

SUMMARY
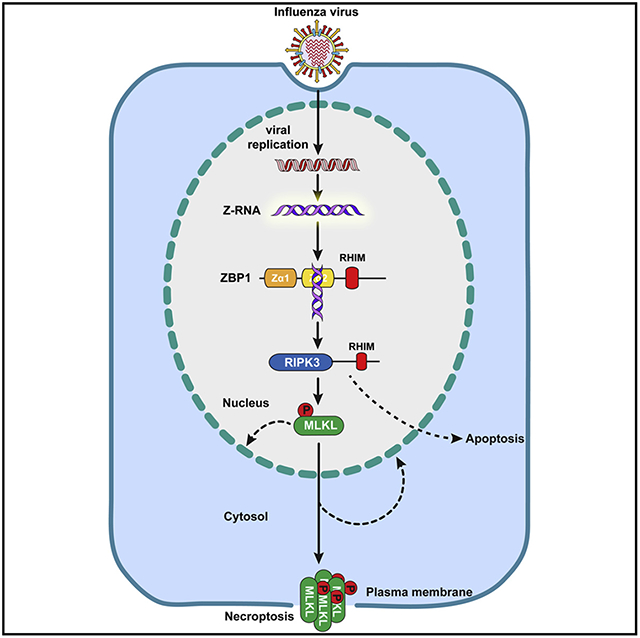
Influenza A virus (IAV) is a lytic RNA virus that triggers receptor-interacting serine/threonine-protein kinase 3 (RIPK3)-mediated pathways of apoptosis and mixed lineage kinase domain-like pseudokinase (MLKL)-dependent necroptosis in infected cells. ZBP1 initiates RIPK3-driven cell death by sensing IAV RNA and activating RIPK3. Here, we show that replicating IAV generates Z-RNAs, which activate ZBP1 in the nucleus of infected cells. ZBP1 then initiates RIPK3-mediated MLKL activation in the nucleus, resulting in nuclear envelope disruption, leakage of DNA into the cytosol, and eventual necroptosis. Cell death induced by nuclear MLKL was a potent activator of neutrophils, a cell type known to drive inflammatory pathology in virulent IAV disease. Consequently, MLKL-deficient mice manifest reduced nuclear disruption of lung epithelia, decreased neutrophil recruitment into infected lungs, and increased survival following a lethal dose of IAV. These results implicate Z-RNA as a new pathogen-associated molecular pattern and describe a ZBP1- initiated nucleus-to-plasma membrane “inside-out” death pathway with potentially pathogenic consequences in severe cases of influenza.
Graphical Abstract
In Brief
Z-RNAs produced by influenza viruses in the nucleus of infected cells are detected by host ZBP1, which activates RIPK3 and MLKL to lead to nuclear envelope rupture and necroptosis, ultimately resulting in neutrophil recruitment and activation in infected tissue.
INTRODUCTION
Influenza A virus (IAV) is a negative-sense RNA virus of the family Orthomyxoviridae. In aquatic birds, the primary IAV reservoir, virus replication occurs within the gastrointestinal tract and is typically asymptomatic. In contrast, mammalian IAV strains reproduce in respiratory tissues and generate symptoms ranging from mild cases of “the flu” to severe, sometimes lethal disease.
We and others have recently described a branched cell death pathway that is active in lung epithelial cells and that accounts for most cell death triggered by replicating IAV in these and other primary cell types (Kuriakose et al., 2016; Nogusa et al., 2016; Thapa et al., 2016). This pathway is initiated when the host protein ZBP1 (also called DAI) senses IAV RNAs and recruits receptor-interacting serine/threonine-protein kinase 3 (RIPK3) (Thapa et al., 2016). RIPK3 then activates two parallel forms of cell death, necroptosis and apoptosis. Necroptosis requires RIPK3 kinase activity and is mediated by mixed lineage kinase domain-like pseudokinase (MLKL). Apoptosis employs RIPK1, Fas-associated protein with death domain (FADD), and caspase-8, and it is independent of RIPK3 kinase activity (Nogusa et al., 2016).
ZBP1 contains two Zα domains. These domains selectively bind left-handed double-helical “Z-form” RNA structures in vitro (Brown et al., 2000; Placido et al., 2007), but whether Z-RNAs are produced during virus infections and serve as activating ligands for ZBP1 is unknown. Here, we show that orthomyxoviruses (IAV and IBV) produce Z-RNAs, and these Z-RNAs activate ZBP1 in infected nuclei. Once activated, ZBP1 stimulates RIPK3, which phosphorylates and activates MLKL in the nucleus. MLKL then triggers disruption of the nuclear envelope and promotes leakage of cellular DNA into the cytosol. Activated MLKL also traffics to the plasma membrane to mediate cell death by necroptosis.
Stimulating MLKL in the nucleus of fibroblasts potently activates neutrophils ex vivo, and Mlkl−/− mice displayed reduced nuclear damage in epithelial cells and dampened neutrophil recruitment upon IAV infection in vivo. Because neutrophils are established drivers of pathology in severe IAV disease (Brandes et al., 2013; Camp and Jonsson, 2017), Mlkl−/− mice also fared significantly better than their wild-type counterparts following a lethal dose of this virus, without notable differences in virus replication rates. These findings provide evidence for Z-RNA as a new PAMP, and delineate a ZBP1-initiated “inside-out” nucleus-to-plasma membrane death signaling pathway with pathogenic consequences during severe influenza infections.
RESULTS
ZBP1 Activates MLKL in the Nucleus of IAV-Infected Cells
IAV (H1N1 strain Puerto Rico/8/1934; hereafter PR8) triggers extensive cytopathic effect (CPE) and death in primary wild-type murine embryo fibroblasts (MEFs) starting at between 8 and 12 h post-infection (p.i.); by 24 h, over 60% of wild-type MEFs are dead (Figure 1A). In the same time frame, however, we observed minimalCPE and >80%–85% viability of similarly infectedZbp1−/− or Ripk3−/− MEFs (Figure 1A). In wild-type MEFs, synthesis of each of the eight IAV negative-sense viral RNA segments (vRNAs) was evident by 2–4 h p.i. and peaked by 8 h p.i. (Figure 1B), preceding production of IAV proteins, which was first seen 4–6 h p.i. (Figure 1C). To extend these studies to human cells, we made HT-29 cells competent for IAV-induced cell death signaling by stably transducing them with FLAG-tagged human ZBP1 (Figure S1A). In these cells, IAV proteins were observed 2–4 h p.i. (Figure S1B). Activation of ZBP1, as measured by phosphorylation of MLKL (pMLKL), was seen in whole-cell extracts 3–6 h p.i. in murine cells (Figure 1C) or 2–4 h p.i. in human cells (Figure S1B), coincident with the onset of virus protein synthesis.
Figure 1. ZBP1 Activates MLKL in the Nucleus of IAV-Infected Cells.
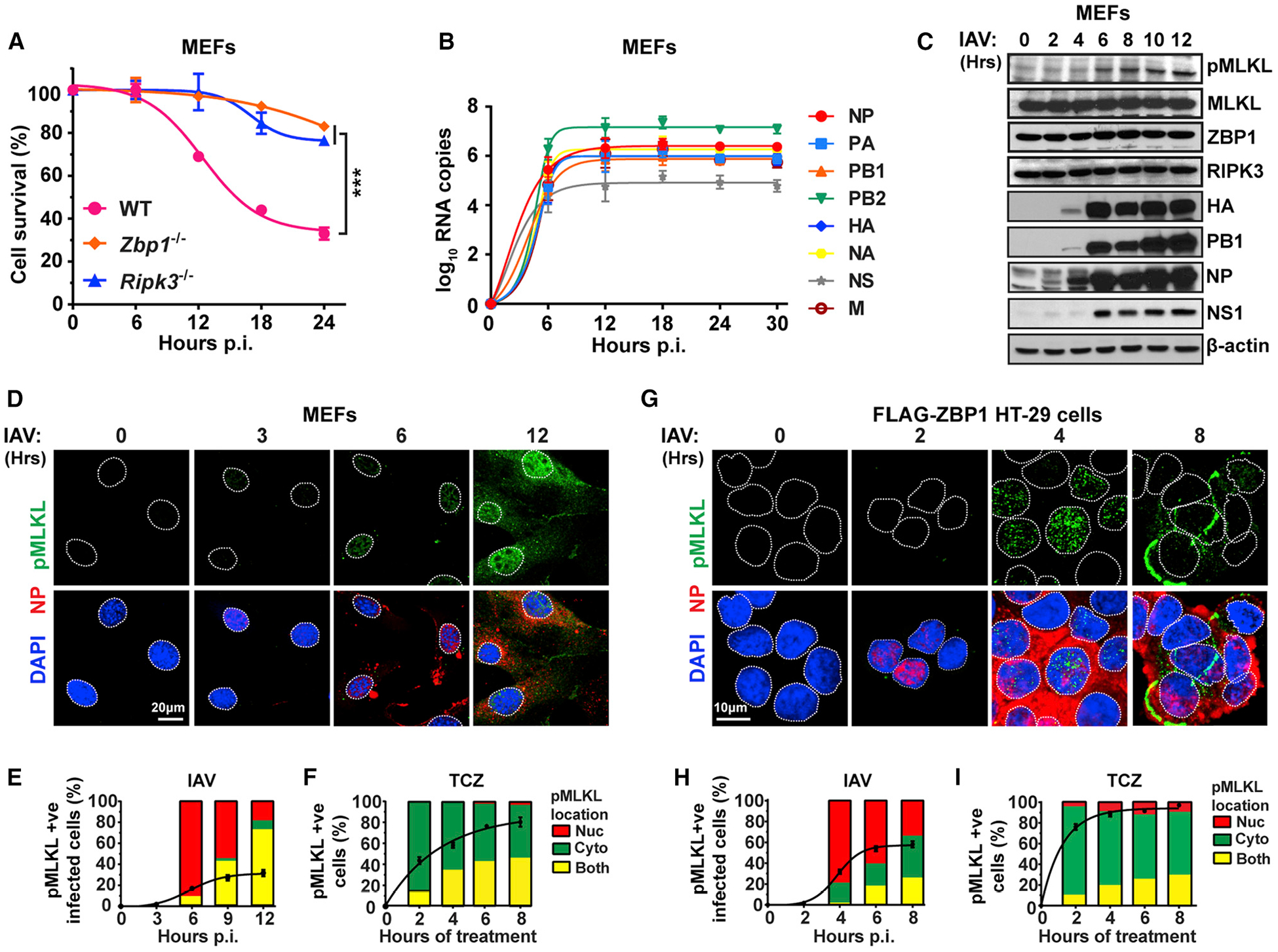
(A) IAV-induced cell death kinetics in primary wild-type (WT), Zbp1−/−, and Ripk3−/− MEFs.
(B) IAV vRNA production kinetics in infected WT MEFs.
(C) Immunoblot analysis of MLKL activation and viral protein synthesis in WT MEFs. (D) Immunofluorescence staining of IAV-infected WT MEFs for phosphorylated MLKL (green) and NP expression (red).
(E and F) Quantification of pMLKL localization in IAV-infected (E) and TCZ-treated (F) WT MEFs.
(G) Immunofluorescence staining of IAV-infected FLAG-ZBP1 expressing HT-29 cells for phosphorylated MLKL (green) and NP expression (red).
(H and I) Quantification of pMLKL localization in IAV-infected (H) and TCZ-treated (I) FLAG-ZBP1 HT-29 cells. Line graphs in (E), (F), (H), and (I) represent the kinetics of pMLKL positivity, and bar graphs show the localization of pMLKL signal. Nuclei are stained with DAPI (blue) and outlined by dashed white lines. TCZ = human or murine TNFα (50 ng/mL) + cycloheximide (250 ng/mL) + zVAD (50 μM). IAV PR8 was used at MOI = 2 in these experiments. Data are representative of at least three independent experiments. Error bars represent mean ± SD. Two-way ANOVA and Tukey’s multiple comparisons test, ***p < 0.0005. Scale bars represent 20 μm (D) and 10 μm (G).
See also Figure S1.
As IAV replicates in the nucleus, we next examined whether activation of necroptosis was initiated in the nucleus. IAV-infected MEFs displayed a distinct pMLKL signal starting at ~6 h p.i.; this signal was first observed in the nucleus, before becoming evident in the cytoplasm and at the plasma membranes of infected cells between 9–12 h p.i. (Figure 1D, quantified in 1E). In contrast, the tumor necrosis factor alpha (TNF-α)-initiated pMLKL signal was first detected in the cytoplasm and only later in the nucleus, as previously reported (Weber et al., 2018; Yoon et al., 2016) (Figures 1F and S1C). Staining IAV-infected HT-29 FLAG-ZBP1 cells for pMLKL showed a readily observable nuclear signal staining by 4 h p.i., followed by cytoplasmic dispersal and plasma membrane localization by 8 h p.i. (Figure 1G, quantified in 1H). In these cells, TNF-α-activated pMLKL was primarily cytoplasmic, detectable within 2 h post-treatment, and seen at the plasma membrane by 4 h (Figures 1I and S1D). These results indicate that IAV-activated ZBP1-RIPK3 necroptosis signaling, unlike canonical TNF-α-induced necroptosis, is initiated in the nucleus.
ZBP1 Senses IAV DVG RNA in the Nucleus
We next tested if ZBP1 sensed IAV RNAs in the nucleus. Our previous RNA sequencing (RNA-seq) results (Thapa et al., 2016) have shown that ZBP1 may preferentially associate with shorter IAV genomic RNAs, including defective viral genomes (DVGs) derived from the PB1, PB2, and PA gene segments, and such DVGs may represent activating ligands for ZBP1. These DVGs are produced when the IAV RNA-dependent RNA polymerase initiates vRNA synthesis, but falls off its genomic template before re-engaging further downstream to complete transcription. Such DVG RNAs nonetheless retain their packaging signals, and are encapsidated into virion-like “defective interfering” (DI) particles (Nayak et al., 1985). IAV PR8 stocks typically contain some fraction of DI particles, but can be grown under conditions that are enriched in, or largely lack, DI particles (Xue et al., 2016); these stocks therefore either have high (HD) or low (LD) levels of DVG RNAs, respectively (Figure 2A). When we infected wild-type MEFs with equivalent amounts of IAV HD or IAV LD virus, we found that IAV HD caused ~60% cell death by 12 h whereas an equivalent inoculum of IAV LD killed <20% of infected cells at this time point (Figure 2B). Only at 18 h p.i. did IAV LD induce cell death to levels that were comparable to IAV HD (Figure 2B), presumably because DVGs began to also accumulate in IAV LD-infected cells (Xue et al., 2016). IAV HD triggered MLKL phosphorylation earlier (Figures 2C and 2D) and more robustly (Figures 2C and 2E) than IAV LD. Because these results strongly indicate that IAV DVGs are dominant ZBP1 activating ligands, we next tested if ZBP1 associated with DVGs in the nucleus of infected cells. We stably reconstituted Zbp1−/− MEFs with FLAG-tagged murine ZBP1, infected these cells with IAV, eluted RNA co-precipitating with either nuclear or cytoplasmic ZBP1, and examined eluates (as well as input lysates) for DVG RNA. We observed PA-segment-derived DVG RNAs in the nuclear input fraction, starting at ~4 h post-infection, and at much lower levels in the cytoplasm later in the course of infection (Figure 2F). Importantly, we were able to readily detect these DVGs co-precipitating with ZBP1 solely in the nuclear fraction (Figure 2F), and when ZBP1-RNA complexes were crosslinked before immunoprecipitation (Figures S4A and S4B). RNA-seq of ZBP1-bound nuclear PA segment-derived RNAs confirmed that the DVG sequences mapped to the 5′ and 3′ termini of the PA segment, demonstrating that they represent internally deleted sub-genomic RNAs (Figure 2G). Together, these results support the idea that ZBP1 senses IAV DVGs and initiates cell death signaling in the nucleus of infected cells.
Figure 2. ZBP1 Senses IAV DVG RNA in the Nucleus.
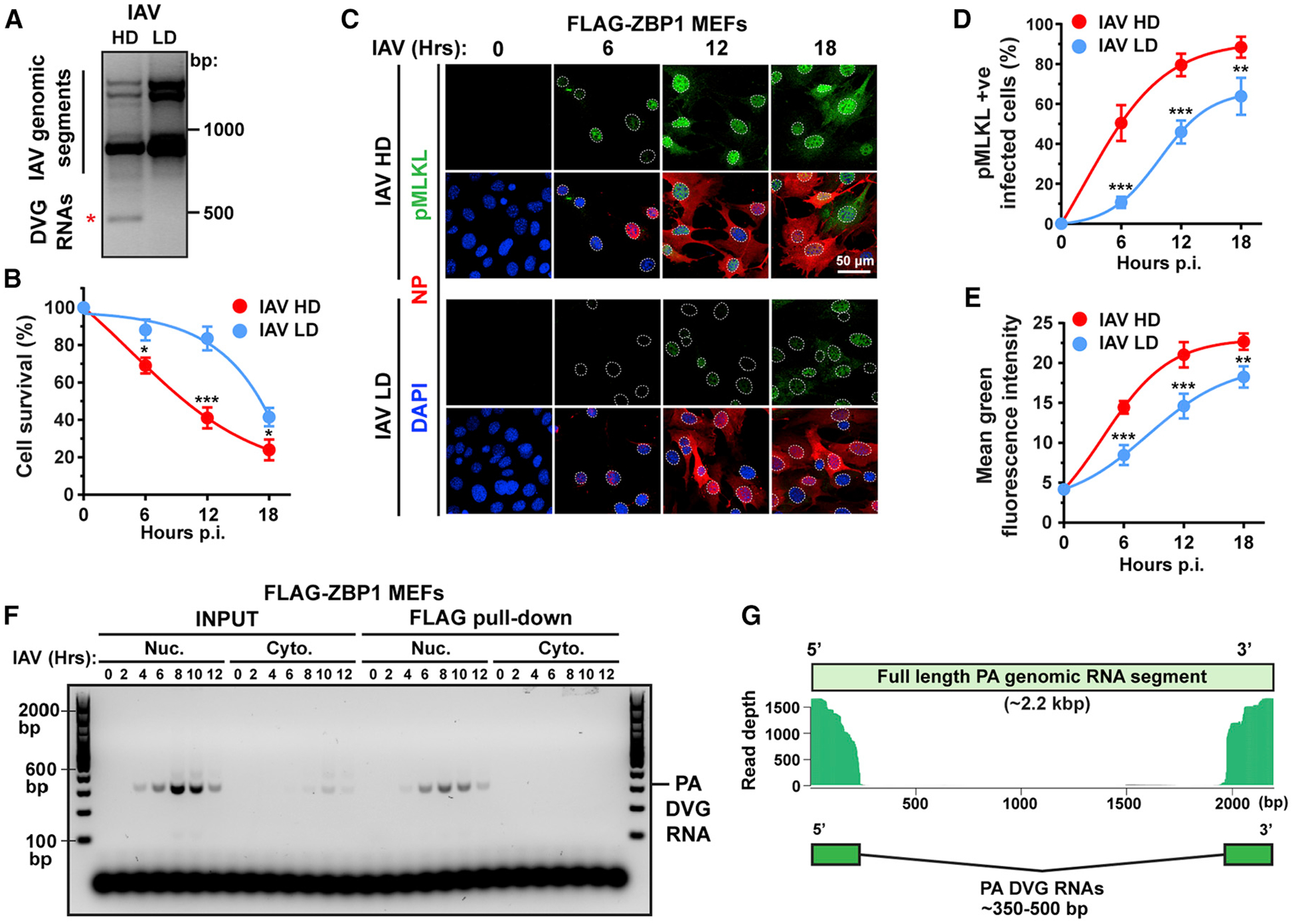
(A) PCR of full-length IAV genomic segments and DVGs in A549 cells infected with IAV PR8 stocks with high (HD) or low (LD) DI particle content. DVGs are indicated by a red asterisk.
(B) Cell death kinetics after IAV HD or LD virus infection of WT MEFs.
(C) Immunofluorescence staining for pMLKL (green) and NP (red) in WT MEFs infected with IAV HD or IAV LD virus. Nuclei are stained with DAPI (blue) and outlined with dashed white lines.
(D) Quantification of pMLKL+ cells as a percentage of infected (NP+) cells.
(E) Quantification of mean fluorescence intensity of the pMLKL signal per cell.
(F) Immortalized Zbp1−/− MEFs stably expressing FLAG-ZBP1 and infected with IAV were separated into nuclear and cytoplasmic fractions at the indicated times p.i. PA segment-derived DVG RNAs in input lysates or eluted from FLAG-ZBP1 pull-downs were determined by PCR.
(G) Paired-end sequencing was performed on PA segment-specific PCR amplicons of IAV RNAs eluted from FLAG-ZBP1 nuclear immunoprecipitates. Sequences with significant overlap are shown mapped to the full-length IAV PR8 PA segment. IAV was used at MOI = 2 in this figure. Data in (A)–(F) are representative of at least three independent experiments. RNA-seq for data in (G) was performed once. Error bars represent mean ± SD. Unpaired Student’s t test, *p < 0.05, **p < 0.005, ***p < 0.0005. Scale bar represents 50 μm.
IAV Produces Z-RNAs in the Nucleus
ZBP1 has two Zα domains, the second of which (Zα2) is essential for sensing IAV RNAs (Thapa et al., 2016). Zα domains bind Z-RNA, but not A-RNA, the right-handed double-stranded RNA (dsRNA) duplex that is structurally very different from Z-RNA (Figure 3A) and that serves as ligand for other dsRNA-binding innate sensors, such as MDA-5 and PKR (Kawai and Akira, 2006). These observations suggest that ZBP1-activating IAV RNAs adopt the Z-conformation, but whether Z-RNAs are produced during virus infections is unknown. A previous study used in situ antibody-based staining to show that Tetrahymena cytoplasm contains Z-RNA, demonstrating that Z-RNAs do exist in nature, and that an immunofluorescence approach to detecting Z-RNA in fixed cells is feasible (Zarling et al., 1987). Although no antibodies to Z-RNA are currently available, Z-RNA and Z-DNA share very similar structures, and several antisera raised to Z-DNA cross-react with Z-RNA in vitro (Hardin et al., 1987, 1988; Zarling et al., 1990). To examine if anti-Z-DNA antisera could also detect Z-RNA in cells, we first synthesized a Z-RNA duplex using a newly described approach in which 2′-O-methyl-8-methyl modification of guanosine nucleosides (m8Gm) allows the stabilization of CG-repeat dsRNA in the Z-conformation (Balasubramaniyam et al., 2018). The introduction of a methyl group at the C8 position strongly favors the syn conformation of guanosine, and modeling this modification in a CG-repeat dsRNA indicates that RNA duplexes containing m8Gm can undergo an A → Z transition with energetically favorable dynamics (Figure 3B). In fact, replacing the majority of guanosines with m8Gm analogs in CG-repeat dsRNAs produces Z-RNAs that are remarkably stable at physiological salt concentrations in vitro (Balasubramaniyam et al., 2018). We therefore synthesized a hairpin CG-repeat Z-RNA in which most guanosines were modified to m8Gm, and, as a control, generated an identical A-RNA hairpin without the m8Gm modification (Figure 3C, top). We then attached a fluorescent (FAM) label to each RNA hairpin and used these RNAs to screen anti-Z-DNA antibodies for their capacity to selectively detect Z-RNA. From this screen, we identified a sheep polyclonal antiserum raised against Z-DNA (hereafter, anti-Z-NA antiserum) that potently and completely retarded the mobility of synthetic m8Gm-containing Z-RNA, but not A-RNA, in an electrophoretic mobility shift assay in vitro (Figure 3C, bottom). Encouraged by this result, we transfected FAM-labeled Z-RNA or A-RNA hairpins into cells and tested if the anti-Z-NA antiserum can detect Z-RNA in cellulo. The anti-Z-NA antiserum produced a specific signal that co-localized with FAM-labeled Z-RNA in almost all (~95%) transfected cells (Figure 3D, quantified in 3E), and with the majority (~80%) of FAM-positive foci in these cells (Figure 3F). Importantly, this antiserum did not detect transfected FAM-labeled A-RNA (Figures 3D–3F). These data demonstrate that the anti-Z-NA antiserum can specifically recognize the presence of Z-RNA in cells.
Figure 3. IAV Produces Z-RNAs in the Nucleus.
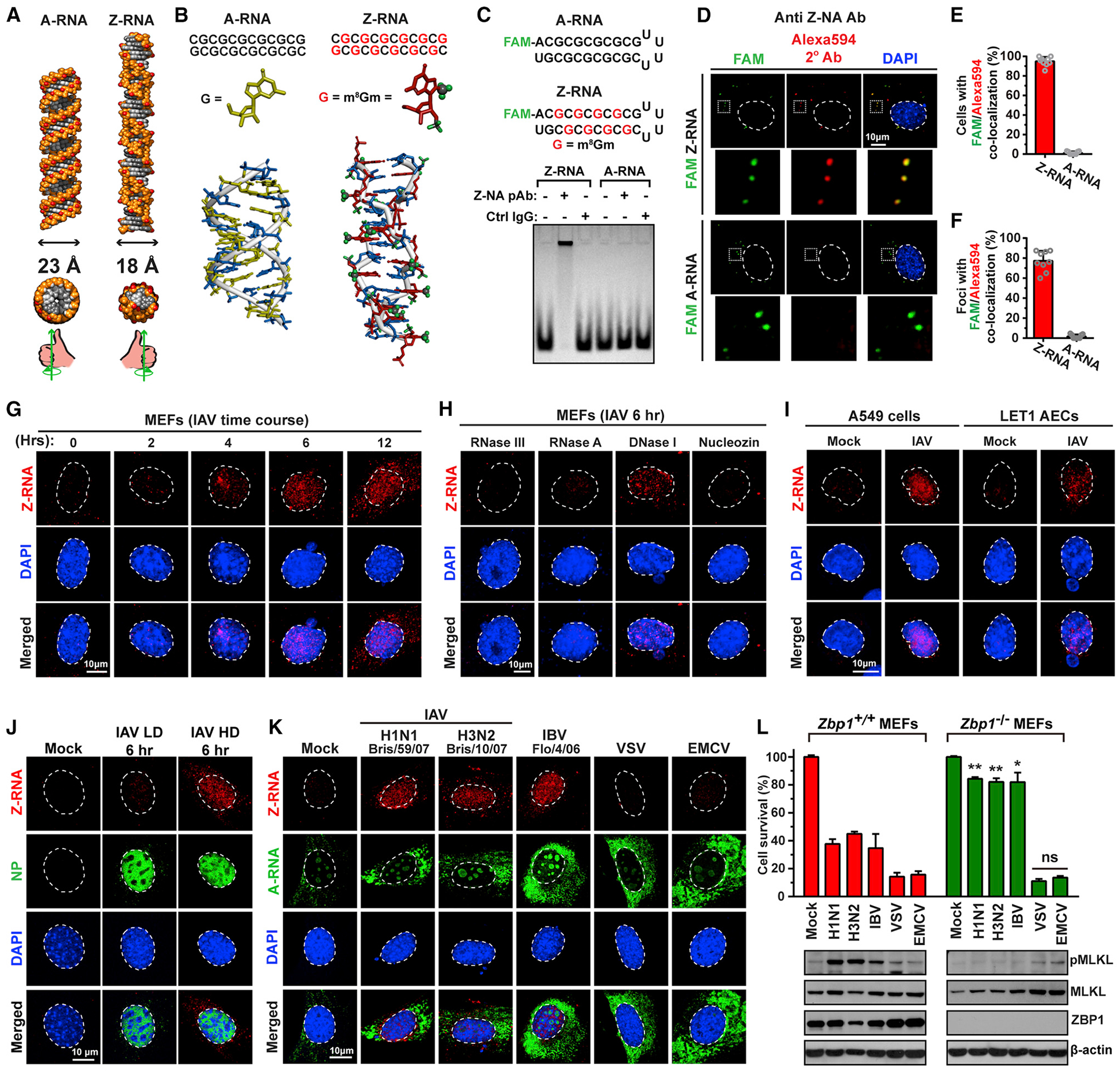
(A) Structures of A-RNA and Z-RNA. Diameters, cross-sections, and “handedness” of each double helix are also depicted.
(B) BIOVIA software generated models of unmodified (left) and m8Gm-modified (right) CG-repeat 12-mer RNA duplexes. Guanosines are shown in yellow and m8Gm-modified guanosine analogs in red, with the 2′-O-methyl and 8-methyl substitutions depicted in green. Cytidine nucleosides are shown in blue.
(C) Schematic representation of FAM-labeled A-RNA and Z-RNA hairpins, with m8Gm-modified guanosine shown in red (top). Electrophoretic mobility shift assay (EMSA) of synthetic A-RNA and Z-RNA in the presence of Z-NA antiserum or an equivalent amount of sheep IgG isotype control (bottom).
(D) WT MEFs transfected with FAM-labeled (green) synthetic Z-RNA (top) or A-RNA (bottom) and fixed 6 h post-transfection were stained with sheep polyclonal anti-Z-NA antiserum, and detected with Alexa594-conjugated secondary antibody (red). Areas selected in the upper images are shown magnified below each image. Nuclei are stained with DAPI (blue) and outlined with dashed lines.
(E) Quantification of cells displaying co-localization of Z-RNA or A-RNA (green) and anti-Z-NA antiserum (red) signals.
(F) Quantification of FAM-positive foci per transfected cell that co-localize with anti-Z-NA antiserum.
(G) Time course of Z-RNA formation in IAV infected MEFs (PR8, MOI = 10). Fixed cells were treated with proteinase K for 40 min before staining.
(H) WT MEFs infected with IAV (PR8, MOI = 10) and fixed at 6 h p.i. were exposed to the indicated nucleases for 45 min, before staining with anti-Z-NA antiserum. IAV RNA polymerase inhibitor nucleozin (20 ng/mL) was added to cells 1 h before infection.
(I) A549 and LET1 airway epithelial cells (AECs) were infected with IAV (PR8, MOI = 10), fixed at 6 h p.i., and stained for Z-RNA.
(J) WT MEFs were infected with IAV HD or LD (MOI = 10) for 6 h and stained with anti-Z-NA antiserum.
(K) WT MEFs were infected with the indicated RNA viruses (MOI = 10), fixed at 6 h p.i., and examined for presence of Z-RNA or A-RNA. Of note, the anti-A-RNA antibody produced modest nucleolar and cytoplasmic signals in uninfected cells. The cytoplasmic signal was perinuclear, punctate, and may represent mitochondrial dsRNAs (Dhir et al., 2018).
(L) Zbp1+/+ and Zbp1−/− MEFs were infected with the indicated RNA viruses (MOI = 2), and viability was determined at 24 h p.i. Immunoblot analysis shows the level of phosphorylated MLKL, total MLKL and ZBP1 after infection. Nuclei are stained with DAPI (blue) and outlined with dashed white lines. Data are representative of at least three independent experiments. Error bars represent mean ± SD. Unpaired Student’s t test, *p < 0.05, **p < 0.005. Scale bars represent 10 μm.
See also Figures S2 and S3.
Using this antiserum, we observed a modest RNase-sensitive nuclear signal in IAV-infected cells, but not in uninfected cells (Figure S2A). A previous study found that IAV-infected cells require protease treatment before IAV RNAs can be detected by antibodies, likely because these RNAs are masked by cellular or viral proteins (Son et al., 2015). We therefore exposed IAV-infected cells to proteinase K for 40 min post-fixation, before staining them with anti-Z-NA antiserum, and were now able to readily detect a nuclear signal that was first seen 2–4 h p.i. and increased in intensity over the 12 h time course of this experiment (Figure 3G, quantified in S2C). The strength of the Z-RNA signal was proportionate to the amount of virus inoculum and correlated with the extent of virus replication, as measured by immunofluorescent detection of nucleoprotein (NP) in the same nuclei (Figure S2B, quantified in S2D). The signal produced by the anti-Z-NA antiserum was sensitive to treatment with RNases III and A, insensitive to DNase I, and abolished by exposure of cells to the IAV replication inhibitor nucleozin (Kao et al., 2010) pre-infection (Figure 3H, quantified to S2E; RNase A and DNase I enzymes are bioactive at the concentrations used in this experiment [Figures S2G–S2I]). Z-RNA staining was also observed in IAV-infected human (A549) and murine (LET1) cell lines derived from airway epithelium (Figure 3I) and was effectively quenched by excess synthetic Z-RNA, but not by equivalent amounts of A-RNA or single-stranded RNA (ssRNA) (Figure S2J, quantified in S2K). Notably, a second antibody (mouse monoclonal Ab, clone Z-22) raised to Z-DNA and previously shown to cross-react with Z-RNA (Zarling et al., 1990) also specifically and robustly bound Z-RNA in vitro (Figure S3A) and readily detected its presence in cells (Figures S3B–S3D). Staining IAV-infected cells with this antibody following proteinase K treatment produced a dose- and time-dependent nuclear signal that was abolished by RNase A treatment and selectively quenched by excess Z-RNA (Figures S3E, S3G, S3I, quantified in S3F, S3H, S3J).
In agreement with the idea that IAV DVGs are a dominant source of Z-RNA, the Z-NA antiserum robustly stained nuclei in MEFs infected with IAV HD, but not in those infected with an equivalent amount of IAV LD, at 6 h p.i. (Figure 3J, quantified in S2F), when activation of MLKL and induction of cell death are evident in IAV HD- but not IAV LD-infected cells. Other orthomyxoviruses, including seasonal strains of IAV and IBV, also produced nuclear Z-RNA (Figure 3K), whereas a rhabdovirus (VSV) or a poliovirus (EMCV) did not generate detectable Z-RNA (Figure 3K). Each of these viruses, however, generated strong cytoplasmic A-RNA signals (Figure 3K), as previously reported (Son et al., 2015). In agreement with the idea that Z-RNAs are activating ligands for ZBP1, only orthomyxoviruses triggered ZBP1-dependent MLKL phosphorylation (Figure 3L, bottom) and cell death (Figure 3L, top) in MEFs. Collectively, these data provide evidence that replicating orthomyxoviruses produce Z-RNAs. They also show that these Z-RNAs are primarily localized to the nucleus, likely of DVG origin, and may function as ZBP1 ligands.
ZBP1 Co-localizes with Z-RNA in the Nucleus following IAV Infection
To test if ZBP1 sensed Z-RNAs in the nucleus, we first examined the cellular location of ZBP1 following infection with IAV. ZBP1 is primarily a cytoplasmic protein in uninfected MEFs, but rapidly accumulates in the nucleus following IAV infection (Figures 4A and 4B). Notably, a deletion mutant of ZBP1 lacking its two Zα domains (ΔZα), and hence incapable of binding viral RNA (Figures S4A and S4B), does not enter the nucleus, suggesting that the mechanism by which ZBP1 is recruited into the nucleus upon IAV infection may involve association with viral or cellular nucleic acids (Figure 4B, quantified in 4C). By the use of limited (20 min) proteinase K treatment, we were able to preserve a significant amount of polypeptide-based nuclear antigenicity but also unmask sufficient Z-RNA for detection with the Z-NA antiserum. Under these conditions, we observed distinct nucleoplasmic areas with co-localization of ZBP1 and Z-RNA (Figure 4D). RIPK3 and MLKL associate with ZBP1, but not mutants of ZBP1 lacking RNA binding (ΔZα) or RIPK3 signaling (RHIM-Amut) capacity upon IAV infection (Figure 4E). Because a significant proportion of the downstream necroptosis signaling components RIPK3 and MLKL are also nuclear (Figure 4A), and because RIPK3 activity was required for MLKL phosphorylation in IAV-infected nuclei (Figure S4C), we next tested if restricting ZBP1 to the nucleus was sufficient to activate necroptosis. By affixing either two tandem nuclear localization sequences (NLS) or nuclear export sequences (NES) onto the N terminus of ZBP1, we were able to, respectively, localize most (>90%) ZBP1 to the nucleus (ZBP1-Nuc) or largely retain this protein in the cytosol (ZBP1-Cyto) (Figure 4F, left). Cells expressing ZBP1-Nuc succumbed to virus with notably faster kinetics and increased magnitude than cells expressing an equivalent amount of wild-type ZBP1 (Figure 4F, right). In contrast, cells expressing ZBP1-Cyto were moderately more resistant to IAV-triggered cell death than controls (Figure 4F, right). Collectively, these results show that ZBP1 is recruited into the nucleus of infected cells, where it co-localizes with IAV Z-RNAs and initiates RIPK3-mediated cell death signaling. It is, however, noteworthy that ZBP1-Cyto remains largely restricted to the cytoplasm even after infection (Figure S4D), indicating that ZBP1 does not need to signal from the nucleus to activate cell death (see Discussion).
Figure 4. ZBP1 Co-localizes with Z-RNA in the Nucleus following IAV Infection.
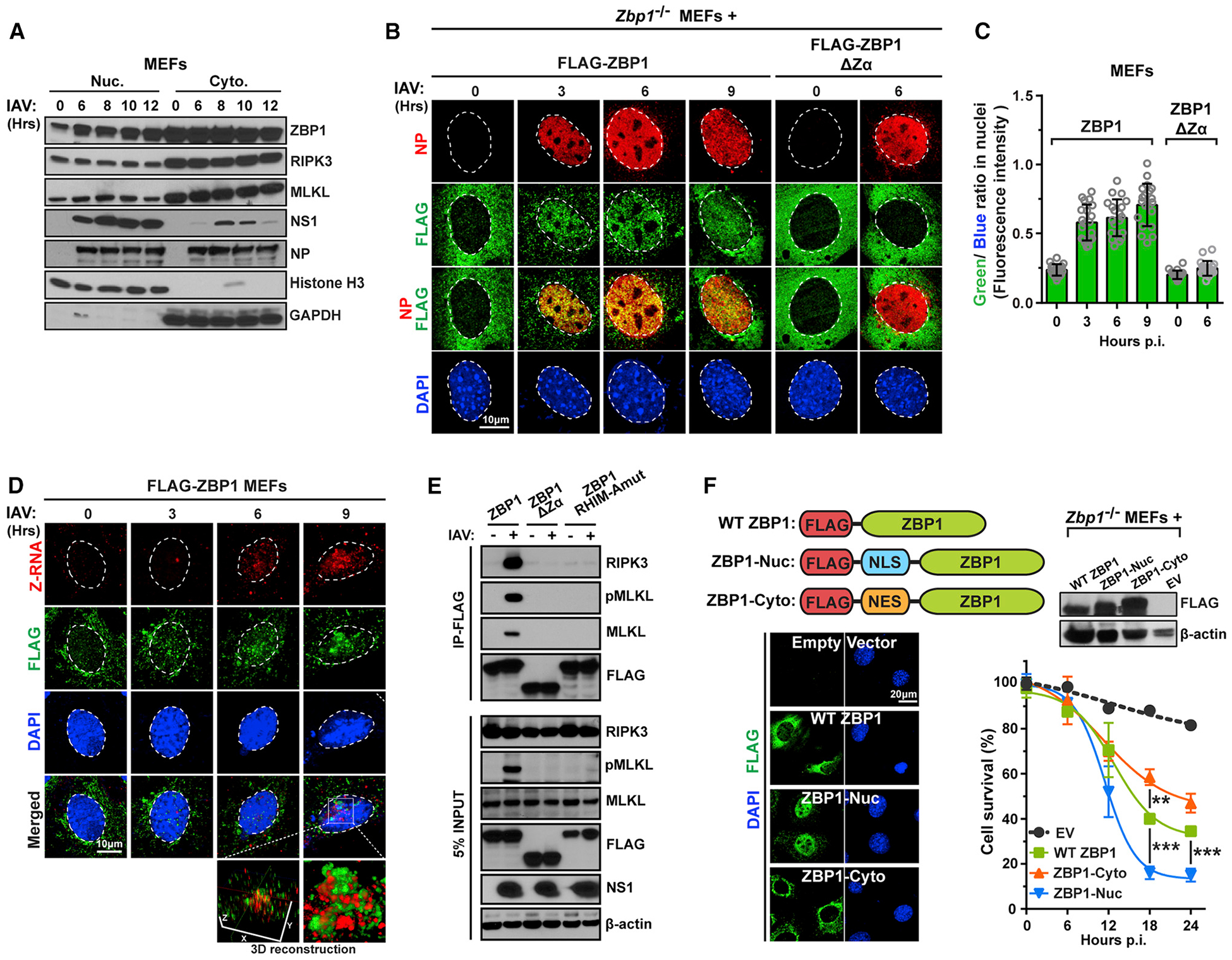
(A) IAV-infected (PR8, MOI = 2) WT MEFs were lysed at the indicated times p.i., separated into nuclear and cytoplasmic fractions, and examined for ZBP1, MLKL, RIPK3, and viral proteins by immunoblotting. Immunoblotting for GAPDH and histone H3 was used to confirm purity of cytoplasmic and nuclear fractions.
(B) Zbp1−/− MEFs stably reconstituted with either FLAG-ZBP1 or a mutant of ZBP1 (ΔZα) lacking both its Zα domains were infected with IAV (PR8, MOI = 10) and evaluated for ZBP1 localization over a time course of 9 h. Virus replication was assessed by co-staining for NP in the same cells.
(C) Quantification of the ratio of FLAG signal (green) and DAPI (blue) in nuclei indicates extent of nuclear accumulation of ZBP1 or mutant ZBP1.
(D) Zbp1−/− MEFs stably expressing FLAG-ZBP1 and infected with IAV (PR8, MOI = 10) were subjected to limited (20 min) proteinase K digestion post-fixation, and co-stained for Z-RNA (red) and FLAG-ZBP1 (green). 3D reconstruction of co-stained nuclei shows areas of Z-RNA:ZBP1 co-localization (yellow).
(E) Zbp1−/− MEFs reconstituted with FLAG-tagged WT ZBP1, ZBP1 lacking its Zα domains (ΔZα) or carrying a four amino acid (IQIG → AAAA) substitution in its first RHIM (RHIM-Amut) were infected with IAV (PR8, MOI = 5), and anti-FLAG immunoprecipitates were examined for RIPK3, MLKL, and FLAG. Whole-cell extract (5% input) was examined in parallel for RIPK3, MLKL, FLAG, and IAV NS1 proteins.
(F) Two tandem NLS or three tandem NES were introduced between the FLAG tag and the N terminus of WT ZBP1 (top left) to produce ZBP1-Nuc and ZBP1-Cyto, respectively. Stably reconstituting Zbp1−/− MEFs with FLAG-tagged ZBP1-Nuc or ZBP1-Cyto either localized ZBP1 to the nucleus or excluded it from the nucleus, respectively, in >90% of cells (bottom left). Expression levels of ZBP1 constructs were confirmed by immunoblotting (top right). Kinetics of IAV-induced death of Zbp1−/− MEFs stably expressing WT ZBP1, ZBP1-Nuc, or ZBP1-Cyto over a 24 h time course (PR8, MOI = 2). Nuclei are outlined with dashed white lines. Data are representative of at least three independent experiments. Error bars represent mean ± SD. Two-way ANOVA coupled with Tukey’s multiple comparisons test, **p < 0.005, ***p < 0.0005. Scale bars represent 10 μm [(B) and (D)] and 20 μm (F).
See also Figure S4.
IAV Triggers MLKL-Dependent Nuclear Envelope Disruption
During the course of these studies, we consistently observed alterations in nuclear morphology, characterized by breaches in the nuclear envelope and protrusion of host DNA into the cytosol, in approximately half of all dying MEFs. The inner and outer nuclear membranes both contain phospholipids, including phosphatidylinositol phosphates that serve as MLKL attractants in the plasma membrane (Dondelinger et al., 2014; Kleinig, 1970; Wang et al., 2014), raising the possibility that these phospholipids might also draw MLKL to nuclear membranes and lead to their rupture during IAV-activated necroptosis. To test if MLKL was responsible for nuclear envelope rupture in IAV-infected cells, we first examined nuclear morphology and nuclear envelope integrity in wild-type MEFs over an 18 h time course of IAV infection. Immunofluorescence imaging of the nuclear lamina protein lamin B1 and the inner membrane protein emerin in uninfected cells showed ovoid nuclei with well demarcated boundaries (Figure 5A). Within 6 h of infection, however, large gaps in lamin B1 staining (Figure 5A, top panels), as well as breaches, involutions, blebs, and other aberrancies in the emerin signal (Figure 5A, middle panels), became evident. Interestingly, DAPI staining for host DNA showed large cytosolic extrusions of herniated genomic material within 6 h p.i. in a significant proportion of infected cells (Figure 5A, bottom panels). Reconstructing images of damaged nuclei in three dimensions showed that DNA appeared to herniate from areas of breached lamina (i.e., absence of lamin B1 staining), and in many cases, these DNA blebs were decorated with emerin (Figure 5B). Quantifying disruptions to the nuclear envelope showed that fully one-half of all dying cells (and approximately one-third of all cells) had ruptured nuclear envelopes and manifested evidence of DNA leakage into the cytosol by 18 h p.i. (Figure 5C). Co-staining infected cells for MLKL and emerin demonstrated co-localization of MLKL with the nuclear envelope in infected wild-type MEFs (Figure 5D). Mlkl−/− MEFs were mostly resistant to IAV-induced membrane damage, but reintroducing MLKL expression in Mlkl−/− MEFs restored to wild-type levels the number of cells with nuclear rupture and DNA leakage (Figures S5A and S5B). These results demonstrate that MLKL can localize to nuclear membranes and is a dominant instigator of nuclear envelope damage during IAV infection.
Figure 5. IAV Triggers MLKL-Dependent Nuclear Envelope Disruption and DNA Leakage.
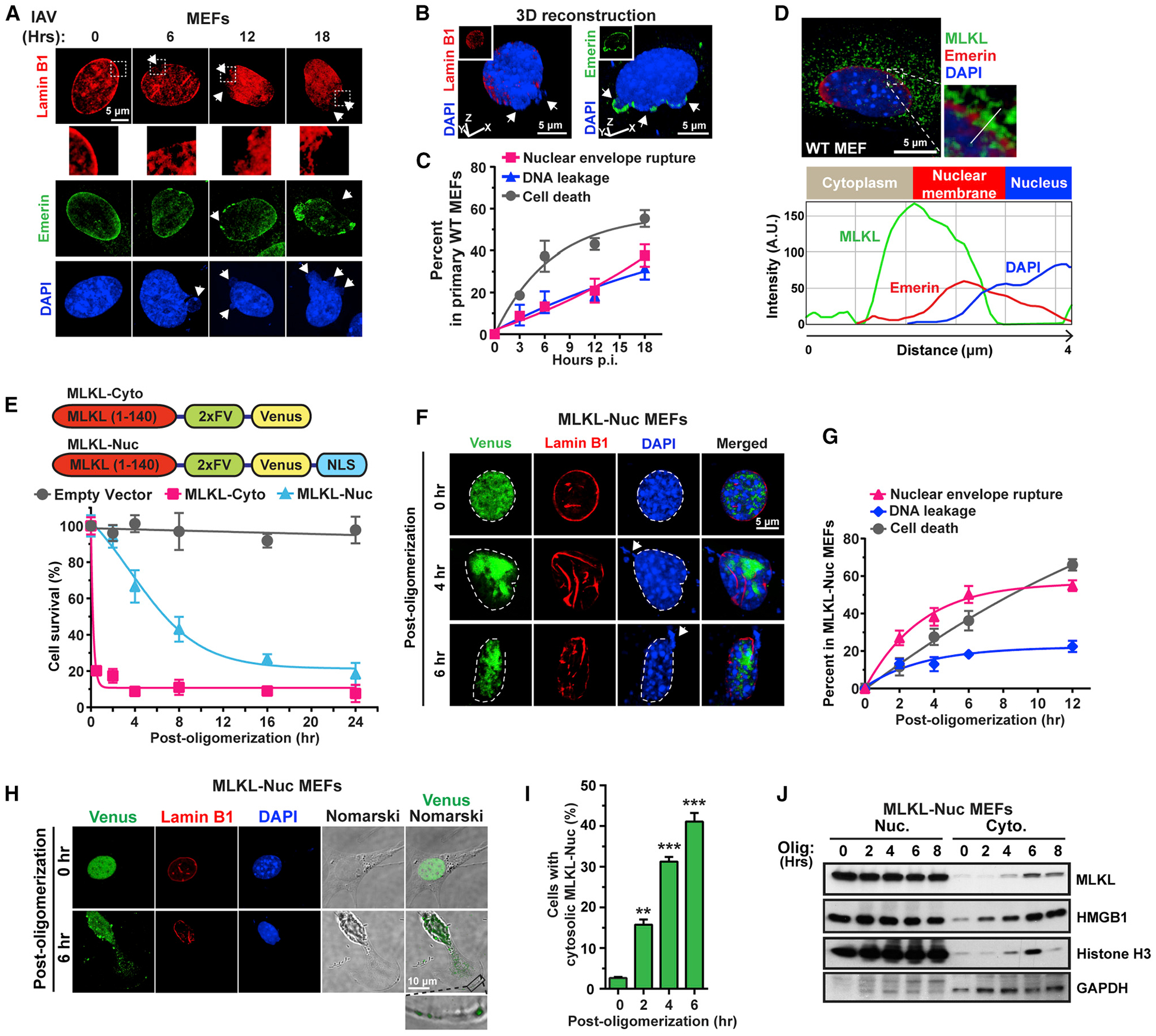
(A) WT MEFs infected with IAV (PR8, MOI = 2) were examined for nuclear envelope integrity using antibodies to lamin B1 and emerin at the indicated times p.i. Arrows point to envelope breaches (lamin B1/emerin staining) and nuclear DNA herniation and leakage (DAPI). Boxed areas from lamin B1 stained nuclei are magnified to show nuclear envelope disruption.
(B) 3D reconstruction of IAV-infected nuclei showing DNA herniating from gaps in the nuclear envelope (upper panels). Arrows indicate areas of herniation.
(C) Kinetics of nuclear envelope breakdown, DNA leakage, and cell death in IAV-infected WT MEFs. (PR8, MOI = 2).
(D) Immunofluorescence imaging of MLKL at nuclear membranes following IAV infection. Multi-channel densitometry along the white line shows localization of MLKL (green) at the nuclear envelope.
(E) Immortalized Mlkl−/− MEFs stably expressing 2xFv-tagged MLKL-Cyto or MLKL-Nuc constructs were examined for cell death at the indicated times post-activation.
(F) Imaging shows aggregation of MLKL-Nuc (green), distortion of nuclear architecture (red), and leakage of DNA into the cytosol (blue) following activation of MLKL-Nuc. Arrows indicate DNA leakage.
(G) Kinetics of nuclear envelope rupture, DNA leakage, and cell death after activation of MLKL-Nuc.
(H) Venus-tagged MLKL-Nuc is seen in the cytoplasm and translocates to the plasma membrane by 6 h post-oligomerization.
(I) Quantification of cells with cytoplasmic MLKL-Nuc at the indicated times post-oligomerization.
(J) Immunoblot analysis of MLKL-Nuc, histone H3, and HMGB1 in nuclear and cytoplasmic fractions from MLKL-Nuc -expressing cells at the indicated times post-oligomerization. Nuclei are outlined with dashed white lines. Data are representative of at least three independent experiments. Error bars represent mean ± SD. Unpaired Student’s t test, **p < 0.005, ***p < 0.0005. Scale bars represent 5 μm ([A], [B], [D], and [F]) and 10 μm (H).
See also Figure S5.
Next, we examined if, in the absence of IAV, activating MLKL in the nucleus is sufficient to trigger nuclear membrane damage. We have previously shown that the N-terminal 140 amino acids of MLKL, comprising its effector helix bundle domain and brace region, is a potent inducer of necroptosis when activated by chemically induced oligomerization (Quarato et al., 2016). By attaching two tandem nuclear localization sequences to this segment of MLKL, we were able to almost wholly restrict its expression to the nucleus and found that nuclear localized MLKL1–140 (hereafter MLKL-Nuc) was able to induce cell death upon activation, albeit with significantly slower kinetics than cytosolic MLKL1–140 (MLKL-Cyto; Figure 5E). Whereas ~80% of MLKL-Cyto-expressing cells were dead by 30 min post-activation, it took 16–24 h for MLKL-Nuc-containing cells to achieve comparable levels of cell death (Figure 5E). MLKL-Nuc in unstimulated cells displayed a diffuse signal that was almost completely contained within the nucleus, but transitioned within 4 h of activation to a pattern of large aggregates, many of which were seen along the nuclear periphery (Figure 5F). Accompanying these alterations in the distribution of MLKL-Nuc were gross distortions in the architecture of the nuclear envelope (Figure 5F). Most dying cells displayed ruptured nuclear envelopes, with detectable herniated DNA in over a third of these cells, by 6 h (Figure 5F). These results are quantified in Figure 5G. Cells expressing activated MLKL-Cyto, in contrast, showed only modest evidence of distorted nuclear architecture and little detectable DNA leakage, at a time point when the extent of cell death was comparable to cells with active MLKL-Nuc (Figures S5C and S5D). By 4–6 h post-activation, MLKL-Nuc was evident in the cytosol and at the plasma membrane (Figures 5H–5J). Also seen in the cytosol of cells harboring activated MLKL-Nuc by 6 h was the nuclear protein HMGB1 (Figure 5J). Treatment of MLKL-Nuc-expressing cells with the nuclear export inhibitor leptomycin B 1 h post-activation was able to retain a significant fraction of MLKL in the nucleus (Figure S5E) and partially protect against both nuclear envelope rupture (Figure S5F) and cell death (Figure S5G).
IAV-Induced Nuclear Envelope Disruption Does Not Require RIPK3-Mediated Apoptosis
ZBP1-RIPK3 signaling activates not only necroptosis, but also apoptosis, via a RIPK1-FADD-caspase-8 axis, in infected cells (Nogusa et al., 2016). To examine if apoptosis was required for nuclear envelope disruption upon activation of ZBP1-RIPK3 cell death signaling, we first confirmed that ZBP1 itself was necessary for nuclear envelope rupture in IAV infected cells. Approximately half of all infected primary wild-type MEFs displayed ruptured nuclear envelopes, but only ~10% of Zbp1−/− MEFs did so; the remaining cells had intact nuclei (Figures 6A and 6B). Reconstituting Zbp1−/− MEFs with wild-type ZBP1, or with deletion mutants of ZBP1 that lack the Zα1 domain (ZBP1 ΔZα1) or the C-terminal third (ZBP1 ΔC) of this protein (Figure S6) but are nonetheless competent for cell-death signaling (Thapa et al., 2016), restored their capacity to undergo nuclear envelope rupture and vent DNA into the cytosol (Figures 6A and 6B). In contrast, reconstituting Zbp1−/−MEFs with mutants of ZBP1 that either fail to bind IAV RNA (ZBP1 Za2mut) or signal to RIPK3 (ZBP1 RHIM-Amut) (Figure S6) prevented both nuclear envelope rupture as well as DNA herniation (Figures 6A and 6B). Exposing wild-type MEFs to the pan-caspase inhibitor zVAD, which blocks IAV-activated apoptosis, did not prevent nuclear envelope rupture or DNA extrusion (Figures 6C and 6D), but pretreating wild-type MEFs with the RIPK3 kinase inhibitor GSK′843, which selectively blocks MLKL activation and necroptosis (Mandal et al., 2014), or with the combination of zVAD and GSK′843, which prevents all IAV-activated cell death (Nogusa et al., 2016), effectively blocked damage to the nuclear membrane (Figures 6C and 6D). Similarly, cells lacking RIPK3 or MLKL were resistant to IAV-triggered nuclear membrane rupture (Figures 6C and 6D), while cells lacking RIPK1 (Ripk1−/− ) or harboring non-cleavable knockin alleles of caspase-8 (Casp8DA; this allele encodes a D387A point mutant of caspase-8) (Kang et al., 2008), and therefore selectively defective in apoptosis signaling downstream of RIPK3 (Nogusa et al., 2016), were as sensitive as wild-type cells to nuclear membrane damage and DNA leakage upon IAV infection (Figures 6C and 6D). In contrast to the necroptosis axis, which is first activated in the nucleus, apoptosis signaling (as measured by staining for activated caspase-3) appears to be a predominantly cytoplasmic event, for reasons that are currently unclear (Figure 6E). Interestingly, MEFs undergoing either death receptor-triggered necroptosis (TCZ, in which the pMLKL signal originates in cytoplasm), or apoptosis (TRAIL + CHX), did not display detectable evidence of nuclear envelope rupture or herniated DNA in the cytosol (Figures 6F and 6G), although both stimuli resulted in misshapen nuclei, and in the case of cells undergoing apoptosis, pronounced nuclear shrinkage (Figure 6F). Thus, apoptosis is not required for nuclear envelope rupture following IAV infection.
Figure 6. RIPK3-Mediated Apoptosis Signaling Is Not Required for IAV-Induced Nuclear Envelope Disruption.
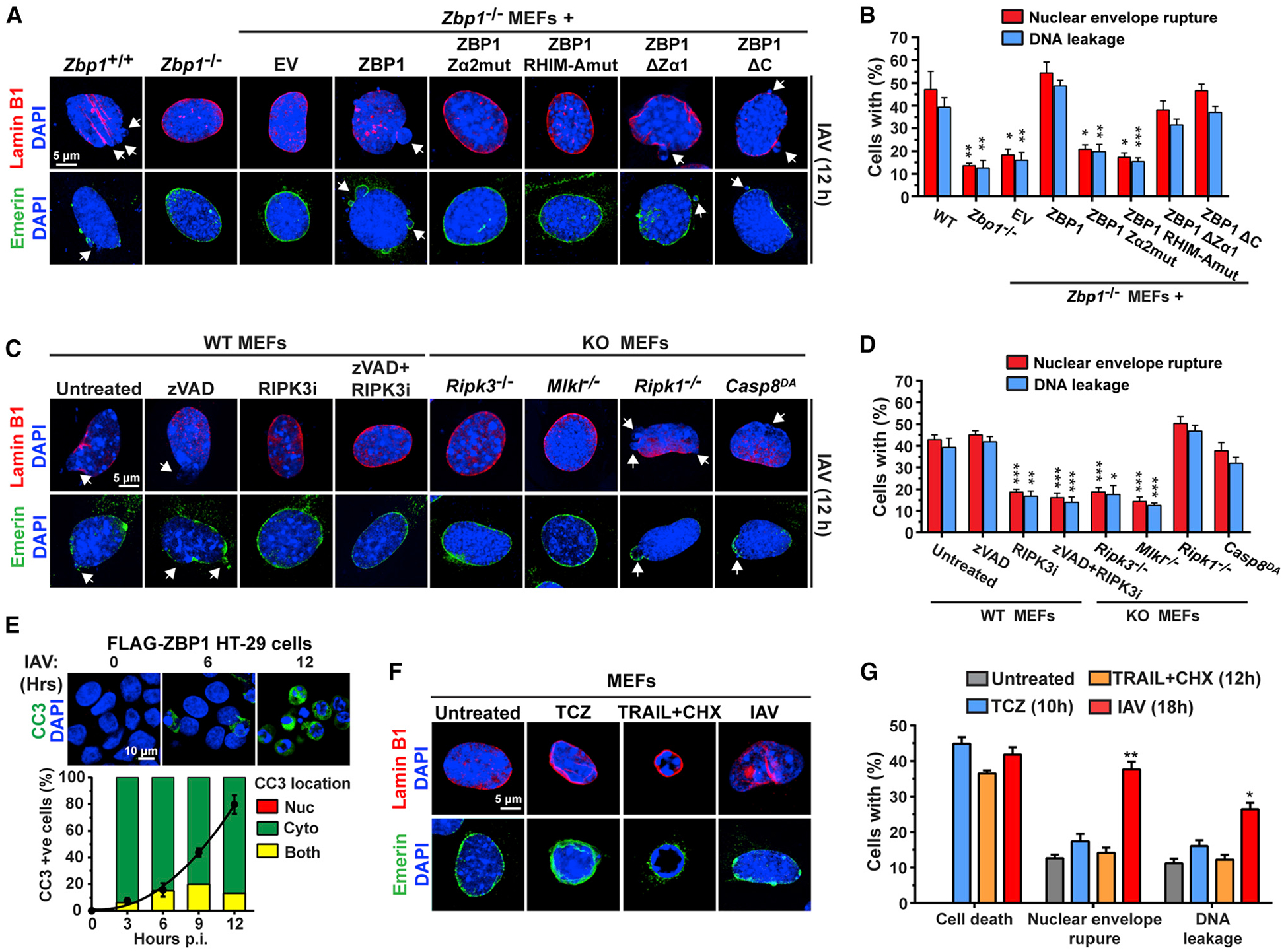
(A) Lamin B1 (red) or emerin (green) staining for nuclear envelope integrity of IAV infected WT or Zbp1−/− MEFs stably reconstituted with the indicated ZBP1 constructs at 12 h p.i.
(B) Quantification of nuclear envelope damage in cells shown in (A).
(C) Lamin B1 (red) or emerin (green) staining for nuclear envelope integrity of IAV-infected WT MEFs treated with zVAD (50 μM), RIPK3 inhibitor (RIPK3i, GSK′843, 5μM) or zVAD + RIPK3 inhibitor, and of IAV-infected Ripk3−/−, Mlkl−/−, Ripk1−/−, and Casp8DA MEFs, at 12 h p.i.
(D) Quantification of nuclear envelope damage of cells shown in (C). Arrows show nuclear envelope breakdown in (A) and (C). PR8 was used at MOI = 2 and cells were fixed at 12 h p.i. for results shown in (A)–(D).
(E) Staining for cleaved caspase 3 (CC3, green) shows activation of apoptosis upon IAV infection (PR8, MOI = 2) is primarily a cytoplasmic event. Line graph shows the kinetics of CC3 positivity, and bars show the localization of the CC3 signal.
(F) Lamin B1 (red) or emerin (green) staining of the nuclear envelope in WT MEFs treated with TCZ, TRAIL (2 μg/mL) plus cycloheximide (250 ng/mL) (TRAIL+CHX) or infected with IAV (PR8, MOI = 2). Cell viability and the percentage of cells with ruptured nuclear envelopes and nuclear DNA leakage were measured at 12 h (TCZ and TRAIL) or 18 h (IAV) post treatment/infection, when cell death was equivalent between the three stimuli. Data are representative of at least three independent experiments. Error bars represent mean ± SD. Unpaired Student’s t test, *p < 0.05, **p < 0.005, ***p < 0.0005. Scale bars represent 5 μm ([A], [C], and [F]) and 10 mm (E).
See also Figure S6.
MLKL Drives Pathogenic Neutrophil Recruitment and Lethality during Severe IAV Disease
To evaluate the role of necroptosis in IAV-triggered inflammatory responses in vivo, we compared disease and survival outcomes in Mlkl−/− mice to their wild-type counterparts, as well as to Zbp1−/− and Ripk3−/− animals. At a modestly lethal dose (EID2500 , ~LD20) of IAV (strain PR8), we found that Mlkl−/− mice were not any more susceptible to lethality than wild-type (C57BL/6J) mice, whereas both Zbp1−/− and Ripk3−/− mice displayed significantly increased rates of mortality (Figure 7A). Because Zbp1−/− and Ripk3−/− animals are deficient in both apoptosis and necroptosis signaling, their increased susceptibility to IAV likely arises from a failure to eliminate infected cells and limit virus spread in pulmonary tissue (Nogusa et al., 2016; Upton et al., 2017). Mlkl−/− mice are deficient only in necroptosis; in these animals, apoptosis signaling is still intact and can effectively mediate virus clearance (Nogusa et al., 2016). Interestingly, when we challenged Mlkl−/− mice with a lethal dose (EID6000) of IAV, they manifested notably better survival outcomes than any of the other genotypes: 40% of Mlkl−/− survived at this dose of virus and made full recoveries by 3 weeks p.i. (Figure 7B). Expectedly, all Zbp1 −/− and Ripk3−/− mice succumbed within 2 weeks of infection at this dose (Figure 7B). We did not find significant differences in either lung virus burden (Figure 7C) or CD8+ T cell-driven adaptive immune responses to IAV (Figure 7D) between Mlkl−/− mice and their littermate controls (Mlkl+/+). The gating strategy used to identify IAV-specific CD8+ T cells is shown in Figure S7A. We did, however, observe that lungs from Mlkl−/− mice had notably fewer epithelial cells with disrupted nuclei, compared to similarly infected lungs from wild-type animals (Figure 7E).
Figure 7. MLKL Drives Pathogenic Neutrophil Recruitment and Lethality during Severe IAV Disease.
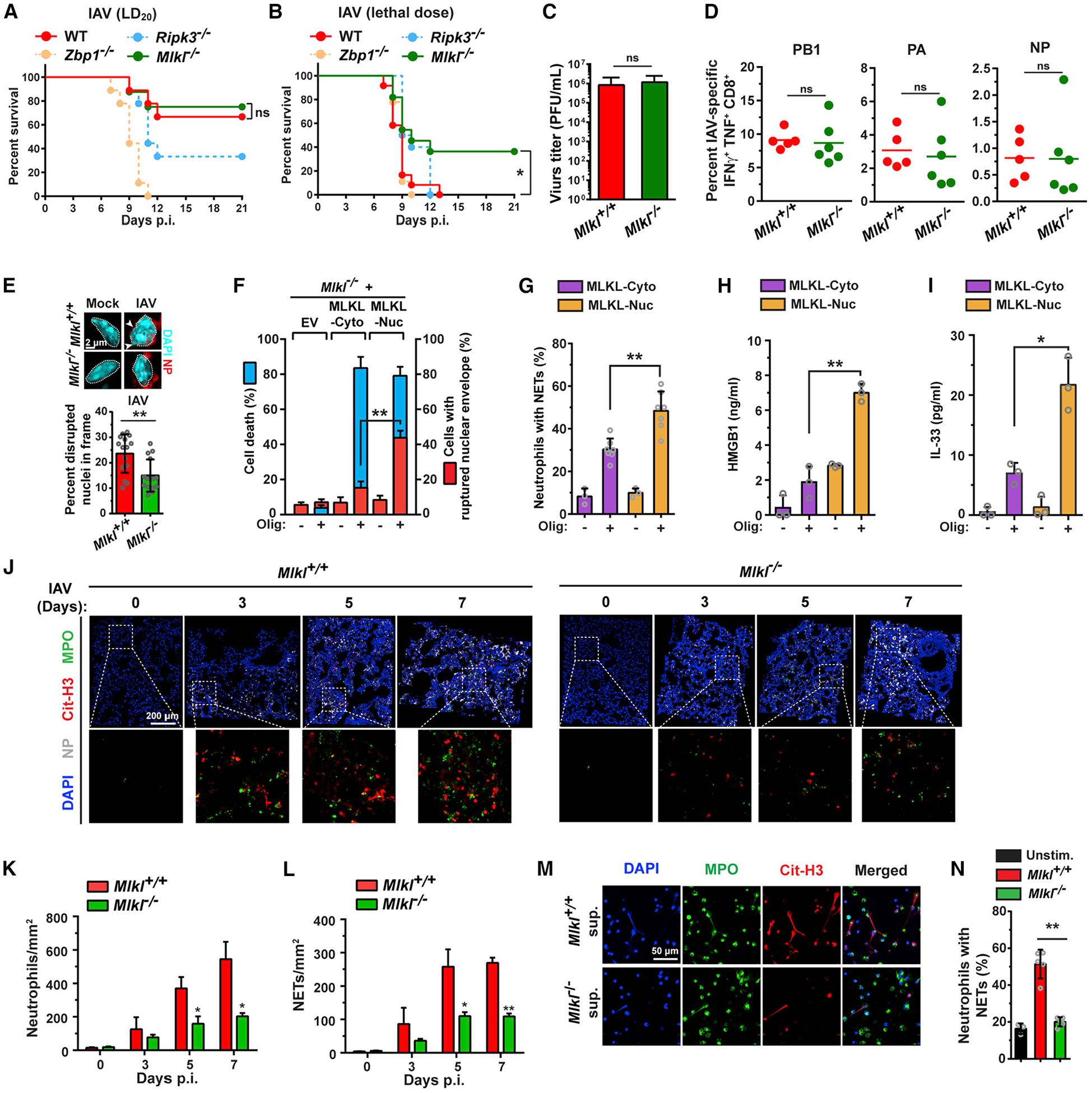
(A) Survival analysis of age- and sex-matched WT (C57BL/6J) (n = 9), Zbp1−/− (n = 9), Ripk3−/− (n = 9), and Mlkl−/− (n = 8) mice infected with a modestly lethal (~LD20) dose of IAV (PR8, 2,500 EID50/mouse intranasally [i.n.]).
(B) Survival analysis of age- and sex-matched WT (C57BL/6J) (n = 12), Zbp1−/− (n = 9), Ripk3−/− (n = 10), and Mlkl−/− (n = 11) mice infected with a lethal dose of IAV (PR8, 6000 EID50/mouse i.n.).
(C) Virus titers from lungs of Mlkl+/+ and Mlkl−/− mice infected with PR8 determined by plaque assay (n = 11 mice/genotype) at 6 days p.i.
(D) Percentage of IAV PB1, PA, NP peptide-stimulated poly-functional IFNγ+TNFα+ CD8+ T cells in BAL fluid from Mlkl+/+ (n = 5) and Mlkl−/− mice (n = 6) at 8 days p.i.
(E) Quantification of epithelial cells with disrupted or ruptured nuclei in Mlkl+/+ and Mlkl−/− lungs on day 7 p.i. (PR8, 6000 EID50, i.n.). Morphology of representative nuclei is shown above the graph. Arrows indicate areas of nuclear DNA herniation in Mlkl+/+ epithelial cells. Nuclei are outlined with dashed white lines.
(F) Quantification of cell death (blue) and nuclear envelope rupture (red) in Mlkl−/− MEFs stably expressing either MLKL-Cyto or MLKL-Nuc at the indicated times post-activation.
(G) Quantification of NETs by staining for citrinullated histone H3 after stimulation of neutrophils with supernatants from MLKL-Cyto and MLKL-Nuc MEFs that were either exposed or not exposed to oligomerizer.
(H and I) Protein levels of HMGB1 (H) and IL-33 (I) in supernatants of MLKL-Cyto and MLKL-Nuc MEFs after exposure to oligomerizer.
(J) Immunofluorescence staining for neutrophils (MPO, green), NETs (citrinullated histone H3, red), and NP (white) in lung sections from IAV (PR8, 6,000 EID50)-infected mice at the indicated days p.i. shows influx of neutrophils is both delayed and significantly diminished in IAV-infected Mlkl−/− lungs, compared to Mlkl+/+ controls.
(K and L) Quantification of neutrophil infiltration (K) and NET formation (L) in Mlkl+/+ and Mlkl−/− lungs on the indicated days p.i.
(M) Supernatants for IAV-infected Mlkl+/+, but not from similarly infected Mlkl−/− MEFs, trigger NET formation in challenged neutrophils ex vivo.
(N) Quantification of NETs after stimulation of neutrophils with supernatants from uninfected or from IAV-infected Mlkl+/+ and Mlkl−/− MEFs. IAV PR8 was used at MOI = 2 in M. Error bars represent mean ± SD. Log-rank (Mantel-Cox) test and unpaired Student’s t test, *p < 0.05, **p < 0.005. Scale bars represent 2 μm (E), 200 μm (J), and 50 μm (M).
See also Figure S7.
The nucleus contains immunogenic damage-associated molecular pattern molecules (DAMPs), including HMGB1 and DNA itself, that when released into the extracellular space, can recruit innate cells, such as macrophages and neutrophils, and stimulate an inflammatory response. Because neutrophils, in particular, are important drivers of IAV pathogenesis (Brandes et al., 2013; Camp and Jonsson, 2017), and because we had noticed that both HMGB1 and DNA were released into the cytosol following MLKL-mediated nuclear rupture (Figures 5A– 5C and 5J), we next evaluated the effect of activating MLKL in the nucleus of cells on the capacity of these dying cells to stimulate neutrophils ex vivo. Activation of both MLKL-Cyto and MLKL-Nuc kill MEFs, albeit with different kinetics, as we have shown earlier (Figure 5E). By 10 min post-addition of oligomerizer, ~80% of cells expressing MLKL-Cyto were dead, whereas MLKL-Nuc killed a similar proportion of cells only ~18 h after activation (Figure 7F). Notably, however, over 50% of dying cells expressing MLKL-Nuc showed evidence of nuclear envelope damage, while <20% of dying cells with MLKL-Cyto manifested disrupted nuclei (Figure 7F). We collected supernatants from cells expressing either MLKL-Cyto or MLKL-Nuc at a time when death in each case was ~80% (i.e., at 10 min or 18 h post-oligomerizer, respectively), used these supernatants to stimulate primary cultures of neutrophils, and quantified neutrophil extracellular traps (NETs) as a readout of neutrophil activation. While supernatants from untreated cells triggered NETs in <10% of primary murine neutrophils, those from dying cells expressing active cytoplasmic MLKL-Cyto induced NET formation in 30% of neutrophils (Figures 7G and S7B). Supernatants from MLKL-Nuc cells, however, induced NETs in a significantly greater proportion (~50%) of challenged neutrophils (Figures 7G and S7B). These supernatants also contained significantly higher levels of the nuclear DAMPs HMGB1 (Figure 7H) and interleukin (IL)-33 (Figure 7I). Collectively, these results demonstrate that MLKL-activated nuclear envelope rupture during IAV infection releases nuclear DAMPs, serves as a potent stimulator of neutrophils in cell culture, and by promoting the recruitment and activation of neutrophils into the infected lung, may contribute to the severity of IAV disease in vivo.
In line with these findings, we found a remarkable attenuation (by ~50%) in the degree of neutrophil infiltration (MPO+ cells; Figure 7J, quantified in 7K) and consequent formation of NETs, (measured by staining for citrinullated-H3) in Mlkl−/− lungs between days 3 and 7 after infection with a lethal dose of IAV (Figure 7J, quantified in 7L). Supernatants from IAV-infected Mlkl+/+ MEFs induced NET formation in ~50% of neutrophils when added to cultures of these cells ex vivo, whereas supernatants from similarly infected Mlkl−/− MEFs activated NET formation in <20% of neutrophils, comparable to those from uninfected controls (Figure 7M, quantified in 7N). These results are suggestive of an unanticipated role for MLKL signaling in driving pathogenesis during severe IAV infections, with potentially important clinical ramifications.
DISCUSSION
In this report, we show that IAV and other orthomyxoviruses produce Z-RNAs and propose that these Z-RNAs serve as activating ligands for ZBP1. For many years, Z-RNA duplexes were not believed to form under natural conditions (Athanasiadis, 2012; Brown et al., 2000), but prior to the current study, several lines of evidence supported the idea that Z-RNAs do occur in nature: Z-RNAs have been detected in Tetrahymena (Zarling et al., 1987); proteins containing Zα domains, most notably ADAR1 (Samuel, 2011) and ZBP1 (Kuriakose et al., 2016; Thapa et al., 2016), respond to RNA viruses; Zα domains associate with Z-RNA in vitro (Brown et al., 2000; Placido et al., 2007); and Zα domains can convert A-RNA to Z-RNA under near-physiological conditions (Brown et al., 2000). Our results now demonstrate that newly formed IAV RNA duplexes may adopt the Z-confirmation. We suggest these Z-RNA structures arise as a consequence of torsional stress induced by negative supercoiling during viral replication, as has been shown to occur with dsDNA during cellular transcription (Rich and Zhang, 2003; Wittig et al., 1991). The primary source of ZBP1-activating ligands appears to be Z-RNAs derived from DVGs, rather than from full-length genomes. We currently have no reason to believe that DVG RNAs are any more likely than full-length genomic RNA duplexes to form Z-RNA structures during replication. DVG RNAs may provide a dominant source of Z-RNA ligands for ZBP1 simply because, compared to full-length genomic RNAs, DVG RNAs are often produced in great excess during virus replication and are improperly packaged for encapsidation, making them more accessible to sensing by innate host defense mechanisms. Whether IAV RNAs may additionally undergo an A → Z transition upon interaction with ZBP1, or whether covalent modifications to IAV RNAs facilitate Z-RNA formation, remains to be seen. Notably, while right-handed RNA (A-RNA) and DNA (B-DNA) duplexes are structurally different from each other— for which reason protein domains that bind to B-DNA do not associate with A-RNA and vice versa—the left-handed Z-form duplexes of RNA and DNA have very similar structures. Indeed, Za domains can bind both Z-DNA and Z-RNA in vitro (Brown et al., 2000) raising the intriguing possibility that proteins with Za domains may represent bifunctional sensors capable of recognizing both Z-RNA and Z-DNA duplexes to initiate host innate-immune responses.
Is replication in the nucleus required for Z-RNA formation or activation of ZBP1? Orthomyxoviruses are almost unique among RNA viruses in that they replicate in the nucleus of the host cell. Other viruses known to activate ZBP1 (e.g., MCMV and HSV-1/2) (Mocarski et al., 2014; Upton et al., 2017) are DNA viruses that also replicate in the nucleus, whereas two cytoplasmic RNA viruses, VSV and EMCV, do not produce detectable Z-RNA or activate ZBP1. Although these results may suggest that nuclear replication is required for production of Z-RNA (or perhaps Z-DNA, in the case of DNA viruses) and/or activation of ZBP1, they do not rule out the existence of scenarios in which Z-RNA formation and ZBP1 activation occurs outside the nucleus. Indeed, multiple lines of evidence support this possibility, including the observations that (1) Z-RNA was first detected in the cytosol of eukaryotic cells (Zarling et al., 1987); (2) ZBP1 excluded from the nucleus still activates cell death upon IAV infection, albeit with slower kinetics that wild-type ZBP1 (Figure 4F); (3) ZBP1 has been implicated in sensing DNA from damaged mitochondria (Szczesny et al., 2018); (4) the Zα domain-containing proteins ADAR1 and PKZ operate in the cytoplasm (Rich and Zhang, 2003), where they limit the replication of cytoplasmic RNA viruses such as measles virus (ADAR1) or inhibit mRNA translation (PKZ); and (5) poxviruses replicate in the cytoplasm, but encode Zα domain containing proteins (e.g., Vaccinia virus E3L).
Once activated in the nucleus, ZBP1 initiates “inside-out” (i.e., nucleus-to-cytoplasm) cell death signaling in response to IAV. Shortly after activation of ZBP1, approximately one-third of infected cells display gross MLKL-dependent nuclear abnormalities, including rupture of the nuclear envelope and DNA herniation into the cytosol. These events are also seen when MLKL is artificially activated in the nucleoplasm, demonstrating that MLKL can directly impinge on the nuclear membrane to induce its rupture. This finding, while unanticipated, is not surprising, as the inner and outer nuclear membranes both contain phospholipids previously shown to mediate MLKL recruitment to the plasma membrane during conventional necroptosis. The mechanism by which active MLKL exits the nucleus and mediates nuclear membrane is currently unclear, although our results from use of the nuclear export inhibitor leptomycin B suggest that a significant proportion of MLKL nuclear egress is controlled by conventional nuclear pore-dependent mechanisms. Notably, leptomycin B-treated cells display distorted nuclear morphology but manifest significantly less evidence of nuclear envelope rupture, suggesting that cytosolic MLKL is a significant contributor to nuclear membrane damage during IAV-induced necroptosis. It will be interesting to determine if breaching of the nuclear envelope is also controlled from the nucleoplasmic side, and if MLKL can exit the nucleus from these breaches. It is also unclear why only some IAV-infected cells show evidence of nuclear envelope rupture, while others do not. Speculatively, ESCRT III-driven membrane repair mechanisms, shown to operate during MLKL-mediated plasma membrane lysis (Gong et al., 2017), and known to maintain nuclear integrity in other contexts (Isermann and Lammerding, 2017; Vietri et al., 2016), may protect nuclei in the IAV-infected cells that display morphologically intact nuclear envelopes. Intriguingly, phosphorylated MLKL in also observed the nucleus during TNF-α-initiated necroptosis (Weber et al., 2018; Yoon et al., 2016), but in these cells, the nuclear envelope, while distorted, is rarely breached, and DNA leakage into the cytosol is not typically seen. We note, however, that HMGB1 can rapidly exit the nuclei of cells undergoing TNF-α-stimulated necroptosis (Murai et al., 2018), suggesting that widespread nuclear envelope damage may not be required for HMGB1 release from necroptotic nuclei. In these cells, the subset of MLKL that transits through the nucleus (Weber et al., 2018; Yoon et al., 2016) may promote release of HMGB1 and other nuclear DAMPs without triggering extensive nuclear envelope damage.
Programmed cell death is an effective mechanism of IAV clearance that not only eliminates infected cells to limit virus spread but also serves to catalyze adaptive immune responses. However, when cell death is unrestrained (Sanders et al., 2013), or when the mode of cell death is primarily necrotic (Rodrigue-Gervais et al., 2014), then injury and severe illness ensue, despite virus clearance. Such severe pathology is observed in mouse models, in which destruction of airway epithelia is a common during lethal IAV infection (Brandes et al., 2013; Kash et al., 2006; Sanders et al., 2011, 2013), and in humans, in whom bronchioalveolar necrosis is a characteristic feature of IAV-induced acute respiratory distress syndrome (ARDS) (Korteweg and Gu, 2008; Mauad et al., 2010). Moreover, necroptotic cell debris is potently immunogenic (Yatim et al., 2017) and can contribute to neutrophil recruitment and activation. Because neutrophils have been shown to trigger a pathogenic feed-forward inflammatory response during lethal IAV infections (Brandes et al., 2013; Narasaraju et al., 2011), and because the nucleus contains numerous DAMPs, including DNA itself, which are capable of provoking deleterious inflammatory responses when released from ruptured nuclei (Roh and Sohn, 2018), our data implicate MLKL-mediated nuclear rupture and necroptosis as a driver of IAV virulence. Given that necroptosis is largely dispensable for virus clearance as long as RIPK3-induced apoptosis (Nogusa et al., 2016) and other effector functions are preserved, these findings identify necroptosis blockade (e.g., by RIPK3 kinase inhibitors) as a potential therapeutic entry point in severe cases of influenza.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Siddharth Balachandran (Siddharth.balachandran@fccc.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J mice were purchased at The Jackson Laboratory. Zbp1−/− (Ishii et al., 2008), Ripk3−/− (Newton et al., 2004), Mlkl−/− (Murphy et al., 2013), and Ripk1−/− (Kelliher et al., 1998) have been previously described. Casp8DA knock-in mice, in which both endogenous caspase8 alleles have been replaced with mutant alleles encoding a D387A point mutant of caspase-8 (Kang et al., 2008; Philip et al., 2016), were generated in-house, and will be described elsewhere (manuscript in preparation). All mice were bred and maintained in specific pathogen-free (SPF) facilities at the Fox Chase Cancer Center and St. Jude Children’s Research Hospital. For all in vivo experiments, mice were separated into experimental groups on the basis of genotype at 10–12 weeks of age. Mixed cohorts of female and male mice were used for all experiments to exclude gender effects. All animal procedures were performed according to the protocols approved by the Committee on Use and Care of Animals at these institutions.
Cell lines and primary MEF cultures
LET1 AECs were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Hyclone), 1 mM sodium pyruvate, 1x GlutaMAX (Thermo Fisher Scientific), and 1% penicillin/streptomycin (Thermo Fisher Scientific) and have been described before (Rosenberger et al., 2014). A549 and HT-29 cells were obtained from the American Type Culture Collection (ATCC). A549 cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 1x GlutaMAX, and 1% penicillin/streptomycin. HT-29 cells were maintained in DMEM supplemented with 10% fetal bovine serum, 1x GlutaMAX, and 1% penicillin/streptomycin. Primary MEFs were generated in-house from E14.5 embryos and used within five passages in experiments. MEFs were maintained in DMEM supplemented with 15% FBS, 1 mM sodium pyruvate, 1x GlutaMAX, and 1% penicillin/ streptomycin. All cells were maintained at 37°C and 5% CO2. A549 and HT-29 cells are human cell lines of male and female origin, respectively. The sex of embryos was not determined before generation of primary MEF cultures.
Viruses
All IAV and IBV strains were propagated by allantoic inoculation of embryonated hen’s eggs with diluted (1:106) seed virus. IAV PR8 LD and HD stocks were prepared as previously described (Xue et al., 2016). Virus titers were determined as 50% egg infectious dose (EID50) and by plaque assay on Madin-Darby Canine Kidney (MDCK) cells. VSV (Indiana strain) and EMCV were obtained from the ATCC.
METHOD DETAILS
Generation of cell lines by retroviral transduction
ZBP1-Cyto and ZBP1-Nuc were produced by inserting three tandem copies of the nuclear export sequence LELLEDLTL (ZBP1-Cyto), or two tandem copies of the c-Myc-derived nuclear localization sequence PAAKRVKLD (ZBP1-Nuc), between the FLAG tag and the N terminus of ZBP1. Other ZBP1 mutants have been described before (Thapa et al., 2016). FLAG-ZBP1 constructs were cloned into the pQCXIH retroviral expression vector and retroviruses pseudotyped with VSV glycoprotein were produced in 293T cells according to the manufacturer’s instructions (Retro-X System, Clontech). Retrovirus containing supernatants from these cells were collected 48 hr post-transfection, and used to transduce immortalized Zbp1−/− MEFs. Cell populations stably expressing FLAG-ZBP1 constructs were obtained by selection in hygromycin. Immortalized Mlkl−/− cells inducibly expressing MLKL or 2xFv-tagged MLKL mutants were produced using the retroviral Tet-On 3G System, as described before (Quarato et al., 2016). MLKL-Cyto (MLKL NBB140-2xFv-Venus) has been described earlier (Quarato et al., 2016). MLKL-Nuc was generated by affixing two tandem copies of the SV40 Large T Ag-derived nuclear localization sequence PKKKRKV to the C terminus of MLKL NBB140-2xFv-Venus.
Immunofluorescence microscopy
Cells were plated on 8-well glass slides (EMD Millipore), and allowed to adhere for at least 24 hr before use in experiments. Following treatment or virus infection, cells were fixed in freshly-prepared 4% (w/v) paraformaldehyde, permeabilized in 0.2% (v/v) Triton X-100, blocked with MAXblock Blocking Medium (Active Motif), and incubated overnight with primary antibodies at 4°C. After three washes in PBS, slides were incubated with fluorophore-conjugated secondary antibodies for 1 hr at room temperature. Following an additional three washes in PBS, slides were mounted in ProLong Gold antifade reagent (Thermo Fisher Scientific) and imaged by confocal microscopy on a Leica SP8 instrument. Fluorescence intensity was quantified using Leica LAS X software. In some cases (such as for detection of Z-RNA and A-RNA in virus-infected cells), cells were subjected to proteinase K treatment (0.008 U/mL) for 20–40 min at 37°C post-fixation. When required, RNase A (1mg/mL), RNase III (50 U/mL) or DNase I (25 U/mL) was used for 1 hr at 37°C after proteinase K treatment. Primary antibodies were used at the following dilutions for immuno-fluoresecence studies: MPO (1:500), Cit-H3 (1:500), phosphorylated murine MLKL (1:7000), phosphorylated human MLKL (1:500), IAV NP (1:1000), Z-NA (polyclonal Ab 1:200; monoclonal Ab 1:200), A-RNA (1:50), B-DNA (1:500), FLAG (1:1000), lamin B1 (1:1000), emerin (1:1000), total MLKL (1:500), Venus (1:500), cleaved caspase 3 (1:500).
Detection of ZBP1-bound nuclear DVGs
Immortalized Zbp1−/− stably reconstituted with FLAG-ZBP1 were infected with IAV, scraped into PBS, washed three times in PBS, and placed in fractionation buffer (10 mM PIPES pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EDTA, 1 mM DTT) on ice for 10 min. Cells were separated into cytoplasmic and nuclear fractions by centrifugation at 5,000g for 5 min. Supernatants (i.e., the cytoplasmic extracts) were decanted and nuclear pellets were washed five times in fractionation buffer. Nuclear extracts were prepared by lysis of these pellets in nuclear lysis buffer (10 mM PIPES pH 6.8, 100 mM NaCl, 300mM Sucrose, 3 mM MgCl2, 1 mM EDTA, 1 mM DTT). Nuclear and cytoplasmic extracts obtained in this manner were incubated with anti-FLAG agarose bead slurry (Sigma) with rotation overnight at 4°C. Agarose beads were collected by centrifugation, were washed five times with wash buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM MgCl2, 0.05% NP40), resuspended in DNase digestion buffer containing DNase I (50 U/mL) and incubated at 37°C for 20 min. Beads were sedimented by centrifugation, washed once with wash buffer, and resuspended in wash buffer. 5%–10% of each sample was saved for immunoblot analysis to confirm purity of fractions. Samples were then treated with proteinase K at 55°C for 30 min. Tri-reagent (Sigma) was added to each sample, and RNA was harvested according to the manufacturer’s instructions. For PCR detection, RNA was reverse transcribed into cDNA using Reverse Transcriptase Kit (Thermo Fisher Scientific) and universal IAV genomic vRNA primers. PCR was then performed for detecting PA-derived DVGs, using the following primers: (Fwd) 5′- TATTCGTCTCAGGGAGCGAAAGCAGGTAC −3′; (Rev) 5′- ATATCGTCTCGTATTAGTAGAAACAAGGTACTT-3′. For RNA-Seq analysis, nucleic acid eluted from nuclear FLAG-ZBP1 immunoprecipitates was used as template for PCR with a set of semi-degenerate primers designed to target the ends of the PA segment. After 25 rounds of PCR, the product was separated by gel electrophoresis, and the visible band at approximately 300–500 bp was excised and purified. The size-selected template was subsequently prepared for library preparation (HyperPrep Kit, Kapa Biosystems) and sequenced on the Illumina MiSeq platform (250 bp paired-end sequencing). Adapters and low-quality bases were removed from the resulting reads using Trim Galore (Babraham Bioinformatics), and reads were then mapped to the full PR8 Influenza genome using Bowtie2 local alignment. To exclude off-target PCR products, improperly paired reads and pairs with a mapping-inferred insert size less than 600 bp were filtered prior to visualization. Properly paired reads mapping to the PA segment with an inferred insert size greater than 600 bp were visualized using the R Sushi package.
Immunoprecipitation and immunoblotting
Immortalized Zbp1−/− MEFs stably reconstituted with FLAG-tagged WT or mutant ZBP1 were lysed in IP lysis buffer (Thermo Fisher Scientific, cat#87787) supplemented with protease and phosphatase inhibitor (Thermo Fisher Scientific, cat#78444). Cell lysates were incubated on ice for 10 min, and briefly sonicated to shear chromatin, then cleared by high speed centrifugation (20,000g, 10 min) at 4°C. After saving 5% of the total cell lysate for input, the extracts were subjected to immunoprecipitation with anti-FLAG M2 affinity gel, according to the manufacturer’s instructions (Sigma, cat#FLAGIPT1). Resin was eluted with 3xFLAG peptide and the supernatants subjected to immunoblot analysis as described before (Chen et al., 2013). Primary antibodies were used at the following dilutions: IAV HA (1:3000), IAV PB1 (1:2000), IAV NP (1:2000), IAV NS1 (1:4000), phosphorylated murine MLKL (1:2000), phosphorylated human MLKL (1:1000), total MLKL (1:2000), ZBP1 (1:2000), RIPK3 (1:2000), FLAG (1:2000), Histone H3 (1:4000), HMGB1 (1:2000), GAPDH (1:4000) and β-actin (1:2000).
fRIP-qPCR
Formaldehyde crosslinked immunoprecipitation (fRIP) was performed as previously described (Zhao et al., 2018). Briefly, immortalized Zbp1−/− MEFs stably reconstituted with FLAG-tagged WT or mutant (ΔZα) ZBP1 were infected with IAV and separated into cytoplasmic and nuclear fractions by centrifugation at 5,000g for 5 min in fractionation buffer (10 mM PIPES pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EDTA, 1 mM DTT). Isolated nuclei were cross-linked with 1% formaldehyde in PBS with slow rotation at room temperature for 10 min, and unreacted formaldehyde was neutralized with 0.3 M glycine for 5 min. Cells were washed twice with PBS and resuspended in RIPA buffer on ice for 10 min. Soluble cell extracts were used for immunoprecipitation with anti-FLAG M2 affinity gel, according to the manufacturer’s instructions (Sigma). Resin was eluted with 3xFLAG peptide and protein-RNA crosslinks were reversed by incubation in buffer containing 100 mM Tris, pH 8.0, 10 mM EDTA, 1% SDS, and 2 mM DTT at 70°C for 45 min. RNA was recovered in TRIzol and extracted in phenol:chloroform:isoamyl alcohol [25:24:1 (vol/vol)] followed by ethanol precipitation. RNA was reverse transcribed into cDNA using SuperScript IV VILO Master Mix (Thermo Fisher Scientific). Standard PCR and qPCR were performed to detect PA DVGs using the Platinum SuperFi PCR Master Mix (Thermo Fisher Scientific) and SYBR green (Thermo Fisher Scientific), respectively, using the following primers: Standard PCR PA-(Fwd) 5′- GATGATTGTCGAGCTTGCGG 3′; PA-(Rev) 5′- TCCCAGGTTCAAGGTTGTCC −3′; qPCR PA-(Fwd) 5′- GCTTCTTATCGTTCAGGCTCTT −3′; PA-(Rev) 5′- GGGATCATTAATCAGGCACTCC −3′
Molecular Modeling of m8Gm-containing RNA
Models of CG-repeat dsRNA 12-mers containing unmodified guanosine or m8Gm guanosine analogs were generated using BIOVIA Discovery Studio 4.5 software, and molecular dynamics simulations performed by employing the Standard Dynamics Cascade protocol in the same software. The five best conformations generated by these simulations were then energy minimized, and the conformation with the lowest energy was selected for display.
Isolation and activation of neutrophils ex vivo
Bone marrow from femurs and tibias of 8-week old C57BL/6J mice was isolated and cells pelleted in sterile Ca2+/Mg2+-free HBSS containing penicillin/streptomycin. Cells were then resuspended in ACK (Ammonium-Chloride-Potassium) Lysing Buffer (Thermo Fisher Scientific) for the lysis of red blood cells and washed twice with 1xHBSS. Neutrophils were separated from mononuclear cells by layering 2 mL of the cell suspension onto 3 mL of an 81% Percoll cushion (GE Healthcare). The cell suspension was then overlaid with 3 mL of 62% Percoll, followed by centrifugation at 2,500g for 20 min at 4°C. The middle layer enriched for neutrophils was washed twice in 1xHBSS, and cells were resuspended in serum-free DMEM. Isolated neutrophils were transferred into 8-well glass slides containing 200 μL of serum-free DMEM and activated by co-culturing with an equal volume of supernatant from treated/infected cells for 24 hr. Cells were then fixed and processed for immunofluorescence staining.
Electrophoretic mobility shift assay
FAM-labeled RNAs (1 μM) and antibodies (10 μM) were incubated in EMSA binding buffer (10 mM Tris-HCl pH 7.0, 100 mM NaCl, 5 mM dithiothreitol, 10 μg/mL BSA, and 10% (v/v) glycerol) for 1 hr at room temperature. Samples were then separated by non-denaturing 4% polyacrylamide gel electrophoresis in 1xTBE (Tris/borate/EDTA) buffer containing 20 mM NaCl at 80 V and 4°C for 2 hr. The gel was imaged on a phosphorimager (LAS-3000, Fujifilm).
ELISA
5×106 MLKL-Cyto and MLKL-Nuc MEFs were cultured in 60 mm cell culture dishes in 1.5 mL DMEM/10% FBS per dish. Following exposure to oligomerizer, supernatants were collected and subjected to ELISA per the manufacturer’s instructions.
In vivo IAV studies
Age- (8–12 week old) and sex-matched mice were anesthetized with Avertin (2,2,2-tribromoethanol) or 3% isoflurane and infected intranasally with virus (PR8) inoculum diluted in endotoxin-free saline. Mice were either monitored for survival over a period of 21 days or sacrificed at defined time points for analysis of histology and virus replication. Mice losing > 30% body weight were considered moribund and euthanized by gradual CO2 asphyxiation. Titration of virus was conducted by standard plaque assay of diluted lung homogenates on monolayers of MDCK cells, and plaques were scored after three days of incubation. To assess IAV- specific CD8+ T cells, bronchoalveolar lavage (BAL) was performed on infected mice, and cells in BAL fluid were pelleted by centrifuging at 500g for 5 min. 1×105 BAL cells were plated for each condition, and cells were stimulated in complete RPMI for 4 hr at 37°C with 1 μM of each of the virus-specific peptides in the presence of brefeldin A and monesin. The following virus-specific peptides were used for stimulation: PB1703 – 711, PA224 – 233, and NP366 – 374. Following stimulation, cells were washed and then blocked using TruStain FcX block (1:100) (Biolegend). Cell surface staining was then performed for 30 min at room temperature using the following antibodies/dyes and dilutions: Ghost Violet 510 Viability Dye (1:100), anti-mouse CD45-APC/Fire 750 (1:100), anti-mouse CD3-PE (1:100), anti-mouse CD8a-APC (1:200), and anti-mouse CD4-FITC (1:200). Cells were then washed, fixed, and permeabilized using Cytofix/Cytoperm solution (BD Pharmigen) according to the manufacturer’s instructions. Cells were stained with anti-mouse IFN-γ-PE/Cy7 (1:100) and anti-mouse TNFα-BV785 (1:100) for 30 min on ice and then analyzed on an LSRII Fortessa machine (BD Biosciences).
QUANTIFICATION AND STATISTICAL ANALYSIS
Curve fitting and statistical analyses were performed with GraphPad Prism 6.0 software, using either unpaired Student’s t test for comparison between two groups or two-way ANOVA followed by Tukey’s test for comparisons between multiple (> 2) groups. Significance of in vivo survival data was determined by the log-rank (Mantel-Cox) test. Information on replicates, sample size (e.g., number of animals per group), precision measures (mean ± SD), statistical methods and significance are indicated in Figure Legends.
DATA AND CODE AVAILABILITY
RNA-sequencing data of FLAG-ZBP1 immunoprecipitates in Figure 2 were deposited in BioProject: PRJNA603378. All data produced by this study are included in the manuscript or available from the corresponding author on request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-mouse MLKL (phospho-S345),Clone EPR9515(2) | Abcam | Cat#Ab196436; RRID: AB_2687465 |
| Mouse monoclonal anti-mouse MLKL (phospho-S345) | Rodriguez et al., 2016 | N/A |
| Rabbit monoclonal anti-human MLKL (phospho-S358), Clone EPR9514 | Abcam | Cat#ab187091; RRID: AB_2619685 |
| Rabbit monoclonal anti-human MLKL (phospho-S358), Clone 6B4 | MYBioSource | Cat#MBS9419709; RRID: AB_2820287 |
| Rat monoclonal anti-MLKL, clone 3H1 | EMD Millipore | Cat#MABC60; RRID: AB_2820284 |
| Mouse monoclonal anti-ZBP1, Clone Zippy-1 | AdipoGen | Cat#AG-20B-0010; RRID: AB_2490191 |
| Rabbit polyclonal anti-RIPK3 | ProSci | Cat#2283; RRID: AB_203256 |
| Rabbit polyclonal anti-Influenza A virus PB1 | GeneTex | Cat#GTX125923; RRID: AB_2753122 |
| Mouse monoclonal anti-Influenza A virus NP, Clone AA5H | Bio-Rad | Cat#MCA400; RRID: AB_2151884 |
| Rabbit polyclonal anti-Influenza A virus NP | GeneTex | Cat#GTX125989; RRID: AB_11168364 |
| Rabbit polyclonal anti-Influenza A virus NS1 | GeneTex | Cat#GTX125990; RRID: AB_11170327 |
| Rabbit polyclonal anti-Influenza A virus Hemagglutinin (HA) | US Biological | Cat#I7651-41; RRID: AB_2820288 |
| Mouse monoclonal anti-β-actin, Clone mAbGEa | GeneTex | Cat#GTX80809; RRID: AB_626122 |
| Rabbit polyclonal anti-Histone H3 | Cell Signaling Technology | Cat#9715; RRID: AB_331563 |
| Mouse monoclonal anti-GAPDH, 1E6D9 | Proeintech | Cat#60004–1-Ig; RRID: AB_2107436 |
| Rabbit monoclonal anti-FLAG | Cell Signaling Technology | Cat#14793; RRID: AB_2572291 |
| Mouse monoclonal anti-FLAG, Clone 5A8E5 | GenScript | Cat#A00187; RRID: AB_1720813 |
| Sheep polyclonal anti-Z-NA | Novus Biologicals | Cat#NB100–749; RRID: AB_10003363 |
| Mouse monoclonal anti-Z-NA, clone Z22 | Absolute Antibody | Cat#Ab00783–3.0; RRID: AB_2820286 |
| Mouse monoclonal anti-dsRNA, clone 9D5 | EMD Millipore | Cat#3361; RRID: AB_2820285 |
| Rabbit polyclonal anti-Lamin B1 | Abcam | Cat#ab16048; RRID: AB_10107828 |
| Rabbit polyclonal anti-Emerin | Abcam | Cat# ab40688; RRID: AB_2100059 |
| Goat polyclonal anti-Venus | MyBioSource | Cat#MBS448126 |
| Rabbit polyclonal anti-HMGB1 | Cell Signaling Technology | Cat#3935; RRID: AB_2295241 |
| Rabbit monoclonal anti-Cleaved Caspase-3 | Cell Signaling Technology | Cat#9664; RRID: AB_2070042 |
| Goat polyclonal anti-Myeloperoxidase (MPO) | R&D systems | Cat#AF3667; RRID: AB_2250866 |
| Rabbit polyclonal anti-Histone H3 (citrulline R2+R8+R17) | Abcam | Cat#ab5103; RRID: AB_304752 |
| Sheep IgG | Thermo Fisher Scientiric | Cat#31243; RRID: AB_243595 |
| Mouse IgG | Thermo Fisher Scientiric | Cat#31903; RRID: AB_10959891 |
| Rat monoclonal anti-CD45-APC/Fire750, Clone 30-F11 | BioLegend | Cat#103154; RRID: AB_2572116 |
| Rat monoclonal anti-CD3-PE, Clone 17A2 | BioLegend | Cat#100206; RRID: AB_312663 |
| Rat monoclonal anti-CD8a-APC, Clone 53–6.7 | BioLegend | Cat#100712; RRID: AB_312751 |
| Rat monoclonal anti-CD4-FITC, Clone GK1.5 | BioLegend | Cat#100406; RRID: AB_312691 |
| Rat monoclonal anti-TNFa-BV785, Clone MP6-XT22 | BioLegend | Cat#506341; RRID: AB_2565951 |
| Rat monoclonal anti-IFNγ-PE/Cy7 | BD Biosciences | Cat#557649; RRID: AB_396766 |
| Bacterial and Virus Strains | ||
| Influenza A/Puerto Rico/8/1934 (H1N1) | This paper | N/A |
| Influenza A/Brisbane/59/2007 (H1N1) | This paper | N/A |
| Influenza A/Brisbane/10/2007 (H3N2) | This paper | N/A |
| Influenza B/Florida/4/2006 | ATCC | ATCC VR-1804 |
| Vesicular stomatitis virus (Indiana strain) | ATCC | ATCC VR-1238 |
| Encephalomyocarditis virus | ATCC | ATCC VR-1762 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Murine TNF-α | R&D Systems | 410-MT |
| Human TNF-α | R&D Systems | 210-TA |
| Murine TRAIL | R&D Systems | 1121-TL |
| Cycloheximide | Sigma | 01810 |
| zVAD-FMK | Bachem | N-1510 |
| RNase A | Thermo Fisher Scientific | EN0531 |
| RNase III | Thermo Fisher Scientific | AM2290 |
| DNase I | Thermo Fisher Scientific | 90083 |
| Proteinase K | NEW ENGLAND BioLabs | P8107 |
| Leptomycin B | LC Laboratories | L-6100 |
| B/B Homodimerizer | Clontech | 635059 |
| Nucleozin | EMD Millipore | 492905 |
| Doxycycline | Clontech | 631311 |
| RIPK3 inhibitor | GlaxoSmithKline | GSK’843 |
| IAV PB1703 – 711 peptide | This paper | N/A |
| IAV PA224 – 233 peptide | This paper | N/A |
| IAV NP366 – 374 peptide | This paper | N/A |
| Critical Commercial Assays | ||
| Mouse IL-33 ELISA kit | R&D Systems | M3300 |
| Mouse HMGB1 ELISA kit | MYBioSource | MBS722248 |
| SuperScript IV VILO Master Mix | Thermo Fisher Scientific | 11756050 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | A25742 |
| FLAG Immunoprecipitation kit | Sigma | FLAGIPT1 |
| Deposited Data | ||
| RNA seq data in Figure 2 | This paper | BioProject ID PRJNA603378 |
| Experimental Models: Cell Lines | ||
| Human cell line: HT-29 | ATCC | ATCC HTB-38 |
| LET1 AECs | Rosenberger et al., 2014 | N/A |
| Human cell line: A549 | ATCC | ATCC CCL-185 |
| Immortalized mouse embryonic fibroblasts | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: zbp1−/− | Ishii et al., 2008 | N/A |
| Mouse: ripk3−/− | Newton et al., 2004 | N/A |
| Mouse: mlkl−/− | Murphy et al., 2013 | N/A |
| Mouse: ripk1−/− | Kelliher et al., 1998 | N/A |
| Mouse: casp8DA | This paper | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides | ||
| Poly (I:C) LMW | InvivoGen | tlrl-picw |
| ISD (interferon stimulatory DNA) | InvivoGen | tlrl-patn |
| Synthetic m8Gm-Z-RNA: ACGCGCGCGCGCGCGCGCGUUUUCGCGCGCGCGCGCGCGCGU | This paper | N/A |
| Synthetic A-RNA: ACGCGCGCGCGCGCGCGCGUUUUCGCGCG CGCGCGCGCGCGU | This paper | N/A |
| IAV PA primer-1 (Fwd): TATTCGTCTCAGGGAGCGAAAGCAGGTAC | This paper | N/A |
| IAV PA primer-1 (Rev): ATATCGTCTCGTATTAGTAGAAACAAGGTACTT | This paper | N/A |
| IAV PA primer-2 (Fwd): GATGATTGTCGAGCTTGCGG | This paper | N/A |
| IAV PA primer-2 (Rev): TCCCAGGTTCAAGGTTGTCC | This paper | N/A |
| IAV PA qPCR primer (Fwd): GCTTCTTATCGTTCAGGCTCTT | This paper | N/A |
| IAV PA qPCR primer (Rev): GGGATCATTAATCAGGCACTCC | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: FLAG-ZBP1 | Thapa et al., 2016 | N/A |
| Plasmid: FLAG-NLS-ZBP1 (ZBP1 Nuc) | This paper | N/A |
| Plasmid: FLAG-NES-ZBP1 (ZBP1 Cyto) | This paper | N/A |
| Plasmid: FLAG-ZBP1 ΔZα | Thapa et al., 2016 | N/A |
| Plasmid: FLAG-ZBP1 RHIM-A-mut | Thapa et al., 2016 | N/A |
| Plasmid: FLAG-ZBP1 ΔZα1 | Thapa et al., 2016 | N/A |
| Plasmid: FLAG-ZBP1 ΔC | Thapa et al., 2016 | N/A |
| Plasmid: MLKL(1–140)-2xFV-Venus (MLKL-Cyto) | Quarato et al., 2016 | N/A |
| Plasmid: MLKL(1–140)-2xFV-Venus-NLS (MLKL-Nuc) | Quarato et al., 2016 | N/A |
| Software and Algorithms | ||
| BIOVIA Discovery Studio 4.5 | BIOVIA | https://www.3dsbiovia.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 6 | GraphPad Software | https://www.graphpad.com |
Highlights.
Replicating influenza A virus (IAV) produces Z-RNAs
IAV Z-RNAs are sensed by host ZBP1 in the nucleus
ZBP1 activates MLKL in the nucleus, triggering nuclear envelope rupture
MLKL-induced nuclear rupture and necroptosis drive IAV disease severity
ACKNOWLEDGMENTS
We are grateful to Charles Hardin and David Stollar for advice on Z-RNA studies and to Glenn Rall for comments on the manuscript. This work was supported by NIH (CA168621, CA190542, AI135025, AI144400, and P30CA006927 to S.B.; AI44828 and CA231620 to D.R.G.; AI134862 and AI137062 to C.B.L.; and AI135709 to J.W.U.). P.G.T. was supported by NIH (AI121832) and the St. Jude Center of Excellence for Influenza Research and Surveillance (SJCEIRS) NIAID (HHSN272201400006C).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Athanasiadis A (2012). Zalpha-domains: at the intersection between RNA editing and innate immunity. Semin. Cell Dev. Biol 23, 275–280. [DOI] [PubMed] [Google Scholar]
- Balasubramaniyam T, Ishizuka T, Xiao CD, Bao HL, and Xu Y (2018). 2′-O-Methyl-8-methylguanosine as a Z-Form RNA Stabilizer for Structural and Functional Study of Z-RNA. Molecules 23, 2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes M, Klauschen F, Kuchen S, and Germain RN (2013). A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154, 197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BA 2nd, Lowenhaupt K, Wilbert CM, Hanlon EB, and Rich A (2000). The zalpha domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc. Natl. Acad. Sci. USA 97, 13532–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JV, and Jonsson CB (2017). A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol 8, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Nogusa S, Thapa RJ, Shaller C, Simmons H, Peri S, Adams GP, and Balachandran S (2013). Anti-CD70 immunocytokines for exploitation of interferon-g-induced RIP1-dependent necrosis in renal cell carcinoma. PLoS ONE 8, e61446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI, Duffy D, Tamby C, et al. (2018). Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. [DOI] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, and Green DR (2017). ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin CC, Zarling DA, Puglisi JD, Trulson MO, Davis PW, and Tinoco I Jr. (1987). Stabilization of Z-RNA by chemical bromination and its recognition by anti-Z-DNA antibodies. Biochemistry 26, 5191–5199. [DOI] [PubMed] [Google Scholar]
- Hardin CC, Zarling DA, Wolk SK, Ross WS, and Tinoco I Jr. (1988). Characterization of anti-Z-RNA polyclonal antibodies: epitope properties and recognition of Z-DNA. Biochemistry 27, 4169–4177. [DOI] [PubMed] [Google Scholar]
- Isermann P, and Lammerding J (2017). Consequences of a tight squeeze: Nuclear envelope rupture and repair. Nucleus 8, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, and Akira S (2008). TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729. [DOI] [PubMed] [Google Scholar]
- Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, and Wallach D (2008). Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J. Immunol 181, 2522–2532. [DOI] [PubMed] [Google Scholar]
- Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, et al. (2010). Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol 28, 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, García-Sastre A, et al. (2006). Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443, 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2006). Innate immune recognition of viral infection. Nat. Immunol 7, 131–137. [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, and Leder P (1998). The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8, 297–303. [DOI] [PubMed] [Google Scholar]
- Kleinig H (1970). Nuclear membranes from mammalian liver. II. Lipid composition. J. Cell Biol 46, 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korteweg C, and Gu J (2008). Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans. Am. J. Pathol 172, 1155–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti T-D (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol 1, aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. (2014). RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauad T, Hajjar LA, Callegari GD, da Silva LF, Schout D, Galas FR, Alves VA, Malheiros DM, Auler JO Jr., Ferreira AF, et al. (2010). Lung pathology in fatal novel human influenza A (H1N1) infection. Am. J. Respir. Crit. Care Med 181, 72–79. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, and Daley-Bauer LP (2014). True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J. Immunol 192, 2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai S, Yamaguchi Y, Shirasaki Y, Yamagishi M, Shindo R, Hildebrand JM, Miura R, Nakabayashi O, Totsuka M, Tomida T, et al. (2018). A FRET biosensor for necroptosis uncovers two different modes of the release of DAMPs. Nat. Commun 9, 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, and Chow VT (2011). Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol 179, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak DP, Chambers TM, and Akkina RK (1985). Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr. Top. Microbiol. Immunol 114, 103–151. [DOI] [PubMed] [Google Scholar]
- Newton K, Sun X, and Dixit VM (2004). Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol 24, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al. (2016). RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 20, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, et al. (2016). Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog. 12, e1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placido D, Brown BA 2nd, Lowenhaupt K, Rich A, and Athanasiadis A (2007). A left-handed RNA double helix bound by the Z alpha domain of the RNA-editing enzyme ADAR1. Structure 15, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, and Green DR (2016). Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol. Cell 61, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich A, and Zhang S (2003). Timeline: Z-DNA: the long road to biological function. Nat. Rev. Genet 4, 566–572. [DOI] [PubMed] [Google Scholar]
- Rodrigue-Gervais IG, Labbé K, Dagenais M, Dupaul-Chicoine J, Champagne C, Morizot A, Skeldon A, Brincks EL, Vidal SM, Griffith TS, and Saleh M (2014). Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe 15, 23–35. [DOI] [PubMed] [Google Scholar]
- Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death & Differentiation 23, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JS, and Sohn DH (2018). Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 18, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger CM, Podyminogin RL, Askovich PS, Navarro G, Kaiser SM, Sanders CJ, McClaren JL, Tam VC, Dash P, Noonan JG, et al. (2014). Characterization of innate responses to influenza virus infection in a novel lung type I epithelial cell model. J. Gen. Virol 95, 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE (2011). Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411, 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders CJ, Doherty PC, and Thomas PG (2011). Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res. 343, 13–21. [DOI] [PubMed] [Google Scholar]
- Sanders CJ, Vogel P, McClaren JL, Bajracharya R, Doherty PC, and Thomas PG (2013). Compromised respiratory function in lethal influenza infection is characterized by the depletion of type I alveolar epithelial cells beyond threshold levels. Am. J. Physiol. Lung Cell. Mol. Physiol 304, L481–L488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son KN, Liang Z, and Lipton HL (2015). Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol 89, 9383–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Marcatti M, Ahmad A, Montalbano M, Brunyánszki A, Bibli SI, Papapetropoulos A, and Szabo C (2018). Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci. Rep 8, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, et al. (2016). DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 20, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Shubina M, and Balachandran S (2017). RIPK3-driven cell death during virus infections. Immunol. Rev 277, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri M, Stenmark H, and Campsteijn C (2016). Closing a gap in the nuclear envelope. Curr. Opin. Cell Biol 40, 90–97. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, and Wang X (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Weber K, Roelandt R, Bruggeman I, Estornes Y, and Vandenabeele P (2018). Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig B, Dorbic T, and Rich A (1991). Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc. Natl. Acad. Sci. USA 88, 2259–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Chambers BS, Hensley SE, and López CB (2016). Propagation and Characterization of Influenza Virus Stocks That Lack High Levels of Defective Viral Genomes and Hemagglutinin Mutations. Front. Microbiol 7, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim N, Cullen S, and Albert ML (2017). Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol 17, 262–275. [DOI] [PubMed] [Google Scholar]
- Yoon S, Bogdanov K, Kovalenko A, and Wallach D (2016). Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 23, 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling DA, Calhoun CJ, Hardin CC, and Zarling AH (1987). Cytoplasmic Z-RNA. Proc. Natl. Acad. Sci. USA 84, 6117–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling DA, Calhoun CJ, Feuerstein BG, and Sena EP (1990). Cytoplasmic microinjection of immunoglobulin Gs recognizing RNA helices inhibits human cell growth. J. Mol. Biol 211, 147–160. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ye X, Dunker W, Song Y, and Karijolich J (2018). RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun 9, 4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data of FLAG-ZBP1 immunoprecipitates in Figure 2 were deposited in BioProject: PRJNA603378. All data produced by this study are included in the manuscript or available from the corresponding author on request.