

ABSTRACT
T cell immunoglobulin and mucin domain-3 (TIM-3) is an immune checkpoint that regulates normal immune responses but can be exploited by tumor cells to evade immune surveillance. TIM-3 is primarily expressed on immune cells, particularly on dysfunctional and exhausted T cells, and engagement of TIM-3 with its ligands promotes TIM-3-mediated T cell inhibition. Antagonistic ligand-blocking anti-TIM-3 antibodies have the potential to abrogate T cell inhibition, activate antigen-specific T cells, and enhance anti-tumor immunity. Here we describe M6903, a fully human anti-TIM-3 antibody without effector function and with high affinity and selectivity to TIM-3. We demonstrate that M6903 blocks the binding of TIM-3 to three of its ligands, phosphatidylserine (PtdSer), carcinoembryonic antigen cell adhesion-related molecule 1 (CEACAM1), and galectin 9 (Gal-9). These results are supported by an atomic resolution crystal structure and functional assays, which demonstrate that M6903 monotherapy enhanced T cell activation. This activation was further enhanced by the combination of M6903 with bintrafusp alfa, a bifunctional fusion protein that simultaneously blocks the transforming growth factor-β (TGF-β) and programmed death ligand 1 (PD-L1) pathways. M6903 and bintrafusp alfa combination therapy also enhanced anti-tumor efficacy in huTIM-3 knock-in mice, relative to either monotherapy. These in vitro and in vivo data, along with favorable pharmacokinetics in marmoset monkeys, suggest that M6903 as a monotherapy warrants further pre-clinical assessment and that M6903 and bintrafusp alfa may be a promising combination therapy in the clinic.
KEYWORDS: TIM-3, PtdSer, Gal-9, CEACAM1, antagonistic antibody
Introduction
In recent years, immune checkpoint inhibitors have become one of the most promising classes of molecules for cancer treatment. T Cell Immunoglobulin and Mucin Domain-3 (TIM-3) is an immune checkpoint that regulates normal immune responses but, like other immune checkpoints, can be exploited by tumor cells to evade immune surveillance. TIM-3 was first identified as a molecule selectively expressed on interferon (IFN)-γ–producing CD4+ T helper 1 (Th1) and CD8+ T cytotoxic 1 (Tc1) cells1 and has subsequently been found to be expressed on the surface of other immune cell types, including natural killer (NK) cells, monocytes, tumor-associated dendritic cells (TADCs), and T regulatory cells (Tregs).2–4 In the absence of ligand binding, TIM-3 association with downstream factors facilitates T cell activation, while ligand binding leads to TIM-3-mediated T cell inhibition.4 Several TIM-3 ligands have been reported, including galectin-9 (Gal-9),5 phosphatidylserine (PtdSer),6 carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1),7 and high-mobility group box 1 protein (HMGBl).8
TIM-3 regulates T cell exhaustion in human and mouse tumor infiltrating lymphocytes (TILs), 2,9 and blockade of TIM-3 in vivo can reverse tumor-induced T cell exhaustion.2 TIM-3 is also upregulated on intratumoral TIM-3+FOXP3+ Tregs, which express high levels of Treg effector molecules and are suggested to promote a dysfunctional phenotype in CD8+ TILs.10 Upon engagement with ligand, TIM-3 can suppress T cell and NK cell responses5 and induce cell death in TIM-3+ Th1 cells.11 Additionally, the interaction of TIM-3 with Gal-9 on T cells can promote myeloid-derived suppressor cells (MDSC) proliferation.11 Blocking TIM-3 interactions with its ligands may inhibit these immunosuppressive phenotypes and enhance T cell responses in the tumor microenvironment (TME).
It has been shown that functionally efficacious anti-murine and anti-human TIM-3 antibodies interfere with binding to both PtdSer and CEACAM1,12 although none of the anti-TIM-3 antibodies tested by Sabatos-Peyton et al. (2017) interfered with the binding of TIM-3 and Gal-9. However, Gal-9 has been hypothesized to be trafficked by TIM-3 and play an important role in TIM-3 signaling.12,13 Gal-9 also has a role in the inhibition of T cell responses4 and may increase tumorgenicity via T cell dysfunction.9,14 Therefore, anti-tumor efficacy could potentially be improved if the interaction of TIM-3 with Gal-9 were blocked in addition to the interaction of TIM-3 with PtdSer and CEACAM1. Indeed, it has been shown that Gal-9 knockout (KO) mice are relatively resistant to acute myeloid leukemia (AML) relative to WT mice, and when treated with anti-PD-L1, 100% of the Gal-9 KO mice survived tumor-free through 80 days post-inoculation.14 Furthermore, over-expression of both Gal-9 and PD-L1 have been shown in patients with chronic B cell leukemia,15 further supporting the notion that a Gal-9 blocking anti-TIM-3 antibody may have added benefit, especially in combination with anti-PD-1/anti-PD-L1 treatment.
Co-blockade of TIM-3 and PD-1 has demonstrated remarkable synergy in enhancing tumor growth inhibition (TGI) relative to monotherapies in preclinical mouse tumor models,9,16 and, in lung adenocarcinoma patients, TIM-3 expression correlated with PD-1 expression and was positively associated with worse relapse free survival and overall survival.17 In addition, TIM-3 is often upregulated on anti-PD-1-bound T cells in mouse lung cancer models and in human lung cancer patients that failed to respond to anti-PD-1 treatment or that acquired adaptive resistance following anti-PD-1 treatment.18 In mouse models, the addition of an anti-TIM-3 antibody at the time of anti-PD-1 failure increased the median survival and increased IFN-γ production,18 suggesting a TIM-3-mediated mechanism in the failure of anti-PD-1 treatment. TIM-3 expression is often associated with co-expression of PD-1 on exhausted CD8+ TILs, and TIM-3+/PD-1+ CD8+ T cells represent a more severely exhausted phenotype compared with TIM-3−/PD-1+ CD8+ T cells,9 suggesting that PD-1/PD-L1 blockade may be a promising combination partner for anti-TIM-3 therapy. Phase I/II trials are currently evaluating the combination of anti-TIM-3 and anti-PD-1 in the clinic. Phase I studies sponsored by Eli Lilly and Company19 (LY3321367 + LY3300054, NCT03099109), Tesaro20 (TSR-022 + TSR-042, NCT02817633), and Novartis Pharmaceuticals21 (MBG453 + spartalizumab, NCT03099109), have reported that the anti-TIM-3 and anti-PD-1 combinations are well tolerated with manageable safety profiles and show signs of clinical activity.
In a murine lung fibrosis model, mice treated with anti-TIM-3 had more bleomycin-induced fibrosis, α smooth muscle actin (αSMA) expression, and collagen deposition in the lungs compared with control treated animals.22 While proinflammatory cytokine production was not increased in this model, anti-TIM-3 treated mice had increased TGF-β1 expression, suggesting that the enhanced fibrosis, αSMA, and collagen could be TGF-β1-mediated.22 Therefore, inhibition of TGF-β signaling may be a beneficial addition to anti-TIM-3 treatment as well.
Bintrafusp alfa (also known as M7824) is an innovative first-in-class bifunctional fusion protein composed of the extracellular domain of the TGF-βRII receptor to function as a TGF-β “trap” fused to a human IgG1 antibody blocking PD-L1. Bintrafusp alfa is designed to simultaneously target both PD-L1 and TGF-β to relieve these two negative regulatory pathways of immunosuppression. It has demonstrated potent anti-tumor activity in nonclinical models23 and is in clinical development in multiple solid tumor indications. Bintrafusp alfa therefore represents another promising combination partner for anti-TIM-3 antibodies. Here we describe a novel anti-TIM-3 antibody, M6903, and its ability to block the interaction of TIM-3 with its ligands, to enhance T cell activation, and to improve bintrafusp alfa-induced inhibition of tumor growth in the MC38 mouse tumor model.
Methods and materials
M6903, bintrafusp alfa, and anti-PD-L1
M6903 is a fully human anti-TIM-3 monoclonal IgG2 antibody that was derived from human immunoglobulin (Ig) transgenic rats (OmniRat®) immunized with His-tagged recombinant human TIM-3 extracellular domain (ECD) protein. Following B-cell cloning, parental anti-TIM-3 antibodies were screened for binding to human and non-human primate TIM-3 ECD by ELISA and flow cytometry analysis. A clone with high affinity for human and non-human primate TIM-3 was optimized to reduce chemical instability and post-translational modifications, limit challenges with manufacturability, and reduce effector function. The optimized anti-TIM-3 antibody was termed M6903. Bintrafusp alfa is a bifunctional fusion protein composed of the extracellular domain of the TGF-βII receptor to function as a TGFβ “trap” fused to a human IgG1 antibody blocking PD-L1.23 The anti-PD-L1 IgG1 antibody used as a control in the T cell activation assays was previously described.23 The isotype control is a human IgG1 or IgG2 that lacks a specific target. Exact dose and treatment schedules for each experiment are listed in the figure legends.
Cell lines
MC38 colon carcinoma cells were obtained from BioVector NTCC, Inc or Shunran Shanghai Biological Technology Co., Ltd and were maintained in Dulbecco’s Modified Eagle’s medium (DMEM; Cellgro, 10-013-CVR) supplemented with 10% heat inactivated fetal bovine serum (FBS; Excell, FSP500). Cells were passaged before in vivo implantation and adherent cells were harvested with trypsin-EDTA treatment. Daudi (human B lymphoblast cell line, CCL-213) and Jurkat cells (human peripheral blood T lymphoblast cell line, clone E6-1) were purchased from the American Type Culture Collection (ATCC). Daudi cells were cultured in RPMI 1640 (Life Technologies, 11875–093) with 10% FBS (Corning/Cellgro, 35-011-CV, 35-015-CV) and 100X GlutaMAX (Gibco, 35050–061). Jurkat cells were cultured in RPMI with 10% FBS.
Affinity determination by SPR or ELISA
To determine the affinity of M6903 to TIM-3 orthologues from various species via SPR, goat anti-human-IgG Fc polyclonal antibody (Jackson Labs, 109-005-098) was immobilized on CM5 chips (GE Healthcare, BR-1006-68). Flow rate throughout was 30 μL/minute. Anti-TIM-3 antibodies were captured at 0.5 and 1.0 µg/mL for 120 seconds. TIM-3 ECD orthologues from different species were either purchased (Human Novoprotein C356, marmoset Novoprotein NP10506, murine Sino Biologicals 51152), or in the case of ECD from cynomolgus monkey, expressed in Expi293 F cells (ThermoFisher, A14527), and purified by immobilized metal affinity chromatography followed by size exclusion chromatography. ECDs were diluted from 0–100 nM. Association was measured for 180 seconds, followed by dissociation in buffer (HEPES buffered saline with EDTA and P20, GE Healthcare, BR-1006-69) for 600 or 900 seconds. Biacore T200 software version 1.0 was used to fit sensorgrams to a 1:1 Langmuir binding model to derive association (ka) and dissociation (kd) kinetic constants. The equilibrium dissociation constant (KD) was determined as the ratio of kinetic constants.
To test for binding of M6903 to rat TIM-3 protein, binding of M6903 to human TIM-3, rat TIM-3, and an unrelated control protein CD47 was measured by ELISA. Briefly, 96-well plates were coated with huTIM-3-His6 (Novoprotein, C356), ratTIM-3-His6 (Sino Biological, EM1/MB12NO0106), and huCD47-His6 (EMD Serono, O12-23-2), each protein at 1 µg/mL. After blocking with 3% bovine serum albumin (BSA), an 11-point 1:3 dilution series from 6 µg/mL or 1 µg/mL starting concentration for each antibody was incubated in duplicate for 1 hr at room temperature. Following washing steps, the bound antibodies were incubated for 1 hr at room temperature with a Peroxidase AffiniPure F(ab’)₂ Fragment Goat Anti-Human IgG, Fcγ fragment specific antibody (Jackson ImmunoResearch Laboratories, 109-036-098), and detection was performed using the TMB HRP Substrate solution (BioFx Lab, TMBW-1000-01). Reagent validation of coating with human TIM-3-His6, rat TIM-3-His6, and human CD47-His6 was performed with a commercial mouse HRP-anti-His Tag (R&D, MAB050 H) antibody and a commercial rabbit polyclonal anti-human/mouse/rat TIM-3 antibody (LifeSpan Bioscience, LS‑C748494), which were detected with the appropriate detection reagent for the commercial primary antibody. As a human isotype control antibody, anti-HEL isotype control antibody was included for comparison to binding by M6903.
Inhibition of Gal-9 binding to TIM-3 by M6903
Inhibition of the Gal-9/TIM-3 interaction was assayed using recombinant human Gal-9 protein (R&D Systems, 2045-GA) coated at 2 µg/mL onto ELISA assay plates and then blocked with 3% BSA (Sigma, A-3912). Recombinant Human TIM-3-Fc Chimera protein (R&D Systems, 2365-TM), which contains full ECD of TIM-3, was biotinylated with EZ-Link Sulfo-NHS-LC-Biotinylation kit (Thermo Scientific, 21327). Biotinylated-human TIM-3-Fc (huTIM-3-Fc-biotin) was added to Gal-9 coated assay plates at a final concentration of 0.5 µg/mL huTIM-3-Fc-biotin in assay buffer and detected with Streptavidin Tag Peroxidase-conjugated antibody (Jackson ImmunoResearch, 016-030-084) and TMB HRP Substrate (BioFX/SurModics IVD, TMBS-1000-01). To test antibodies for inhibition of Gal-9 binding to TIM-3, huTIM-3-Fc-biotin was mixed with antibodies in an 11-point 1:3 dilution series from 50 µg/mL starting concentration in quadruplicate for each antibody and incubated 50 minutes at room temperature. Following this incubation step, the huTIM-3-Fc-biotin + antibody mixtures were added to the Gal-9 coated ELISA assay plates and incubated for 75 minutes at room temperature, then washed and developed with detection reagents and optical density (OD) (450 nm) was read.
Inhibition of TIM-3 binding to PtdSer by M6903
To evaluate the ability of M6903 to block the interaction of TIM-3 with its ligand PtdSer, a blocking assay was performed using apoptotic Jurkat cells. Jurkat cells (1 × 107 cells) were treated with Staurosporine (Sigma, S6942-200 μL, 2 μg/mL) for 18 hours to induce apoptosis; untreated Jurkat cells were used as a negative control. Apoptotic cells from each well were treated with 5% human AB serum (Valley Biomedical, HP1022) at room temperature for 1 hour. rhTIM-3-Fc AF647 (R&D Systems, 2365-TM; final concentration 2 μg/mL) was pre-incubated with 1:3 serial dilutions of either M6903 or an anti-HEL IgG2 isotype control (EMD Serono, A15-125-1, starting at 60 μg/mL and diluting to 0.027 μg/mL final concentration) on ice for 30 minutes. Apoptotic Jurkat cells were then incubated with either the mixture of rhTIM-3-Fc AF647 and M6903 or the mixture of rhTIM-3-Fc AF647 and the isotype control for 30 minutes on ice. Cells were washed once with 5% human AB serum in PBS (Life technologies, 20012–027, 20012–043) and resuspended in 5% human AB serum in PBS with 7AAD (BD Pharmingen, 51-68981E), for viability analysis. TIM-3-Fc AF647 positive cells were analyzed by flow cytometry and the median fluorescence intensity (MFI) of AF647 was compared between cells treated with the isotype control and those treated with M6903.
Inhibition of TIM-3 binding to CEACAM1 by M6903
Inhibition of the interaction between TIM-3 and CEACAM1 was detected using an ELISA-based assay in the presence of 10 mM CaCL2 (Sigma, C3306-100 G). In a flat bottom 96-well MaxiSorp plate, 0.5 µg rhTIM-3-IgG1-Fc in 1xTBS (Thermo Scientific, 28358) with 10 mM CaCL2 (Ca2+) was added to each well and the plate was incubated at 4°C overnight. The plate was then blocked in 2% BSA (SeraCare Life Sciences, AP-4510-01) TBS (Ca2+) at room temperature with shaking for 1 hour. After the supernatant was removed, M6903 or isotype control (anti-HEL IgG2) in TBS (Ca2+) was added to each well and the plate was incubated at room temperature for 1 hour, and then CEACAM1-poly His (ARCO, CE1-H5220) in TBS (Ca2+) was added to each well and the plate was incubated at room temperature for 1 hour with shaking. The plate was then washed and anti-6XHis-HRP (Biolegend, 652504) in TBS (Ca2+) was added to each well followed by incubation at room temperature for 1 hour with shaking. After washing, TMB was added to each well followed by shaking the plate at room temperature for 20 minutes and adding stop solution to each well. The OD (450 nm and 570 nm) was read immediately using EnVision plate reader.
Hotspot mapping
To determine the hotspot residues involved in M6903 binding to TIM-3, selected residues were mutated to alanine, expressed in HEK cells, and purified by affinity chromatography. Variants were characterized for overall stability by observing the monodispersity and calculating the percent monomer on analytical size exclusion chromatography (SEC). Additionally, to ensure that mutations did not result in gross protein misfolding, the affinity of the mutant was checked with several anti-TIM-3 antibodies unrelated to M6903, including 27.12E12,24 ABTIM3-hum03,25 and mab15.26 The binding affinity of M6903 for wild-type and each mutant was determined using SPR. Binding hotspots, residues that contribute most to the binding energy, were identified through evaluation of the change in Gibbs free energy (ΔΔG) of binding upon mutation using the formula ΔΔG = -RT*ln(KDTIM−3/KDmutant), where R is the gas constant and T is the temperature in Kelvin.
Co-crystallization of TIM-3 with M6903 Fab
To obtain the co-crystal structure of TIM-3 with M6903, human TIM-3 was expressed in E. coli inclusion bodies, refolded by rapid dilution in redox buffer, and purified by SEC. The Fab fragment of M6903 was expressed as a polyhistidine-tagged construct in Expi293 F cells and purified by immobilized metal affinity chromatography. TIM-3 ECD and Fab were incubated at a 2:1 ratio on ice, followed by size exclusion purification of the complex. Crystal screening of the complex was performed at 4°C using the hanging drop vapor diffusion method. X-ray diffraction data were collected at the SWISS LIGHT SOURCE (SLS, Villigen, Switzerland) using cryogenic conditions and the structure of the complex solved by molecular replacement (Supplementary Table 1).
Antigen specific T cell assay (CEF assay)
To determine the effects of M6903 on T cell activation, a peptide pool (AnaSpec, AS-61036) of 32 HLA class I-restricted T cell epitopes, a collection of viral antigens from cytomegalovirus [C], Epstein Barr virus [E] and influenza virus [F] was purchased from AnaSpec and used to stimulate human peripheral blood mononuclear cells (PBMCs). T cell activation and IFN-γ production were measured following the co-treatment of cells with immune-modulatory antibodies, such as M6903, anti-PD-L1, and bintrafusp alfa. In this assay, human PBMCs (Research Blood Components) were thawed and resuspended before being stimulated with CEF peptide pool (final concentration 40 μg/mL) in AIM V plus 5% human AB serum and treated with antibodies. For the dose-response monotherapy study, cells were treated with seven 1- to 4-fold serial dilutions, beginning with 20,000 ng/mL and diluting to 4.88 ng/mL, of M6903 for 6 days. For combination studies, cells were treated with four 1- to 5-fold serial dilutions of M6903, from 10,000 ng/mL to 80 ng/mL, in combination with 10 µg/mL bintrafusp alfa (EMD Serono, APTA5A01/MSB0011359 C) or isotype control (anti-HEL IgG1 or inactive anti-PD-L1, EMD Serono) for 4 days. Additionally, anti-PD-L1 was used a control as previously described23 (EMD Serono, PDZC003/MSB0010718 C). For the single dose study, cells were treated with 10 μg/mL isotype control, M6903, bintrafusp alfa, or a combination of M6903 + bintrafusp alfa, with or without 2 ng/mL rhTGF-β1 (R&D Systems, 7754-BH/CF) for 4 days. IFN-γ in supernatant was measured by an IFN-γ ELISA kit according to manufacturer’s instructions (R&D Systems, DY285B). Data were collected using OD450 on Envision plate reader.
Allo-antigen specific T cell assay (Daudi Allo assay)
An allo-antigen specific T cell assay, the Daudi Allo assay, was used to further examine the effects of M6903 on T cell activation. The Daudi Allo assay is a one-way mixed lymphocyte reaction (MLR) assay using primary human PMBCs and the Burkitts lymphoma Daudi cell line to measure T cell activation via an IFN-γ ELISA. Daudi cells were irradiated with 3000 Rad (MDS Nordion, Gammacell® 40 Exactor), washed, and diluted to 4 × 105 cells/mL. T cells were isolated from human PBMCs (Research Blood Components) using the Pan T cell kit (Miltenyi Biotec, 130-094-131) according to manufacturer instructions and diluted to 2 × 106 cells/mL in 10% RPMI. Cells were co-cultured by plating 2 × 105 T cells and 2 × 104 irradiated Daudi cells per well in a 96-well plate with reconstituted human IL-2 (R&D Systems, 202-IL-050, 50 μL of 40 IU/mL) to a total volume of 200 μL (day 0). On days 2, 4, and 6 of the 7-day incubation period, 50 μL of media was removed and replaced with 50 μL of fresh 40 UI/mL IL-2 media. On day 7, T cells were harvested from all wells and resuspended in 2 × 106 cells/mL in 10% RPMI. Additional Daudi cells were then irradiated, washed, and resuspended to 1 × 106 cells/mL. Cells were then plated at a 2:1 effector T cells: Daudi cells ratio.
To test the effects of M6903 on T cell activation compared with isotype control, serial dilutions of M6903 or anti-HEL IgG2 isotype control were added to the co-cultured T cells and Daudi cells. The antibody titrations were setup as seven 1- to 4-fold serial dilutions, beginning with 20,000 ng/mL and diluting to 4.88 ng/mL. After 48 hours, supernatant was harvested and a human IFN-γ ELISA (R&D Systems) was performed, according to manufacturer instructions.
To determine the effect of combination of M6903 with bintrafusp alfa, serial dilutions of M6903 were setup as four 1- to 10- fold serial dilutions, beginning with 10,000 ng/mL and diluting to 10 ng/mL. To these M6903 dilutions, 10 μg/mL of hIgG1 isotype control, anti-PD-L1, or bintrafusp alfa was added, and this antibody mix was incubated with co-cultured T cells and Daudi cells. After 48 hours, supernatant was harvested and a human IFN-γ ELISA (R&D Systems) was performed, according to manufacturer instructions.
Superantigen stimulation assay (SEB assay)
The effects of M6903 on T cell activation were then measured in an assay using superantigen Staphylococcal enterotoxin B (SEB, Toxin Technologies, BT202red) to stimulate human PBMCs and activate T cells. Cryopreserved human PBMCs (Research Blood Components) were thawed and resuspended to 1 × 106 cells/mL. The PBMCs were stimulated with SEB (Toxin Technologies) at a final concentration of 100 ng/mL SEB and incubated for 9 days with the following antibodies (10 μg/mL); M6903, anti-PD-L1, bintrafusp alfa, a combination of M6903 and anti-PD-L1, a combination of M6903 and bintrafusp alfa, or IgG1 or IgG2 isotype controls. On day 9, the cells were washed once with media, and incubated for an additional 2 days in fresh media containing 100 ng/mL SEB and 10 μg/mL of antibodies. On day 11, the supernatant was harvested for IFN-γ ELISA (R&D Systems) analysis. Data were collected using OD450 on an Envision plate reader (PerkinElmer).
Gal-9 binding to TIM-3
To test the binding of Gal-9 to TIM-3, recombinant human Gal-9 (R&D Systems, 2045-GA-050) or murine Gal-9 (R&D Systems, 3535-GA-050) were diluted to 1 μg/mL and coated on ELISA plates and incubated overnight at 4°C. Plates were washed and blocked, then hu-TIM-3-ECD-Fc-His (Novoprotein, CD71) was added in serial dilutions of 0.125, 0.25, 0.5, 1, 2, and 4 μg/mL and incubated for 2 hrs at room temperature. After washing, a secondary mouse anti-His-HRP detection antibody (R&D Systems, MAB050 H) was incubated on the plates for 1 hr at room temperature, followed by washes and detection reagents. Data were collected using OD450 on a Bio-Tek plate reader.
In vivo mouse studies
In order to measure anti-tumor efficacy of M6903 alone or in combination with bintrafusp alfa, female humanized TIM-3 knock-in transgenic mice (hu-TIM-3 KI) (C57BL/6 background, 6–8 weeks old) were obtained from Beijing Biocytogen Co., Ltd. Mice were housed with ad libitum access to food and water in pathogen-free facilities at the Animal center of Beijing Biocytogen Co., Ltd. All animal care and experimental procedures were approved and conducted according to guidelines issued by the Institutional Animal Care and Use Committee of EMD Serono Research and Development Institute.
Hu-TIM-3 KI mice were subcutaneously injected with MC38 tumor cells (5 x 105) in the right flank (day −10) and assigned to treatment groups (n = 10 mice per group) using stratified randomization when the average tumor volume reached approximately 50 mm3 (day 0). Treatments were as follows: isotype control (inactive anti-PD-L1 antibody, EMD Serono, A09-275-1/MSB0010715 H-1 – 20 mg/kg, i.v on days 0, 3, 6), bintrafusp alfa (24 mg/kg, i.v. on days 0, 3, 6), isotype control + M6903 (10 mg/kg, i.p. on days 0, 3, 6, 9, 12, 15, 18), bintrafusp alfa + M6903. All antibody dilutions for in vivo treatments were prepared with sterile PBS (Gibco, 20012027). Tumor volume and body weight were measured twice weekly and mice were sacrificed if their tumor size reached 2000 mm3. Tumor growth inhibition (TGI) was calculated for each group using the formula: TGI (%) = [1-(Ti-T0)/(Vi-V0)] ×100; where Ti is the average tumor volume of a treatment group on a given day, T0 is the average tumor volume of the treatment group on the first day of treatment, Vi is the average tumor volume of the vehicle control group on the same day as Ti, and V0 is the average tumor volume of the vehicle group on the first day of treatment. The results shown are from a representative study that was repeated with similar results.
Binding of M6903 to monkey primary cells
To determine the binding ability of M6903 to TIM-3 on nonhuman primate T cells, flow cytometry analysis was performed. Human PBMCs were prepared from buffy coat blood (Research Blood Components), cynomolgus and marmoset monkey PBMCs were prepared from whole blood (Worldwide Primates, Inc.) using a Ficoll gradient, and rhesus monkey PBMCs were purchased (BioIVT). Human, cynomolgus, marmoset, and rhesus PBMCs were treated with 20 ng/mL staphylococcal enterotoxins A (SEA; Sigma, S9399, Toxin Technologies, AR101red) for 5, 6, 6, or 5 days, respectively. Naive control PBMCs (untreated with SEA) were also analyzed.
For flow cytometry analysis, SEA treated and untreated PBMCs were harvested, resuspended in FACS buffer with normal mouse IgG (Life Technologies, 10400 C) and anti-HEL hIgG antibody (60 μg/mL), and incubated on ice for 30 minutes. Human, cynomolgus, and rhesus PBMCs were then stained with anti-CD4 (BioLegend, 317410 (human and rhesus); BD Biosciences, 550630) and anti-CD8 (BioLegend, 301006 (human and rhesus); Beckman Coulter, 6603861) fluorophore-conjugated antibodies, and marmoset PBMCs were stained with anti-CD3 fluorophore-conjugated antibodies (BD Biosciences, 556611). In addition to these T cell markers, PBMCs were also incubated with a 1 μg biotinylated anti-HEL IgG1 control (EMD Serono, A11-122-3 labeled in-house), biotinylated anti-LAG (EMD Serono, 4290D11, labeled in-house), or biotinylated anti-TIM-3 IgG1 antibody (produced in transiently transfected HEK293 cells, EMD Serono, 3903E11 (VH1.2-VL1.3) labeled in-house) for 45 minutes, followed by streptavidin-APC (eBioscience, 17–431782) for 30 minutes. 7AAD was used as a viability marker for flow cytometry gating. Cells were gated on viable cells and the expression of TIM-3 was analyzed as histograms for each T cell population.
Expression of TIM-3 in untreated monkey cells was also determined on B cells (CD20+; anti-human CD20 PE, Beckman Coulter, 6603446), NK cells (CD3−/CD56+; anti-human CD56-PE, BD Biosciences, 340363), monocytes (CD14+; anti-human CD14-FITC, BioLegend, 301804), mDCs (Lin1−, HLA-DR+ (anti-HLA-DR-PerCP, Biolegend, 307630), CD123− (anti-CD123-PE-Cy7, BD Biosciences, 560826), CD11c+ (anti-CD11c-PB, Biolegend, 301626)) and pDCs (Lin1−HLA-DR+CD123+CD11c−). Lin markers included anti-CD3-FITC (BD Pharmingen, 556611), anti-CD20-FITC (BD Pharmingen, 556632), anti-CD14-FITC (BD Pharmingen, 557153), anti-CD16-FITC (Biolegend, 302006).
Quantitative PCR
To measure mRNA expression of various genes before and after SEA activation, human and cynomolgus monkey PBMCs were treated with 20 ng/mL SEA for 6 days. Naive control PBMCs (untreated with SEA) and SEA-treated PBMCs were harvested and RNA was extracted from these samples with RNeasy mini kit (Qiagen, 74106). Synthesis of cDNA was done with SuperScript VILO cDNA Synthesis Kit (Life Technologies, 11754–050). Taqman PCR with primers (Life Technologies, 4370048) of β-actin (ACTB; Mf04354341_g1), TIM-3 (Mf02850235_m1), LAG-3 (Mf02841705_g1), PD-L1 (Mf02865485_m1), TIGIT (Mf02887844_m1) was run on QuantStudio 12 K Flex machine (Life Technologies). Using β-actin as the reference gene, relative gene expression was calculated as the level of mRNA expression of individual genes relative to that of β-actin which was set to 1. Calculations were done as follows: ΔCt = CtAvg GOI - CtAvg ACTB, where ACTB is the reference gene, GOI is gene of interest (e.g., TIM-3, LAG-3, PD-L1, TIGIT), and relative gene expression = 2ΔCt.
Target occupancy assay
To determine the target occupancy (TO) of M6903 on monocytes, serial dilutions of M6903 (0, 0.64, 3.2, 16, 80, 400, 2000, and 10000 ng/mL) were incubated with human whole blood samples (Research Blood components) for 1 hour, and the unoccupied TIM-3 on CD14+ monocytes was measured by flow cytometry with an APC conjugated anti-human CD366 TIM-3 antibody (anti-TIM-3 (2E2), Biolegend, 345012) which is known to compete with M6903 for TIM-3 binding.
Specifically, an anti-TIM-3 (2E2) antibody mix was prepared by adding anti-CD14-PE (BioLegend, 301806) and anti-TIM-3 (2E2). For the isotype control, mouse IgG1-APC (BioLegend, 400120) was added instead of the anti-TIM-3(2E2) antibody. The appropriate solution was added to each tube of human whole blood and cells were incubated in the dark for 30 minutes. Red blood cell (RBC) lysis was performed using 1X RBC lysis buffer (eBioscience, 00-4300-54). The tubes were then vortexed gently and incubated in the dark for 10 minutes. The cells were then washed and resuspended in FACS buffer containing 7-AAD. Flow cytometry analysis was performed using a BD-Caliber gated on CD14+ cells and analyzed as follows. Percentage of target occupancy (TO %) was calculated using this formula, TO (%) = (1-(Dt-Ct)/(D0-C0))*100, where Dt = MFI value of TIM-3 staining at a certain concentration of M6903. Ct = MFI value of isotype control the same concentration of M6903 as Dt. D0 = MFI values of TIM-3 staining in the absence of M6903. C0 = MFI values of isotype control in the absence of M6903. Post-data analysis was performed using CellQuest Pro software. Batch data analysis was performed to record MFI values of anti-TIM-3(2E2) staining and the ΔMFI and percent TO were calculated.
Single-dose intravenous (iv) administration pharmacokinetics (PK) study in marmoset monkeys
The PK study was performed by Covance GmbH in compliance with necessary guidelines or recommendations. All procedures complied with the German Animal Welfare Act and were approved by the local Institutional Animal Care and Use Committee. Purpose-bred marmoset monkeys (Callithrix jacchus) from the University of Münster or Covance Preclinical Services GmbH were selected to provide eight healthy animals (2 males and 2 females per dosing group). The animals were 2–3 years old and weighed >300 g each. After arrival, animals were acclimated to study procedures for a period of at least 2 weeks and an animal health assessment was performed by a veterinarian before start of the pre-dose phase to confirm the suitability of every animal for the study. Animals were housed in a climate-controlled room in same sex pairs or groups, given water ad libitum, and offered a variety of food twice daily. Where possible, animals were assigned to treatment groups based on existing social groups and stratified body weights.
M6903 was administered as 0.2, 1, 5, or 20 mg/kg doses in 5 mL/kg vehicle (10 mM histidine, 8% trehalose, pH 5.5, 0.05% Tween20), based on individual body weight. Blood samples (0.1 mL) were collected from all animals at day −7 of the pre-dose phase, at 1, 6, 24, 48, 72, and 96 hrs post-dose (day 1) time points, and on days 8 (168 hrs), 15 (336 hrs), 22 (504 hrs), and 29 (672 hrs) of the dosing phase. Serum was obtained from blood samples and a qualified sandwich ELISA assay based on MSD technology was used for quantification of M6903 in marmoset serum.
Statistical analyses
Statistical analyses were performed using GraphPad Prism Software, version 8.0.1. Ligand binding data are presented graphically with mean ± standard deviation (SD) by symbols. Non-linear best fit lines were generated for all plots using a Sigmoid dose-response equation. T cell activation assay data are presented as mean ± SD. Non-linear best fit lines were also generated, and p-values were calculated. For the CEF, Daudi, and SEB assays, t-tests were performed with Welch’s correction for comparisons with combination therapy. Tumor volume data are presented graphically as mean ± standard error of the mean (SEM) by symbols or as individual mice by lines. To assess differences in tumor volumes between treatment groups, a two-way analysis of variance (ANOVA) was performed followed by Tukey’s multiple comparison test. To compare mRNA expression between human and cynomolgus monkey stimulated PBMCs or to compare unstimulated and stimulated PBMCs, unpaired t-tests were performed.
Results
M6903 is an anti-TIM-3 antibody that binds to human TIM-3 with high affinity
M6903 is a fully human anti-TIM-3 monoclonal IgG2 antibody. The kinetics and affinity of M6903 for several TIM-3 orthologues were evaluated by surface plasmon resonance (SPR) technology or enzyme-linked immunosorbent assay (ELISA). While M6903 bound with high affinity to human and non-human primate TIM-3, it lacked affinity for murine or rat TIM-3 (Table 1 and Supplementary Figure 1).
Table 1.
M6903 bound to human and non-human primate TIM-3, but not to mouse or rat TIM-3. Kinetic constants for binding of M6903 to orthologues of TIM-3 were determined by SPR using a Langmuir 1:1 binding model for human, cynomolgus, marmoset, and mouse, and the equilibrium binding constant was derived from the ratio of the kinetic constants. Rat TIM-3 was tested by ELISA. Mouse and rat TIM-3 were measured using pre-optimized versions of the antibodies with identical variable region sequence and IgG1 allotype. “No binding” indicates values were not above background. Constants shown are the average of two measurements.
| Species | ka (M−1 s−1) |
kd (s−1) |
KD (nM) |
|---|---|---|---|
| Human | 1.9*105 | 4.3*10−4 | 2.3 |
| Cynomolgus | 1.2*105 | 1.4*10−3 | 11.8 |
| Marmoset | 1.8*105 | 2.0*10−3 | 10.8 |
| Mouse | No binding | ||
| Rat | No binding | ||
M6903 inhibits TIM-3 binding to Gal-9, PtdSer, and CEACAM1
To determine the ability of M6903 to block the interaction of TIM-3 with its ligands, ELISA and flow cytometry analyses were utilized. In a competition-based ELISA, M6903, but not the isotype control antibody, partially inhibited huTIM-3-biotin binding to recombinant human Gal-9 (rhGal-9) in a concentration-dependent manner (Figure 1a), with an IC50 of 7.46 ± 0.052 nM. Calculating the percent blocking revealed that M6903 blocked up to 55% of the TIM-3/Gal-9 binding signal that was obtained in the absence of antibody.
Figure 1.
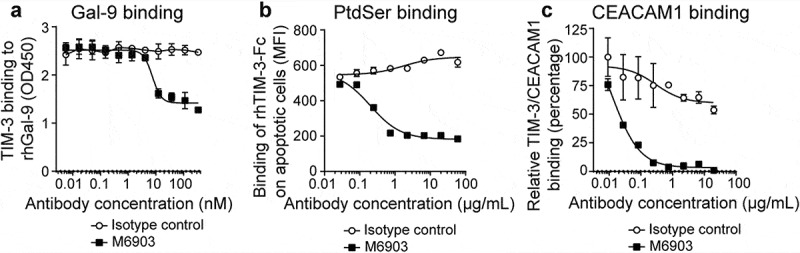
M6903 inhibited TIM-3 binding to Gal-9, PtdSer, and CEACAM1. a) Binding of huTIM-3-Fc biotin (0.5 μg/mL) to plate-bound Gal-9 (2 μg/mL) was evaluated via ELISA after pre-incubation with serial dilutions of M6903 or isotype control (nM). b) Staurosporine (2 μg/mL, 18 hrs) induced apoptosis in Jurkat cells, leading to exposure of surface PtdSer. Apoptotic Jurkat cells were then incubated with rhTIM-3-Fc AF647 pre-incubated with various concentrations of M6903 or isotype control (μg/mL), and the binding of rhTIM-3-Fc AF647 on the Jurkat cells was measured by flow cytometry (MFI). c) Binding of recombinant His-tagged CEACAM1 to plate-bound huTIM-3-Fc (0.5 μg/well) was evaluated via ELISA after pre-incubation with serial dilutions of M6903 or isotype control (μg/mL). a-c) Non-linear best fit lines were generated for all plots using a Sigmoid dose-response equation and mean and standard deviation (SD) are presented.
In a flow cytometry-based binding assay, apoptotic Jurkat cells were used as a source for PtdSer, as the induction of Jurkat cell apoptosis by Staurosporine led to PtdSer exposure on the cell membrane of these cells. Binding of TIM-3 with PtdSer was then measured by incubating apoptotic Jurkat cells with fluorescence-conjugated recombinant human TIM-3-Fc (rhTIM-3 AF647) and analyzing the median fluorescence intensity (MFI). Pre-incubation of rhTIM-3 AF647 with M6903 led to reduced binding of rhTIM-3 AF647 to apoptotic Jurkat cells, whereas pre-incubation with an isotype control had no effect on rhTIM-3 AF647 binding (Figure 1b). Therefore, M6903 was able to efficiently block the interaction between TIM-3 and PtdSer in a dose-dependent manner, with an IC50 of 4.44 ± 3.12 nM (0.666 ± 0.467 μg/mL).
Finally, in an ELISA binding assay, binding of TIM-3 with CEACAM1 was measured by incubating soluble recombinant His-tagged CEACAM1 protein with plate-bound recombinant human TIM-3-Fc (rhTIM-3-Fc). Pre-incubation of plate-bound rhTIM-3 with M6903 reduced binding of rhTIM-3-Fc to His-tagged CEACAM1 in a dose-dependent manner, with an IC50 of 0.353 ± 0.383 nM (0.053 ± 0.057 μg/mL), whereas pre-incubation with an isotype control had no effect on the binding (Figure 1c). We were unable to experimentally confirm the binding of HMGB1 with TIM-3 and therefore were unable to directly assess whether M6903 could block this interaction. Taken together, these in vitro assays demonstrate that M6903 can efficiently block the interaction of TIM-3 with three of its ligands.
The crystal structure of M6903 bound to TIM-3 explains the affinity and specificity of binding
To gain a greater understanding of how M6903 binds to TIM-3 and its mechanism of action, we solved the crystal structure of the Fab form of M6903 bound to human TIM-3 (Figure 2, Supplementary Table 1). A total of 925 Å2 on the surface of TIM-3 is buried in the interaction with M6903 (yellow surface in Figure 2a). A close examination of the binding interface reveals that proline 59 (P59) and phenylalanine 61 (F61) form extensive van der Waals interactions with the complementarity determining regions (CDRs) of M6903 (Figure 2b); indeed, mutation to alanine reveals that these two residues are hotspots for binding (Supplementary Table 2). Glutamic acid 62 (E62) is a third hotspot (Supplementary Table 2) that forms a hydrogen bond to the backbone of M6903 (Figure 2b). The total binding free energy contributed by these hotspot residues explains the high affinity binding to human TIM-3 that is observed by SPR analysis (Table 1).
Figure 2.
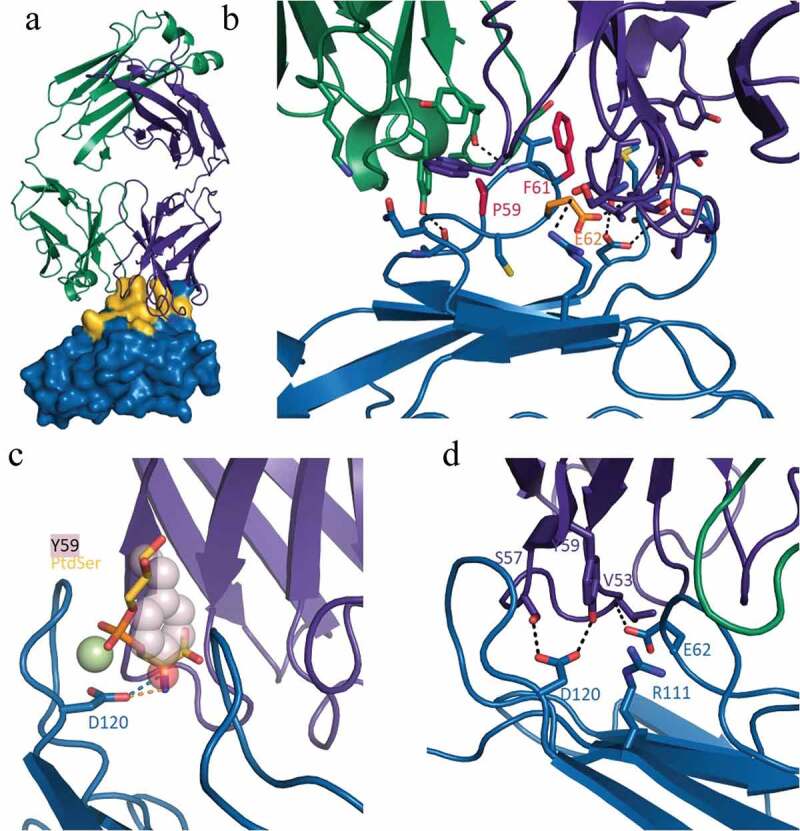
Crystal structure of human TIM-3 in complex with M6903. X-ray diffraction data were collected using cryogenic conditions, and the structure of the Fab complexed with TIM-3 was solved by molecular replacement. a) Overview of the Fab portion of M6903 (heavy chain in purple, light chain in green cartoon representation) bound to TIM-3 shown as a surface representation (blue). Extensive contacts made on TIM-3 are highlighted in yellow. The majority of the contact occurs with the heavy chain and the third complementarity determining region of the light chain (CDR-L3) of M6903. b) The epitope hotspot residues of TIM-3 are highlighted in red (most important, P59 and F61) and orange (E62). The residues form extensive hydrophobic and electrostatic interactions to M6903. c) The polar head group of PtdSer (yellow and orange sticks) and the coordinating calcium ion (light green) have been modeled into the structure of M6903 bound TIM-3 by superposition with the structure of murine TIM-3 (3KAA27). The binding site of PtdSer coincides with the placement of Y59 (pink spheres) of the heavy chain from M6903. Hydrogen bonds from D120 on TIM-3 to PtdSer or M6903, respectively, are shown in orange and blue. d) The polar interactions of M6903 with the CEACAM1 binding residues of TIM-3 are shown with dashed lines.
Our structure explains the pattern of orthologue cross-reactivity observed in SPR analysis (Table 1). The three hotspot residues that are responsible for high affinity binding to human TIM-3 are conserved in cynomolgus monkey and marmoset TIM-3 sequences (Supplementary Figure 2), explaining the cross-reactivity. In contrast, the F61 is replaced by serine or leucine, respectively, in mouse and rat (Supplementary Figure 2), where an alanine mutation was shown to result in a large loss of binding energy. Furthermore, in mouse, V60 is replaced by a larger tryptophan in mouse, which would sterically prevent binding of M6903. Thus, the crystal structure and hotspot mapping of the epitope fully explains the species selectivity of M6903.
The structure of M6903 bound to TIM-3 also helps explain its mechanism of action and competition with ligands. PtdSer was shown to bind a metal ion-dependent ligand binding site on TIM-3;27,28 in our structure, tyrosine 59 of M6903 extends into this site and occludes the cavity that normally binds the PtdSer head group (Figure 2c), thereby explaining the competition observed biochemically (Figure 1b). While no high-resolution structure of the CEACAM1/TIM-3 complex is available, Huang et al. (2015) demonstrated through mutational analysis that residues E62, R111, and D120 contribute significantly to that interaction.7 Our structure reveals that these residues interact closely with the heavy chain of M6903, and D120 forms direct hydrogen bonds to the paratope (Figure 2d). This clearly demonstrates that M6903 binding prevents CEACAM1 binding by direct competition for overlapping binding sites. Finally, it has been reported that HMGB1 binds TIM-3 at residue 62.8 Although we were unable to experimentally confirm HMGB1 binding to TIM-3 and therefore unable to test for the competition directly, based on Chiba et al.’s assertion that residue 62 is a hotspot for HMGB1 binding8 and our observation that residue 62 is a hotspot for M6903 binding, we would expect M6903 to displace HMGB1.
M6903 enhances T cell activation in vitro as a monotherapy or in combination with bintrafusp alfa
To evaluate the impact of M6903 on T cell activation, M6903 was tested in multiple in vitro assays. The effect of M6903 was first assessed in a primary T cell assay that specifically measures CEF (human cytomegalovirus [C], Epstein-Barr virus [E], and influenza virus [F]) viral antigen-specific T cell recall responses in human peripheral blood mononuclear cells (PBMCs) from healthy volunteer donors who were likely previously infected with at least one of these three common viruses. CEF assays demonstrated that M6903 monotherapy significantly increased IFN-γ production relative to isotype control, with an EC50 of 6.7 ± 8.7 nM (1 ± 1.3 μg/mL) (Figure 3a). The combination of M6903 with bintrafusp alfa, but not anti-PD-L1, further enhanced IFN-γ production relative to the combination of M6903 and isotype control (Figure 3b), suggesting that bintrafusp alfa further enhances T cell activation, consistent with previous observations.23
Figure 3.
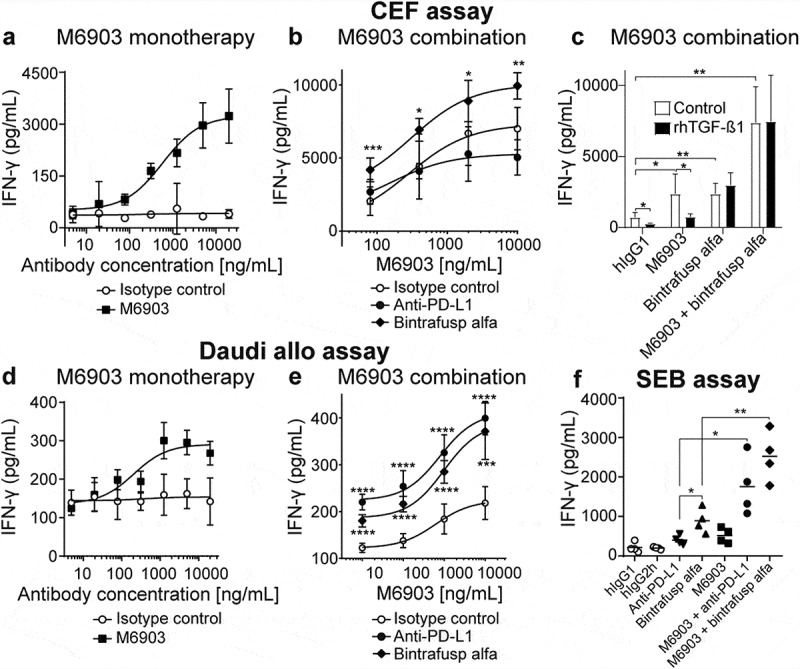
M6903 increased T cell activation as a monotherapy or in combination with bintrafusp alfa in three different in vitro assays. a-c) CEF assay – PBMCs were treated with 40 μg/mL CEF viral peptide pool and cultured (a) in the presence of serial dilutions of M6903 or isotype control, (b) in the presence of serial dilutions of M6903 combined with either 10 μg/mL isotype control, anti-PD-L1, or bintrafusp alfa, or (c) with 2 ng/mL rhTGF-β1 or without rhTGF-β1 (termed ‘control’) in the presence of hIgG1 isotype control (10 μg/mL), bintrafusp alfa (10 μg/mL), or a combination of M6903 and bintrafusp alfa. IFN-γ levels in the supernatant were measured via ELISA after 4, 6, or 4 days, respectively. (a-b) Non-linear regression analysis was performed and mean and SD are presented; results were calculated from multiple experiments. (b) *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 denote significant differences between bintrafusp alfa and isotype control groups. (c) T-tests were performed and mean and SD are presented. *p ≤ 0.05, **p ≤ 0.01 denotes significant difference between treatment groups. d-e) Daudi allogenic one-way MLR assay – irradiated Daudi cells and human T cells were co-cultured with (d) serial dilutions of M6903 or isotype control or (e) serial dilutions of M6903 combined with either 10 μg/mL isotype control, anti-PD-L1, or bintrafusp alfa. IFN-γ levels in the supernatant were measured via ELISA. Non-linear regression analysis was performed and mean and SD are presented; results were calculated from multiple experiments. (e) ***p ≤ 0.001 and ****p ≤ 0.0001 denote significant differences between anti-PD-L1 or bintrafusp alfa and isotype control groups. (f) SEB assay – Human PBMCs were treated with 100 ng/mL SEB in the presence of 10 μg/mL M6903 or isotype control alone or in combination with anti-PD-L1 or bintrafusp alfa for 9 days; the cells were then re-stimulated with SEB and the same antibodies. After 2 days, IFN-γ levels in the supernatant were measured via ELISA. T-tests were performed with Welch’s correction, and mean and SD are presented. *p ≤ 0.05, **p ≤ 0.01 denotes significant difference between treatment groups.
Certain assays have shown that targeting PD-(L)-1 can enhance T cell activation induced by TIM-3 blockade.9,16,29–31 However, since we did not observe enhanced IFN-γ production with M6903 and anti-PD-L1 combination therapy, the CEF assay may not be the best indicator of PD-(L)-1 blockade-induced IFN-γ production, and TGF-β may contribute significantly in such an assay. In order to confirm the contribution of TGF-β, recombinant human TGF-β1 (rhTGF-β1) was added in the CEF assay and its effect on IFN-γ production and M6903 activity was assessed (Figure 3c). The addition of rhTGF-β1 significantly reduced IFN-γ production in hIgG1-treated PBMCs (p = .0112). Although M6903 monotherapy significantly increased IFN-γ relative to isotype control (p = .0314), the addition of rhTGF-β1 to M6903 treatment reverted IFN-γ production back to baseline levels (p = .0181). Bintrafusp alfa monotherapy also increased levels of IFN-γ relative to isotype control (p = .0020), but these levels were unaffected by the addition of rhTGF-β1 (p > .05) (Figure 3c). Indeed, bintrafusp alfa and M6903 combination therapy significantly increased IFN-γ production relative to isotype control (p = .0004) and the rhTGF-β1 did not reduce these levels (p > .05), suggesting that bintrafusp alfa was able to revert the suppressive effect that rhTGF-β1 had on M6903 treatment. Therefore, the effect of bintrafusp alfa in this CEF assay may be primarily due to TGF-β sequestration.
In a Daudi Allo assay, IFN-γ production was evaluated after human allogenic reactive T cells were co-cultured with freshly irradiated Daudi cells and treated with M6903 antibody or isotype control. M6903 dose-dependently enhanced IFN-γ production in these cells compared to the isotype control, with an EC50 = 0.77 ± 0.78 nM (116 ± 117 ng/mL) (Figure 3d). Combination with bintrafusp alfa significantly enhanced the effect of M6903 on IFN-γ production, and thus T cell activation (Figure 3e). The combination with anti-PD-L1 significantly enhanced the effect of M6903 on IFN-γ production in a manor comparable to bintrafusp alfa (Figure 3e), suggesting TGF-β may not contribute significantly in such an assay.
Finally, IFN-γ production was measured in an SEB assay in which human PBMCs were activated by exposure to superantigen Staphylococcal enterotoxin B (SEB), which activates CD4+ T cells nonspecifically via cross-linking T cell receptor (TCR) and MHC class II molecules. M6903 and bintrafusp alfa were incubated with SEB either as monotherapies or in combination. Although bintrafusp alfa and M6903 monotherapies were able to increase IFN-γ production in SEB-stimulated T cells relative to human IgG controls (p = .0154 and p = .0440), combination of M6903 and bintrafusp alfa significantly increased IFN-γ production relative to bintrafusp alfa (p = .0078) or M6903 (p = .0054) monotherapies (Figure 3f). The combination of M6903 with anti-PD-L1 significantly increased IFN-γ production relative to anti-PD-L1 (p = .0340) or M6903 (p = .0403) monotherapies (Figure 3f). However, bintrafusp alfa monotherapy further enhanced IFN-γ production relative to anti-PD-L1 monotherapy (p = .0434), suggesting that the ability of bintrafusp alfa to sequester TGF-β and its ability to saturate PD-L1 both contribute to increased T cell activation in this assay.
M6903 enhances anti-tumor activity of bintrafusp alfa in hu-TIM-3 KI mice
Because M6903 does not cross react with the murine TIM-3, mouse TIM-3 knockout and human TIM-3 knock-in (hu-TIM-3 KI) mice (Beijing Biocytogen, China) were utilized to evaluate the pharmacological activity of M6903 alone or in combination with bintrafusp alfa in a subcutaneous MC38 colorectal carcinoma model in vivo. These mice express the extracellular domain of human TIM-3, while the rest of the receptor is murine. Hu-TIM-3 KI mice, therefore, retain an intact murine immune system, allowing the role of M6903 effector function in anti-tumor activity to be investigated. Given that human TIM-3 binds both human and murine Gal-9 in dose-dependent manners (Supplementary Figure 3), hu-TIM-3 mice also allow the humanized TIM-3 to maintain functional signaling in response to endogenous murine ligands.
Relative to isotype control, M6903 monotherapy did not induce significant tumor growth inhibition (TGI = 10.3%, p = .3213, day 28) (Figure 4). No anti-drug antibodies (ADA) were detected in the plasma of hu-TIM-3 KI mice treated with 4 doses of M6903 given every 3 days (data not shown). In contrast, bintrafusp alfa monotherapy resulted in significant anti-tumor activity in this model (TGI = 44.5%, p < .0001) (Figure 4), consistent with previously reported findings.23 The combination of M6903 with bintrafusp alfa further enhanced anti-tumor activity (TGI = 66.7%) relative to M6903 monotherapy (p < .0001, day 28) or bintrafusp alfa monotherapy (p = .0016, day 28) (Figure 4).
Figure 4.
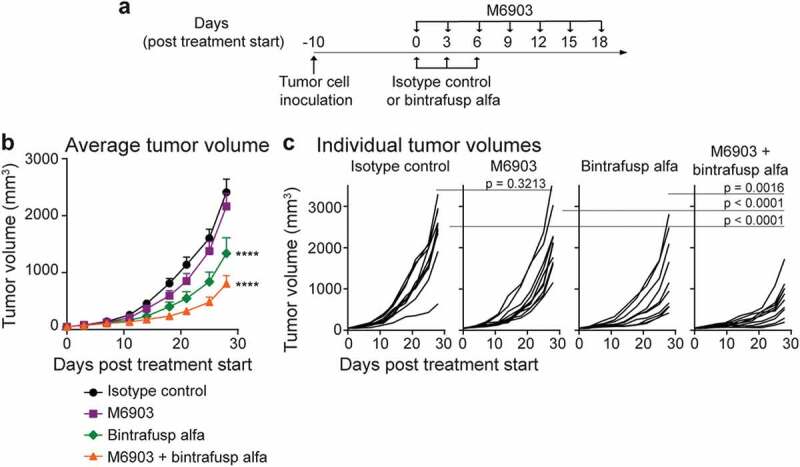
Combination of M6903 with bintrafusp alfa inhibits MC38 tumor growth in hu-TIM-3 KI mice. a-c). Hu-TIM-3 KI mice were inoculated with MC38 (5 x 105 cells) subcutaneously (s.c.) in the flank (day -10) and when average tumor volume reached approximately 50 mm3 (day 0), treated (n = 10 mice/group) with isotype control (20 mg/kg i.v.; day 0, 3, 6), M6903 (10 mg/kg i.p.; day 0, 3, 6, 9, 12, 15, 18), bintrafusp alfa (24 mg/kg i.v.; day 0, 3, 6) or M6903 + bintrafusp alfa. a) Schematic showing the schedule for treatment administrations. Tumor volumes were measured twice weekly and presented as (b) mean ± standard error of the mean (SEM) or (c) individual tumor volumes. P-values were calculated by two-way repeated measures ANOVA with Tukey’s posttest. ****p ≤ 0.0001 denotes significant differences relative isotype control.
M6903 binds to human and non-human primate immune cells
We next investigated the binding of M6903 to human and non-human primate primary immune cells, as these studies may provide a basis for selecting relevant species for additional nonclinical M6903 studies. The ability of M6903 to bind to primary immune cells from human, macaque (cynomolgus and rhesus) monkeys, and marmoset monkeys was evaluated using flow cytometry and biotinylated M6903. In untreated PBMCs, M6903 showed notable binding to human T cells, but not cynomolgus, rhesus, or marmoset monkey T cells (Supplementary Figure 4a). After activation of PBMCs with the superantigen Staphylococcal enterotoxin A (SEA), the increase in M6903 binding to human and marmoset CD4+ T cells was relatively greater than the increase seen in cynomolgus or rhesus monkey CD4+ T cells (Supplementary Figure 4b), and M6903 had relatively greater binding to human, marmoset, and rhesus monkey CD8+ T cells than it did to cynomolgus monkey CD8+ T cells (Supplementary Figure 4c). To rule out a lack of response to SEA stimulation in the T cells of macaque monkeys, activated T cells were stained with biotinylated anti-LAG-3 (lymphocyte-activation gene 3) and analyzed by flow staining. Comparable LAG-3 staining suggested that T cells across species responded well to SEA stimulation (Supplementary Figure 4b-c). Taken together, these results suggest that the binding profile of M6903 on activated human and marmoset T cells is different from that on cynomolgus or rhesus monkey T cells.
To further understand the regulation of TIM-3 expression in activated T cells from human and cynomolgus monkeys, quantitative PCR (q-PCR) was utilized to compare TIM-3 expression at the mRNA level before (day 0) and after (day 6) SEA stimulation. To further confirm the responsiveness to SEA stimulation in the T cells of cynomolgus monkeys, expression of three other key checkpoints, PD-L1, LAG3, and TIGIT (T cell immunoreceptor with Ig and ITIM domains), were compared at the mRNA level before and after SEA stimulation. T cell activation significantly increased TIM-3 expression in human PBMCs (p = .032, day 0 vs. day 6), whereas, there was no significant increase in TIM-3 in cynomolgus monkey PBMCs (p = .0878, day 0 vs. day 6) (Supplementary Figure 5). Indeed, the relative expression of TIM-3 was significantly lower in cynomolgus monkey PMBCs compared with human PBMCs after SEA stimulation (p = .0029), suggesting a difference in the regulation of TIM-3 in human and cynomolgus monkey PBMCs. However, other immune checkpoint genes, PD-L1, LAG3, and TIGIT, were significantly increased with SEA treatment in cynomolgus monkey PBMCs (p = .0086, p = .0011, and p = .0018, respectively, day 0 vs. day 6) (Supplementary Figure 5), confirming that these cells responded well to SEA stimulation. Taken together, these data suggest that there may be a transcriptional difference between TIM-3 expression in human and cynomolgus activated PBMCs, and this may lead to less TIM-3 protein for M6903 to bind with on cynomolgus T cells compared with human or marmoset T cells.
In addition to T cell analysis, binding of M6903 to other immune cells was also tested with flow cytometry analysis using biotinylated M6903. M6903 bound to cynomolgus and rhesus monkey B cells, while no notable binding was seen on human or marmoset monkey B cells (Supplementary Figure 6a). In addition, M6903 bound to human NK cells and monocytes, but not to marmoset, cynomolgus, or rhesus monkey NK cells (Supplementary Figure 6b) or monocytes (Supplementary Figure 6c). These data suggest that M6903 has a differential binding profile across human and non-human primate species. However, considering the central role played by T cells regarding TIM-3 biology, the fact that humans and marmoset monkeys share a relatively similar pattern of M6903 binding on activated T cells may provide a basis for species selection for future M6903 nonclinical studies.
M6903 efficiently saturated TIM-3 in vitro in human whole blood and demonstrated expected pharmacokinetics in marmoset monkeys
The target occupancy (TO) of M6903 to the cell surface of CD14+ monocytes from human whole blood samples was then assessed via flow cytometry analysis. The samples were incubated with serial dilutions of M6903 followed by anti-TIM-3(2E2)-APC, which has been shown to compete with M6903 in binding to TIM-3 on CD14+ monocytes and therefore measured TIM-3 unoccupied by M6903. As expected, TO percentage positively correlated with increased concentrations of M6903 with an average EC50 of 0.74 ± 0.57 nM (111.1 ± 85.6 ng/mL) across 10 donors (Figure 5a). The highest concentrations of M6903 saturated TIM-3 on the cell surfaces.
Figure 5.
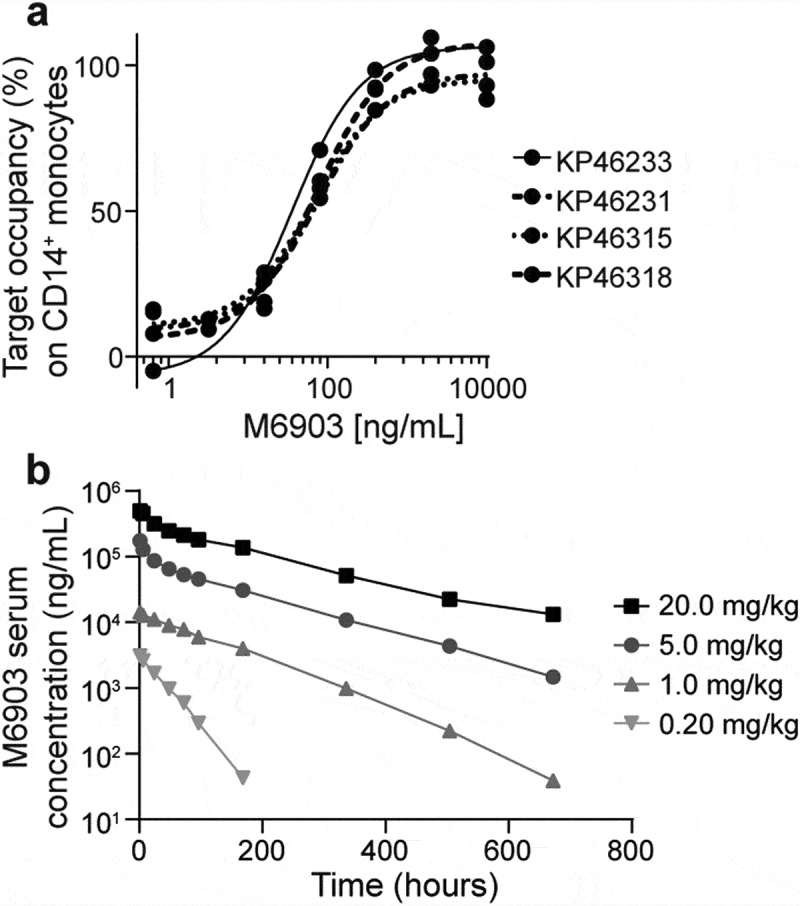
M6903 efficiently saturated TIM-3 in vitro in human whole blood and demonstrated expected pharmacokinetics in marmoset monkeys. a) TO increased with increased concentrations of M6903. Serial dilutions of M6903 were incubated with fresh human whole blood for 1 hour. The unoccupied TIM-3 on CD14+ cells was measured by flow cytometry with anti-TIM-3 (2E2)-APC, which competes with M6903 for TIM-3 binding. The graph shows 4 representative donors (KP46318, KP46315, KP46231 and KP46233) out of the 10 total donors. b) M6903 PK profile in marmoset monkeys in vivo following a single iv administration. The graph shows serum concentrations (ng/mL) of M6903 at 0, 1, 6, 24, 48, 72, 96, 168, 336, 504, and 672-hour time points after dosing with 0.20, 1.0, 5.0, or 20.0 mg/kg doses.
To evaluate the pharmacokinetics (PK) of M6903 in a relevant model, marmoset monkeys (2 males and 2 females per dose group) were dosed with a single iv slow bolus dose of 0.2 mg/kg, 1 mg/kg, 5 mg/kg, or 20 mg/kg of M6903. The additional assessment of toxicity was based on clinical observations, body weights, and pathology evaluations. Following the single iv administration of M6903, in 0.2 mg/kg and 1 mg/kg dose groups, 4 out of 8 animals (50%) showed absorption-like profiles during the first 24 hours. In the rest of the animals, serum concentration declined with monophasic trend. Nonlinear PK were observed with slower elimination at high doses. The individual serum concentrations decreased with a biphasic trend at the dose levels of 5 mg/kg and 20 mg/kg for all animals. A steep distribution phase was followed by a smoother elimination phase from about 24 hours onwards (Figure 5b). No relevant differences in clearance (CL) were observed after increasing the dose from 5 mg/kg to 20 mg/kg (Supplementary Table 3), indicating that the nonlinear component of CL was saturated at doses above 5 mg/kg.
The half-life (t1/2) increased with the dose of M6903 (Supplementary Table 3). M6903 was generally quantifiable in serum up to the last sampling time (672 hours) in animals dosed 1, 5, or 20 mg/kg (Figure 5b). The exposure to M6903 (AUC0-∞) increased more than dose proportionally from 0.2 mg/kg to 20 mg/kg. Target mediated drug disposition (TMDD) is likely the cause of the observed nonlinear PK, which is saturated at the dose levels greater than 5 mg/kg in marmosets. Volume of distribution was close to or slightly higher than plasma volume in all dose groups (Supplementary Table 3), indicating distribution limited to plasma and interstitial space as expected for an antibody.
Discussion
In this study, we characterize M6903, a fully human anti-TIM-3 antibody that binds with high affinity to human TIM-3. Although the functional efficacy of anti-murine and anti-human TIM-3 antibodies has been linked with their ability to interfere with binding to both PtdSer and CEACAM1, but not with binding to Gal-9,12 we demonstrate, through atomic resolution crystal structure and functional assays, that M6903 can disrupt the interaction of TIM-3 with not only PtdSer and CEACAM1 but also Gal-9. M6903 is therefore the first published anti-TIM-3 antibody that blocks TIM-3 binding to its 3 putative ligands and shows confirmed functional activity by inducing T cell activation. T cell activation and anti-tumor activity were further enhanced by the combination of M6903 with the bifunctional fusion protein bintrafusp alfa, suggesting that M6903 and bintrafusp alfa may be promising combination partners in the clinic.
Direct evidence of the binding of M6903 to human TIM-3 is provided by our co-crystal structure, which is the first publication of the structure of human TIM-3 in complex with a therapeutic antibody. A comparison of this structure to available murine (2OYP) and human (6DHB) TIM-3 crystal structures reveals a conserved structure in the IgV domain (RMSD 1.11 Å and 1.19 Å across C-alpha atoms, respectively), especially in the regions of regular secondary structure (core RMSD 0.54 Å and 0.73 Å, respectively) with most variation occurring in the BC loop. The loops that form the PtdSer binding site are structurally conserved across the two murine structures (apo structure 2OYP;32 and PtdSer bound structure 3KAA27), the high resolution apo human structure (6DHB33), as well as our human TIM-3 structure. Also conserved are the strands that form the CEACAM1 binding site. Interestingly, it appears that efficacious anti-TIM-3 antibodies elicit their functional effect through either direct competition with the ligands, as is the case with M6903, RMT3-23, and F38.2E2,12 or through indirect interactions, presumably allosteric inhibition, as is the case with 5D12 and B8.2C12.12
Although the results from Sabatos-Peyton et al. (2017) suggest that blocking PtdSer and CEACAM1 is important for anti-tumor efficacy of anti-TIM-3 antibodies,12 they do not discount a role for Gal-9 in TIM-3 function. Gal-9 contains two tandem carbohydrate recognition domains (CRDs), which are hypothesized to act as a bridge to bring together adjacent TIM-3-ligand complexes.12 This may help explain the important, although possibly indirect, role for Gal-9 in TIM-3 signaling. It has been shown that Gal-9 binding triggers a signaling cascade, which leads to inhibition of T cell signaling4,12 and induces cell death in TIM-3+ Th1 cells.5 Gal-9 is also involved in recruiting TIM-3 to interfaces between lymphocytes and antigen presenting cells (an immunological synapse), which can lead to inhibition of proximal T cell signaling.12,34 In human AML cells, Gal-9 and TIM-3 form a stable complex and it is hypothesized that TIM-3 may act as a trafficker for Gal-9, which subsequently inhibits the anti-tumor activity of cytotoxic T cells and NK cells.13 Therefore, inhibition of the TIM-3/Gal-9 interaction and the autocrine loop they form may increase the anti-tumor activity of NK and T cells. T cell expression of TIM-3 has also been shown to promote CD11b+Gr-1+ MDSCs in a Gal-9-dependent manner.11 Thus, in addition to the critical roles of PtdSer and CEACAM1 in functionally efficacious anti-TIM-3 antibodies as explored by Sabatos-Peyton et al. (2017), Gal-9 also plays an important role in the inhibition of anti-tumor immunity and immune escape,13,35–37 and blocking the interaction of Gal-9 with TIM-3 may be important in improving the anti-tumor efficacy of anti-TIM-3 antibodies.
M6903 and F38.2E2 share overlapping epitopes, yet M6903 is unique among the described antibodies in its demonstrated ability to compete with Gal-9 binding to TIM-3. As the epitope of F38.2E2 has only been resolved to medium resolution using hydrogen-deuterium exchange, it is not possible to precisely define the differences in binding. However, due to the incredible diversity of the antibody response, it is plausible that F38.2E2 binds with a different orientation and to a different set of residues on TIM-3 compared with M6903. The data shown in Figure 1a suggest a model in which M6903 sterically hinders the binding of Gal-9. TIM-3 is known to be heavily glycosylated5 with several predicted N- and O-linked glycosylation sites. It has been shown that Gal-9 interacts with non-glycosylated TIM-3 with nanomolar affinity (Kd = 2.8 × 10−8 M),38 and Gal-9 binding to TIM-3 may be further strengthened by the interaction of Gal-9 with glycosylated TIM-3.35 The N- and C-terminal CRDs of Gal-9 have both been shown to bind with sub- to low-micromolar affinity to glycans.39,40 We propose a model in which Gal-9 binds bivalently to TIM-3 in several different modes, only some of which M6903 can compete with. The tandem CRDs of Gal-9 can bind to two sites in one TIM-3 molecule, as shown in Figure 6, or act as a bridge between TIM-3 molecules to form multimeric complexes.12 Either of these configurations may be sterically hindered by M6903 via proximity to either one of the glycan binding sites or to a protein-protein binding site in the IgV domain (Figure 6). As the affinity of M6903 is several orders of magnitude higher than the affinity of Gal-9 for glycans, this competition is expected to be efficient, even though it may affect only a subset of bound Gal-9 molecules. Although it is still unclear whether Gal-9 needs full interaction with TIM-3, or if certain binding sites are critical for the inhibitory effect, even a partial competition by M6903 may be sufficient to contribute to an anti-tumor immune response. Interestingly, an investigational anti-TIM-3 therapeutic antibody (LY3321367, Eli Lilly & Company) was reported to partially block the TIM-3/GAL-9 interaction and completely blocks the binding of huTIM-3 to PtdSer, but does not block the TIM-3/CEACAM-1 complex.41 It may be that the maximal capability for an anti-TIM-3 monoclonal antibody is only a partial blockade of the TIM-3/Gal-9 interaction, due to the aforementioned multiple binding sites for Gal-9 on TIM-3.
Figure 6.
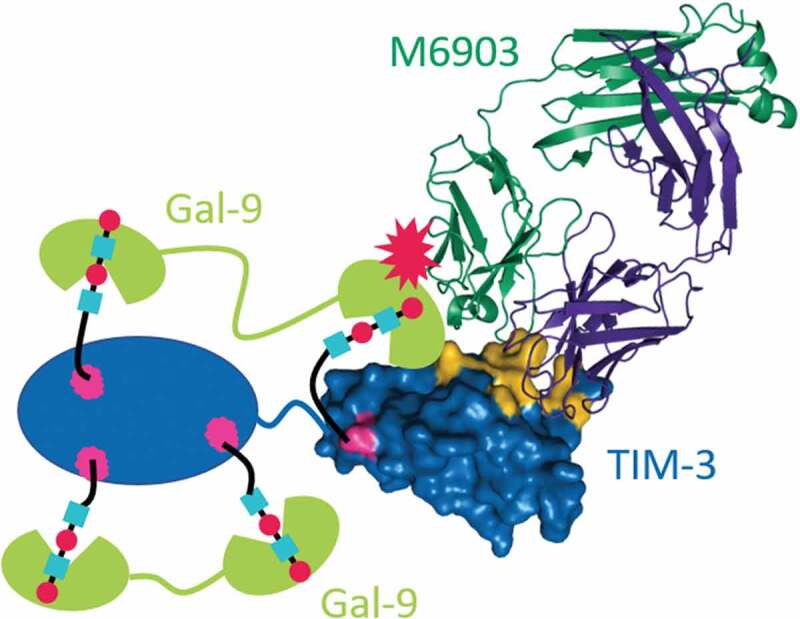
Model for M6903 partial competition with Gal-9 binding. Crystal structure of the Fab of M6903 bound to TIM-3 Ig-V domain (right side of figure) with a depiction of Gal-9 (represented as green cartoons) binding to the glycans on the TIM-3 Ig-V domain and the TIM-3 mucin domain (represented as a blue oval). Glycans are depicted as cyan squares and red circles bound by Gal-9. Coloring as in Fig 2A with Asn-99, as well as three additional predicted sites of glycosylation in the mucin domain colored magenta. Non-glycan mediated interaction to the IgV domain of TIM-3 is also shown schematically, although the exact binding site is unknown.38 Partial competition is represented by a red explosion symbol. The N- or C-terminal CRD of Gal-9 may bind to any of the glycans, and this may explain competition.
Co-expression of TIM-3 and PD-1 by tumor-infiltrating T cells has been reported in different preclinical tumor models and is associated with the exhausted phenotype of the cells.9 In addition, TIM-3 is upregulated in cancer patients resistant to anti-PD-1 treatment,18 and a number of pre-clinical studies have shown substantial anti-tumor activity of TIM-3 blockade in combination with immune checkpoint blockade therapies, including anti-PD-1 and anti-PD-L1.16 These include a study demonstrating significant anti-tumor activity of a surrogate anti-TIM-3 (RMT3-23) antibody alone or in combination with anti-PD-1 therapy in syngeneic tumor models, including the MC38 colon carcinoma model, in WT mice.16 Taken together, these studies suggest that M6903 may be a promising combination partner for anti-PD-1/PD-L1 treatments. The current study extends the anti-tumor potential for TIM-3 blocking with bintrafusp alfa, a first-in-class bifunctional fusion protein that simultaneously targets PD-L1 and TGF-β pathways, inducing superior anti-tumor activity compared with targeting PD-L1 alone.23 In this manuscript, we report that bintrafusp alfa and M6903 combination therapy enhances T cell activation and anti-tumor activity relative to either monotherapy, suggesting that M6903 may be a promising combination partner for anti-PD-1/PD-L1 therapy or bintrafusp alfa in clinic.
The TIM-3 expression pattern and pathway in non-human primates has not yet been fully elucidated. Therefore, understanding the binding of anti-TIM-3 to non-human primate immune cells and identifying similar binding patterns to human immune cells may provide insights into appropriate models for studying TIM-3 biology in human disease. Despite high homology to human HAVCR2 (the gene that encodes TIM-3) and high similarity to the amino acid sequence of human TIM-3, there are little to no antibodies that react with rhesus TIM-3.42 In addition, although rhesus have constitutive expression of TIM-3 on innate immune cells and a higher frequency of B cells that express TIM-3 compared with humans, rhesus have relatively fewer TIM-3 expressing T cells compared with humans.43 Similarly, we report that M6903 binds to monocytes and NK cells in human PBMCs but not to these immune cells in cynomolgus, rhesus, and marmoset monkey PBMCs, suggesting that there are notable differences in TIM-3 expression between human and monkey immune cells. Such contrasts may be due to differences in the immune experience of monocytes and NK cells from human and non-human primates or the regulation of TIM-3 expression in these immune populations. B cells also express TIM-3 in cynomolgus and rhesus monkey PBMCs,43 but not in human or marmoset monkey PBMCs, suggesting possible differences between human and macaques monkeys in the regulation of TIM-3 expression in B cells.
We also report that M6903 binds strongly to both human and marmoset monkey activated T cells, whereas it does not bind as strongly to macaques (cynomolgus and rhesus) monkey T cells. Quantitative PCR of relative mRNA levels also revealed that TIM-3 expression is upregulated in activated human PBMCs, but not in activated cynomolgus monkey PBMCs, whereas PD-L1, LAG3, and TIGIT were upregulated in activated PBMCs from both species. Taken together, it is reasonable to speculate that the regulation of TIM-3, particularly in T cells, might be significantly different between human and macaques monkeys. However, the comparable staining by M6903 on activated human and marmoset T cells confirmed that M6903 is capable of binding marmoset TIM-3 orthologues and suggests that the regulation of TIM-3 in T cells of human and marmoset monkey may be relatively similar or comparable. Considering the central role played by T cells regarding TIM-3 biology, this may have implications in selecting a relevant species for conducting M6903 nonclinical studies.
Following single iv administration of M6903, non-linear PK were observed within the tested dose range of 0.2 to 20 mg/kg in marmosets. No significant differences of CL of M6903 were observed after the dose increase from 5 to 20 mg/kg indicating that the non-linear component of CL was saturated at the doses above 5 mg/kg. The volume of distribution of M6903 was close to or slightly higher than plasma volume of marmosets, as expected for an antibody. The single dose PK results in marmosets and the ex-vivo target occupancy data in human whole blood can be used as a guide to predict the appropriate clinical dose of M6903.
The results reported in this manuscript support the use of M6903 in combination with bintrafusp alfa for the treatment of patients with solid tumors. Although we present the structure of human TIM-3 in complex with M6903 and demonstrate that M6903 blocks the interaction of TIM-3 with 3 of its ligands, further nonclinical and clinical assessment of M6903 is warranted for further elucidation of the mechanism of action of this therapeutic antibody.
Funding Statement
This study was supported by EMD Serono, a biopharmaceutical business of Merck KGaA, Darmstadt, Germany.
Acknowledgments
We thank the study teams at EMD Serono, Billerica, MA, USA, and Merck KGaA, Darmstadt, Germany. We thank Rachel Fontana, Maria Soloviev, Xiubin Gu, Bijan Zakeri, Elisa Bertotti, Jinyang Zhang, Shruti Pratapa, and Smruthi Sahukar for their technical contributions, and Wanping Geng, Angela Lim, Jukka Konola, Alec Gross, Amit Deshpande, Lars Toleikis, and Jacques Moisan for their conceptual contributions and/or supervisory roles in these studies.
Accession Codes
The coordinates for M6903 Fab in complex with TIM-3 have been deposited at the Worldwide Protein Data Bank under the ID 6TXZ.
Disclosure of potential conflicts of interest
D.Z., F.J., R.Z., H.H., V.S., H.W., M.H.J., Q.J., Y.W., M.S., A.P., M.G.D., Q.A., and J.-P.H., are all employees of EMD Serono, which has filed a patent application relating to the subject matter presented in this manuscript. D.Z., X.Z., Q.A., D.N., R.Z., V.S., and C.I. are named as inventors on the pending patent application. The other authors declare that no competing interests exist.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA.. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(536–541). doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 2.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(2175–2186). doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan J, Xu H, Yan Q, Yang S, Duan X, Jiang Z. Tim-3 expression defines regulatory T cells in human tumors. PLoS One. 2013;8(e58006). doi: 10.1371/journal.pone.0058006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(97–111). doi: 10.1111/imr.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(1245–1252). doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 6.Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113(3821–3830). doi: 10.1182/blood-2008-10-185884. [DOI] [PubMed] [Google Scholar]
- 7.Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen B-S, Melum E, Pertel T. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517(386–390). doi: 10.1038/nature13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(832–842). doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(2187–2194). doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth MJ, Anderson AC. TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2(e23849). doi: 10.4161/onci.23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, Cornejo M, Nishi N, Yamauchi A, Quintana FJ. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol. 2010;185(1383–1392). doi: 10.4049/jimmunol.0903275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabatos-Peyton CA, Nevin J, Brock A, Venable JD, Tan DJ, Kassam N, Xu F, Taraszka J, Wesemann L, Pertel T. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology. 2018;7(e1385690). doi: 10.1080/2162402x.2017.1385690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goncalves Silva I, Rüegg L, Gibbs BF, Bardelli M, Fruehwirth A, Varani L, Berger SM, Fasler-Kan E, Sumbayev VV. The immune receptor Tim-3 acts as a trafficker in a Tim-3/galectin-9 autocrine loop in human myeloid leukemia cells. Oncoimmunology. 2016;5(e1195535). doi: 10.1080/2162402x.2016.1195535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, Murphy WJ, Azuma M, Anderson AC, Kuchroo VK. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(4501–4510). doi: 10.1182/blood-2010-10-310425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taghiloo S, Singh G, Mahapatra M, Kumar L, Chandra NC. Upregulation of Galectin-9 and PD-L1 immune checkpoints molecules in patients with chronic lymphocytic leukemia. Asian Pacific J Cancer Preven. 2017;18(2269–2274). doi: 10.22034/apjcp.2017.18.8.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MWL, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71(3540–3551). doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 17.Su H, Xie H, Dai C, Ren Y, She Y, Xu L, Chen D, Xie D, Zhang L, Jiang G. Characterization of TIM-3 expression and its prognostic value in patients with surgically resected lung adenocarcinoma. Lung Cancer. 2018;121(18–24). doi: 10.1016/j.lungcan.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7(10501). doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harding JJ, Patnaik A, Moreno V, Stein M, Jankowska AM, Velez de Mendizabal N, Tina Liu Z, Koneru M, Calvo E. Abstract 12: A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): interim safety, efficacy, and pharmacokinetic findings in advanced cancers. J Clin Oncol. 2019:37. doi: 10.1200/JCO.2019.37.8_suppl.12. [DOI] [Google Scholar]
- 20.Davar D, Boasberg PD, Eroglu Z, Falchook GS, Gainor JF, Hamilton EP, Hecht JR, Luke JJ, Pishvaian M, Ribas A, et al. Abstract O21: A phase 1 study of TSR-022, an anti-TIM-3 monoclonal antibody, in combination with TSR-042 (anti-PD-1) in patients with colorectal cancer and post-PD-1 NSCLC and melanoma. J Immuno Therapy Cancer. 2018;6(Suppl 1):155. doi: 10.1186/s40425-018-0423-x. [DOI] [Google Scholar]
- 21.Curigliano G, Gelderblom H, Mach N, Doi T, Tai W, Forde P, Sarantopoulos J, Bedard P, Lin C-C, Hodi S, et al. Abstract CT183: Phase (Ph) I/II study of MBG453± spartalizumab (PDR001) in patients (pts) with advanced malignancies. Cancer Res. 2019;79(13 Suppl):CT183. doi: 10.1158/1538-7445.AM2019-CT183. [DOI] [Google Scholar]
- 22.Isshiki T, Akiba H, Nakayama M, Harada N, Okumura K, Homma S, Miyake S. Cutting edge: anti-TIM-3 treatment exacerbates pulmonary inflammation and fibrosis in mice. J Immunol. 2017;199(3733–3737). doi: 10.4049/jimmunol.1700059. [DOI] [PubMed] [Google Scholar]
- 23.Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, Yu H, Qin G, Sircar A, Hernández VM. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med. 2018;10(eaan5488). doi: 10.1126/scitranslmed.aan5488. [DOI] [PubMed] [Google Scholar]
- 24.Karsunky HJ, Jiang Y-P. Antibodies that specifically bind to Tim3. United States patent US 8,841,418 B2. 2014.
- 25.Sabatos-Peyton CA, Brannetti B, Harris AS, Huber T, Pietzonka T, Mataraza JM, Blattler WA, Hicklin DJ, Vasquez M, DeKruyff RH, et al. Antibody molecules to tim-3 and uses thereof. United States patent US2015/0218274 A1. 2015.
- 26.Feldman I, Novobrantseva T, Wong J, Phennicie R, Sazinsky S, Sathyanarayanan S. Antibodies that inhibit tim-3: LILRB2Interactions and uses thereof. United States patent US 2016/0200815 A1. 2016.
- 27.DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YLE, Lee -H-H, Karisola P, Pichavant M, Kaplan GG, Umetsu DT. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184(1918–1930). doi: 10.4049/jimmunol.0903059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(172–189). doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, Xue S-A, Behboudi S, Mohammad GH, Pereira SP, Morris EC. Ex vivo PD-L1/PD-1 pathway blockade reverses dysfunction of circulating CEA-Specific T cells in pancreatic cancer patients. Clin Cancer Res. 2017;23(6178–6189). doi: 10.1158/1078-0432.CCR-17-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Zhao X, Li Z, Qi G. Targeting PD-1 and tim-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous dendritic/tumor vaccines. J Immunother (Hagerstown, Md.: 1997). 2016;39(171–180). doi: 10.1097/cji.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 31.Lu X, Yang L, Yao D, Wu X, Li J, Liu X, Deng L, Huang C, Wang Y, Li D. Tumor antigen-specific CD8(+) T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol. 2017;313(43–51). doi: 10.1016/j.cellimm.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, Zencheck WD, Lary JW, Cole JL, Deng H. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. 2007;26(311–321). doi: 10.1016/j.immuni.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 33.Gandhi AK, Kim WM, Sun ZYJ, Huang Y-H, Bonsor DA, Sundberg EJ, Kondo Y, Wagner G, Kuchroo VK, Petsko G. High resolution X-ray and NMR structural study of human T-cell immunoglobulin and mucin domain containing protein-3. Sci Rep. 2018;8(17512). doi: 10.1038/s41598-018-35754-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clayton KL, Haaland MS, Douglas-Vail MB, Mujib S, Chew GM, Ndhlovu LC, Ostrowski MA. T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol. 2014;192(782–791). doi: 10.4049/jimmunol.1302663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goncalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, Varani L, Hussain R, Siligardi G, Ceccone G. The tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine. 2017;22(44–57). doi: 10.1016/j.ebiom.2017.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golden-Mason L, McMahan RH, Strong M, Reisdorph R, Mahaffey S, Palmer BE, Cheng L, Kulesza C, Hirashima M, Niki T. Galectin-9 functionally impairs natural killer cells in humans and mice. J Virol. 2013;87(4835–4845). doi: 10.1128/jvi.01085-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S-I, Niiro H, Yurino A, Miyawaki K, Takenaka K. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17(341–352). doi: 10.1016/j.stem.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 38.Prokhorov A, Gibbs BF, Bardelli M, Rüegg L, Fasler-Kan E, Varani L, Sumbayev VV. The immune receptor Tim-3 mediates activation of PI3 kinase/mTOR and HIF-1 pathways in human myeloid leukaemia cells. Int J Biochem Cell Biol. 2015;59(11–20). doi: 10.1016/j.biocel.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 39.Delaine T, Collins P, MacKinnon A, Sharma G, Stegmayr J, Rajput VK, Mandal S, Cumpstey I, Larumbe A, Salameh BA. Galectin-3-binding glycomimetics that strongly reduce bleomycin-induced lung fibrosis and modulate intracellular glycan recognition. Chembiochem. 2016;17(1759–1770). doi: 10.1002/cbic.201600285. [DOI] [PubMed] [Google Scholar]
- 40.Nielsen MI, Stegmayr J, Grant OC, Yang Z, Nilsson UJ, Boos I, Carlsson MC, Woods RJ, Unverzagt C, Leffler H. Galectin binding to cells and glycoproteins with genetically modified glycosylation reveals galectin-glycan specificities in a natural context. J Biol Chem. 2018;293(20249–20262). doi: 10.1074/jbc.RA118.004636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haidar JN, Antonysamy S, Mathew S, Wu L, Zhang Y, Kearins MC, Shen L, JM Sauder, Schaer D, KE Driscoll, et al. Abstract 2753: the molecular basis of blocking the TIM-3 checkpoint with the LY3321367 mAb in cancer immunotherapy. Cancer Res. 2019;79(2753–2753). doi: 10.1158/1538-7445.am2019-2753. [DOI] [Google Scholar]
- 42.Fujita T, Burwitz BJ, Chew GM, Reed JS, Pathak R, Seger E, Clayton KL, Rini JM, Ostrowski MA, Ishii N. Expansion of dysfunctional Tim-3–expressing effector memory CD8+ T cells during simian immunodeficiency virus infection in rhesus macaques. J Immunol. 2014;193(5576–5583). doi: 10.4049/jimmunol.1400961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amancha PK, Hong JJ, Ansari AA, Villinger F. Up-regulation of Tim-3 on T cells during acute simian immunodeficiency virus infection and on antigen specific responders. AIDS. 2015;29(531–536). doi: 10.1097/QAD.0000000000000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.