

ABSTRACT
When two therapeutic agents are combined in a single formulation, i.e., coformulated, the quality and safety of the individual agents must be preserved. Here we describe an approach to evaluate the quality attributes of two individual monoclonal antibodies (mAbs), designated mAb-A and mAb-B, in coformulation. The mAbs were fractionated from heat-stressed coformulated drug product (DP) by hydrophobic interaction chromatography. Each purified mAb fraction was then compared with mAb-A and mAb-B in their individual formulations from the same drug substance sources used to make the coformulated DP lot, which was subjected to the same stress conditions. Product variants were evaluated and compared by using several analytical tests, including high-performance size exclusion chromatography (HPSEC), reducing and nonreducing gel electrophoresis, ion-exchange chromatography, capillary isoelectric focusing, and peptide mapping with mass spectrometry. Intermolecular interactions in coformulated and photostressed DPs were studied by evaluating aggregates fractionated from coformulated DP by HPSEC. Aggregate fractions of coformulated DP contained dimers, but not coaggregates, of mAb-A or mAb-B. Moreover, extensive assays for higher-order structure and biological interactions confirmed that there was no interaction between the two mAb molecules in the coformulation. These results demonstrate that the two coformulated therapeutic mAbs had the same quality attributes as the individually formulated mAb-A and mAb-B, no new quality attributes were formed, and no physicochemical, intermolecular, or biological interactions occurred between the two components. The approach described here can be used to monitor the product quality of other coformulated antibodies.
KEYWORDS: Quality attributes, monoclonal antibody, combination therapy, coformulated antibodies, hydrophobic interaction chromatography, higher-order structure, intermolecular interactions, degradation pathways, degradation products, physicochemical interactions, forced degradation studies, size variants, charge variants, post translational modification
Introduction
Combination therapies with multiple therapeutic antibodies may improve efficacy without sacrificing safety. Strategies for the development of combination therapies have included coadministration of two or more monoclonal antibodies (mAbs) and coformulation of two or more mAbs into one drug product (DP).1–7 To enable use of coformulated antibodies as a combination therapy, the safety and efficacy of the coformulated product should be similar to those of the individual products when coadministered individually as a combination treatment.8–10 Although the coformulation approach offers several advantages, including reduced medication errors and enhanced convenience for patients, it also increases the complexity of the drug product development process and creates challenges for characterization and control of product quality.11,12 This challenge is exacerbated when the coformulated antibodies have similar physicochemical properties and widely disparate concentrations. Moreover, each of the coformulated antibodies can exhibit heterogeneities in size, charge, and post-translational modifications during manufacturing.9 For these reasons, interactions between component mAbs in a coformulated product need to be characterized and understood.
Even when the individual mAbs in a co-formulated product have completed comprehensive efficacy and safety testing as monotherapy drugs, it is important to conduct stability studies to demonstrate that the risk of interaction between them is low when they are used in combination.8 The characterization of co-formulated antibodies can be challenging due to similarities in the physicochemical properties of combined mAbs,13 especially when concentrations of individual mAbs are significantly different from each other. For example, Cao et al. have discussed the limitations of currently available analytical methods to characterize coformulated antibodies, focusing on characterizing charge variants.14
In this study, we demonstrate an approach to extensively characterize the quality attributes of two mAbs in a coformulated product (referred to herein as Combo). This approach involves combining two mAbs, mAb-A and mAb-B, with mAb-A as the dominant component. Aggregation, fragmentation, post-translational modification (e.g., deamidation and oxidation), and higher order structures were previously identified as critical quality attributes (CQAs) for mAb-A and mAb-B. We conducted studies to identify product quality attributes and to assess the potential interactions of the antibodies when combined. Biological interactions were evaluated by using the potency assays of each component mAb, as well as the physicochemical, intermolecular, and biological interactions between the two components. In addition, forced degradation studies were used to assess the Combo. Forced degradation studies of therapeutic antibodies can provide insight into degradation pathways and the degradation products that may form during a product’s life cycle. The conditions used for forced degradation are chosen based on the likelihood that the products will be exposed to those detrimental conditions during processing, packaging, shipping, and handling. Comprehensive forced degradation studies using heat, pH, light, and chemical oxidation stress were conducted to determine the degradation pathways of the individual mAbs and compared to the Combo. Characterization results of Combo by various analytical techniques were then compared with those of the individual mAb-A and mAb-B and used to assess if there were any interactions or new degradation pathways that could potentially affect safety or efficacy.
Results
Comparison of degradation pathways of Combo, mAb-A, and mAb-B
The degradation pathways of Combo were compared with those of mAb-A and mAb-B when they were subjected to heat, pH, light, and chemical oxidation stress.
Thermal stress
Combo, mAb-A, and mAb-B were subjected to heat stress conditions at 25°C or 40°C for 3 months. Data for samples stressed at 40°C for 3 months are presented throughout this report. Data for samples at 25°C for 3 months had similar profiles and are presented in the Supplementary Material.
The mAb-A and mAb-B were fractionated from heat-stressed Combo by hydrophobic interaction chromatography (HIC), resulting in earlier elution of mAb-A (Figure 1). Cofractionation of the mAb-A fraction into the mAb-B fraction occurred because of the greater concentration of mAb-A over that of mAb-B and a limitation of the HIC separation method, in which late-eluting species tend to demonstrate co-elution of early-eluting species due to a chromatographic tailing effect. Despite this effect, the presence of low-level mAb-A in the mAb-B fraction was not expected to have significant interference in subsequent tests, and if it were to occur, it could have been taken into account. Each fraction was compared with individual mAb-A and mAb-B from the same drug substance sources used to make the Combo, which was subjected to the same stress conditions. Product variants were compared by using several analytical tests, including high-performance size exclusion chromatography (HPSEC), reducing and nonreducing gel electrophoresis, ion-exchange chromatography (IEC), capillary isoelectric focusing (cIEF), and peptide mapping with mass spectrometry.
Figure 1.
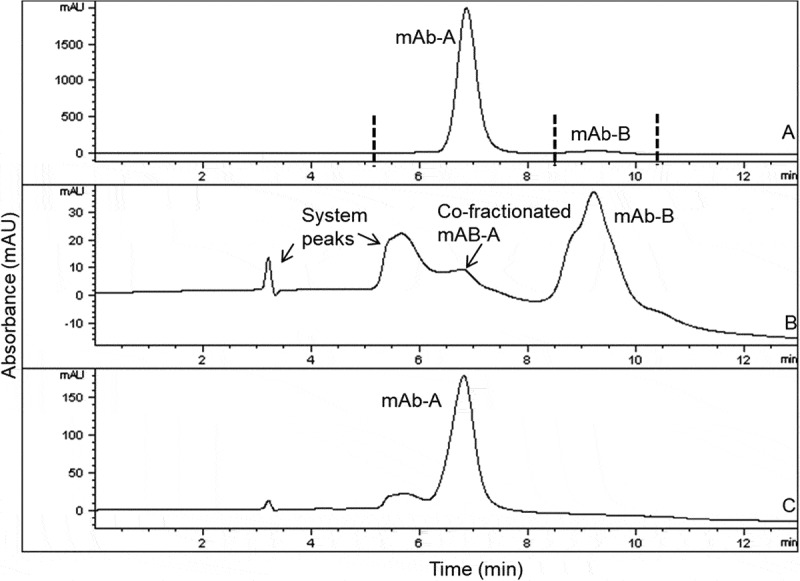
HIC overlays of heat-stressed Combo (profile A) and HIC-fractionated mAb-A (profile C) and mAb-B (profile B). Vertical lines in Profile A indicate fractionation times. Each fraction of HIC-separated Combo (profile A) was reinjected on HIC to verify enrichment (profile B and profile C). Heat-stressed Combo at 40°C for 3 months is shown.
Figure 2 shows overlaid profiles by various assays among size variants of individual mAb-A or mAb-B stressed at 40°C for 3 months (profile A), mAb-A or mAb-B fractionated from heat-stressed Combo under the same conditions (profile B), and Combo (profile C). Analysis by HPSEC (Figure 2(a,b)) showed that the aggregate and fragment peak profiles of mAb-A and mAb-B fractionated from heat-stressed Combo (profile B) were similar to those of heat-stressed mAb-A and mAb-B (profile A), respectively. Based on HPSEC profile comparison of mAb-A and mAb-B in Figure 2, the fragment peak profile of Combo (profile C) was due to mAb-A fragmentation. mAb-B had an insignificant contribution to the fragments in Combo, as it showed no fragment peak even under heat stress conditions.
Figure 2.
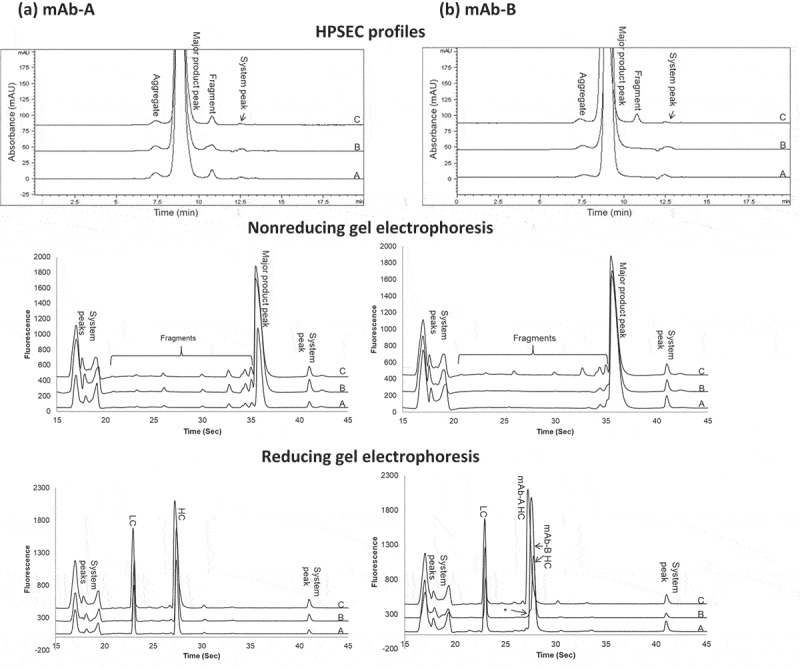
Comparison of size variant profiles for (a) mAb-A and (b) mAb-B. Profiles A, mAb-A, and mAb-B; profiles B, mAb-A, and mAb-B fractionated by HIC from Combo; profiles C, Combo by HPSEC and gel electrophoresis (nonreducing and reducing). HC = heavy chain; LC = light chain.
Analysis by both nonreducing and reducing gel electrophoresis (Figure 2(a,b)) showed that the peak profiles of mAb-A and mAb-B fractionated from heat-stressed Combo (profile B) were similar to those of heat-stressed mAb-A and mAb-B (profile A), respectively. Furthermore, the peak profiles of Combo (profile C) were the same as those detected in mAb-A. The contribution of mAb-B peaks to Combo was insignificant due to the low levels of mAb-B degradation under stress conditions and to the low concentration of mAb-B in Combo. The shoulder peak, indicated with an asterisk to the left of the mAb-B heavy chain in Figure 2(b) (Reducing gel electrophoresis), corresponded to the mAb-A heavy chain caused by the small amount of mAb-A cofractionated with mAb-B from Combo during HIC fractionation (see Figure 1).
The charge variants in Combo were the same as those in mAb-A and mAb-B, with no new species identified in Combo. The IEC and cIEF (Figure 3) charge variant profiles of mAb-A and mAb-B fractionated from Combo (profile B) had the same peaks and a similar profile as individual mAb-A and mAb-B (profile A). The heat-stressed mAb-B IEC prepeaks (Figure 3(b), IEC profile and mAb-B area enlarged) and cIEF acidic peaks (Figure 3(b), cIEF) were slightly higher in mAb-B fractionated from Combo (profile B; 70.9% of total peak area by cIEF) than in mAb-B (profile A; 51.4% of total peak area by cIEF). This result was expected because Combo was formulated at a higher pH than was mAb-B, which led to an increased rate of deamidation in mAb-B that showed at either prepeaks of IEC or at acidic peaks of cIEF. Analysis by peptide mapping with mass spectrometry of stressed materials (25°C and 40°C for 3 months; Table 1) showed that deamidation was the major degradation pathway contributing to the charge variants in Combo and occurred at the same sites as in mAb-A and mAb-B when stressed individually. These comprehensive analytical test results showed that mAb-A and mAb-B fractionated from Combo were similar to, and had the same peaks as, mAb-A and mAb-B.
Figure 3.
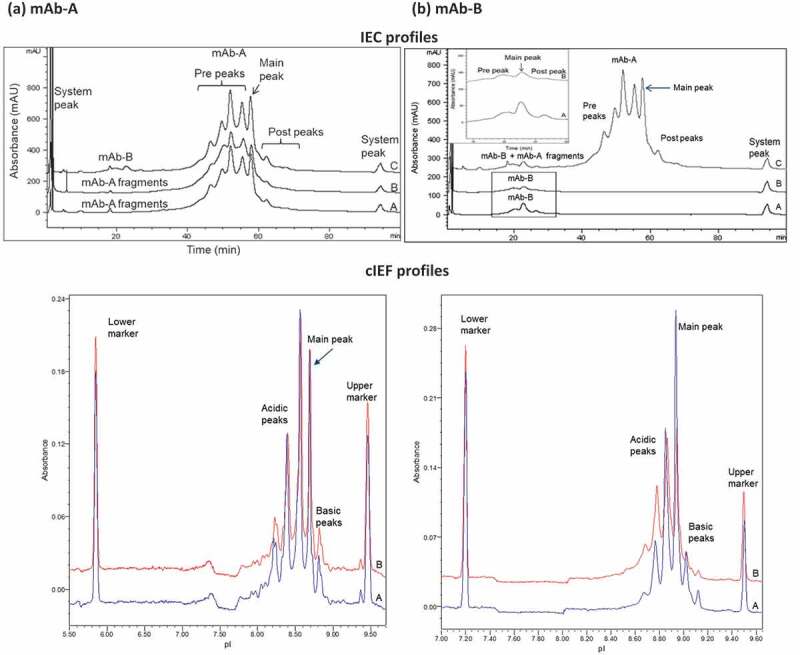
Comparison of charge variant profiles for (a) mAb-A and (b) mAb-B. Profiles A, mAb-A and mAb-B; profiles B, mAb-A and mAb-B fractionated by HIC from COMBO; profiles C, Combo by IEC and cIEF. Rectangular area is magnified in inset of (b) IEC profiles.
Table 1.
Deamidation in Combo, mAb-A and mAb-B fractions by HIC, and mAb-A and mAb-B.
| Deamidation (%) |
||||
|---|---|---|---|---|
| Condition | Sample Description | HC Asn-388, Asn-393 | HC Asn-329 | mAb-B LC Asn-30 |
| 25°C | Combo | 4.3 | 5.3 | 18.6 |
| mAb-A | 3.5 | 5.6 | NA | |
| mAb-A fraction by HIC | 3.8 | 6.6 | NA | |
| mAb-B | 4.2 | ND | 14.3 | |
| mAb-B fraction by HIC | 3.9 | ND | 13.4 | |
| 40°C | Combo | 8.8 | 47.4 | 60.0 |
| mAb-A | 8.7 | 48.3 | NA | |
| mAb-A fraction by HIC | 8.6 | 48.3 | NA | |
| mAb-B | 6.9 | 8.8 | 33.6 | |
| mAb-B fraction by HIC | 9.2 | 10.8 | 45.9 | |
HC = heavy chain; LC = light chain; NA = not applicable; ND = not detected.
When two mAbs are combined, aggregates may be either homogenous (composed of the same mAb) or heterogenous (composed of both mAbs, coaggregates). Whereas homogenous aggregates are commonly observed in single therapeutic mAbs, heterogenous aggregates can form in the coformulated product and become a new product variant that is not present in the individual products. To characterize the composition of the aggregates in Combo, a surface plasmon resonance (SPR) binding assay was developed to detect the presence of coaggregates of mAb-A and mAb-B in Combo. In this method, an anti-mAb-A antibody was immobilized to a sensor chip so that test samples containing mAb-A would bind to the chip. An anti-mAb-B antibody was then used to bind any existing mAb-B present as a coaggregate with mAb-A. A positive control for the test was prepared by using HPSEC to fractionate aggregates of a 1:1 mixture of mAb-A and mAb-B subjected to extreme stress conditions at 50°C for 33 days. No response was observed in the SPR analysis of aggregates fractionated by HPSEC from either nonstressed Combo or heat-stressed Combo at 40°C for 3 months (Figure 4). This result confirmed the absence of coaggregates in Combo, indicating that the aggregates were similar to those in mAb-A and mAb-B.
Figure 4.
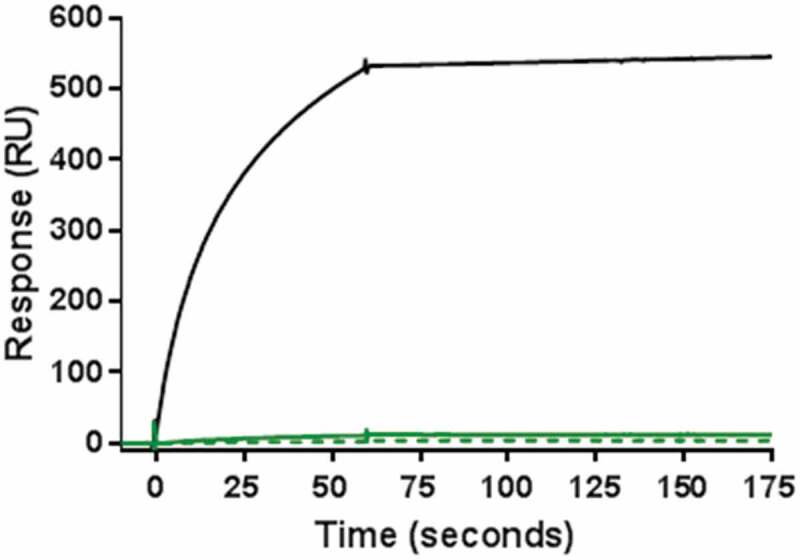
Results of SPR binding assay for detection of coaggregates of mAb-A and mAb-B. Positive control of coaggregates fractionated by HPSEC was obtained from a 1:1 mixture of mAb-A and mAb-B incubated at 50°C for 33 days (black solid line). Shown are aggregate fractions by HPSEC from nonstressed Combo (green solid line) and Combo incubated at 40°C for 3 months (green dotted line).
Effect of pH
To examine the effect of pH on the degradation pathway, Combo and mAb-A and mAb-B were each formulated at pH 5.5 and pH 6.5 with the same excipients used in the Combo formulation buffer and then stored at 25°C and 40°C for 3 months. The product variants of Combo were then compared with those in mAb-A and mAb-B in their respective formulations. The level of major product peaks determined by HPSEC and reducing and nonreducing gel electrophoresis in Combo was similar to that in mAb-A (data not shown). The contribution of mAb-B aggregates and fragments to Combo was insignificant due to the low levels of mAb-B impurities formed under stress conditions and the low level of mAb-B in Combo. Combo had no new charge variants over pH 5.5–6.5. Post-translational modifications (deamidation) in heat-stressed Combo and in mAb-A and mAb-B over a pH range of 5.5–6.5 occurred at the same sites, indicating that there were no new modification sites in Combo (Table 2(a) showed pH 6.5 data).
Table 2.
Observed post-translational modifications of Combo, mAb-A and mAb-B under stress conditions.
| Sample | (a) Deamidation sites under pH stress at 6.5 |
(b) Methionine oxidation sites under chemical oxidation conditions |
(c) Tryptophan oxidation sites under photostress conditions |
|||||
|---|---|---|---|---|---|---|---|---|
| HC Asn-388, Asn-393 | mAb-B LC Asn-30 | HC Met-256 | HC Met-432 | HC Met-362 | mAb-B HC Met-401 | mAb-B HC Trp-52 | mAb-A HC Trp-102 | |
| Combo | 8.8 | 31.0 | 80.0 | 38.7 | 42.2 | 55.2 | 12.2 | 61.2 |
| mAb-A | 8.2 | NA | 79.9 | 39.7 | 45.4 | NA | NA | 63.6 |
| mAb-B | 8.8 | 30.6 | 60.9 | 41.3 | 22.0 | 56.9 | 10.9 | NA |
HC: heavy chain; LC: light chain; NA = not applicable.
Chemical oxidation
Combo and mAb-A and mAb-B were spiked with hydrogen peroxide to a final concentration of 34 ppm and incubated at room temperature for 7 days in the dark to induce protein oxidation. Analysis by peptide mapping demonstrated that methionine oxidation was the predominant degradation pathway for these products when exposed to oxidative stress. The oxidation sites detected in Combo (Met-256, Met-362, Met-401, and Met-432) were the same as in mAb-A and mAb-B (Table 2(b)). No new oxidation sites were identified in Combo.
Photostress
Combo and mAb-A and mAb-B were exposed to 1.2 × 106 Lux hours of cool white light and more than 200 watt hours/m2 of UV light. In a subsequent comparison, product variants were evaluated by using several analytical tests, including HPSEC, nonreducing, and reducing gel electrophoresis, and peptide mapping with mass spectrometry (data not shown). The results showed that Combo profiles were similar to and had the same peaks as the mAb-A and mAb-B. Aggregates analyzed by HPSEC and fragments evaluated by reducing and nonreducing gel electrophoresis were predominantly from degradation of mAb-A in Combo. Formation of dimer species was the major degradation pathway for Combo under photostress conditions. Oxidation was another major degradation pathway and occurred at the same sites in Combo as in mAb-A and mAb-B. Oxidation of two tryptophan sites in Combo was compared with those in mAb-A and mAb-B (Table 2(c)). The product-related impurities and charge variants in Combo were the same as those in mAb-A and mAb-B, and no new product-related impurities were formed in Combo.
Characterization of molecular interactions in Combo
For elucidation of molecular interactions, Combo and mAb-A and mAb-B were characterized by spectroscopic and calorimetric tests, including differential scanning calorimetry (DSC), far ultraviolet (UV) circular dichroism (CD), near UV CD, and Fourier-transform infrared spectroscopy (FTIR). The experimentally determined Combo profiles were compared with the Combo reconstructed profiles that were expected for a noninteracting system, calculated from the experimental profiles of mAb-A and mAb-B in Combo formulation buffer. The calculation is shown in the following equation:
where a and b are the mixing ratios of mAb-A and mAb-B, respectively, in Combo.
Combo had the expected combined contributions of the individual mAb-A and mAb-B secondary and tertiary structures when tested by DSC, far UV CD, near UV CD, and FTIR (Figure 5). The reconstructed profiles of Combo, calculated for noninteracting molecules, was consistent with the experimental profiles. The Combo profiles were also similar to that of mAb-A, the predominant component in Combo. Characterization studies assessing the secondary and tertiary structures of Combo showed that there was no interaction between mAb-A and mAb-B and that Combo contained the expected combined contributions of the individual mAb-A and mAb-B higher-order structures.
Figure 5.
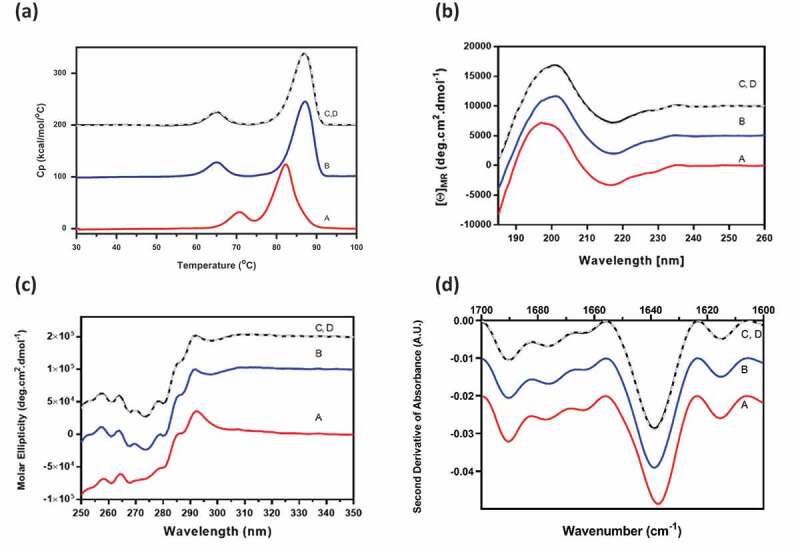
Higher-order structure profiles of experimental mAb-B and mAb-A, Combo, and reconstructed Combo by (a) DSC, (b) far UV CD, (c) near UV CD, and (d) FTIR. Profile A, mAb-B (red); profile B, mAb-A (blue); profile C, experimental Combo (black); profile D, reconstructed Combo (gray).
Intermolecular interactions of Combo
For probing the presence of intermolecular interactions, Combo and mAb-A and mAb-B were each dialyzed into the Combo formulation buffer. The intermolecular interactions between mAb-A and mAb-B in Combo were assessed by using several analytical tests, including analytical ultracentrifugation (AUC), isothermal titration calorimetry (ITC), and dynamic light scattering (DLS).
Analytical ultracentrifugation
Reversible self-association of proteins results in changes in sedimentation profiles due to association of the monomers into higher-order species as the concentration is increased.15–17 Similarly, interaction (binding) between two different antibodies is also expected to result in changes in sedimentation profiles as a function of total concentration or ratio of species. To assess interactions in Combo, we compared the AUC sedimentation profiles of mAb-A, Combo (a:b mixtures of mAb-A and mAb-B), and a positive control of intermolecular interactions (a:b mixtures of mAb-A and anti-mAb-A antibody) (Figure 6(a)). AUC profiles of mAb-A and Combo were similar, indicating that there were no interactions between mAb-A and mAb-B in Combo. In contrast, the addition of anti-mAb-A to mAb-A resulted in a new peak between sedimentation coefficients 6 S and 8 S, corresponding to the interactions of mAb-A and anti-mAb-A antibody. The positive control demonstrated that this method could detect intermolecular interactions at the nominal a:b ratio of interacting molecules and confirmed the absence of interactions in Combo.
Figure 6.
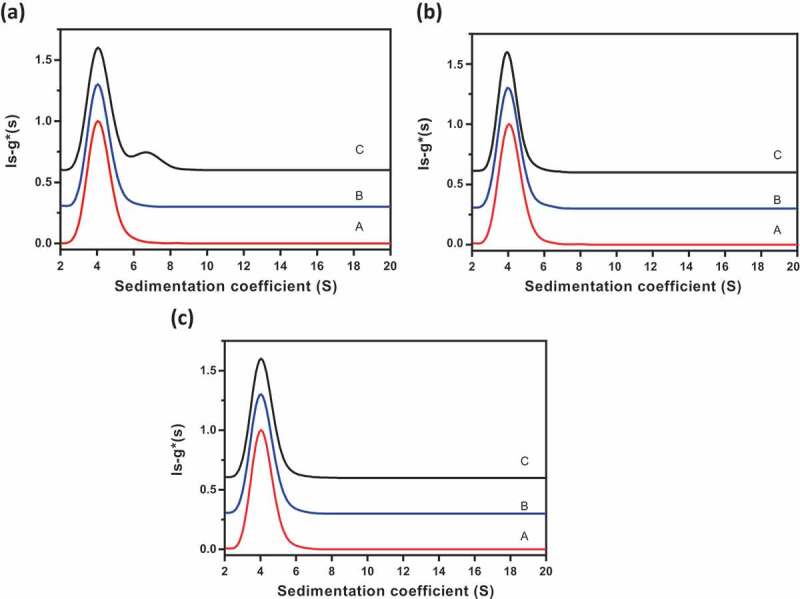
AUC profiles for evaluating intermolecular interactions of mAb-A and mAb-B in Combo. (a) Comparison of AUC profiles of mAb-A, Combo, and a positive control of intermolecular interactions (a:b mixtures of mAb-A and anti-mAb-A antibody). Profile A, mAb-A (red); profile B, Combo (blue); profile C, positive control (a:b mixtures of mAb-A and anti-mAb-A antibody; black). (b) Impact of Combo concentration on ls-g*(s) sedimentation profile. Profile A, Combo at 0.2 mg/mL (red); profile B, Combo at 0.5 mg/mL (blue); profile C, Combo at 2 mg/mL (black). (c) Impact of mAb-A:mAb-B ratio on ls-g*(s) sedimentation profile. Profile A, a:b mixture of mAb-A and mAb-B (red); profile B, 4:1 mixture of mAb-A and mAb-B (blue); profile C, 1:1 mixture of mAb-A and mAb-B (black).
Additional studies were performed to assess interactions in Combo as a function of Combo concentration and ratio of mAb-A to mAb-B. The sedimentation profiles were similar for three concentrations of Combo (0.2, 0.5, and 2.0 mg/mL) and for three ratios of mAb-A and mAb-B (a:b, 4:1, and 1:1), respectively (Figure 6(a,c)). These results further demonstrate that there were no interactions between mAb-A and mAb-B in Combo.
Isothermal titration calorimetry
ITC can provide enthalpy change data due to bimolecular interactions. The stronger the reaction between two molecules, the larger the enthalpy change. Figure 7 shows the real-time calorimetric titration curves of mAb-A and mAb-B (upper panel) and the integrated enthalpy change for each injection of the corresponding titration (lower panel). Titration of mAb-B in mAb-A resulted in very small positive but uniform peaks due to the heat of dilution. This indicates that there was no significant change in enthalpy when more mAb-B was titrated in mAb-A. A positive protein–protein interaction would result in larger and stoichiometric heat changes.18 Taken together, the ITC data suggest that there was no stoichiometric and/or nonspecific binding interaction between mAb-A and mAb-B.
Figure 7.
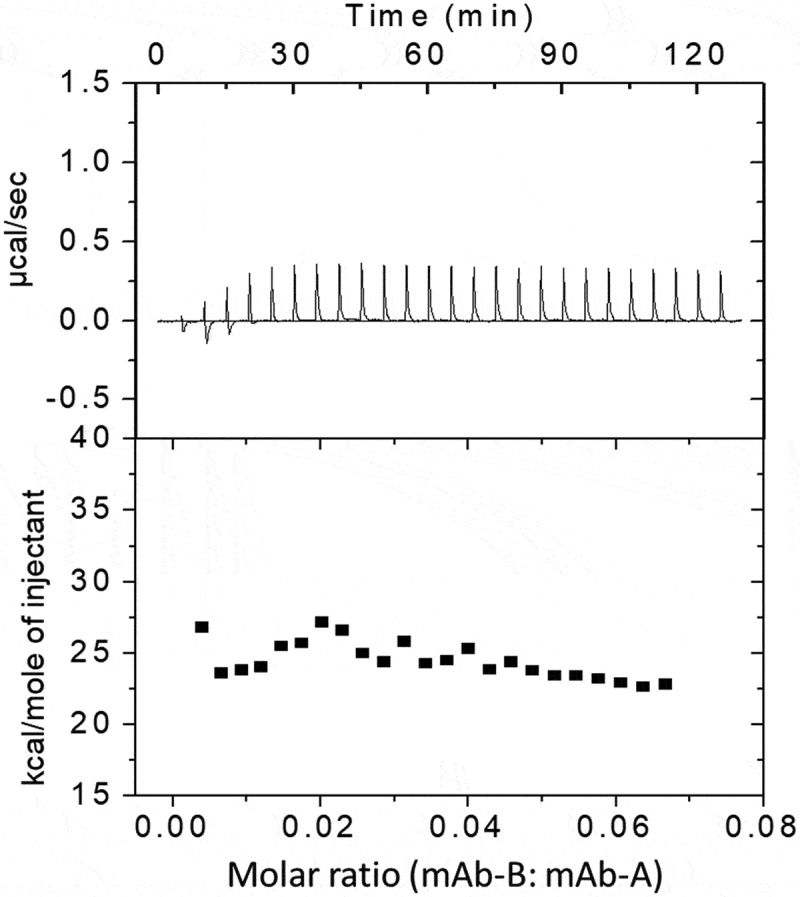
ITC profiles by titration of mAb-B in mAb-A
Dynamic light scattering
Nonspecific and noncovalent interactions (electrostatic, ionic, or hydrophobic) between two protein molecules can result in reversible association, especially at high concentrations. This may not result in significant heat changes in ITC experiments. DLS can detect such protein-protein associations by measuring the hydrodynamic size of the molecules.16,17 When mAb-A and mAb-B existed as monomers, the theoretical average diameter in Combo was calculated to be 13.4 nm (Table 3). Conversely, if mAb-A and mAb-B associate with each other, the theoretical diameter of the Combo could range from 16.9 nm (minimum) to 26.9 nm (maximum). Experimentally determined average hydrodynamic diameters by DLS for mAb-A, mAb-B, and Combo are shown in Table 3. The diameter for Combo was equivalent to the theoretical average monomer diameter (13.4 nm), indicating that there was no protein-protein association. Furthermore, as the concentration of the proteins increased from 1 to 40 mg/ml, there was no significant change in diameter, indicating that there was no self-association propensity between mAb-A:mAb-A, mAb-B:mAb-B, or mAb-A:mAb-B, even at higher concentrations (Figure 8). An intermolecular interaction study showed that there were no interactions between mAb-A and mAb-B in Combo.
Table 3.
Comparison of hydrodynamic diameters for monomer and dimers in Combo.
| Theoretical average diameters (nm) |
Experimental average diameters (nm) |
||||
|---|---|---|---|---|---|
| Combo as monomer | Combo as dimer |
mAb-A | mAb-B | Combo | |
| Minimum | Maximum | ||||
| D3 = | D3 = D1 + D2 | D1 | D2 | D3 | |
| 13.4 | 16.9 | 26.9 | 13.5 ± 1.1 | 13.4 ± 0.5 | 14.4 ± 1.5 |
Figure 8.
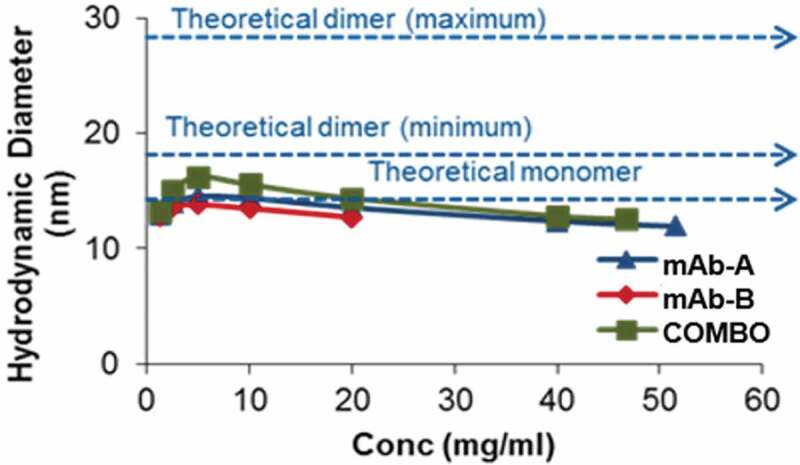
Comparison of average hydrodynamic diameters by DLS with increasing protein concentrations.
Biological interactions
Biological activity, target binding characteristics, and effector functions were evaluated by cell-based potency assays, a binding assay with SPR, and assessment of Fc effector functions with antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity assays. Results of these analyses indicated that the biological properties of mAb-A and mAb-B in Combo were similar to those of mAb-A and mAb-B, respectively (Figure 9). These results indicate that there were no biological interactions between mAb-A and mAb-B in Combo.
Figure 9.
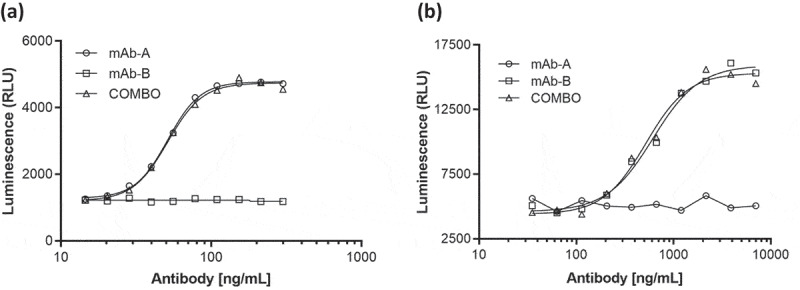
Representative dose–response curves for (a) mAb-A and (b) mAb-B bioassay of Combo and mAb-A and mAb-B.
Discussion
Coformulated antibodies as a combination therapy can have synergistic effects that can improve the efficacy of therapeutics, provided that there are no additional safety concerns as compared with individual mAbs.7 It is therefore important to conduct a full characterization of coformulated antibodies to evaluate physicochemical, intermolecular, and biological interactions. In the ideal case, there would be no interactions between coformulated mAbs that would result in new product variants not present in individual mAbs. If interactions or new product variants are detected then these must be evaluated for their potential impacts on safety and efficacy.
Characterization of coformulated antibodies can be challenging due to similarities in physicochemical properties, especially in combinations in which the concentrations of the component mAbs are significantly different. Systematic characterization is key to identify CQAs to be monitored in coformulated mAbs and to justify the use of platform methods. In this study, coformulated antibodies composed of a:b mixtures of mAb-A and mAb-B, in which the concentration of the mAb-A component was much greater than that of mAb-B, was fully characterized for degradation pathways, intermolecular interactions, and biological interactions. These characterization results were then compared with those of the individual mAb-A and mAb-B. The results showed that Combo had the same critical quality attributes as the individual components, mAb-A and mAb-B, and that no new CQAs were formed. Furthermore, there were no physicochemical, intermolecular, or biological interactions between the two components. These characterization results can be leveraged to establish control strategies and to address the limitations of analytical methods for analysis of coformulated mAbs.
The approaches described in this study for evaluating CQAs, determining higher-order structure, investigating the presence of intermolecular interactions and biological interactions can be employed for other coformulated antibodies. In order for this approach to be successful, the mAbs in coformulated product should be fractionated by a high-resolution method that completely separates the individual components and maintains their properties to ensure that characterization results of fractionated mAbs are representative of the mAbs in the co-formulated product. The HIC methodology used in this study can be employed for other mAbs. Other non-denaturing separation techniques such as ion-exchange chromatography, affinity chromatography, or multi-dimensional separations can also be explored. The characterization methods used for mAb-A, mAb-B and coformulated product are applicable for other IgG combinations.
Materials and methods
Chemicals, recombinant proteins, and combination proteins
mAb-A and mAb-B are full-length IgG1 and IgG2 antibodies produced by AstraZeneca (Gaithersburg, MD). Combo was made by combining mAb-A and mAb-B at various ratios. Trypsin was obtained from Promega (Madison, WI). Dithiothreitol (no-weight format) was obtained from Pierce Protein Biology (Rockford, IL). Urea (OmniPur), water (OmniSolve, high-pressure liquid chromatography [HPLC] and spectrophotometry grade), and acetonitrile (OmniSolve, HPLC, and spectrophotometry grade) were obtained from EMD Serono (Billerica, MA). Iodoacetamide (OneQuant) was obtained from G-Biosciences (St. Louis, MO). Trifluoroacetic acid in flame-sealed, 1-mL ampules was obtained from Sigma-Aldrich (St. Louis, MO).
HIC for fractionation of mAb-A and mAb-b from Combo
An HIC column (7.5 mm × 7.5 cm, 10-µm TSKgel Ether-5PW; Tosoh, Tokyo, Japan) was installed on an Agilent 1200 HPLC system (Agilent, Santa Clara, CA) capable of delivering at least two mobile phases simultaneously and equipped with an automatic fraction collector. Mobile phase A was 1.8 M sodium sulfate and 0.1 M sodium phosphate, pH 7.0, and mobile phase B was 0.1 M sodium phosphate, pH 7.0. Flow rate was 0.75 mL/min, and the column was maintained at ambient temperature. Samples were loaded at 100% mobile phase A and eluted with a linear gradient of mobile phase B between 50% and 100% over 5 min. Mobile phase B was then maintained at 100% for 4 min before the column was regenerated with 100% mobile phase A for 6 min. Fraction collection time was adjusted to ensure minimal contamination of mAb-A in the mAb-B fraction. Buffer exchange was performed at 4°C, using either 20-mL or 500-µL spin columns (30 x 103 molecular weight cutoff) into Combo formulation buffer (without polysorbate 80). Protein concentration was confirmed with a NanoDrop microvolume spectrometer (Thermo Fisher Scientific, Waltham, MA).
High-performance size exclusion chromatography
To compare the size exclusion chromatography profiles of samples, an injection volume was adjusted to load 250 μg onto a TSK-gel G3000SWXL column (7.8 mm × 30 cm; Tosoh) at ambient temperature. The sample was eluted isocratically with a mobile phase composed of 0.1 M sodium phosphate, 0.1 M sodium sulfate, and 0.05% sodium azide, pH 6.8, at a flow rate of 1.0 mL/min. Eluted protein was detected with UV absorbance at 280 nm.
For aggregate fractionation, injection volume was adjusted to load 1000 μg of each sample onto a TSK-gel G3000SWXL column (7.8 mm × 30 cm; Tosoh) at ambient temperature. Collected aggregate fractions were buffer exchanged to Combo formulation buffer (without polysorbate 80) and concentrated.
Nonreducing and reducing gel electrophoresis
Samples were diluted to 4 mg/mL in 1 × phosphate-buffered saline and then diluted 1:1 in either reducing sample buffer containing 6% (vol/vol) of 1 M dithiothreitol or nonreducing sample buffer containing 6% (vol/vol) of 1 M N-ethylmaleimide. Reduced and nonreduced samples were denatured for 5 min and 1 min, respectively, on a heating block at 80°C, and the protein ladder was heated at 100°C for 5 min. After denaturation, samples and protein ladder were spun at 13,200 rpm to cool and collect condensate. Six microliters of each sample and protein ladder were then diluted with 84 μL of ultrapure water. After chip priming, 6 μL of either diluted sample or protein ladder was loaded in wells of a Protein LabChip (PerkinElmer, Waltham, MA), which was loaded with gel dye and destain solutions and then placed in an Agilent 2100 Bioanalyzer and read using Agilent Technologies 2100 Expert software (Agilent).
IEC for charge profiling
IEC was used to measure the charge heterogeneity of mAb-A and mAb-B, mAb-A and mAb-B fractionated from Combo by HIC, and Combo. An Agilent 1200 liquid chromatography system with a binary or quaternary pump (Agilent) was used. Samples were injected onto an analytical ProPac WCX-10 column (4 × 250 mm; Thermo Fisher) connected to a ProPac WCX-10 G guard column (4 × 50 mm; Thermo Fisher) at a column temperature of 25°C. Peaks were eluted in a salt gradient from 30% to 100% (solvent B) over 80 min with mobile phase A composed of 20 mM sodium phosphate, pH 6.0, and mobile phase B composed of 20 mM sodium phosphate and 100 mM sodium chloride, pH 6.0, at a flow rate of 1.0 mL/min. The eluted protein was detected by UV absorbance at 220 nm with a diode array detector to assess charge heterogeneity. Results are reported as percentage of acidic peaks (prepeaks), percentage of main peak, and percentage of basic peaks (postpeaks) of mAb-A and mAb-B.
cIEF for charge profiling
Samples were diluted to 0.3 mg/mL in a master mix containing 0.35% methylcellulose solution; 4% Pharmalyte, pH 3–10; 0.005% pI marker 5.85; and 0.005% pI marker 9.46. The samples were then loaded onto an iCE280 analyzer (Protein Simple, San Jose, CA) and focused at 1500 V for 1 min, followed by 3000 V for 5 min. The protein was detected under UV at 280 nm, and the resulting electrophoresis profiles were analyzed with the use of Empower software (Waters, Milford, MA).
Tryptic peptide mapping with mass spectrometry for post-translational modifications
Peptide mapping was used to verify primary sequences and evaluate post-translational modifications. Samples were diluted to 10 mg/mL in water mixed with a denaturing buffer composed of 1.3 M guanidine; 8 M urea; 130 mM Tris, pH 8.0; and 500 mM dithiothreitol and incubated at 37°C for 30 min. Iodoacetamide was added for alkylation, and the mixture was incubated in the dark at room temperature for 30 min. The reduced and alkylated samples were diluted in 100 mM Tris buffer, pH 7.5, and digested with trypsin at 37°C for 3 h. The digestion was quenched with trifluoroacetic acid before liquid chromatography–mass spectrometry analysis.
SPR binding assay
SPR detection was performed on a Biacore T200 optical biosensor instrument, using a CM5 sensor chip. An anti-mAb-A antibody, prepared in-house, was immobilized by using an amine coupling strategy on two discrete sensor surfaces. One of the surfaces served as a reference surface, and the other served as an experimental surface. Each assay cycle consisted of an injection of size exclusion chromatography–fractionated aggregate of Combo over the experimental surface, followed by an injection of anti-mAb-B antibody, prepared in-house, over reference and experimental surfaces. Reference and experimental sensor surfaces were regenerated at the end of each cycle. Data collected from the reference surface were subtracted from those from the experimental surface to account for any nonspecific binding to anti-mAb-A. For each Combo sample tested, an assay cycle consisting of an injection of sample diluent was included to account for any nonspecific response due to sample diluent.
Dynamic light scattering
DLS was used to determine the molecular size of protein samples by measuring the hydrodynamic radius. DLS experiments were performed with a 384-well plate DynaPro DLS instrument equipped with a 633-nm laser (Wyatt Technology, Goleta, CA). The scattered light was monitored at 173° to the incident beam, and autocorrelation functions were generated by using a digital auto-correlator. mAb-A, mAb-B, and Combo samples were serially diluted from their stock solutions to 1.25 mg/mL, using appropriate formulation buffers, and the samples were filtered with a 0.22-µm filter. Each sample was loaded in triplicate onto a plate and centrifuged at 2000 rpm for 2 min to remove any air bubbles. Data were collected with 10 five-second acquisitions per sample at a temperature of 25°C.
Differential scanning calorimetry
DSC was used to monitor the thermal unfolding transitions of a protein and to evaluate the thermal stability of the protein molecule in a specific formulation. A MicroCal VP-Capillary system (Malvern Panalytical, Westborough, MA) was used to evaluate conformational stability and thermal transitions of mAb-A, mAb-B, and Combo samples. The protein solution of 1 mg/mL was gradually heated over a temperature range of 20–110°C, using a temperature ramp of 90°C/h. The reported DSC thermogram was calculated by subtracting a baseline, using a linear-connect method. Thermal melting transitions were observed as endothermic peaks in the DSC thermogram. Melting temperatures were determined for individual melting transitions.
Isothermal titration calorimetry
ITC was used to measure the thermodynamic properties of protein–protein interactions. The observable signal for ITC was heat evolved (negative peak) or absorbed (positive peak) upon complex formation. ITC experiments were performed on a MicroCal VP-ITC calorimeter (Malvern Panalytical). The sample cell was filled with mAb-A, and the syringe was filled with mAb-B (titrant). The titration schedule included the addition of 10 µL of mAb-B per injection, with 25 injections spaced at 5-min intervals. The titration syringe was stirred at 300 rpm, and the sample cell was maintained at 25°C. Reference titration was carried out by injecting mAb-B into mAb-A buffer alone in the calorimetric cell, and heat of dilution was subtracted from the protein ligand titration data.
Analytical ultracentrifugation
AUC sedimentation velocity was used to measure purity, fragmentation, and aggregation. Species with different sizes were separated on the basis of their sedimentation behavior under a strong centrifugal field. Samples and reference buffer were loaded into 12-mm double-sector cells with Epon centerpieces and sapphire windows, placed into an An50-Ti rotor, and installed into a ProteomeLab XL-I centrifuge set to 20°C (Beckman Coulter, Brea, CA). A rotor speed of 42,000 rpm was used to collect UV scans at 280 nm at a radial resolution of 0.002 cm over a range of 5.9–7.2 cm. Reversible self-association of proteins results in a characteristic shift to higher values in the weight-average sedimentation coefficient distribution as the concentration is increased.15 In a similar manner, new peaks representing complexes can be detected by AUC as a function of concentration and ratio of proteins if they interact to form associated species. The SEDFIT software program (National Institutes of Health, Bethesda, MD) was used to generate ls-g*(s) profiles, from which the concentration dependence of the weight-average apparent sedimentation coefficient was determined.
Far UV and near UV CD
Far UV CD was used to determine protein secondary structure. Samples were diluted with 10 mM phosphate buffer to a working concentration of 0.15 mg/mL before being placed into a 1-mm quartz cuvette (Starna Scientific, Ilford, UK). Far UV CD spectra in the range of 180–260 nm were collected with a J-815 instrument (Jasco, Easton, MD) under optimized conditions (standard sensitivity range of 100 millidegrees, with 0.5-nm data pitch, 20-nm/min scanning rate, and 8-s integration time). Buffer blanks were recorded and subtracted from all sample spectra to correct for instrument, cuvette, and baseline effects. For each sample, two replicates, each consisting of four independent scans, were averaged for the final reported result.
Near UV CD was used to assess protein tertiary structure. Samples were diluted with formulation buffer to a working concentration of 1 mg/mL before being placed into a 1-cm quartz cuvette (Starna Scientific, Ilford, UK). Near-UV CD spectra in the range of 250–350 nm were collected on a J-815 instrument (Jasco) under optimized conditions (standard sensitivity range, 100 millidegrees, with 0.5-nm data pitch, 10-nm/min scanning rate, and 16-s integration time). Buffer blanks were recorded and subtracted from all sample spectra to correct for instrument, cuvette, and baseline effects. For each sample, two replicates, each consisting of four independent scans, were averaged for the final reported result.
Fourier-transform infrared spectroscopy
FTIR spectroscopy was used to determine protein secondary structure motifs, including β-sheets, α-helices, and random coils. FTIR spectra in the range of 3000–1000 cm–1 were collected on a Tensor II spectrometer (Bruker, Billerica, MA) in transmission mode with samples loaded into an infrared-compatible liquid cell (AquaSpec transmission cell; Bruker). Spectra were acquired at a resolution of 4 cm–1 under optimized conditions. A blank spectrum of formulation buffer was recorded under identical conditions. The amide I region from 1700 to 1600 cm–1 was processed with second derivative, baseline correction, and area normalization.
Funding Statement
This work was supported by AstraZeneca.
Acknowledgments
We thank Ramin Samadani for contributions to the development of the HIC method and Douglas Johnson, Jack Yang, LeeAnn Machiesky, Carrie Sowers, Anthony Shannon, Rinku Baid, and Murali Bilikallahalli for their support in testing of samples. We also appreciate thoughtful discussions with Shihua Lin on binding assays and bioassays. Editorial support was provided by Deborah Shuman.
Disclosure of potential conflicts of interest
All authors are or were employees of AstraZeneca at the time that this research was performed, and have stock and/or stock options or interests in the company.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Baselga J, Swain SM.. CLEOPATRA: a phase III evaluation of pertuzumab and trastuzumab for HER2-positive metastatic breast cancer. Clin Breast Cancer. 2010;10:489–12. doi: 10.3816/CBC.2010.n.065. [DOI] [PubMed] [Google Scholar]
- 2.Havelund S, Ribel U, Hubalek F, Hoeg-Jensen T, Wahlund PO, Jonassen I. Investigation of the physico-chemical properties that enable co-formulation of basal insulin degludec with fast-acting insulin aspart. Pharm Res. 2015;32:2250–58. doi: 10.1007/s11095-014-1614-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt C. The benefits of immunotherapy combinations. Nature. 2007;552:S67–S69. doi: 10.1038/d41586-017-08702-7. [DOI] [PubMed] [Google Scholar]
- 4.Kaplon H, Reichert JM. Antibodies to watch in 2018. mAbs. 2018;10:183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagasse HA, Alexaki A, Simhadri VL, Katagiri NH, Jankowski W, Sauna ZE, Kimchi-Sarfaty C. Recent advances in (therapeutic protein) drug development. F1000Res. 2017;6:113. doi: 10.12688/f1000research.9970.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koefoed K, Steinaa L, Soderberg JN, Kjaer I, Jacobsen HJ, Meijer PJ, Haurum JS, Jensen A, Kragh M, Andersen PS, et al. Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. mAbs. 2011;3:584–95. doi: 10.4161/mabs.3.6.17955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasmussen SK, Naested H, Muller C, Tolstrup AB, Frandsen TP. Recombinant antibody mixtures: production strategies and cost considerations. Arch Biochem Biophys. 2012;526:139–45. doi: 10.1016/j.abb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 8.US Food and Drug Administration . Guidance for industry: codevelopment of two or more new investigational drugs for use in combination. Center for drug evaluation and research. Rockville (MD): US Food and Drug Administration; 2013. [Google Scholar]
- 9.Cho IH, Lee N, Song D, Jung SY, Bou-Assaf G, Sosic Z, Zhang W, Lyubarskaya Y. Evaluation of the structural, physicochemical, and biological characteristics of SB4, a biosimilar of etanercept. mAbs. 2016;8:1136–55. doi: 10.1080/19420862.2016.1193659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glover ZW, Gennaro L, Yadav S, Demeule B, Wong PY, Sreedhara A. Compatibility and stability of pertuzumab and trastuzumab admixtures in i.v. infusion bags for coadministration. J Pharm Sci. 2013;102:794–812. doi: 10.1002/jps.23403. [DOI] [PubMed] [Google Scholar]
- 11.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer. 2017;5:16. doi: 10.1186/s40425-017-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alt N, Zhang TY, Motchnik P, Taticek R, Quarmby V, Schlothauer T, Beck H, Emrich T, Harris RJ. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals. 2016;44:291–305. doi: 10.1016/j.biologicals.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Haverick M, Mengisen S, Shameem M, Ambrogelly A. Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications. mAbs. 2014;6:852–58. doi: 10.4161/mabs.28693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao M, De Mel N, Shannon A, Prophet M, Wang C, Xu W, Niu B, Kim J, Albarghouthi M, Liu D, et al. Charge variants characterization and release assay development for co-formulated antibodies as a combination therapy. mAbs. 2019;11:489–99. doi: 10.1080/19420862.2019.1578137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem. 2003;320:104–24. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 16.Wei JY, Bou-Assaf GM, Houde D, Weiskopf A. Technical decision-making with higher-order structure data: detecting reversible concentration-dependent self-association in a monoclonal antibody and a preliminary investigation to eliminate it. J Pharm Sci. 2015;104:3984–89. doi: 10.1002/jps.24616. [DOI] [PubMed] [Google Scholar]
- 17.Esfandiary R, Hayes DB, Parupudi A, Casas-Finet J, Bai S, Samra HS, Shah AU, Sathish HA. A systematic multitechnique approach for detection and characterization of reversible self-association during formulation development of therapeutic antibodies. J Pharm Sci. 2013;102:3089–99. doi: 10.1002/jps.23654. [DOI] [PubMed] [Google Scholar]
- 18.Velázquez-Campoy AOH, Nezami A, Muzammil S, Freire E. Isothermal titration calorimetry. Current Protoc Cell Biol. 2004;23:17.18.11–17.18.24. doi: 10.1002/0471143030.cb1708s23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.