Foundation (P2EZP3_148742; P300P3_151154), the European Molecular Biology Organization (ALTF 1123-2013), and the Human Frontiers in Science Program (LT000089/2014-L). R.H. was also supported by NIH funding for the UCSF-Gladstone Institute of Virology and Immunology Center for AIDS Research (CFAR; P30-AI027763). J.D.G., A.D.F., L.A.B., and D.C.C. were supported by NIH grant P50 GM082250. J.F.H. is supported by amfAR grant 109504-61-RKRL with funds raised by generationCURE, NIH grant K22 AI136691, a supplement from the NIH-supported Third Coast CFAR SP0029591. N.J.K. was supported from NIH grants P50 GM082250, U19 AI106754, P01 HL089707, P01 CA177332, U19 AI118610, R01 AI120694 and P01 AI063302. J.W.H. was supported by RO1 AG011085. B.A.S. was supported by R37GM069530, ALSAC/St. Jude, and the Max Planck Society. A.M. was supported in part by the NIH (NIGMS P50 GM082250 and NIDA DP2 DA042423). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub, has received funding from the Innovative Genomics Institute (IGI) and is a member of the Parker Institute for Cancer Immunotherapy (PICI).The Marson lab has received a gift from Gilead to support HIV cure research. The proteomics work was carried out in the Thermo Fisher Scientific Mass Spectrometry Facility for Disease Target Discovery at the J. David Gladstone Institutes. The virus experiments were performed in the BSL3 facility at the at the J. David Gladstone Institutes. We thank members of the Krogan lab for helpful advice and comments and Mike Shales for help with figure design.
Footnotes
DECLARATION OF INTERESTS
A.M. is a co-founder of Arsenal Biosciences, Spotlight Therapeutics, and Sonoma Biotherapeutics. A.M. serves as on the scientific advisory board of PACT Pharma and was a former advisor to Juno Therapeutics The Marson laboratory has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi and a gift from Gilead.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Albin JS, Anderson JS, Johnson JR, Harjes E, Matsuo H, Krogan NJ, Harris RS, 2013. Dispersed sites of HIV Vif-dependent polyubiquitination in the DNA deaminase APOBEC3F. J. Mol. Biol. 425, 1172–1182. doi: 10.1016/j.jmb.2013.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, and Jinek M (2014). In vitro enzymology of Cas9. Methods Enzymol. 546, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso T, Lane WS, Conaway JW, Conaway RC, 1995. Elongin (SIII): a multisubunit regulator of elongation by RNA polymerase II. Science 269, 1439–1443. [DOI] [PubMed] [Google Scholar]
- Batra J, Hultquist JF, Liu D, Shtanko O, Von Dollen J, Satkamp L, Jang GM, Luthra P, Schwarz TM, et al. (2018). Protein interaction mapping identifies RBBP6 as a negative regulator of Ebola virus replication. Cell 175, 1917–1930.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binning JM, Smith AM, Hultquist JF, Craik CS, Caretta Cartozo N, Campbell MG, Burton L, La Greca F, McGregor MJ, Ta HM, Bartholomeeusen K, Peterlin BM, Krogan NJ, Sevillano N, Cheng Y, Gross JD, 2018. Fab-based inhibitors reveal ubiquitin independent functions for HIV Vif neutralization of APOBEC3 restriction factors. PLoS Pathog. 14, e1006830. doi: 10.1371/journal.ppat.1006830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B, 2014. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168–e168. doi: 10.1093/nar/gku936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen MML, Lampert F, Barnes CA, Soste M, Piwko W, Peter M, 2016. CRL4(WDR23)-Mediated SLBP Ubiquitylation Ensures Histone Supply during DNA Replication. Mol. Cell 62, 627–635. doi: 10.1016/j.molcel.2016.04.017 [DOI] [PubMed] [Google Scholar]
- Chen S-H, Jang GM, Huttenhain R, Gordon DE, Du D, Newton BW, Johnson JR, Hiatt J, Hultquist JF, Johnson TL, Liu Y-L, Burton LA, Ye J, Reichermeier KM, Stroud RM, Marson A, Debnath J, Gross JD, Krogan NJ, 2018. CRL4AMBRA1 targets Elongin C for ubiquitination and degradation to modulate CRL5 signaling. EMBO J. 37, e97508. doi: 10.15252/embj.201797508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin Z-Y, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras A-C, Nesvizhskii AI, 2011. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73. doi: 10.1038/nmeth.1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Liu G, Mellacheruvu D, Tyers M, Gingras A-C, Nesvizhskii AI, 2012. Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr Protoc Bioinformatics Chapter 8, Unit8.15–8.15.23. doi: 10.1002/0471250953.bi0815s39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Chang C-Y, Clough T, Broudy D, Killeen T, MacLean B, Vitek O, 2014. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. doi: 10.1093/bioinformatics/btu305 [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cronin J, Zhang X-Y, Reiser J, 2005. Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther 5, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, et al. (2015). Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 57, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrincat MA, Zhang J-G, Willson TA, Silke J, Connolly LM, Simpson RJ, Alexander WS, Nicola NA, Kile BT, Hilton DJ, 2007. Ankyrin repeat and suppressors of cytokine signaling box protein asb-9 targets creatine kinase B for degradation. J. Biol. Chem. 282, 4728–4737. doi: 10.1074/jbc.M609164200 [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CAP, 2009. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434. doi: 10.1146/annurev.biochem.78.101807.093809 [DOI] [PubMed] [Google Scholar]
- Djakbarova U, Marzluff WF, Köseoğlu MM, 2016. DDB1 and CUL4 associated factor 11 (DCAF11) mediates degradation of Stem-loop binding protein at the end of S phase. Cell Cycle 15, 1986–1996. doi: 10.1080/15384101.2016.1191708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove KK, Kemp HA, Di Bona KR, Reiter KH, Milburn LJ, Camacho D, Fay DS, Miller DL, and Klevit RE (2017). Two functionally distinct E2/E3 pairs coordinate sequential ubiquitination of a common substrate in Caenorhabditis elegans development. Proc. Natl. Acad. Sci. USA 114, E6576–E6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Olszewski JL, Schuermann JP, Kurinov I, Miller DJ, Nourse A, Alpi AF, Schulman BA, 2013. Structure of HHARI, a RING-IBR-RING ubiquitin ligase: autoinhibition of an Ariadne-family E3 and insights into ligation mechanism. Structure 21, 1030–1041. doi: 10.1016/j.str.2013.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M, Zhang W, Gross AM, Von Dollen J, Johnson JR, Franks-Skiba KE, Swaney DL, Johnson TL, Jang GM, et al. (2018). Multiple routes to oncogenesis are promoted by the human papillomavirus-host protein network. Cancer Discov. 8, 1474–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberley ED, Mosadeghi R, Deshaies RJ, 2012. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. J. Biol. Chem. 287, 29679–29689. doi: 10.1074/jbc.M112.352484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enchev RI, Scott DC, da Fonseca PCA, Schreiber A, Monda JK, Schulman BA, Peter M, Morris EP, 2012. Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep 2, 616–627. doi: 10.1016/j.celrep.2012.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Scrima A, Böhm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, Iwai S, Gut H, Sugasawa K, Thomä NH, 2011. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147, 1024–1039. doi: 10.1016/j.cell.2011.10.035 [DOI] [PubMed] [Google Scholar]
- Fujii K, Munshi UM, Ablan SD, Demirov DG, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO, 2009. Functional role of Alix in HIV-1 replication. Virology 391, 284–292. doi: 10.1016/j.virol.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervaix A, West D, Leoni LM, Richman DD, Wong-Staal F, Corbeil J, 1997. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. U.S.A. 94, 4653–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson TJ, Seiler M, Veitia RA, 2013. The transience of transient overexpression. Nat. Methods 10, 715–721. doi: 10.1038/nmeth.2534 [DOI] [PubMed] [Google Scholar]
- Goff SP, 2007. Host factors exploited by retroviruses. Nat. Rev. Microbiol. 5, 253–263. doi: 10.1038/nrmicro1541 [DOI] [PubMed] [Google Scholar]
- Harper JW, Tan M-KM, 2012. Understanding cullin-RING E3 biology through proteomics-based substrate identification. Mol. Cell Proteomics 11, 1541–1550. doi: 10.1074/mcp.R112.021154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu T-H, Spindler KR, 2012. Identifying host factors that regulate viral infection. PLoS Pathog. 8, e1002772. doi: 10.1371/journal.ppat.1002772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF, Schulman BA, 2009. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol. Cell 33, 483–495. doi: 10.1016/j.molcel.2009.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Schulman BA, 2005. Expression, purification, and characterization of the E1 for human NEDD8, the heterodimeric APPBP1-UBA3 complex. Meth. Enzymol. 398, 9–20. doi: 10.1016/S0076-6879(05)98002-6 [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA, 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA, 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Huh K, Zhou X, Hayakawa H, Cho J-Y, Libermann TA, Jin J, Harper JW, Munger K, 2007. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 81, 9737–9747. doi: 10.1128/JVI.00881-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Hiatt J, Schumann K, McGregor MJ, Roth TL, Haas P, Doudna JA, Marson A, Krogan NJ, 2019. CRISPR-Cas9 genome engineering of primary CD4+ T cells for the interrogation of HIV-host factor interactions. Nat Protoc 14, 1–27. doi: 10.1038/s41596-018-0069-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS, 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234. doi: 10.1128/JVI.05238-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Schumann K, Woo JM, Manganaro L, McGregor MJ, Doudna J, Simon V, Krogan NJ, Marson A, 2016. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep 17, 1438–1452. doi: 10.1016/j.celrep.2016.09.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhain R, Soste M, Selevsek N, Röst H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Niméus-Malmström E, Rinner O, Aebersold R, 2012. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med 4, 142ra94–142ra94. doi: 10.1126/scitranslmed.3003989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D'Orso I, Fernandes J, Fahey M, Mahon C, O'Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ, 2011a. Global landscape of HIV-human protein complexes. Nature 481, 365–370. doi: 10.1038/nature10719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ, 2011b. Vif hijacks CBF-ß to degrade APOBEC3G and promote HIV-1 infection. Nature 481, 371–375. doi: 10.1038/nature10693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean Beltran PM, Federspiel JD, Sheng X, and Cristea IM (2017). Proteomics and integrative omic approaches for understanding host-pathogen interactions and infectious diseases. Mol. Syst. Biol 13, 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Côté RG, Martens L, Quinn AF, Taylor CF, Derache W, Hermjakob H, Apweiler R, 2006. PRIDE: a public repository of protein and peptide identifications for the proteomics community. Nucleic Acids Res. 34, D659–63. doi: 10.1093/nar/gkj138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane JR, Stanley DJ, Hultquist JF, Johnson JR, Mietrach N, Binning JM, Jónsson SR, Barelier S, Newton BW, Johnson TL, Franks-Skiba KE, Li M, Brown WL, Gunnarsson HI, Adalbjornsdóttir A, Fraser JS, Harris RS, Andrésdóttir V, Gross JD, Krogan NJ, 2015. Lineage-Specific Viral Hijacking of Non-canonical E3 Ubiquitin Ligase Cofactors in the Evolution of Vif Anti-APOBEC3 Activity. Cell Rep 11, 1236–1250. doi: 10.1016/j.celrep.2015.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsall IR, Duda DM, Olszewski JL, Hofmann K, Knebel A, Langevin F, Wood N, Wightman M, Schulman BA, Alpi AF, 2013. TRIAD1 and HHARI bind to and are activated by distinct neddylated Cullin-RING ligase complexes. EMBO J. 32, 2848–2860. doi: 10.1038/emboj.2013.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, Gross JD, 2013. CBFß Stabilizes HIV Vif to Counteract APOBEC3 at the Expense of RUNX1 Target Gene Expression. Mol. Cell 49, 632–644. doi: 10.1016/j.molcel.2012.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Weng S, Matyskiela M, Zheng X, Fang W, Wood S, Surka C, Mizukoshi R, Lu C-C, Mendy D, Jang IS, Wang K, Marella M, Couto S, Cathers B, Carmichael J, Chamberlain P, Rolfe M, 2018. UBE2G1 governs the destruction of cereblon neomorphic substrates. Elife 7, 6085. doi: 10.7554/eLife.40958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, 2016. Interplay between the virus and the ubiquitin-proteasome system: molecular mechanism of viral pathogenesis. Curr Opin Virol 17, 1–10. doi: 10.1016/j.coviro.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Jacobs EY, Greco TM, Mohammed KD, Tong T, Keegan S, Binley JM, Cristea IM, Fenyö D, Rout MP, Chait BT, Muesing MA, 2016. HIV-host interactome revealed directly from infected cells. Nat Microbiol 1, 16068. doi: 10.1038/nmicrobiol.2016.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon C, Krogan NJ, Craik CS, Pick E, 2014. Cullin E3 ligases and their rewiring by viral factors. Biomolecules 4, 897–930. doi: 10.3390/biom4040897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D, 2004. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18, 2861–2866. doi: 10.1101/gad.1249904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrashidi KM, Elwell CA, Verschueren E, Johnson JR, Frando A, Von Dollen J, Rosenberg O, Gulbahce N, Jang G, et al. (2015). Global mapping of the inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe 18, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sandrin V, McCullough J, Katsuyama A, Baci Hamilton I, Sundquist WI, 2011. ESCRT-III protein requirements for HIV-1 budding. Cell Host & Microbe 9, 235–242. doi: 10.1016/j.chom.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder LCF, Ooms M, Majdak S, Smedresman J, Linscheid C, Harari A, Kunz A, Simon V, 2010. Moderate influence of human APOBEC3F on HIV-1 replication in primary lymphocytes. J. Virol. 84, 9613–9617. doi: 10.1128/JVI.02630-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SK, Lima CD, 2013. Structure of a ubiquitin E1-E2 complex: insights to E1-E2 thioester transfer. Mol. Cell 49, 884–896. doi: 10.1016/j.molcel.2013.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn BH, Netter Z, Johnson JR, Von Dollen J, Jang GM, Johnson T, Ohol YM, Maher C, Bell SL, et al. (2018). An Mtb-human protein-protein interaction map identifies a switch between host antiviral and antibacterial responses. Mol. Cell 71, 637–648.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pery E, Sheehy A, Nebane NM, Brazier AJ, Misra V, Rajendran KS, Buhrlage SJ, Mankowski MK, Rasmussen L, White EL, Ptak RG, Gabuzda D, 2015. Identification of a novel HIV-1 inhibitor targeting Vif-dependent degradation of human APOBEC3G protein. J. Biol. Chem. 290, 10504–10517. doi: 10.1074/jbc.M114.626903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce NW, Lee JE, Liu X, Sweredoski MJ, Graham RLJ, Larimore EA, Rome M, Zheng N, Clurman BE, Hess S, Shan S-O, Deshaies RJ, 2013. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 153, 206–215. doi: 10.1016/j.cell.2013.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage HR, Kumar GR, Verschueren E, Johnson JR, Von Dollen J, Johnson T, Newton B, Shah P, Horner, et al. (2015). A combined proteomics/genomics approach links Hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 57, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma JM, Liu X, Reichermeier KM, Moradian A, Sweredoski MJ, Hess S, Deshaies RJ, 2017. Composition and Regulation of the Cellular Repertoire of SCF Ubiquitin Ligases. Cell 171, 1326–1339.e14. doi: 10.1016/j.cell.2017.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F, 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. doi: 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Kamura T, Shirata N, Murata T, Kudoh A, Iwahori S, Nakayama S, Isomura H, Nishiyama Y, Tsurumi T, 2009. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 5, e1000530. doi: 10.1371/journal.ppat.1000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler M, Würfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Münch J, Kirchhoff F, 2003. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J. Virol. 77, 10548–10556. doi: 10.1128/JVI.77.19.10548-10556.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, Marson A, 2015. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. U.S.A. 112, 10437–10442. doi: 10.1073/pnas.1512503112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Rhee DY, Duda DM, Kelsall IR, Olszewski JL, Paulo JA, de Jong A, Ovaa H, Alpi AF, Harper JW, Schulman BA, 2016. Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell 166, 1198–1214.e24. doi: 10.1016/j.cell.2016.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Sviderskiy VO, Monda JK, Lydeard JR, Cho SE, Harper JW, Schulman BA, 2014. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell 157, 1671–1684. doi: 10.1016/j.cell.2014.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, Johnson JR, Von Dollen J, Ramage HR, et al. (2018). Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell 175, 1931–1945.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PS, Wojcechowskyj JA, Eckhardt M, Krogan NJ, 2015. Comparative mapping of host-pathogen protein-protein interactions. Curr. Opin. Microbiol. 27, 62–68. doi: 10.1016/j.mib.2015.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. doi: 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Eckels J, Taylor GK, Shulman NJ, Stergachis AB, Joyner SA, Yan P, Whiteaker JR, Halusa GN, Schilling B, Gibson BW, Colangelo CM, Paulovich AG, Carr SA, Jaffe JD, MacCoss MJ, MacLean B, 2014. Panorama: a targeted proteomics knowledge base. J. Proteome Res. 13, 4205–4210. doi: 10.1021/pr5006636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH, 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9, 1404–1407. doi: 10.1038/nm945 [DOI] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW, 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403. doi: 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley DJ, Bartholomeeusen K, Crosby DC, Kim DY, Kwon E, Yen L, Cartozo NC, Li M, Jäger S, Mason-Herr J, Hayashi F, Yokoyama S, Krogan NJ, Harris RS, Peterlin BM, Gross JD, 2012. Inhibition of a NEDD8 Cascade Restores Restriction of HIV by APOBEC3G. PLoS Pathog. 8, e1003085. doi: 10.1371/journal.ppat.1003085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC, 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12, 591–601. doi: 10.1038/nri1236 [DOI] [PubMed] [Google Scholar]
- Tang Q, Wu P, Chen H, Li G, 2018. Pleiotropic roles of the ubiquitin-proteasome system during viral propagation. Life Sci. 207, 350–354. doi: 10.1016/j.lfs.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz P-A, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus H-J, Albar JP, Martinez-Bartolomé S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H, 2014. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226. doi: 10.1038/nbt.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votteler J, Sundquist WI, 2013. Virus budding and the ESCRT pathway. Cell Host & Microbe 14, 232–241. doi: 10.1016/j.chom.2013.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL, Holton JM, Schulman BA, 2003a. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell 12, 1427–1437. [DOI] [PubMed] [Google Scholar]
- Walden H, Podgorski MS, Schulman BA, 2003b. Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for NEDD8. Nature 422, 330–334. doi: 10.1038/nature01456 [DOI] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM, 2015. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. doi: 10.1126/science.aac7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Kawakami E, Shoemaker JE, Lopes TJS, Matsuoka Y, Tomita Y, Kozuka-Hata H, Gorai T, Kuwahara T, Takeda E, Nagata A, Takano R, Kiso M, Yamashita M, Sakai-Tagawa Y, Katsura H, Nonaka N, Fujii H, Fujii K, Sugita Y, Noda T, Goto H, Fukuyama S, Watanabe S, Neumann G, Oyama M, Kitano H, Kawaoka Y, 2014. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host & Microbe 16, 795–805. doi: 10.1016/j.chom.2014.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissing S, Galloway NLK, Greene WC, 2010. HIV-1 Vif versus the APOBEC3 cytidine deaminases: an intracellular duel between pathogen and host restriction factors. Mol. Aspects Med. 31, 383–397. doi: 10.1016/j.mam.2010.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WF, Kohu K, Chiba T, Sato T, Satake M, 2011. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology 132, 157–164. doi: 10.1111/j.1365-2567.2010.03381.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Kovacev J, Pan Z-Q, 2010. Priming and extending: a UbcH5/Cdc34 E2 handoff mechanism for polyubiquitination on a SCF substrate. Mol. Cell 37, 784–796. doi: 10.1016/j.molcel.2010.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhu W, Nhan T, Toth JI, Petroski MD, Wolf DA, 2013. CAND1 controls in vivo dynamics of the cullin 1-RING ubiquitin ligase repertoire. Nat Commun 4, 1642. doi: 10.1038/ncomms2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu X-F, 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302, 1056–1060. doi: 10.1126/science.1089591 [DOI] [PubMed] [Google Scholar]
- Zemla A, Thomas Y, Kedziora S, Knebel A, Wood NT, Rabut G, Kurz T, 2013. CSN-and CAND1-dependent remodelling of the budding yeast SCF complex. Nat Commun 4, 1641. doi: 10.1038/ncomms2628 [DOI] [PubMed] [Google Scholar]
- Zhang W, Du J, Evans SL, Yu Y, Yu X-F, 2011. T-cell differentiation factor CBF-ß regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481, 376–379. doi: 10.1038/nature10718 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Generation of CUL5-CBFß-ELOB Protein Interaction Network, Related to Figure 2
(A) SAINT protein interaction network scoring for CUL5, CBFß, and ELOB in the context of HIV wt infection.
(B) SAINT protein interaction network scoring for CUL5, CBFß, and ELOB in the context of Vif-deficient HIV infection.
(C) SAINT protein interaction network scoring for CUL5, CBFß, and ELOB in the context of mock infection.
(D) MSstats analysis for targeted proteomics validation of CUL5 interactors.
(E) MSstats analysis for targeted proteomics validation of ELOB interactors.
(F)MSstats analysis for targeted proteomics validation of CBFß interactors.
Table S2. Vif-dependent changes on CUL5-CBFß-ELOB Protein Interaction Network, Related to Figure 3
(A) MSstats results for Vif-dependent changes on CUL5 interactors.
(B) MSstats results for Vif-dependent changes on ELOB interactors.
(C) MSstats results for Vif-dependent changes on CBFß interactors.
Table S3. Protein interactor scoring for ARIH2 using CompPASS, Related to Figure 7.
Table S4. List of oligonucleotides used in this study, Related to STAR Methods.
Data Availability Statement
Shotgun proteomics data access.
RAW data and database search results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009012 (Jones et al., 2006; Vizcaíno et al., 2014). The data can be accessed using this link: https://www.ebi.ac.uk/pride/archive/projects/PXD009012.
Targeted proteomics data access.
Raw data and SRM transition files can be accessed, queried, and downloaded via Panorama (https://panoramaweb.org/) (Sharma et al., 2014) using the following link https://panoramaweb.org/project/UCSF%20-%20Krogan%20Lab/CRL5_HIV_interactome/begin.view?.