

ABSTRACT
The Transient Receptor Potential Melastatin 8 (TRPM8) ion channel is an important sensor of environmental cold temperatures. Cold- and menthol-induced activation of this channel requires the presence of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. This review discusses recent findings on the role of PI(4,5)P2 and G-proteins in the modulation of TRPM8 upon receptor activation. We will also summarize knowledge on the role of PI(4,5)P2 in Ca2+ dependent desensitization/adaptation of TRPM8 activity, and recent advances in the structural basis of how this lipid binds to TRPM8.
KEYWORDS: TRPM8, PIP2, phosphoinositides, GPCR, G-alpha-q
Introduction
Peppermint oil, and its main active compound, menthol has been known to evoke a cooling sensation for centuries [1]. The primary menthol receptor, the Transient Receptor Potential Melastatin 8 (TRPM8) channel was identified independently by two laboratories in 2002 [2,3]. This channel is expressed in the sensory neurons of the Dorsal Root Ganglia (DRG) and the Trigeminal Ganglia (TG), and it is activated by cold, and chemical agonists such as menthol, icilin and WS12 [4,5]. TRPM8 is a Ca2+ permeable nonselective cation channel; its activation depolarizes neurons and induces a Ca2+ signal. Genetic deletion of TRPM8 in mice revealed that this channel is important for sensing moderately cold temperatures [6–8]. Later studies showed that the channel is also involved in pathological cold sensation [9,10].
TRPM8 activity decreases in the continuous presence of menthol in an extracellular Ca2+ dependent manner [2]. This phenomenon was initially termed desensitization [2], and later referred to as adaptation [11]; here we will use these two terms interchangeably. This decreased activity was proposed to be caused by Ca2+ dependent activation of a phospholipase C (PLC) isoform by influx of Ca2+ through the channel, and the subsequent reduction in the levels of the plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], which is an important cofactor for TRPM8 activity [12]. This mechanism is widely supported by work from several laboratories; here we will briefly discuss the evidence for the involvement of PI(4,5)P2 in channel desensitization, as well as the recent structural advances in the molecular mechanism of PI(4,5)P2 activation of TRPM8.
PLC activation by G-Protein Coupled Receptors (GPCRs) also inhibits TRPM8 activity. Many of the Gq-coupled receptors in DRG neurons are activated by proinflammatory mediators such as bradykinin, prostaglandins, histamine and extracellular ATP. Activation of these receptors is known to sensitize the heat- and capsaicin-activated Transient Receptor Potential Vanilloid 1 (TRPV1) channels, leading to increased sensitivity to heat (thermal hyperalgesia) [13–15]. Inhibition of TRPM8 by Gq-coupled receptor activation was proposed to contribute to thermal hyperalgesia in inflammation [16]. The focus of this review will be to discuss the involvement of PI(4,5)P2, and alternative mechanisms in GPCR-mediated inhibition of TRPM8, discussing in detail two recent publications that suggested seemingly contradictory conclusions [17,18].
PI(4,5)P2 is a specific direct regulator of TRPM8 activity
PI(4,5)P2 constitutes ~1% of the lipids in the inner leaflet of the plasma membrane. It is generated by the sequential phosphorylation of phosphatidylinositol (PI) to PI(4)P and then to PI(4,5)P2, catalyzed by phosphatidylinositol 4-kinase (PI4K) and phosphatidylinositol 4-phosphate 5-kinase (PIP5K) enzymes respectively (Figure 1). PI(4,5)P2 and PI(4)P can also be further phosphorylated by phosphoinositide 3-kinase (PI3K) enzymes to PI(3,4,5)P3 and PI(3,4)P2 respectively. PLC enzymes convert PI(4,5)P2 into diacylglycerol (DAG), which activates Protein Kinase C (PKC) and inositol 1,4,5 trisphosphate (IP3), which releases Ca2+ from the endoplasmic reticulum. PLCβ enzymes are activated by G-protein coupled receptors that couple to Gαq, while PLCγ-s are activated by receptor tyrosine kinases [19]. All PLC isoforms require some Ca2+ for activity; PLCδ enzymes are the most Ca2+ sensitive isoforms, and they can be activated by Ca2+ alone in a cellular environment (Figure 1).
Figure 1.
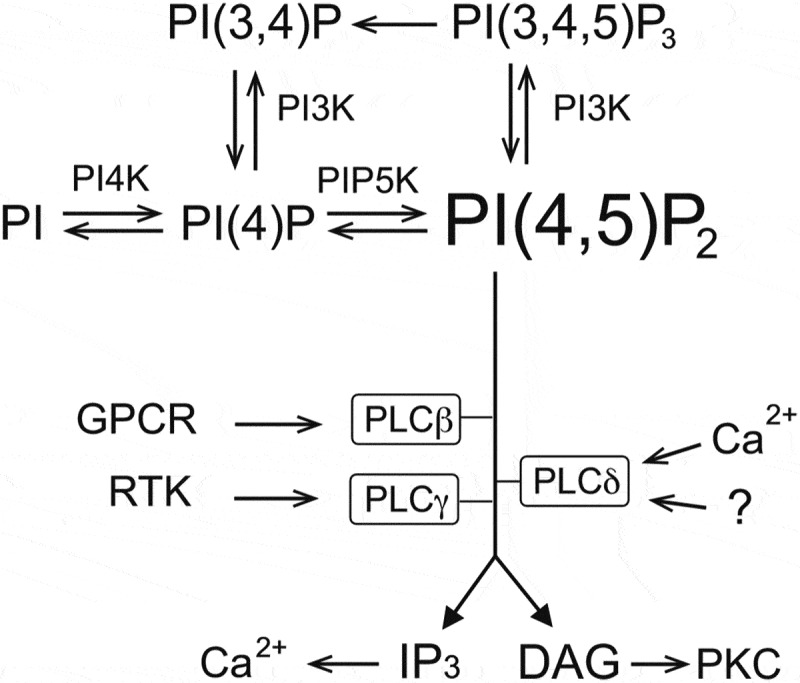
Phosphoinositides metabolism, see main text for details.
There are multiple lines of evidence indicating that PI(4,5)P2 is required for menthol- and cold-induced activity of TRPM8; most of these experiments have been discussed in detail before [20], we briefly review them here. First, two laboratories independently demonstrated that TRPM8 activity shows a spontaneous decrease (rundown) in excised inside out patches [12,21], which is a common property of PI(4,5)P2 dependent ion channels, and it is generally attributed to dephosphorylation of PI(4,5)P2 to PI(4)P and PI [22]. Rundown was accelerated by the application of poly-Lys, which chelates PI(4,5)P2 [12] and was delayed by a phosphatase inhibitor cocktail, that prevents dephosphorylation of PI(4,5)P2 [21]. More importantly, channel activity was restored in excised patches by the application of PI(4,5)P2 [12,21]; while other phosphoinositides, such as PI(4)P, PI(3,4)P2 and PI(3,4,5)P3 were much less effective, exerting 10–30% of the effect of PI(4,5)P2 [12]. TRPM8 activity was also restored by the application of MgATP in excised inside out patches, and its effect was eliminated by blocking PI4K activity in the patch [23], signifying that MgATP exerted its effect via replenishing PI(4,5)P2. The purified TRPM8 protein reconstituted in planar lipid bilayers also required PI(4,5)P2 for cold- or menthol-induced activity [24]. Similar to excised patches, PI(4)P, PI(3,4)P2 and PI(3,4,5)P3 were less effective than PI(4,5)P2 in planar lipid bilayers [24]. The ability of PI(4,5)P2 to support the activity of the purified TRPM8 protein in planar lipid bilayers demonstrates that it exerts its effect by binding to the channel directly.
The recent cryoEM structure of TRPM8 with PI(4,5)P2 and icilin (and WS12) gives a detailed molecular understanding of how the lipid binds to TRPM8 (Figure 2). Positively charged residues from three regions of the same subunit (TRP domain, S4-S5 linker and preS1 segment), and one additional cytoplasmic region (MHR4) of an adjacent subunit form the lipid binding site in the TRPM8-PI(4,5)P2 (6nr3) structure [25]. Consistent with being a PI(4,5)P2 contact residue, neutralization of the positively charged R998 residue in the proximal C-terminal TRP domain of the rat TRPM8 (R997 in flycatcher isoform used for structure determination) was shown earlier to reduce the apparent affinity of PI(4,5)P2 in excised inside out patch clamp experiments [12]. Neutralization of the R1008 residue (R1007 in flycatcher) also resulted in a right shift of the PI(4,5)P2 dose response [12]. This TRP domain residue however is not in contact with PI(4,5)P2 in the cryoEM structure (6nr2), but rather it interacts with the menthol analogue WS12 [25]. Consistent with this, the menthol dose response in the R1008Q mutant was also right shifted [12]. Menthol was shown to increase the apparent affinity of the channel for PI(4,5)P2 [12] therefore the right shift in the PI(4,5)P2 dose response in the R1008Q mutant was likely due to an indirect effect of reduced menthol affinity.
Figure 2.
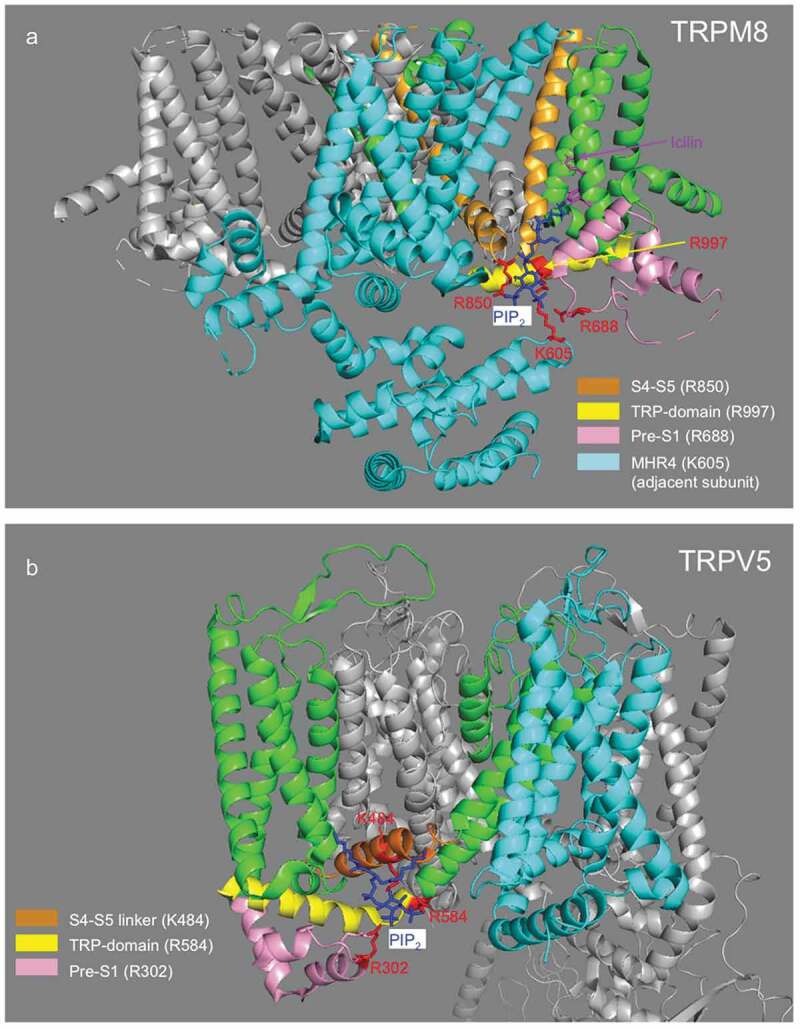
Structure of TRPM8 and TRPV5 with PI(4,5)P2. (a). Structure of TRPM8 with PI(4,5)P2 and icilin (6nr3) from Yin et al [25]. PI(4,5)P2 is colored blue, interacting residues are colored red. Parts of the channel where the interacting residues located are also color-coded: S4-S5 orange, TRP-domain yellow, Pres-S1 segment purple. The remaining parts (S6 and S1-S3) of the same subunit where these residues are located are colored green. The entire adjacent subunit is labeled cyan, the MHR4 region of this subunit contains an additional PI(4,5)P2 interacting residue; most other cytoplasmic parts have been removed for clarity. (b). Structure of TRPV5 with PI(4,5)P2 (6dmu) from Hughes et al [27]. Color-coding is similar to that in panel A.
Interestingly, the cryoEM structure of TRPM8 in the combined presence of PI(4,5)P2 and icilin or WS12 was still in a closed state [25], pointing to the possibility of missing additional factors, such as polyphosphate [26]. This is in sharp contrast to TRPV5 cryoEM structure, which showed an open configuration in the presence of PI(4,5)P2 [27]. While the proximal C-terminus, the S4-S5 linker and the pre-S1 segment contributed to PI(4,5)P2 binding in both structures, the location of the lipid was different in TRPM8 and TRPV5 (Figure 2).
The role of PI(4,5)P2 in channel desensitization/adaptation
Many Ca2+ permeable ion channels undergo Ca2+ dependent desensitization i.e. decreased activity in the continuous presence of the activator. For TRPM8 it was shown that the mechanism of desensitization is the activation of a Ca2+ sensitive PLC isoform by Ca2+ influx through the channel, and the concurrent decrease in PI(4,5)P2 levels (Figure 3). This model is based on the following findings. 1. As described in detail before, TRPM8 activity requires the presence of PI(4,5)P2. 2. Activation of TRPM8 in the presence of extracellular Ca2+ evokes a decrease in PI(4,5)P2 levels [11,12,23]. The dominant PLC isoform that is activated by Ca2+ influx is PLCδ4; genetic deletion of this enzyme resulted in increased cold- and menthol-induced current is DRG neurons [28]. 3. A decrease in PI(4,5)P2 levels by voltage activated or chemically activated lipid phosphatases (independent of PLC activation) is sufficient to inhibit TRPM8 activity [11,23,29]. 4. Interfering with PI(4,5)P2 depletion by supplying excess PI(4,5)P2 through the whole cell patch pipette decreased desensitization [23].
Figure 3.
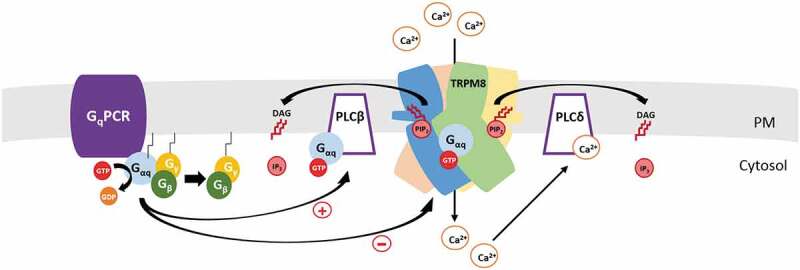
Regulation of TRPM8 by PI(4,5)P2 and G-proteins During GPCR activation, the Gαq subunit stimulates PLCβ isoforms that induce a decrease of plasma membrane PI(4,5)P2 levels which limits TRPM8 activity. Gαq also directly binds to the channel and inhibits its activity (left side of cartoon). Ca2+ entering through the channel stimulates PLCδ isoforms, which leads to a decrease in PI(4,5)P2 levels and inhibits channel activity (right side of cartoon). See main text for details.
There are two lines of evidence suggesting a role of PLC-mediated PI(4,5)P2 depletion in temperature adaptation in vivo. First, an elegant study by the Gereau laboratory showed that withdrawal of the hind paw from cold stimuli does not depend on the absolute temperature of the stimuli, but rather on the temperature difference between the cold stimulus applied and the temperature of the glass surface the mice were standing on [30]. In other words, mice adapt to the temperature of their environment. This adaptation was significantly reduced by genetic deletion of TRPM8 and by intraplantar injection of the PLC inhibitor U73122 [30]. These data indicate that PLC mediated desensitization of TRPM8 plays a role in vivo in temperature adaptation. Second, as describe before, the main PLC isoform activated by Ca2+ influx in DRG neurons was identified to be PLCδ4. PLCδ4−/- mice showed increased sensitivity of evaporative cold, but not to noxious heat and to mechanical stimuli [28].
TRPM8 inhibition by Gq-coupled receptors; the roles of PI(4,5)P2, Gαq, and PKC
Activation of Gq-coupled receptors, that activate PLCβ isoforms, such as the bradykinin B2 receptor, was also shown to inhibit TRPM8 activity. In contrast to the general agreement on the involvement of PI(4,5)P2 depletion in Ca2+ dependent desensitization, the involvement of this lipid in receptor-induced TRPM8 inhibition has been debated.
Both publications that originally described PI(4,5)P2 dependence of TRPM8 activity found that activation of cell surface receptors that couple to PLC inhibited TRPM8 activity in expression systems, and both papers attributed it to PI(4,5)P2 depletion [12,21]. Recombinant TRPM8 channels expressed in Xenopus oocytes were inhibited by activation or the receptor tyrosine kinase PDGF receptor that couples to PLCγ [12]. Mutations in the PDGFR that prevent PLC activation eliminated TRPM8 inhibition. Mutation of putative PI(4,5)P2 interacting residues in TRPM8 that decreased apparent affinity for PI(4,5)P2 increased the level of inhibition [12]. Consistent with these results, Liu et al found that activation of both NGF receptors that activate PLCγ and M1 muscarinic receptors that activate PLCβ inhibited menthol-induced TRPM8 currents [21].
A publication shortly after these two papers showed that menthol-induced Ca2+ responses in DRG neurons were inhibited by bradykinin, and the effect was eliminated by the PKC inhibitor Bisindolylmaleimide (BIM) [31]. High concentrations of PKC activating phorbol esters, PDBu and PMA (1 μM) inhibited TRPM8 activity, and both the effects of phorbol esters and bradykinin were inhibited by the protein phosphatase inhibitor okadaic acid, therefore it was concluded that TRPM8 is inhibited by PKC-mediated dephosphorylation upon bradykinin receptor activation [31].
A later publication postulated a novel alternative mechanism of inhibition by direct binding of Gαq to TRPM8, and challenged the roles of both PKC and PI(4,5)P2 depletion [16]. The key findings of this manuscript supporting inhibition by direct binding of Gαq are the following. The authors replaced a small segment of Gαq with that of Gαi and found that this chimera (Gαqiq) was deficient in activating PLC. Coexpression of a constitutively active mutant (Q209 L) of this Gαqiq chimera was sufficient by itself to inhibit TRPM8 activity. The authors also found that Gαq binds to TRPM8, and application of purified Gαq to excised inside out patches inhibited TRPM8 activity in the presence of PI(4,5)P2 [16]. A follow up publication found that Gα11 was much less efficient than Gαq in inhibiting TRPM8 [32], despite that Gα11 is similarly efficient to Gαq in stimulating PLC.
Zhang et al also found that the PKC inhibitor BIM did not reduce the inhibitory effect of bradykinin, and PMA did not inhibit TRPM8 activity, arguing against the role of PKC [16]. The authors listed two arguments against the involvement of PI(4,5)P2. 1. The PLC inhibitor U73122 had no effect on bradykinin-induced inhibition of TRPM8, but it partially reduced the inhibitory effect of activating Gq-coupled (H1R) histamine receptors. 2. Mutations in putative PI(4,5)P2 binding residues in the TRP domain did not eliminate histamine-induced inhibition of TRPM8 [16]. Mutation of these residues however is expected to increase inhibition, by reducing apparent affinity for PI(4,5)P2 [12], and indeed the histamine-induced inhibition of the K995Q mutant was somewhat larger than that observed in wild-type channels [16].
A subsequent paper by Liu et al provided evidence that decreased levels of PI(4,5)P2 are involved in receptor-induced inhibition of TRPM8 activity [17]. This work demonstrated that PI(4,5)P2 levels decrease in DRG neurons upon activation of endogenous Gq-coupled receptors using two different fluorescence-based PI(4,5)P2 sensors. The PI(4,5)P2 decrease in TRPM8 positive neurons was more pronounced than that observed in TRPM8 negative neurons. To stimulate endogenous Gq-coupled receptors a mixture of pro-inflammatory agonists was used, as no single receptor was expressed in all TRPM8 positive neurons. Consistent with earlier data, PI(4,5)P2 levels also decreased in a recombinant system upon activation of Gq-coupled receptors. This decrease was larger in the presence of extracellular Ca2+ than in its absence, which correlated well with the larger inhibition in the presence of extracellular Ca2+ compared to nominally Ca2+ free conditions. Inclusion of PI(4,5)P2 in the whole cell patch pipette decreased inhibition by Gq-coupled receptor activation both in DRG neurons and in an expression system [17]. Cumulatively, these data provided strong support for the involvement of PI(4,5)P2 depletion in TRPM8 inhibition upon activation of Gq-coupled receptors.
Liu et al [17] also confirmed that coexpressing the constitutively active Q209 L-Gαqiq construct used by Zhang et al [16] inhibited menthol-induced TRPM8 activity. Co-transfecting Q209 L-Gαqiq with TRPM8 also rendered the channel more sensitive to inhibition by decreasing PI(4,5)P2 levels by the voltage activated phosphoinositide phosphatase ciVSP [17]. Overall Liu et al concluded that the decrease of PI(4,5)P2 and direct binding of Gαq synergize in inhibiting TRPM8 activity (Figure 3).
Another paper by Zhang [18], published at the same time as Liu et al [17] made a case that PI(4,5)P2 depletion is not involved in bradykinin receptor mediated inhibition of TRPM8, and proposed that direct inhibition by Gαq is the sole mechanism. The key findings of the article [18] are the following: Genetic deletion of Gαq eliminated inhibition of TRPM8 in DRG neurons, but potentiation of TRPV1 currents remained intact after bradykinin receptor activation. Mutating Gαq interacting residues in TRPM8 eliminated inhibition by bradykinin receptor activation, but the channel was still inhibited by depleting PI(4,5)P2 with a chemically inducible phosphatase. Additionally, co-expression of the bradykinin B2 receptor rendered the channel resistant to inhibition by chemically induced depletion of PI(4,5)P2 [18]. In our hands, co-expression of B2 bradykinin receptors did not interfere with inhibition of TRPM8 activity by decreasing PI(4,5)P2 using the voltage dependent lipid phosphatase ciVSP (Figure 4). This shows that the ability of the B2 receptor to interfere with inhibition of TRPM8 by decreasing PI(4,5)P2 may be specific to the chemically inducible phosphatase system.
Figure 4.
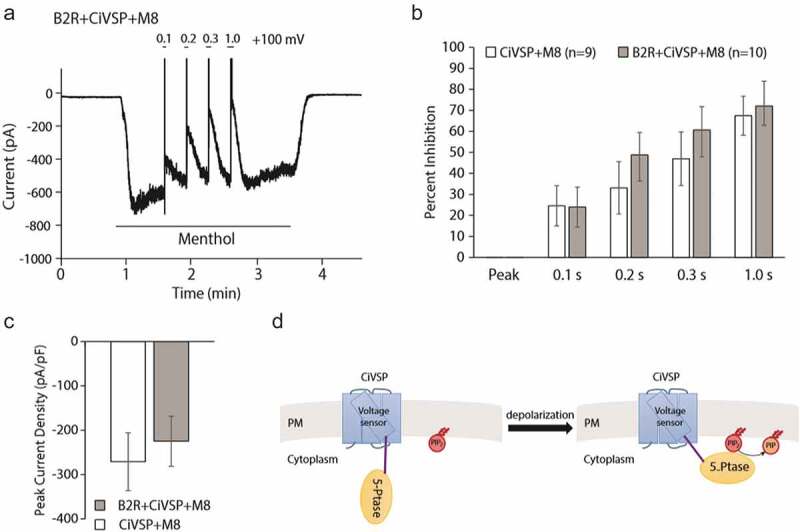
Bradykinin receptor co-expression does not interfere with TRPM8 inhibition by the voltage sensitive phosphoinositide phosphatase ciVSP. (a). Representative trace showing menthol-induced TRPM8 activity in HEK293 cells transfected with TRPM8, ciVSP, and B2 bradykinin receptors. The holding potential was −60 mV, channel activity was evoked by the application of 500 μM menthol in the absence of extracellular Ca2+ to avoid desensitization, ciVSP was activated by depolarizing pulses to 100 mV for 0.1, 0.2, 0.3 and 1 s. The experiment was performed the same way as shown in Figure 10 of Liu et al [17]. (b). Summary of the data, channel inhibition by depolarizing voltage pulses is shown for cells transfected with B2 receptors, TRPM8 and ciVSP, and for cells transfected with TRPM8 and ciVSP. (c). Summary of current densities (d). Cartoon explaining ciVSP.
The proposed exclusive role of inhibition by Gαq was specific to B2 bradykinin receptors, as mutating Gαq binding residues did not eliminate TRPM8 inhibition by H1 histamine receptors, and TRPM8 was inhibited by chemically induced PI(4,5)P2 depletion in the presence of H1 histamine receptors [18].
Conclusions
The plasma membrane phospholipid PI(4,5)P2 is an important cofactor for TRPM8. The recent cryoEM structure of TRPM8 with PI(4,5)P2 provides molecular level understanding of how this lipid binds to TRPM8. Ca2+ influx through TRPM8 activates a Ca2+ sensitive PLCδ isoforms leading to decreased PI(4,5)P2 levels and concurrently decreased channel activity. This Ca2+ dependent desensitization leads to adaptation to lower temperatures in vivo. Upon activation of Gq-coupled receptors, both decreased PI(4,5)P2 levels and direct binding of Gαq plays a role in channel inhibition in a synergistic fashion, but the relative contributions of the individual pathways is likely receptor specific.
Funding Statement
This work was supported by the National Institute of General Medical Sciences [GM093290]; National Institute of General Medical Sciences [GM131048]; National Institute of Neurological Disorders and Stroke [NS055159].
Disclosure Statement
No potential conflict of interest was reported by the authors.
References
- [1].Eccles R. Menthol and related cooling compounds. J Pharm Pharmacol. 1994;46:618–630. [DOI] [PubMed] [Google Scholar]
- [2].McKemy DD, Neuhausser WM, Julius D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature. 2002;416:52–58. [DOI] [PubMed] [Google Scholar]
- [3].Peier AM, Moqrich A, Hergarden AC, et al. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108:705–715. [DOI] [PubMed] [Google Scholar]
- [4].Almaraz L, Manenschijn JA, de la Pena E, et al. Trpm8. Handb Exp Pharmacol. 2014;222:547–579. [DOI] [PubMed] [Google Scholar]
- [5].McKemy DD. TRPM8: the cold and menthol receptor. In: Liedtke WB, Heller S, editors TRP ion channel function in sensory transduction and cellular signaling cascades. Boca Raton (FL); 2007;13:1–30 [PubMed] [Google Scholar]
- [6].Bautista DM, Siemens J, Glazer JM, et al. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature. 2007;448:204–208. [DOI] [PubMed] [Google Scholar]
- [7].Colburn RW, Lubin ML, Stone DJ Jr., et al. Attenuated cold sensitivity in TRPM8 null mice. Neuron. 2007;54:379–386. [DOI] [PubMed] [Google Scholar]
- [8].Dhaka A, Murray AN, Mathur J, et al. TRPM8 is required for cold sensation in mice. Neuron. 2007;54:371–378. [DOI] [PubMed] [Google Scholar]
- [9].Knowlton WM, Daniels RL, Palkar R, et al. Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS One. 2011;6:e25894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ongun S, Sarkisian A, McKemy DD. Selective cold pain inhibition by targeted block of TRPM8-expressing neurons with quaternary lidocaine derivative QX-314. Commun Biol. 2018;1:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Daniels RL, Takashima Y, McKemy DD. Activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J Biol Chem. 2009;284:1570–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rohacs T, Lopes CM, Michailidis I, et al. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci. 2005;8:626–634. [DOI] [PubMed] [Google Scholar]
- [13].Cesare P, McNaughton P. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci U S A. 1996;93:15435–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Moriyama T, Higashi T, Togashi K, et al. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain. 2005;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tominaga M, Wada M, Masu M. Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci U S A. 2001;98:6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang X, Mak S, Li L, et al. Direct inhibition of the cold-activated TRPM8 ion channel by Gαq. Nat Cell Biol. 2012;14:851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu L, Yudin Y, Nagwekar J, et al. G alphaq sensitizes TRPM8 to inhibition by PI(4,5)P2 depletion upon receptor activation. J Neurosci. 2019;39:6067–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang X. Direct Gαq gating is the sole mechanism for TRPM8 inhibition caused by Bradykinin receptor activation. Cell Rep. 2019;27(3672–3683):e3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kadamur G, Ross EM. Mammalian phospholipase C. Annu Rev Physiol. 2013;75:127–154. [DOI] [PubMed] [Google Scholar]
- [20].Rohacs T. Phosphoinositide regulation of TRP channels. Handb Exp Pharmacol. 2014;233:1143–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu B, Qin F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2005;25:1674–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hilgemann DW, Ball R. Regulation of cardiac Na+-Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273:956–959. [DOI] [PubMed] [Google Scholar]
- [23].Yudin Y, Lukacs V, Cao C, et al. Decrease in phosphatidylinositol 4,5-bisphosphate levels mediates desensitization of the cold sensor TRPM8 channels. J Physiol. 2011;589:6007–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zakharian E, Cao C, Rohacs T. Gating of transient receptor potential melastatin 8 (TRPM8) channels activated by cold and chemical agonists in planar lipid bilayers. J Neurosci. 2010;30:12526–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yin Y, Le SC, Hsu AL, et al. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science. 2019;363:eaav9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zakharian E, Thyagarajan B, French RJ, et al. Inorganic polyphosphate modulates TRPM8 channels. PLoS One. 2009;4:e5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hughes TET, Pumroy RA, Yazici AT, et al. Structural insights on TRPV5 gating by endogenous modulators. Nat Commun. 2018;9:4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yudin Y, Lutz B, Tao YX, et al. Phospholipase C delta4 regulates cold sensitivity in mice. J Physiol. 2016;594:3609–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Varnai P, Thyagarajan B, Rohacs T, et al. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol. 2006;175:377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brenner DS, Golden JP, Vogt SK, et al. A dynamic set point for thermal adaptation requires phospholipase C-mediated regulation of TRPM8 in vivo. Pain. 2014;155:2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Premkumar LS, Raisinghani M, Pingle SC, et al. Downregulation of transient receptor potential melastatin 8 by protein kinase C-mediated dephosphorylation. J Neurosci. 2005;25:11322–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li L, Zhang X. Differential inhibition of the TRPM8 ion channel by Galphaq and Galpha 11. Channels (Austin). 2013;7:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]