

Abstract
Crimean Congo hemorrhagic fever virus (CCHFV) causes an acute disease with the potential of a fatal outcome. The virus is prevalent in about 30 countries. Clinical symptoms of infection commonly include fever, myalgia, and hemorrhages. Levels of liver enzymes are raised, and bleeding markers are often increased. A role of inflammatory cytokines in the pathogenesis has been suggested, and CCHFV employs a range of passive and active mechanisms to avoid induction of the antiviral type I interferons. Here, we review the most recent findings on the molecular pathogenesis and the interaction of CCHFV with the type I interferon and cytokine responses and discuss implications for pathogenesis.
Keywords: Crimean Congo Hemorrhagic fever virus, Pathogenesis, Cytokines, Interferons, CCHFv
1. The Bunyaviridae family
The family Bunyaviridae is one of the largest virus groups comprising over 350 arthropod and rodent borne viruses [1]. The virions are enveloped and spherical with a diameter of approximately 100 nm. The virus family is divided into five genera, Orthobunyavirus, Phlebovirus, Hantavirus, Nairovirus and Tospovirus.
The genome of the bunyaviruses consists of three single-stranded RNA segments of negative polarity, designated as the small (S), medium (M) and large (L) segment [1], [2], [3]. The S segment codes for a nucleocapsid (N) protein, and for some members of the family also for a non-structural protein (NSs) [4], [5], [6], [7], [8]. The M segment encodes a precursor for the two envelope glycoproteins Gn and Gc, and in some cases also a non-structural protein (NSm) [9], [10], [11], [12]. The L segment of the Bunyaviridae family codes for the RNA-dependent RNA polymerase. The terminal nucleotides of these RNA segments are partially complementary in sequence and predicted to form a stable pseudo-circular “panhandle” structure.
The viral envelope glycoproteins Gn and Gc interact with specific receptors on the host cell. After attachment, the virus enters the cell by receptor-dependent endocytosis. Replication of bunyaviruses takes place in the cytoplasm. The N protein interacts with newly synthesized viral RNAs forming the ribonucleocapsids which, in turn, interact with the cytoplasmic part of Gn and Gc. These processes trigger the budding of virions into the Golgi compartment [1], [2].
2. Crimean Congo hemorrhagic fever virus
The Nairovirus genus includes 34 described viruses divided into seven different serotypes. Crimean Congo hemorrhagic fever virus (CCHFV) is the medically most important member of this genus [13].
CCHFV is the etiological agent of a human disease characterized by fever, prostration, severe hemorrhages and death. The first documented outbreaks were recorded in 1944 and 1945, where 200 cases of a severe hemorrhagic disease occurred in Russia. Later, the russian virus was shown to be antigenically identical to a virus isolated from a febrile patient in the Congo [13]. Nowadays, CCHFV is known to be widely distributed throughout large areas of sub-Saharan Africa, the Balcans, Northern Greece, European Russia, Pakistan, the Xinjiang province of Northwest China, the Arabian Peninsula, Turkey, Iraq and Iran [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24]. CCHFV is the second most widespread arbovirus of medical importance after Dengue virus, [25], [26]. CCHFV also infects animals, but these remain asymptomatic as shown for cattle, sheep, goats, camels and hares. Antibodies against CCHFV have been demonstrated in sera of horses, goats, and cattle in endemic areas [25], [26]. The virus can be transmitted to humans through ticks of the genus Hyalomma, in particular Hylomma marginatum marginatum [25], [26]. CCHFV has also been isolated from other Hyalomma species. Human infection also occurs by contact with blood or tissue material from infected animals or humans. In addition, person-to-person transmission can occur via bloody vomit, body fluids or by aerosol from patients in advanced stages of disease [27], [28], [29]. Therefore, risk groups are found among professions with contact to infected animals (e.g. livestock breeders, abattoir workers) or infected humans (e.g. health care workers) [30].
3. Disease and pathogenesis
Humans are the only known host that develops disease after infection with CCHFV. The infection usually results in a severe hemorrhagic fever. The disease progression is rapid and can be subdivided into four different stages (Fig. 1 ) [13]: incubation, pre-hemorrhagic, hemorrhagic and the convalescence phase. The incubation period ranges from a few days up to 1 week, the length most probably depending on the transmission route and the amount of inoculum. The pre-hemorrhagic phase usually initiates with fever, myalgia, dizziness, headache and vomiting and ends on average after 3 days. The hemorrhagic fever phase is short and characterized by epistaxis, bleeding from the gastrointestinal system, urinary and respiratory tract and also skin bleeding ranging from petechiae to ecchymoses. Other symptoms include enlarged spleen and liver which approximately 30% of patients experience. The average mortality rate is 30% but can be as high as 70% [31], [32], [33] The severity of disease has been shown to correlate to the amount of virus in the blood (up to 109 genome equivalents/ml blood) [34], [35]. Furthermore, It has been shown that antibody responses have a highly significant inverse correlation with viral loads.
Fig. 1.
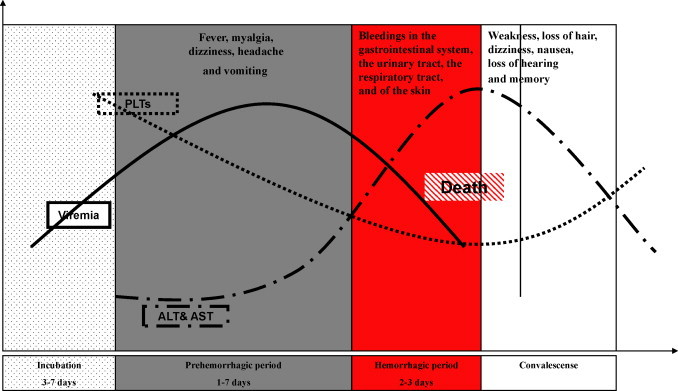
Clincal course of CCHF: The disease progression can be subdivided into four different stages with different symptoms; incubation, pre-hemorrhagic, hemorrhagic and the convalescence phase. Laboratory analyses demonstrate the elevated liver enzymes aspartate and alanine aminotransferase (AST and ALT) as well as low platelets count (PLTs) in the fatal cases.
Convalescence starts on average at 0–20 days post onset of illness. This phase is characterized by weakness, loss of hair, dizziness, nausea, loss of hearing, and loss of memory. It should be mentioned that duration and symptoms in these different phases vary significantly between individuals (Fig. 1).
The pathogenesis of CCHF is only poorly characterized due to several reasons such as (i) infections occur sporadically and in areas where facilities are limited for performing complete autopsies, (ii) virus handling requires biosafety level 4 (BSL-4) containment laboratories, and (iii) a lack of available animal models of disease.
The limited knowledge about CCHF pathogenesis is mostly derived from blood analyses and liver biopsies of patients, using materials e.g. from small outbreaks in Turkey [36], [37], [38], [39], [40]. The most comprehensive study involving 50 CCHF patients from South Africa was undertaken by Swanepoel et al. [31]. In their report, they describe that cerebral hemorrhage, severe anemia, severe dehydration, and shock associated with prolonged diarrhea, lung edema, and pleural effusion are the factors causing fatal outcome. Almost all patients who died developed multiple organ failure.
In the fatal cases, platelet counts can be extremely low from an early stage of illness on. Increases in aspartate and alanine aminotransferase (AST and ALT) levels in the serum, prolongation of prothrombin and partial thromboplastin times have also been observed [31].
4. Innate immunity – the interferon system
The most efficient and rapid host response against viruses consists of the production of type I interferons (IFN-α/β), an essential part of the antiviral innate immune system. As far as it is known, all nucleated cells of the mammalian body are able to synthesize and secrete type I IFNs. The mode of induction and the type of IFN being secreted, however, can differ among cell types. Secreted IFNs stimulate neighbouring cells to express potent antiviral proteins [41], [42], [43]. Besides their role as antiviral messengers, IFNs posses a wide range of other biological activities including inhibition of cell proliferation, regulation of apoptosis, and, importantly, immunomodulation [44], [45]. IFN production triggered by the first contact with the viral intruder slows down or even stops virus multiplication, buys the organism time, and helps to establish an adaptive immune response.
Type I IFNs are classified according to their amino acid sequence and comprise a large number (at least 13) of IFN-α subtypes and a single IFN-β [43], as well as some additional family members [46], [47], [48]. Expression patterns, i.e. which IFNs will be synthesized at which time point, mostly depend on the particular cell type.
4.1. IFN induction
Epithelial cells, fibroblasts and neurons mainly secrete IFN-β as an initial response to infection but switch to IFN-α during the subsequent amplification phase of the IFN response [49], [50]. In contrast, dendritic cells, which play an important role in immunosurveillance and provide an interface between innate and adaptive immunity, directly produce high levels of IFN-α subtypes [51].
IFN induction in fibroblasts occurs mainly by an intracellular pathway (Fig. 2 ). Hallmark molecules of RNA viruses such as double-stranded (ds) RNA and 5′-triphosphorylated single-stranded (ss) RNA trigger a signalling chain which activates IFN-β gene expression [52], [53], [54], [55]. Two RNA helicases, RIG-I and MDA-5, are the main intracellular receptors of viral RNA [56], [57], [58]. RIG-I and MDA-5 recognize different and non-overlapping sets of viruses, suggesting a degree of specificity in RNA recognition [59]. Indeed, it was recently found that RIG-I has the unique ability to bind the triphosphate groups on the 5′-end of uncapped viral ssRNA [53], [54], [55]. Moreover, RIG-I binds to short dsRNA molecules whereas MDA-5 activation is apparently more dependent on long dsRNA structures [60]. The binding of a viral RNA to RIG-I and MDA-5 induces a signalling chain which eventually results in the phosphorylation of the transcription factor IRF-3 [61]. IRF-3 is a member of the IFN regulatory factor (IRF) family [62] and plays a central role in the activation of the IFN-β promoter. Phosphorylated IRF-3 homo-dimerises and moves into the nucleus where it initiates IFN-β mRNA synthesis. This first-wave IFN triggers expression of a related factor, IRF-7, which in fibroblasts is only present in low amounts [63]. Expression and activation of IRF-7 leads to a positive-feedback loop that initiates the synthesis of several IFN-α subtypes as the second-wave IFNs. In addition, the transcription factors NF–κB (activated by RIG-I, MDA-5 and the dsRNA-dependent kinase PKR) and AP-1 (activated by stress-induced Jun kinase) are triggered by viral replication to enhance IFN gene expression [64], [65]
Fig. 2.
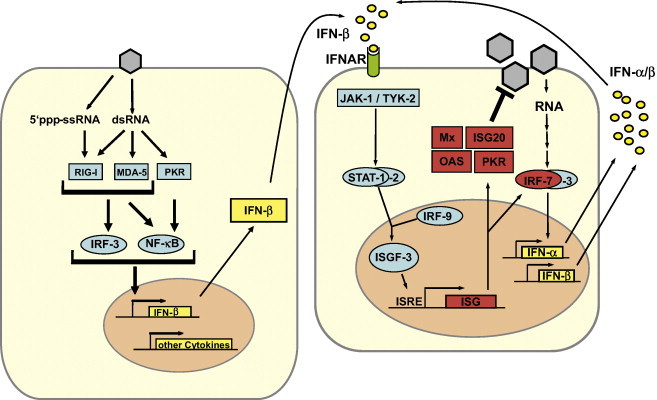
Type I IFN induction, signaling, and action. Left panel: 5′ triphosphate ssRNA and dsRNA, two hallmark by-products of virus replication, bind and activate the pattern recognition receptors RIG-I, MDA5 and PKR. A signal transduction chain involving various adapters and kinases (see text) culminates in activation of the transcription factors IRF-3 and NF-kB which, in turn, cooperatively transactivate the promoter of the IFN-β gene and other cytokine genes. Right panel: IFN-β binds to the type I IFN receptor (IFNAR) and activates the expression of numerous ISGs via the JAK/STAT pathway. IRF-7 amplifies the IFN response by inducing expression of several IFN subtypes. Mx, ISG20, OAS and PKR are examples of proteins with antiviral activity.
Myeloid dendritic cells (mDCs) [66] and, most prominently, plasmacytoid dendritic cells (pDCs) [51] are the main IFN producers of the lymphatic system. mDCs can sense dsRNA by the classic intracellular pathway [66] and, in addition, by the endosomal toll-like receptor (TLR) 3 [67] pDCs predominantly monitor RNA virus infections by the endosomal TLR7 and TLR8 which recognize ssRNA [68]. Activated TLRs signal through different intracellular adaptor molecules to induce IRF- and NF–κB-dependent IFN transcription (Fig. 2).
4.2. IFN signaling
All IFN-α/β subtypes bind to and activate a common type I IFN receptor which is present on virtually all host cells [69]. Binding of IFN-α/β leads to conformational changes in the intracellular parts of the receptor, which activate the so-called JAK-STAT signalling pathway. The signal transducer and activator of transcription (STAT) proteins are latent cytoplasmic transcription factors which become phosphorylated by the Janus kinase (JAK) family members JAK-1 and TYK-2 [70], [71]. Phosphorylated STAT-1 and STAT-2 recruit a third factor, IRF-9 (also called p48), to form a complex known as IFN stimulated gene factor 3 (ISGF-3). The ISGF-3 heterotrimer translocates to the nucleus and binds to IFN-stimulated response elements (ISRE) in the promoter regions of IFN-stimulated genes (ISGs), thereby inducing their transcription.
4.3. IFN effector proteins
IFN-α/β activate the expression of more than 300 IFN-stimulated genes (ISGs) which have antiviral, antiproliferative, and immunomodulatory functions [72]. IFN-induced proteins include enzymes, transcription factors, cell surface glycoproteins, cytokines, chemokines and a large number of factors with unknown function. Up to now, only a few proteins with antiviral activity have been characterized in detail. These are the Mx GTPases, the protein kinase R (PKR), the 2′,5′ oligoadenylate synthetases (2–5 OAS)/RNaseL system, the RNA-specific adenosine deaminase 1 (ADAR 1), viperin, and the products of the ISG56 (p56) and ISG20 genes. Mx proteins belong to the superfamily of dynamin-like large GTPases and have been discovered as mediators of genetic resistance against orthomyxoviruses in mice. The human MxA protein blocks replication of the infecting virus soon after cell entry by targeting and missorting viral ribonucleoprotein particles [73], [74]. PKR, 2–5 OAS and ADAR are constitutively expressed in a latent, inactive form. Basal mRNA levels are upregulated by IFN-α/β and these enzymes need to be activated by viral dsRNA. PKR is also activated by ssRNA containing a 5′ triphosphate group and a short stem-loop [75]. PKR is a serine-threonine kinase that phosphorylates the alpha subunit of the eukaryotic translation initiation factor eIF2 [76], thus blocking translation of cellular and viral mRNAs. The 2–5 OAS catalyses the synthesis of short 2′,5′ oligoadenylates [77] that activate the latent endoribonuclease RNaseL which in turn degrades both viral and cellular RNAs [78]. ADAR 1 catalyzes the deamination of adenosine on target dsRNAs to yield inosine. As a result the secondary structure is destabilized due to a change from an AU base pair to the less stable IU base pair and mutations accumulate within the viral genome [42]. Viperin is localized at the ER membrane and impairs the formation of lipid rafts which are need by some viruses for replication or budding [79], [80]. P56 binds the eukaryotic initiation factor 3e (eIF3e) subunit of the eukaryotic translation initiation factor eIF3. It functions as an inhibitor of translation initiation at the level of eIF3 ternary complex formation and is likely to suppress viral RNA translation [81]. ISG20 is an IFN-induced 3′,5′ exonuclease that specifically degrades ssRNA in vitro. In cell culture, expression of ISG20 leads to a reduction of vesicular stomatitis virus (VSV), influenza virus and retrovirus replication [82].
5. CCHFV: The protective power of the IFN system
Type I IFNs are of increasing interest as antiviral compounds [83], [84], [85], [86], [87], [88], [89], [90], and CCHFV is one of the newest additions to the list of IFN-sensitive viruses [91]. However, no clinical studies exist so far which address the effect of IFNs against any viral hemorrhagic fever including CCHF. More progress was achieved with respect to the ISGs affecting CCHFV replication.
MxA significantly contributes to the antiviral activity of IFN against CCHFV, similar to what was observed for other members of the Bunyaviridae [91], [92], [93], [94], [95], [96]. MxA was found to colocalize and interact with CCHFV nucleocapsid protein in the perinuclear region of infected cells, an interaction which is most likely responsible for the inhibition of virus replication. As a similar MxA-N interaction is known from other bunyaviruses [97], [98], it is likely that all bunyaviruses may be restricted in their intracellular growth by MxA, and probably by the same mechanism. However, by using an siRNA approach to deplete cells from MxA, we could demonstrate that MxA is not the sole player in IFN-induced antiviral activity against CCHFV [91]. Our most recent studies indicate a contribution of ISG20 and PKR (manuscripts in preparation). Thus, apparently, there are several ISGs with antiviral activity against CCHFV, which most probably act in concert to combat the infection.
6. IFN escape mechanisms of CCHFV
Despite being strongly inhibited by type I IFNs, CCHFV still runs its devastating course in the human host. Estimates of CCHFV mortality range from a few percent to up to 70% [13], [32], [99]. Likely explanations for this discrepancy are that virus-induced IFN has no therapeutic, but only a prophylactic effect, or that production of IFN by infected cells is inefficient. In fact, recent data show that both these mechanisms seem to be in place. We have shown that IFN has no significant activity against an already established CCHFV infection [100]. Treatment of cells with 1000 U/ml IFN at 1 h after infection is ineffective, whereas the same dose applied before infection results in a significant inhibition of CCHFV replication. This indicates that CCHFV strongly counteracts IFN signalling, and experiments are underway to clarify this issue. In addition, CCHFV appears to counteract the induction of IFN synthesis by several independent strategies. Firstly, it is conceivable that, similar to other negative-strand RNA viruses and bunyaviruses [52], CCHFV does not produce significant amounts of the IFN inducer molecule dsRNA. Our ongoing studies are aimed at testing this assumption. Secondly, we found that the important RIG-I signaling pathway which is triggered by viral ssRNA carrying a 5′ triphosphate is not activated by the CCHFV genome [101]. Whereas the ssRNA genome of the related Rift Valley fever virus (RVFV) contains a 5′ triphosphate group and induces IFN induction via RIG-I, the CCHFV genome contains a RIG-I-neutral 5′monophosphate. Most likely, CCHFV cleaves off the 5′ triphosphate group during genome replication in order to escape recognition by RIG-I. In line with this, IFN induction by CCHFV-infected cells is weak and occurs relatively late [100]. Members of the family Bunyaviridae therefore appear to have evolved two radically different strategies to inhibit the RIG-I pathway. Whereas RVFV inhibits IFN induction by expressing a dedicated anti-IFN factor, the non-structural protein NSs [102], [103]. CCHFV removes the RIG-I ligand from its genome. Other bunyaviruses fall into the same categories, e.g. La Crosse virus expresses an NSs protein blocking IFN induction [4], but Hantaan virus contains a monophosphate on its genome 5′-end [101], [104].
Nonetheless, IFN-activating structures other than RNA seem to be present in CCHFV particles, and the virus possesses also at least one anti-IFN factor. Experiments using UV irradiation revealed that the IRF-3-dependent ISG56 gene is weakly activated by UV-inactivated CCHFV particles at an early time point after infection [100]. IRF-3-activated ISG56 upregulation by enveloped viral particles is described to occur by an unknown, TLR- and RIG-I-independent, signal pathway [105], [106]. In cells infected with intact virus, by contrast, ISG56 upregulation occurred with a delay of 14 h. In line with this, intact virus delays the activation of IRF-3, assayed by measuring its nuclear translocation, by approximately 20 h [100]. This indicates that CCHFV expresses a factor which counteracts IRF-3 activation and hence ISG56 upregulation. The factor is not known so far, but sequence analyses suggest that CCHFV has the capacity to encode an additional gene on the S segment (data not shown). The corresponding protein product has yet to be identified. Besides this, it has been demonstrated that the L protein of CCHFV contains an ovarian tumor (OTU) domain allowing innate immune evasion [107]. This domain is also present on the L protein of the related Nairoviruses Dugbe virus and Nairobi sheep disease virus, but not of the phylogenetically more distant Phlebovirus RVFV. The OTU domain represents a superfamily of Ubiquitin (Ub)-deconjugating proteases found in prokaryotes, eukaryotes, and viruses. Ub and interferon-stimulated gene product 15 (ISG15) are short proteins which are covalently conjugated to other proteins and mediate innate antiviral responses. Frias-Staheli et al. showed that the OTU domain-containing proteases from CCHFV and related Nairoviruses deconjugate Ub and ISG15 from cellular target proteins [107]. Moreover, expression of a viral OTU domain-containing protein antagonizes the antiviral effects of ISG15 and inhibits NF-κB-dependent signaling.
Thus, CCHFV employs a range of strategies to evade and counteract the innate immune reponse (Fig. 3 ). RIG-I recognition is circumvented by removal of the 5′ triphosphate group from the viral genome, IRF-3 activation mediated by a particle recognition pathway is delayed by a yet to be identified viral factor, and NF-kappaB activation and the antiviral action of ISG15 are downregulated by the OTU domain on the L protein.
Fig. 3.
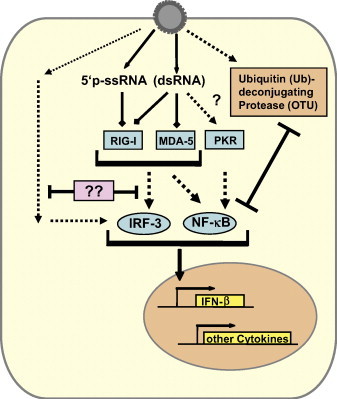
Countermeasures of CCHFV to escape the type I IFN response. The 5′-ends of the CCHFV ssRNA genome do not contain the triphosphate group characteristic for negative-strand RNA viruses, but rather are monophosphorylated. The recognition receptor RIG-I does not bind to 5′monophosphorylated ssRNA and is hence unable to trigger an IFN response. Like other negative-strand RNA viruses, CCHFV most likely does not produce significant amounts of dsRNA, thus rendering the RIG-I, MDA5 and PKR pathways inactive. PKR is possibly nonetheless activated, either by trace amounts of dsRNA or by cell stress resulting from infection. Contact with UV-inactivated CCHFV particles triggers IRF-3 by an unknown mechanism and is countered in productively infected cells by a yet to be identified viral factor. CCHFV L segment codes for an OTU domain Ubiquitin (Ub) – deconjugating protease, which antagonizes the antiviral effects of ISG15 and inhibits NF-kB-dependent signaling.
7. CCHFV pathogenesis and cytokine responses
Viral strategies to antagonize the innate immune response are usually not perfect and mostly delay rather than completely suppress the IFN system [41], [58] A common suggested feature of agents causing hemorrhagic fever is the deregulation of host immune responses by combating and attacking cells involved in initiation of antiviral responses. The existing knowledge concerning CCHF emanates from autopsies and clinical findings. The primary pathophysiological events appear to be leakage of erythrocytes and plasma through the vasculature into tissues [13]. Endothelial damage can contribute to coagulopathy by deregulated stimulation of platelet aggregation, which in turn activates the intrinsic coagulation cascade, ultimately leading to clotting factor deficiency causing hemorrhages. For CCHFV, vascular leakage may be caused either by destruction of endothelial cells or by a disruption of the tight junctions which constitute the endothelial barrier between cells. Moreover, it is unclear whether these events are a direct consequence of infection or whether virus-induced host factors cause the endothelial dysfunction [108]. However, in an epithelial cell line model CCHFV neither caused disruption of tight junctions nor necrosis or apoptosis of cells [109]. This could suggest that the hemorrhages and coagulation disturbances may be caused indirectly, possibly by high levels of proinflammatory cytokines. In fact, for other hemorrhagic fever viruses like Ebola and Dengue virus there is a correlation between the strength of the proinflammatory response, vascular leakage, and disease severity [84], [90], [110], [111], [112]. Key players in disease progression are the cytokines interleukin (IL)-10, IL-1, IL-6 and tumour necrosis factor (TNF)-α [111]. It has been shown that yellow fever patients with fatal outcome had high levels of IL-1, IL-6 and TNF-α [113]. In another study, early elevated levels of IL-6 and IL-1 beta in Ebola virus-infected patient correlated with non-fatal outcome, whereas release of IL-10 and high levels of neopterin and IL-RA in the plasma correlated with fatal outcomes [114] . Recently, Ergonul and coworkers demonstrated significantly higher level of IL-6 and TNF-α in CCHFV patients with fatal outcome compared to the non-fatal cases [38]. Another observation was a correlation between high levels of IL-6 and TNF-α to the onset of disseminated intravascular coagulation (DIC) in patients. However, elevated levels of IL-10 were not observed in these patients and the levels of IL-10 were negatively correlated to the DIC scores. In another study, Papa et al. confirmed that high TNF-α levels are correlated with severity of CCHF disease, but they suggested that IL-6 could be found in both mild and severe cases [115]. The only fatal case in this study had high levels of both IL-6 and TNF-α compared to non-fatal cases. Another interesting observation is elevated levels of Neopterin in patients with Dengue fever or Ebola hemorrhagic fever [114], [116]. Neopterin derivates are produced by macrophages and dendritic cells upon stimulation by IFNs [117]. Neopterin is a useful tool to assess the intensity of cell-mediated immune response [117] and a recent study indeed noted a correlation between elevated levels of neopterin in CCHFV patients and disease severity [39].
All these results suggest that capillary fragility, a common feature of CCHF, is most probably due to multiple host-induced mechanisms in response to CCHFV infection. Endothelial damage would cause the characteristic rash and contribute to hemostatic failure. Interestingly, some authors have noticed similarities between various viral hemorrhagic fevers and the septic shock caused by severe bacterial infections [99].
8. Concluding remarks
Research on CCHFV is hampered by several constraints. Firstly, work with the virus requires BSL-4 containment facilities, the highest security standard possible. Secondly, there is no animal model which mimics human disease, and thirdly, CCHF cases occur so sporadically that clinical samples are rare. Using the available systems, however, some of the most pressing questions on CCHFV should be addressed in the near future:
-
-
What is the molecular pattern eliciting IRF-3 activation by CCHFV? Treatment of fibroblasts with UV-irradiated virus particles activates IRF-3. As the genome of CCHFV lacks a 5′ triphosphate group to trigger RIG-I and IRF-3 [101], other structures, e.g. the viral enveloped particles or the nucleocapsids must be recognized by the infected cell.
-
-
Which TLRs are involved in CCHFV recognition and cytokine induction in vivo?
-
-
Can DCs be infected, and what is their role in the cytokine response to CCHFV?
-
-
Is there an NSs-like gene suppressing IFN and cytokine induction? For bunyaviruses of the genera Orthobunyavirus and Phleboviruses, e.g. La Crosse virus, Bunyamwera virus, and Rift Valley fever virus, a non-structural protein encoded on the S segment (NSs) is known to strongly suppress transcriptional activation of IFNs and other inducible genes [4], [5], [118], [119]. Moreover, some members of the Hantavirus genus also encode an NSs protein which weakly supresses IFN induction [8], [120]. Moreover, for Hantaviruses it is known that the glycoproteins can interfere with IFN induction [121], [122]. The delay of IRF-3 activation observed in productively infected cells indicates that CCHFV encodes proteins or domains dedicated to counteract IFN induction.
-
-
Does CCHFV interfere with IFN signalling, and if so, which viral gene product is responsible? IFN is effective if given in advance, but not if given at 1 h after infection [100]. It is therefore conceivable that CCHFV is capable of shutting down the JAK/STAT pathway.
-
-
Which ISGs contribute to the inhibition of CCHFV by IFNs? Other ISGs besides MxA and perhaps PKR and ISG20 may be efficient in blocking various steps of CCHFV replication.
-
-
What is the role of the OTU domain in pathogenesis? There is currently no reverse genetics system available to generate infectious viruses from cloned cDNA plasmids. Mutational inactivation of the OTU domain, rescue of the respective virus and a phenotypical comparison with the wild-type virus can reveal the contribution of the OTU domain to virus replication in cell culture and in vivo.
-
-
How does the virus cause vascular leakage? As it has been mentioned, the endothelium can be targeted in two ways – indirectly by virus-mediated host-derived factors that cause endothelial dysfunction, and/or directly by virus infection in endothelial cells. Recent studies demonstrate that increased levels of IL-6 and TNF-α are correlated with disease severity [38], suggesting that the endothelial damage may be due to a “cytokine storm”. To date, there is no experimental studies investigating whether the virus replication cycle per se or whether it is cytokines, which damage the tight junctions of endothelial cells.
Answering these questions would certainly improve our knowledge of CCHFV pathogenesis and open new ways for the prevention and treatment of the hemorrhagic fever caused by this highly virulent pathogen.
Acknowledgements
Our own work described in the text was supported by grants from the Swedish Medicine Council (To A.M, K2007-56X-20349-01-3) and from the Deutsche Forschungsgemeinschaft (to F.W., We 2616/5-1).
Biographies

Friedemann Weber received his M.Sc. in 1993 from the Department of Microbiology and his Ph.D. in 1997 from the Department of Virology at the University of Freiburg, Germany. He was an EMBO Long Term Postdoctoral Fellow at the Institute of Virology in Glasgow, UK, and is currently a Professor of Virology at the University of Freiburg. In 2003, he has received the Milstein Young Investigator Award from the International Society for Interferon and Cytokine Research (ISICR) and in 2004 the Heine-Medin Medal of the European Society for Clinical Virology. In 2007 he was awarded the “Löffler-Frosch-Preis” of the Gesellschaft für Virologie. He is interested in the innate immune responses to highly pathogenic RNA viruses such as SARS-Coronavirus and bunyaviruses. The particular focus is on interferon-inducing viral structures, their intracellular receptors, and the viral escape strategies. He has published over 50 research papers, review articles and book chapters.

Ali Mirazimi received his B.Sc. in 1994 from the Karolinska Institute, and his Ph.D in 2000 from the Department of Microbiology, Tumour biology and Cell biology at Karolinska Institute. Short after, he received a junior lecturer position at Karolinska Institute. At 2002 he received a position as Molecular virologist at Swedish Institute for Infectious Disease control. At 2006 he achieved Associate professor position at Karolinska Institute. Today, he is the head of the BSL-4 Virology Unit at Centre for Microbiological Preparedness at Swedish Institute for Infectious disease. His group is interested to understand the molecular pathogenesis of emerging viruses. The major focus is the role of Innate Immunity for controlling the viral replication cycle. He is also interested to investigate the virus-host cell interaction and virus biology. His group is also interested in developing the diagnostic tools for detection of BSL-4 agents.
References
- 1.Nichol S. Bunyaviruses. In: Knipe D., Howley P., editors. Fields virology. Lippincott Williams and Wilkins; Philadelpia: 2001. pp. 1603–1633. [Google Scholar]
- 2.Elliott R.M. In: The Bunyaviridae. Elliott R.M., editor. Plenum Press; New York: 1996. [Google Scholar]
- 3.Elliott R.M., Schmaljohn C.S., Collett M.S. Bunyaviridae genome structure and gene expression. Curr Top Microbiol Immunol. 1991;169:91–141. doi: 10.1007/978-3-642-76018-1_4. [DOI] [PubMed] [Google Scholar]
- 4.Blakqori G., Delhaye S., Habjan M., Blair C.D., Sánchez-Vargas I., Olson K.E. La Crosse bunyavirus nonstructural protein NSs serves to suppress the type I interferon system of mammalian hosts. J Virol. 2007 May;81(10):4991–4999. doi: 10.1128/JVI.01933-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le May N., Dubaele S., Proietti De Santis L., Billecocq A., Bouloy M., Egly J.M. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell. 2004;116(4):541–550. doi: 10.1016/s0092-8674(04)00132-1. [DOI] [PubMed] [Google Scholar]
- 6.Kormelink R., Kitajima E.W., De Haan P., Zuidema D., Peters D., Goldbach R. The nonstructural protein (NSs) encoded by the ambisense S RNA segment of tomato spotted wilt virus is associated with fibrous structures in infected plant cells. Virology. 1991;181(2):459–468. doi: 10.1016/0042-6822(91)90878-f. [DOI] [PubMed] [Google Scholar]
- 7.Fuller F., Bhown A.S., Bishop D.H. Bunyavirus nucleoprotein, N, and a non-structural protein, NSS, are coded by overlapping reading frames in the S RNA. J Gen Virol. 1983;64(Pt 8):1705–1714. doi: 10.1099/0022-1317-64-8-1705. [DOI] [PubMed] [Google Scholar]
- 8.Jääskeläinen K.M., Kaukinen P., Minskaya E.S., Plyusnina A., Vapalahti O., Elliott R.M. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J Med Virol. 2007;79(10):1527–1536. doi: 10.1002/jmv.20948. [DOI] [PubMed] [Google Scholar]
- 9.Bergeron E., Vincent M.J., Nichol S.T. Crimean-Congo hemorrhagic fever virus glycoprotein processing by the endoprotease SKI-1/S1P is critical for virus infectivity. J Virol. 2007;81(23):13271–13276. doi: 10.1128/JVI.01647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontana J., López-Montero N., Elliott R.M., Fernández J.J., Risco C. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cell Microbiol. 2008 doi: 10.1111/j.1462-5822.2008.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altamura L.A., Bertolotti-Ciarlet A., Teigler J., Paragas J., Schmaljohn C.S., Doms R.W. Identification of a novel C-terminal cleavage of Crimean-Congo hemorrhagic fever virus PreGN that leads to generation of an NSM protein. J Virol. 2007;81(12):6632–6642. doi: 10.1128/JVI.02730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollitt E., Zhao J., Muscat P., Elliott R.M. Characterization of Maguari orthobunyavirus mutants suggests the nonstructural protein NSm is not essential for growth in tissue culture. Virology. 2006;348(1):224–232. doi: 10.1016/j.virol.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 13.Ergonul O. Crimean-Congo haemorrhagic fever. Lancet Infect Dis. 2006;6(4):203–214. doi: 10.1016/S1473-3099(06)70435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunster L., Dunster M., Ofula V., Beti D., Kazooba-Voskamp F., Burt F. First documentation of human Crimean-Congo hemorrhagic fever, Kenya. Emerg Infect Dis. 2002;8(9):1005–1006. doi: 10.3201/eid0809.010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drosten C., Minnak D., Emmerich P., Schmitz H., Reinicke T. Crimean-Congo hemorrhagic fever in Kosovo. J Clin Microbiol. 2002;40(3):1122–1123. doi: 10.1128/JCM.40.3.1122-1123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nabeth P., Thior M., Faye O., Simon F. Human Crimean-Congo hemorrhagic fever, Senegal. Emerg Infect Dis. 2004;10(10):1881–1882. doi: 10.3201/eid1010.040586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nabeth P., Cheikh D.O., Lo B., Faye O., Vall I.O., Niang M. Crimean-Congo hemorrhagic fever, Mauritania. Emerg Infect Dis. 2004;10(12):2143–2149. doi: 10.3201/eid1012.040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.el-Azazy O.M., Scrimgeour E.M. Crimean-Congo haemorrhagic fever virus infection in the western province of Saudi Arabia. Trans R Soc Trop Med Hyg. 1997;91(3):275–278. doi: 10.1016/s0035-9203(97)90072-9. [DOI] [PubMed] [Google Scholar]
- 19.Williams R.J., Al-Busaidy S., Mehta F.R., Maupin G.O., Wagoner K.D., Al-Awaidy S. Crimean-congo haemorrhagic fever: a seroepidemiological and tick survey in the Sultanate of Oman. Trop Med Int Health. 2000;5(2):99–106. doi: 10.1046/j.1365-3156.2000.00524.x. [DOI] [PubMed] [Google Scholar]
- 20.Burney M.I., Ghafoor A., Saleen M., Webb P.A., Casals J. Nosocomial outbreak of viral hemorrhagic fever caused by Crimean Hemorrhagic fever-Congo virus in Pakistan, January 1976. Am J Trop Med Hyg. 1980;29(5):941–947. doi: 10.4269/ajtmh.1980.29.941. [DOI] [PubMed] [Google Scholar]
- 21.Papa A., Bino S., Llagami A., Brahimaj B., Papadimitriou E., Pavlidou V. Crimean-Congo hemorrhagic fever in Albania, 2001. Eur J Clin Microbiol Infect Dis. 2002;21(8):603–606. doi: 10.1007/s10096-002-0770-9. [DOI] [PubMed] [Google Scholar]
- 22.Papa A., Christova I., Papadimitriou E., Antoniadis A. Crimean-Congo hemorrhagic fever in Bulgaria. Emerg Infect Dis. 2004;10(8):1465–1467. doi: 10.3201/eid1008.040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheikh A.S., Sheikh A.A., Sheikh N.S., Rafi-U-Shan, Asif M., Afridi F. Bi-annual surge of Crimean-Congo haemorrhagic fever (CCHF): a five-year experience. Int J Infect Dis. 2005;9(1):37–42. doi: 10.1016/j.ijid.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Papa A., Ma B., Kouidou S., Tang Q., Hang C., Antoniadis A. Genetic characterization of the M RNA segment of Crimean Congo hemorrhagic fever virus strains, China. Emerg Infect Dis. 2002;8(1):50–53. doi: 10.3201/eid0801.010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoogstraal H. The epidemiology of tick-borne Crimean-Congo hemorrhagic fever in Asia, Europe, and Africa. J Med Entomol. 1979;15(4):307–417. doi: 10.1093/jmedent/15.4.307. [DOI] [PubMed] [Google Scholar]
- 26.Watts D.M., Kziasek T.G., Linthicum K.J., Hoogstraal H. Crimean congo hemorrahgic fever. In: Monath T.P., editor. The arboviruses: epidemiology and ecology. CRC Press; Boca Raton, Fl, USA: 1988. p. 177260. [Google Scholar]
- 27.van de Wal B.W., Joubert J.R., van Eeden P.J., King J.B. A nosocomial outbreak of Crimean-Congo haemorrhagic fever at Tygerberg Hospital. Part IV. Preventive and prophylactic measures. S Afr Med J. 1985;68(10):729–732. [PubMed] [Google Scholar]
- 28.van Eeden P.J., Joubert J.R., van de Wal B.W., King J.B., de Kock A., Groenewald J.H. A nosocomial outbreak of Crimean-Congo haemorrhagic fever at Tygerberg Hospital. Part II. Management of patients. S Afr Med J. 1985;68(10):718–721. [PubMed] [Google Scholar]
- 29.Swanepoel R., Shepherd A.J., Leman P.A., Shepherd S.P. Investigations following initial recognition of Crimean-Congo haemorrhagic fever in South Africa and the diagnosis of 2 further cases. S Afr Med J. 1985;68(9):638–641. [PubMed] [Google Scholar]
- 30.Chinikar S., Persson S.M., Johansson M., Bladh L., Goya M., Houshmand B. Genetic analysis of Crimean-Congo hemorrhagic fever virus in Iran. J Med Virol. 2004;73(3):404–411. doi: 10.1002/jmv.20106. [DOI] [PubMed] [Google Scholar]
- 31.Swanepoel R., Gill D.E., Shepherd A.J., Leman P.A., Mynhardt J.H., Harvey S. The clinical pathology of Crimean-Congo hemorrhagic fever. Rev Infect Dis. 1989;11(Suppl 4):S794–S800. doi: 10.1093/clinids/11.supplement_4.s794. [DOI] [PubMed] [Google Scholar]
- 32.Schwarz T.F., Nsanze H., Ameen A.M. Clinical features of Crimean-Congo haemorrhagic fever in the United Arab Emirates. Infection. 1997;25(6):364–367. doi: 10.1007/BF01740819. [DOI] [PubMed] [Google Scholar]
- 33.Baskerville A., Satti A., Murphy F.A., Simpson D.I. Congo-Crimean haemorrhagic fever in Dubai: histopathological studies. J Clin Pathol. 1981;34(8):871–874. doi: 10.1136/jcp.34.8.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wölfel R., Paweska J.T., Petersen N., Grobbelaar A.A., Leman P.A., Hewson R. Virus detection and monitoring of viral load in Crimean-Congo hemorrhagic fever virus patients. Emerg Infect Dis. 2007;13(7):1097–1100. doi: 10.3201/eid1307.070068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duh D., Saksida A., Petrovec M., Ahmeti S., Dedushaj I., Panning M. Viral load as predictor of Crimean-Congo hemorrhagic fever outcome. Emerg Infect Dis. 2007;13(11):1769–1772. doi: 10.3201/eid1311.070222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yilmaz M., Aydin K., Akdogan E., Sucu N., Sonmez M., Omay S.B. Peripheral blood natural killer cells in Crimean-Congo hemorrhagic fever. J Clin Virol. 2008;42(4):415–417. doi: 10.1016/j.jcv.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 37.Celikbaş A., Ergönül O., Dokuzoğuz B., Eren S., Baykam N., Polat-Düzgün A. Crimean Congo hemorrhagic fever infection simulating acute appendicitis. J Infect. 2005;50(4):363–365. doi: 10.1016/j.jinf.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 38.Ergonul O., Tuncbilek S., Baykam N., Celikbas A., Dokuzoguz B. Evaluation of serum levels of interleukin (IL)-6, IL-10, and tumor necrosis factor-alpha in patients with Crimean-Congo hemorrhagic fever. J Infect Dis. 2006;193(7):941–944. doi: 10.1086/500836. [DOI] [PubMed] [Google Scholar]
- 39.Onguru P., Akgul E.O., Akinci E., Yaman H., Kurt Y.G., Erbay A. High serum levels of neopterin in patients with Crimean-Congo hemorrhagic fever and its relation with mortality. J Infect. 2008;56(5):366–370. doi: 10.1016/j.jinf.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozkurt Z., Kiki I., Erol S., Erdem F., Yilmaz N., Parlak M. Crimean-Congo hemorrhagic fever in Eastern Turkey: clinical features, risk factors and efficacy of ribavirin therapy. J Infect. 2005 doi: 10.1016/j.jinf.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Haller O., Kochs G., Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology. 2006;344(1):119–130. doi: 10.1016/j.virol.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samuel C.E. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14(4):778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadler A.J., Williams B.R. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8(7):559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hertzog P.J., O’Neill L.A., Hamilton J.A. The interferon in TLR signaling: more than just antiviral. Trends Immunol. 2003;24(10):534–539. doi: 10.1016/j.it.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 45.Le Bon A., Tough D.F. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002;14(4):432–436. doi: 10.1016/s0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 46.van Pesch V., Michiels T. Characterization of interferon-alpha 13, a novel constitutive murine interferon-alpha subtype. J Biol Chem. 2003;278(47):46321–46328. doi: 10.1074/jbc.M302554200. [DOI] [PubMed] [Google Scholar]
- 47.van Pesch V., Lanaya H., Renauld J.C., Michiels T. Characterization of the murine alpha interferon gene family. J Virol. 2004;78(15):8219–8228. doi: 10.1128/JVI.78.15.8219-8228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts R.M., Ezashi T., Rosenfeld C.S., Ealy A.D., Kubisch H.M. Evolution of the interferon tau genes and their promoters, and maternal-trophoblast interactions in control of their expression. Reprod Suppl. 2003;61:239–251. [PubMed] [Google Scholar]
- 49.Delhaye S., Paul S., Blakqori G., Minet M., Weber F., Staeheli P. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA. 2006;103(20):7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marie I., Durbin J.E., Levy D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. Embo J. 1998;17(22):6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colonna M., Krug A., Cella M. Interferon-producing cells: on the front line in immune responses against pathogens. Curr Opin Immunol. 2002;14(3):373–379. doi: 10.1016/s0952-7915(02)00349-7. [DOI] [PubMed] [Google Scholar]
- 52.Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80(10):5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornung V., Ellegast J., Kim S., Brzózka K., Jung A., Kato H. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 54.Pichlmair A., Schulz O., Tan C.P., Näslund T.I., Liljeström P., Weber F. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 55.Plumet S., Herschke F., Bourhis J.M., Valentin H., Longhi S., Gerlier D. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE. 2007;2(3):e279. doi: 10.1371/journal.pone.0000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoneyama M., Onomoto K., Fujita T. Cytoplasmic recognition of RNA. Adv Drug Deliv Rev. 2008;60(7):841–846. doi: 10.1016/j.addr.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 57.Pichlmair A., Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27(3):370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 58.Randall R.E., Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89(Pt 1):1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 59.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 60.Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205(7):1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282(21):15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 62.Paun A., Pitha P.M. The IRF family, revisited. Biochimie. 2007;89(6–7):744–753. doi: 10.1016/j.biochi.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sato M., Suemori H., Hata N., Asagiri M., Ogasawara K., Nakao K. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13(4):539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 64.Chu W.M., Ostertag D., Li Z.W., Chang L., Chen Y., Hu Y., Williams B. JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity. 1999;11(6):721–731. doi: 10.1016/s1074-7613(00)80146-6. [DOI] [PubMed] [Google Scholar]
- 65.Escalante C.R., Shen L., Thanos D., Aggarwal A.K. Structure of NF-kappaB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-beta promoter. Structure. 2002;10(3):383–391. doi: 10.1016/s0969-2126(02)00723-2. [DOI] [PubMed] [Google Scholar]
- 66.Diebold S.S., Montoya M., Unger H., Alexopoulou L., Roy P., Haswell L.E. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424(6946):324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 67.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 68.Uematsu S., Akira S. Toll-like receptor and innate immunity. Seikagaku. 2007;79(8):769–776. [PubMed] [Google Scholar]
- 69.de Weerd N.A., Samarajiwa S.A., Hertzog P.J. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282(28):20053–20057. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- 70.Schindler C., Levy D.E., Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 71.Levy D.E., Darnell J.E., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 72.Der S.D., Zhou A., Williams B.R., Silverman R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95(26):15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haller O., Staeheli P., Kochs G. Interferon-induced Mx proteins in antiviral host defense. Biochimie. 2007;89(6–7):812–818. doi: 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 74.Haller O., Kochs G., Weber F. Interferon, Mx, and viral countermeasures. Cytokine Growth Factor Rev. 2007;18(5–6):425–433. doi: 10.1016/j.cytogfr.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nallagatla S.R., Hwang J., Toroney R., Zheng X., Cameron C.E., Bevilacqua P.C. 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science. 2007;318(5855):1455–1458. doi: 10.1126/science.1147347. [DOI] [PubMed] [Google Scholar]
- 76.Garcia M.A., Meurs E.F., Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89(6–7):799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 77.Hovanessian A.G., Justesen J. The human 2′-5′oligoadenylate synthetase family: unique interferon-inducible enzymes catalyzing 2′-5′ instead of 3′-5′ phosphodiester bond formation. Biochimie. 2007;89(6–7):779–788. doi: 10.1016/j.biochi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 78.Bisbal C., Silverman R.H. Diverse functions of RNase L and implications in pathology. Biochimie. 2007;89(6–7):789–798. doi: 10.1016/j.biochi.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chin K.C., Cresswell P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc Natl Acad Sci USA. 2001;98(26):15125–15130. doi: 10.1073/pnas.011593298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X., Hinson E.R., Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe. 2007;2(2):96–105. doi: 10.1016/j.chom.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 81.Sen G.C., Peters G.A. Viral stress-inducible genes. Adv Virus Res. 2007;70:233–263. doi: 10.1016/S0065-3527(07)70006-4. [DOI] [PubMed] [Google Scholar]
- 82.Degols G., Eldin P., Mechti N. ISG20, an actor of the innate immune response. Biochimie. 2007;89(6–7):831–835. doi: 10.1016/j.biochi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 83.Shresta S., Kyle J.L., Snider H.M., Basavapatna M., Beatty P.R., Harris E. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T- and B-cell-dependent immunity are less critical. J Virol. 2004;78(6):2701–2710. doi: 10.1128/JVI.78.6.2701-2710.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bray M., Geisbert T.W. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int J Biochem Cell Biol. 2005;37(8):1560–1566. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 85.Bray M. Pathogenesis of viral hemorrhagic fever. Curr Opin Immunol. 2005;17(4):399–403. doi: 10.1016/j.coi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 86.Navarro-Sanchez E., Despres P., Cedillo-Barron L. Innate immune responses to dengue virus. Arch Med Res. 2005;36(5):425–435. doi: 10.1016/j.arcmed.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 87.Baize S., Pannetier D., Faure C., Marianneau P., Marendat I., Georges-Courbot M.C. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes Infect. 2006;8(5):1194–1202. doi: 10.1016/j.micinf.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 88.Summerfield A., Alves M., Ruggli N., de Bruin M.G., McCullough K.C. High IFN-alpha responses associated with depletion of lymphocytes and natural IFN-producing cells during classical swine fever. J Interferon Cytokine Res. 2006;26(4):248–255. doi: 10.1089/jir.2006.26.248. [DOI] [PubMed] [Google Scholar]
- 89.Hutchinson K.L., Rollin P.E. Cytokine and chemokine expression in humans infected with Sudan Ebola virus. J Infect Dis. 2007;196(Suppl 2):S357–S363. doi: 10.1086/520611. [DOI] [PubMed] [Google Scholar]
- 90.Bray M., Pilch R. Filoviruses: recent advances and future challenges. Expert Rev Anti Infect Ther. 2006;4(6):917–921. doi: 10.1586/14787210.4.6.917. [DOI] [PubMed] [Google Scholar]
- 91.Andersson I., Lundkvist A., Haller O., Mirazimi A. Type I interferon inhibits Crimean-Congo hemorrhagic fever virus in human target cells. J Med Virol. 2006;78(2):216–222. doi: 10.1002/jmv.20530. [DOI] [PubMed] [Google Scholar]
- 92.Bridgen A., Dalrymple D.A., Weber F., Elliott R.M. Inhibition of Dugbe nairovirus replication by human MxA protein. Virus Res. 2004;99(1):47–50. doi: 10.1016/j.virusres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 93.Frese M., Kochs G., Feldmann H., Hertkorn C., Haller O. Inhibition of bunyaviruses, phleboviruses, and hantaviruses by human MxA protein. J Virol. 1996;70(2):915–923. doi: 10.1128/jvi.70.2.915-923.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kanerva M., Melén K., Vaheri A., Julkunen I. Inhibition of puumala and tula hantaviruses in Vero cells by MxA protein. Virology. 1996;224(1):55–62. doi: 10.1006/viro.1996.0506. [DOI] [PubMed] [Google Scholar]
- 95.Sandrock M., Frese M., Haller O., Kochs G. Interferon-induced rat Mx proteins confer resistance to Rift Valley fever virus and other arthropod-borne viruses. J Interferon Cytokine Res. 2001;21(9):663–668. doi: 10.1089/107999001753124390. [DOI] [PubMed] [Google Scholar]
- 96.Andersson I., Lundkvist A., Haller O., Mirazimi A. Human MxA protein inhibits the replication of Crimean-Congo hemorrhagic fever virus. J Virol. 2004;78(8):4323–4329. doi: 10.1128/JVI.78.8.4323-4329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kochs G., Janzen C., Hohenberg H., Haller O. Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc Natl Acad Sci USA. 2002;99(5):3153–3158. doi: 10.1073/pnas.052430399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Reichelt M., Stertz S., Krijnse-Locker J., Haller O., Kochs G. Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic. 2004;5(10):772–784. doi: 10.1111/j.1600-0854.2004.00219.x. [DOI] [PubMed] [Google Scholar]
- 99.Whitehouse C.A. Crimean-Congo hemorrhagic fever. Antiviral Res. 2004;64(3):145–160. doi: 10.1016/j.antiviral.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 100.Andersson I., Karlberg H., Mousavi-Jazi M., Martínez-Sobrido L., Weber F., Mirazimi A. Crimean-Congo hemorrhagic fever virus delays activation of the innate immune response. J Med Virol. 2008;80(8):1397–1404. doi: 10.1002/jmv.21222. [DOI] [PubMed] [Google Scholar]
- 101.Habjan M., Andersson I., Klingström J., Schümann M., Martin A., Zimmermann P. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE. 2008;3(4):e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Billecocq A., Spiegel M., Vialat P., Kohl A., Weber F., Bouloy M. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J Virol. 2004;78(18):9798–9806. doi: 10.1128/JVI.78.18.9798-9806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bouloy M., Janzen C., Vialat P., Khun H., Pavlovic J., Huerre M. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J Virol. 2001;75(3):1371–1377. doi: 10.1128/JVI.75.3.1371-1377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garcin D., Lezzi M., Dobbs M., Elliott R.M., Schmaljohn C., Kang C.Y. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J Virol. 1995;69(9):5754–5762. doi: 10.1128/jvi.69.9.5754-5762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Collins S.E., Noyce R.S., Mossman K.L. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol. 2004;78(4):1706–1717. doi: 10.1128/JVI.78.4.1706-1717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Paladino P., Cummings D.T., Noyce R.S., Mossman K.L. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol. 2006;177(11):8008–8016. doi: 10.4049/jimmunol.177.11.8008. [DOI] [PubMed] [Google Scholar]
- 107.Frias-Staheli N., Giannakopoulos N.V., Kikkert M., Taylor S.L., Bridgen A., Paragas J. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007;2(6):404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schnittler H.J., Feldmann H. Viral hemorrhagic fever – a vascular disease? Thromb Haemost. 2003;89(6):967–972. [PubMed] [Google Scholar]
- 109.Connolly-Andersen A.M., Magnusson K.E., Mirazimi A. Basolateral entry and release of Crimean-Congo hemorrhagic fever virus in polarized MDCK-1 cells. J Virol. 2007;81(5):2158–2164. doi: 10.1128/JVI.02070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen S.T., Lin Y.L., Huang M.T., Wu M.F., Cheng S.C., Lei H.Y. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008;453(7195):672–676. doi: 10.1038/nature07013. [DOI] [PubMed] [Google Scholar]
- 111.Chen H.C., Hofman F.M., Kung J.T., Lin Y.D., Wu-Hsieh B.A. Both virus and tumor necrosis factor alpha are critical for endothelium damage in a mouse model of dengue virus-induced hemorrhage. J Virol. 2007;81(11):5518–5526. doi: 10.1128/JVI.02575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lei H.Y., Yeh T.M., Liu H.S., Lin Y.S., Chen S.H., Liu C.C. Immunopathogenesis of dengue virus infection. J Biomed Sci. 2001;8(5):377–388. doi: 10.1007/BF02255946. [DOI] [PubMed] [Google Scholar]
- 113.ter Meulen J., Sakho M., Koulemou K., Magassouba N., Bah A., Preiser W. Activation of the cytokine network and unfavorable outcome in patients with yellow fever. J Infect Dis. 2004;190(10):1821–1827. doi: 10.1086/425016. [DOI] [PubMed] [Google Scholar]
- 114.Baize S., Leroy E.M., Georges A.J., Georges-Courbot M.C., Capron M., Bedjabaga I. Inflammatory responses in Ebola virus-infected patients. Clin Exp Immunol. 2002;128(1):163–168. doi: 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Papa A., Bino S., Velo E., Harxhi A., Kota M., Antoniadis A. Cytokine levels in Crimean-Congo hemorrhagic fever. J Clin Virol. 2006;36(4):272–276. doi: 10.1016/j.jcv.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 116.Chan C.P., Choi J.W., Cao K.Y., Wang M., Gao Y., Zhou D.H. Detection of serum neopterin for early assessment of dengue virus infection. J Infect. 2006;53(3):152–158. doi: 10.1016/j.jinf.2005.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wirleitner B., Reider D., Ebner S., Böck G., Widner B., Jaeger M. Monocyte-derived dendritic cells release neopterin. J Leukoc Biol. 2002;72(6):1148–1153. [PubMed] [Google Scholar]
- 118.Thomas D., Blakqori G., Wagner V., Banholzer M., Kessler N., Elliott R.M. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J Biol Chem. 2004;279(30):31471–31477. doi: 10.1074/jbc.M400938200. [DOI] [PubMed] [Google Scholar]
- 119.Le May N., Mansuroglu Z., Léger P., Josse T., Blot G., Billecocq A. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog. 2008;4(1):e13. doi: 10.1371/journal.ppat.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jääskeläinen K.M., Plyusnina A., Lundkvist A., Vaheri A., Plyusnin A. Tula hantavirus isolate with the full-length ORF for nonstructural protein NSs survives for more consequent passages in interferon-competent cells than the isolate having truncated NSs ORF. Virol J. 2008;5:3. doi: 10.1186/1743-422X-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alff P.J., Gavrilovskaya I.N., Gorbunova E., Endriss K., Chong Y., Geimonen E. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J Virol. 2006;80(19):9676–9686. doi: 10.1128/JVI.00508-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Spiropoulou C.F., Albariño C.G., Ksiazek T.G., Rollin P.E. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J Virol. 2007;81(6):2769–2776. doi: 10.1128/JVI.02402-06. [DOI] [PMC free article] [PubMed] [Google Scholar]