

ABSTRACT
Minor changes in the quality of biologically manufactured monoclonal antibodies (mAbs) can affect their bioactivity and efficacy. One of the most important variations concerns the N-glycosylation pattern, which directly affects an anti-tumor mechanism called antibody-dependent cell-meditated cytotoxicity (ADCC). Thus, careful engineering of mAbs is expected to enhance both protein-receptor binding and ADCC. The specific aim of this study is to evaluate the influence of terminal carbohydrates within the Fc region on the interaction with the FcγRIIIa/CD16a receptor in native and label-free conditions. The single mAb molecule comprises variants with minimal and maximal galactosylation, as well as α2,3 and α2,6-sialic acid isomers. Here, we apply native electrospray ionization mass spectrometry to determine the solution-phase antibody-receptor equilibria and by using temperature-controlled nanoelectrospray, a thermal stability of the complex is examined. Based on these, we prove that the galactosylation of a fucosylated Fc region increases the binding to CD16a 1.5-fold when compared with the non-galactosylated variant. The α2,6-sialylation has no significant effect on the binding, whereas the α2,3-sialylation decreases it 1.72-fold. In line with expectation, the galactoslylated and α2,6-sialylated mAb:CD16a complex exhibit higher thermal stability when measured in the temperature gradient from 20 to 50°C. The similar binding pattern is observed based on surface plasmon resonance analysis and immunofluorescence staining using natural killer cells. The results of our study provide new insight into N-glycosylation-based interaction of the mAb:CD16a complex.
KEYWORDS: Glycoengineering, monoclonal antibody, fc gamma receptor, native mass spectrometry, thermal stability
Introduction
Glycosylation linked to asparagine(N)-297 in the Fc region has been recognized as a critical quality attribute (CQA) in the development of therapeutic monoclonal antibodies (mAbs).1 By activating specific Fcγ receptors (FcγR) expressed on a variety of immune effector cells,2 mAbs are known to mediate an anti-tumor mechanism called antibody-dependent cell-mediated cytotoxicity (ADCC) via their Fc. The interaction is highly dependent on the N-oligosaccharides, which modulate binding affinity, and thereby clinical efficacy in antibody-based immunotherapy.3
Typically, N-glycans attached to immunoglobulin G (IgG) have a biantennary structure and are highly heterogeneous with respect to galactose, sialic acid, bisecting N-acetylglucosamine (GlcNAc), and core fucose.4 Moreover, the total glycan composition can change upon certain conditions, such as pregnancy, where elevated levels of galactosylation and sialylation occur.5 Hence, these structures were found to enhance ADCC and anti–inflammatory activities.6,7 Similarly, the binding affinity to FcγRIIIa/CD16a improved 50-fold with a fucose-deficient IgG1.8 Commercially produced mAbs contain mainly high abundant agalactosylated forms, which differentiate anti–inflammatory properties and functionality.9
In the manufacturing process, the optimization effort to obtain mAbs with a desired glycosylation pattern mainly focusses on host cell engineering and cell culture conditions.10–12 However, in-vitro glycoengineering strategies have been developed recently where N-glycans are entirely remodeled on the protein surface.13 In principle, heterogeneous N-glycans are cleaved via an endoglycosidase, leaving only the first N-acetylglucosamine residue. Subsequently, a well-defined oligosaccharide structure is reattached via a glycosynthase mutant using a synthetic N-glycan oxazoline as a substrate. The method was proposed by Manabe et al.13 to prepare a homogenous antibody-drug conjugate to the N-glycan structure of a Fc. Fine tuning of terminal sugars might also be achieved via enzymes such as galactosidase, galactosyltransferase or sialyltransferase to ensure a uniform a/galactose and sialic acid profile.14 This approach increases efficacy and safety of the final product from the health-care perspective, and facilities process development offering host expression flexibility and lower batch-to-batch inconsistency.15
To better understand the impact of N-glycosylation on mAb-FcγR binding affinity, a variety of biophysical analytical tools such as surface plasmon resonance (SPR),16 isothermal titration calorimetry (ITC),17 nuclear magnetic resonance (NMR)18 and X-ray crystallography are used.19 Among these, SPR is often considered the “gold standard” in protein-protein interaction studies. Native mass spectrometry (MS) has emerged in the field of structural biology as an equally powerful technique, providing information on complex stoichiometry, the existence of sub-complexes and the structural arrangement of subcomplexes.20 It can also be used to study protein assemblies, potentially revealing cooperativity and estimations of binding affinities.21 Of particular interest are its simplicity, sensitivity (low sample consumption), and that it does not require labeling or protein immobilization. Proteins and their complexes are transferred into the gas phase using electrospray ionization (ESI), and subsequently the mass-to-charge (m/z) ratios are recorded.22 ESI produces multiply-charged signals with a narrow distribution of lower charge states if proteins are analyzed in “native” buffered aqueous solutions.23 Using low-resolution MS, a nonglycosylated and slightly glycosylated proteins can be easily measured, whereas highly heterogeneous glycoproteins pose an analytical challenge due to overlapping ion signals from naturally existing glycoforms.24 This limitation was highlighted by Wang et al.25 when determining the molecular weight (MW) of epidermal growth factor receptor and CD38. Nevertheless, it was shown that the noncovalent interaction between heterogeneous and high-MW proteins like antibodies can be preserved in the gas phase, revealing the complexes with better resolved charge state. Consequently, this offers a new perspective for the analysis of highly heterogeneous proteins in a native and label-free conditions.
Here, we applied native electrospray ionization mass spectrometry (ESI-MS) to investigate the mAb-CD16a interaction in the gas phase and to understand the role of N-glycans in complex formation. A recombinant extracellular domain of CD16a with multiple glycosylation sites was used as an example of a heterogeneous protein, whereas homogenous glycoengineered mAbs with a low and high galactose profile, as well as variants with α-2,3 and α-2,6-linked sialic acids were developed as high-MW “carrier” proteins. A direct titration approach was used to determine the equilibrium dissociation constant (Kd) for each glycovariant. Subsequently, the complex’s thermal stability was evaluated by employing a home-built temperature-control nanoelectrospray (TCnESI)26 where free mAb dissociation was monitored as a function of temperature. Additionally, our results were validated by microscale thermophoresis (MST), SPR and a cell-based assay using engineered natural killer (NK) cells expressing the high variant (V176) of CD16a receptor.
Results
Enzymatic glycoengineering of mAb glycovariants
The low and high galactose as well as the α-2,3 and α-2,6-linked sialic variants were produced based on an in vitro glycoengineering approach proposed by Thomann et al.14 Starting from the bulk material collected directly from the fed-batch bioreactor, the N-glycans were remodeled using specific enzymes according to the workflow presented in Figure 1. A hypo-galactosylated (G0 F) variant was restructured by use of β-1,4-galactosidase that cleaves the terminal galactose residue, whereas a hyper-galactosylated (G2 F) type was achieved with β-1,4-galactosyltransferase upon availability of nucleotide sugar donor, UDP-galactose. The α-2,3 and α-2,6-sialic acid isomers (ST3 and ST6), predominantly disialylated structures, were obtained with corresponding α-2,3 and α-2,6-sialyltransferese.
Figure 1.
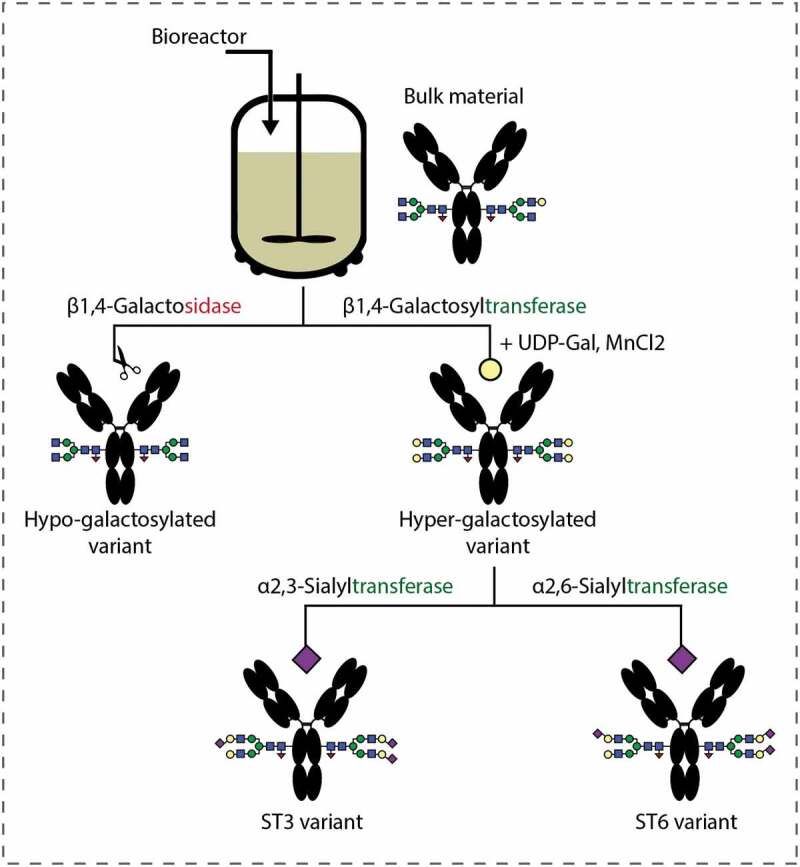
Workflow for producing mAb glycovariants based on an enzymatic in-vitro glycoengineering strategy.
The detailed glycosylation profile of investigated mAbs was determined using a hydrophilic interaction liquid chromatography (HILIC) analysis. Before that, N-glycans were cleaved from the protein backbone surface and labeled with 2-aminobenzamide (2-AB) for a subsequent fluorescence detection. The percentages of N-glycan composition for each respective glycovariant is shown in Table 1. For G0 F and G2 F type, 82.1% and 81.1% of homogenous structures were achieved, respectively. In the case of sialic acid isomers, more than 50% consisted of di-sialic structures attached to both α-1,3-and α-1,6-galactose arms, whereas mono-sialylation reached approximately 17–18%. The total afucosylation counted as a sum of afucosylated structures was comparable for all mutants and oscillated between 6.3–6.7%, thus its influence on Fc binding affinity was not further considered in this study. The MW of intact wild-type (WT) and glycovariants based on native MS is presented in Figure S1.
Table 1.
N-glycan profile determined based on HILIC-HPLC analysis after labeling with 2-AB.
| N-glycan type | mAb glycovariant |
|||
|---|---|---|---|---|
| G0 F | G2 F | ST3 | ST6 | |
| G0 F | 82.1 | 0.2 | 0.4 | 0.4 |
| G1 F | 1.6 | 0.4 | 0.2 | 0.2 |
| G2 F | 0.1 | 81.1 | 1.1 | 1.8 |
| G2S1 F | 0.1 | 0.4 | 16.9 | 17.9 |
| G2S2 F | 0.3 | 0.5 | 58.0 | 54.1 |
| G0 F-N | 1.9 | – | 0.1 | 0.1 |
| G1 F-N | – | 1.8 | 0.4 | 0.5 |
| G1S1 F-N | – | – | 2.5 | 2.5 |
| Afucosylated | 6.5 | 6.7 | 6.7 | 6.3 |
| M5 | 3.1 | 3.7 | 6.3 | 6.5 |
| Minor | 4.3 | 5.2 | 7.5 | 9.7 |
*Afucosylated type as a sum of G0, G1, G2, G0-N, G1-N, G2S1 and G2S2
Heterogeneous CD16a and native MS
While the analysis of high MW homogenous proteins was successfully achieved on time-of-flight (TOF) mass spectrometers, a heterogeneous protein that co-exists in a mixture of a number of glycoforms or proteoforms poses a more difficult challenge. An example is the FcγRIIIa/CD16a receptor in which the extracellular domains are highly glycosylated with five N-glycosylation sites (Figure 2(a)). ESI signals of the truncated protein cannot be resolved due to overlapping ion signals caused by the diversity of glycoforms carrying different charges. As a result, a broad unresolved peak distribution is observed in the mass spectrum, as presented in Figure 2(b). Addition of a charge reducing reagent such as triethylammonium acetate does not improve the spectral resolution significantly. Similarly, a sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) analysis indicates a broad MW range of 45–50 kDa (Figure 2(b)) when compared to standard protein markers. It is interesting to note that when CD16a is incubated with a specific mAb carrying N-linked glycans in the Fc region, the complex can be detected at higher m/z compared to the well-resolved charge states coming from monomer mAb molecule (Figure 2(c)). Conversely, no complex formation is observed for the deglycosylated mAb, suggesting that N-glycan removal impedes interaction and binding to CD16a. A previous study does not suggest a significant change in the secondary and tertiary structure upon PNGase F treatment.28
Figure 2.
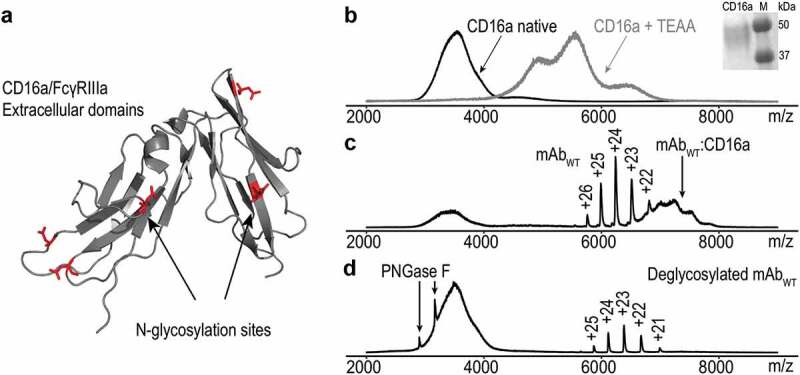
(a) Ribbon diagram of CD16a/FcγIIIa extracellular domains with 5 potential N-glycosylation sites marked in red (PDB: 1E4 J).27 (b) Native ESI spectra of CD16a in 150 mM ammonium acetate solution, pH 7.4 without/with triethylammonium acetate (black and gray line, respectively). Inset: Evaluation of the MW of CD16 using SDS-PAGE. (c) Attaching CD16a to WT mAb results in a high MW CD16a-WT complex indicated by the black arrow. (d) Deglycosylated WT and CD16a do not form a complex when mixed together in solution. Black arrows show charge states of PNGase F used for deglycosylation procedure.
Dissociation constants
To gain insight into the binding of each particular glycovariant in complex with CD16a, we designed titration experiments using native nESI-qTOF MS. The samples were measured at equilibrium with an increasing concentration of CD16a and a constant level of mAb. Subsequently, the complex was quantified by using a linear regression analysis that facilities separating complex from the overlapping mAb charge distribution (Figure 3 A). The one-site binding indicated by the crystal structure and MS for the IgG1–FcγR interaction showed a hyperbolic dependence when bound mAb is plotted against free CD16a concentration.27 The saturation curves and estimated Kd values are shown in Figure 3 B-F. Our results suggest that the N-glycan composition affects binding in favor of galactosylation and α-2,6-sialylation. The G2 F improves the binding affinity 1.5-fold compared with the hypo-galactosylated type. Similarly, α-2,6-sialylation shows a 1.72- fold increase over α-2,3-sialylation. These values agree well with other reports.29,30
Figure 3.

(a) Mass spectrum of CD16a (L) in complex with mAb (RL) measured from 150 mM ammonium acetate solution, pH 7.4 in ESI positive mode. The red dotted line approximated by linear regression indicates the boundary of integrated area between mAb and its complex. Each measurement was repeated three times and the data obtained from titration experiments were fitted with a model equation. Saturation binding curve as a function of free ligand concentration for WT (b), G0 F (c), G2 F (d), ST3 (e) and ST6 (f) glycovariants with estimated Kd values are denoted by the red square line on an x-axis at half of the total mAb associated with CD16a.
Thermal denaturation of mAb:CD16a complex
To further investigate the influence of the specific N-glycan types on complex formation, we performed thermal denaturation experiments by coupling a TCnESI (Figure 4, left panel) with a qTOF MS instrument. All glycovariants saturated with an excess of CD16a created high abundance complex at 20°C. When the temperature is increased to 50°C, the complex disappeared and the mAb-free charge distribution appeared. The abundances of specific mAb and its corresponding complex were then calculated from the relative intensities, and plotted as a function of temperature (Figure 4 A-D). The complex’s melting temperature (Tm) is determined from the intersection point that defines a temperature where half of the complex is denatured. The mAbs with terminal galactosylation and α-2,6 sialylation in complex with CD16a showed higher thermal stability, with a Tm in the range of 50 to 52°C. In contrast, the hypo-galactosylated and α-2,3 sialic variants had a consistently decreased Tm in the range of 42–44°C.
Figure 4.
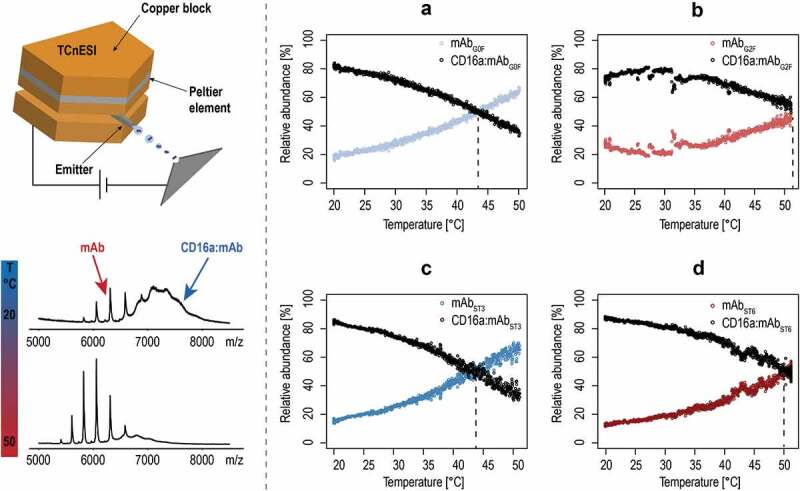
ESI-MS melting experiments. On the left, the schematic drawing of the TCnESI source with a capillary embedded between two copper blocks is shown. A temperature ramp from 20 to 50°C is applied to the CD16a-mAb complex and release of free mAb is monitored. (a–d) Temperature melting curves of CD16a in complex with different glycovariants. Black dots indicate the relative abundance of the complex, whereas colored dots display the mAb relative abundance as a sum of all individual charge states. Dotted lines indicate the melting points of individual complexes.
Subsequently, we used nano-differential scanning fluorimetry (nanoDSF) to determine the thermal stability of each glycovariant. It has been previously demonstrated that antibodies have several transitions points showing the unfolding of their various domains.31,32 We detected two transitions that may be associated first with the melting of the CH2 domain in the Fc and the melting of the antigen-binding fragment, whereas the second point may reflect the unfolding of the CH3 domain. Studies using deglacosylated mAbs indicated that the transition point around 70°C is affected by glycosylation and its variation may reduce the stabilization energy resulting from CH2-CH2 and/or CH2-CH3 inter-domain interaction.28,33 From the overall TI,1 comparison between mAb groups, we observed a statistically significant difference as determined by one-way ANOVA (F(4,10) = 3.43994, p =.04126). However, when considering individual levels using a Tukey post hoc test, no statistically significant difference was found (Figure S2), although, the α-2,3 sialic acid type slightly deviated. Data from the thermal unfolding experiments measured in triplicate are shown in Figure S3.
MST and SPR
To confirm the results from the native MS experiments, we subjected our samples to two independent binding assays, label-dependent MST for Kd determination and label-free SPR to compare the glycovariant binding capacities relative to the WT.
MST is based on differences in the diffusional behavior of substances in their free and complexed state upon application of a temperature gradient, as well as on the temperature-related intensity change (TRIC). Both principles are affected by changes in the microenvironment of an observed receptor upon binding of a ligand. Properties like surface charge, hydration shell, size, and conformation contribute to these changes and correlate with ligand binding affinity to its receptor.34 Typically, low thermal gradients of 2 to 6 K are applied in order to remain at physiologically relevant temperatures. The detection technology used in MST is fluorescence to capture both the diffusional movement of the observed receptor as well as the TRIC phenomenon. Two labeling strategies that attach the dye either via a non-covalent interaction to the His-tag or covalent linking were applied in our study. In the first approach, nonspecific binding of all mAbs to the dye itself as well as to a control His6-peptide was observed. Therefore, 2nd generation RED-NHS dye coupled to lysine residue of CD16a was selected as the final strategy. The mAb:CD16a interaction triggered a significant ligand-induced fluorescence change, suggesting close proximity of the binding interface to the fluorescent tag. A denaturing test used as a control ruled out unspecific binding and confirmed a signal increase only when the ligand was specifically bound to the receptor. The signal-to-noise ratios obtained all exceeded the designated threshold (S/N above 10). Based on that, changes in Kds were detected only for the hypo- and hyper-galactosylated variants (2.5-fold increase) compared to the WT and the two sialic acid-modified mAbs (Table 2). Neither a change in the galactosylation level (G0 F vs. G2 F) nor type of sialic acid (ST3 vs. ST6) affected the binding properties. In addition, we analyzed the data derived from the MST traces despite high initial fluorescence values that may hamper the analysis. With slightly lower S/N ratios, we concluded that all mAbs except G2 F bind equally to CD16a, whereas G2 F shows a 1.5-fold increase (compared to G0 F). The MST binding curves using either the initial fluorescence change or trace analysis plotted against the concentration of the WT and glycovariants are shown in Figures S4 and S5, respectively.
Table 2.
The Kd determination with MST using trace analysis and initial fluorescence change. The CD16a was covalently labeled with the 2nd generation RED-NHS dye and titrated with serial dilutions of WT and glycovariants.
| MST-Trace Analysis |
Initial Fluorescence Change Analysis |
|||
|---|---|---|---|---|
| mAb | Kd [µM] | S/N | Kd [µM] | S/N |
| WT | 2.4 ± 0.6 | 7.7 ± 1.0 | 1.6 ± 0.4 | 10.9 ± 0.6 |
| G0 F | 2.2 ± 0.2 | 8.3 ± 1.2 | 0.6 ± 0.1 | 11.9 ± 1.4 |
| G2 F | 1.5 ± 0.1 | 10.2 ± 0.2 | 0.6 ± 0.1 | 19.6 ± 1.0 |
| ST3 | 2.6 ± 0.4 | 10.9 ± 1.6 | 1.1 ± 0.1 | 13.7 ± 4.1 |
| ST6 | 2.6 ± 0.1 | 8.6 ± 1.2 | 1.2 ± 0.2 | 11.6 ± 2.9 |
Subsequently, the MST data were verified with an SPR experiment using native condition. The strong interaction of the mAbs with the His-tag capturing nitrilotriacetic acid moiety on the sensor surface confirmed the observation made by MST. Thus, we used an anti-His antibody to capture first the His-tagged CD16a and then, all mAbs as analytes were injected in concentrations of 0.75 µM and 1.5 µM. Due to the linear binding behavior in the range of 0.1x to 10x Kd, the equilibrium signal for each antibody could be directly compared as relative binding capacity, refrained from a full kinetic analysis (Figure 5). In contrast to the MST measurements, we were able to deduce a difference in the relative binding capacity between all investigated glycovariants. Similar to the native MS data, ST6 showed a stronger binding affinity to CD16a compared to ST3, thus supporting the observed impact of the 2,6 vs. 2,3 sialic acid linkage. A statistically significant difference for G2 F and G0 F was not observed in SPR.
Figure 5.
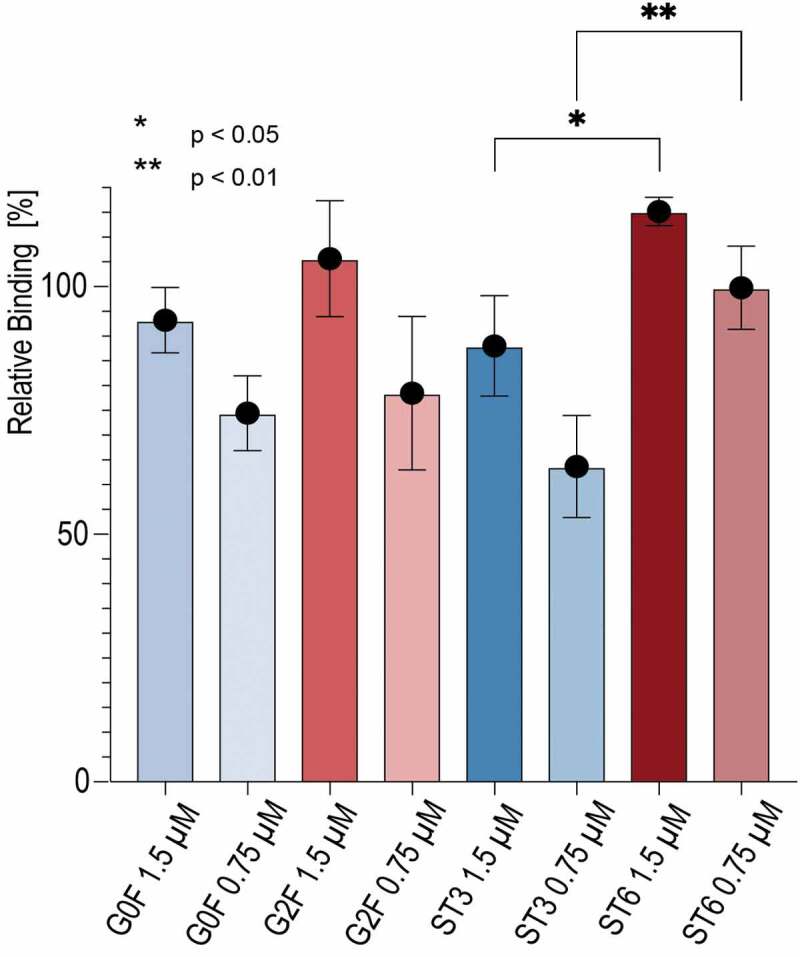
Binding affinities of mAb variants to CD16a determined by surface plasmon resonance (SPR). His-tagged CD16a was immobilized on the chip surface and mAbs as analytes were injected in concentrations of 0.75 µM and 1.5 µM.
CD16.NK-92 cell-line assay
Last, the binding of all glycoengineered mAbs was evaluated with an in-vitro assay using a recombinant NK cell line expressing a high affinity CD16a receptor. The protein-cell interaction was measured by flow cytometry. Hence, glycovariants were applied as primary molecules recognizing CD16a antigen, whereas a phycoerythrin (PE)-conjugated anti-human F(ab’)2 fragment was used to visualize the primary interaction. To exclude unstained cell population and unspecific binding, cells were first incubated either with phosphate-buffered saline (PBS) buffer (control) or only with the detection antibody (negative) (Figure 6 A, gray and dashed line histogram, respectively). The positive cell population was assigned to WT (Figure 6 A, white histogram, solid line). Afterward, a titration experiment was performed with serial mAb dilutions maintaining the same number of cells stained in the same volume in order to compare the glycovariant’s specific binding to NK cells. An example histogram at 100 µg/mL is presented in Figure 6 B. In the concentration-dependent analysis, the onset of binding was observed already below 20 µg/mL with continuous increase above 100 µg/mL (Figure 6 C). It is clear that G2 F and ST6 show the highest mean fluorescence intensity. However, the binding affinity is in favor of G2 F, which differs from our previous results. The potential cause might be the CD16a receptor itself, due to an inherent structural heterogeneity introduced to by the cell line, changes in culture conditions or new formulation excipients. Moreover, mammalian cells are fragile and dead cells or other surface proteins can nonspecifically bind ligands. In contrast, SPR and native MS are performed in pure buffer conditions, which excludes the possibility of any other interactions.
Figure 6.
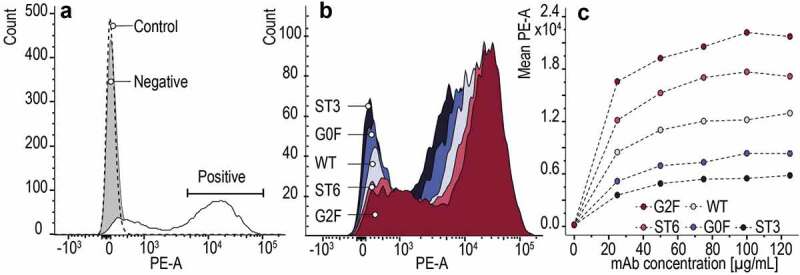
Flow cytometry analysis of mAb’s glycovariants binding to NK-92 cells expressing the CD16a receptor, detected by PE-conjugated to anti-human F(ab’)2 fragment. (a) Binding of WT to the NK-92 cells corresponds to a positive population, and unstained cells (control) with a negative cell population (– primary Ab, + secondary Ab). (b) Comparison of the binding of all glycovariants to the NK cells at the concentration of 100 µg/mL. (c) Concentration-dependent binding of mAb glycovariants to the NK-92 cell line.
Discussion
We propose a novel bioanalytical approach using native MS and a cell-based assay to study Fc-CD16a interactions and to investigate the effect of the antibody’s N-glycosylation pattern on its effector function. Homogenous mAb models were enzymatically glycoengineered by extending or cleaving a core sugar structure with galactose and sialic acid. As indicated by our results, the final product might be redesigned in a precise manner, reducing the need of host cell engineering and variations arising from the manufacturing process. Moreover, this technique offers speedy production with free choice of the expression system,15 although its introduction would certainly require additional processing steps into existing operations, such as monitoring of product purity to exclude the risk of additional contaminants, contributing itself to the overall manufacturing costs and a new regulatory framework.
We showed that native MS can be a powerful tool to characterize the interaction between therapeutic mAbs and their target receptors. By N-glycan remodeling, minor changes in the binding pattern might be observed. As a whole, our results agree with other reports where the binding affinity increases for G2 F and ST6 structures as opposed to G0 F and ST3.7,35 For example, Houde et al.35 have shown a significantly higher binding to the FcγRIIIa when the mAb was non-fucosylated, but hypergalactosylated in comparison to the non-fucosylated and agalactosylated form. Moreover, in the crystal structures a total of 6 H-bonds between sugar and amino acid residues have been spotted for a galactose glycan type, whereas only two H-bonds were seen in the truncated version. Thus, it seems that the absence of a terminal galactose in the α1-6 arm destabilized the stretch from 244 to 247 amino acid residues in the CH2 domain.36 On the contrary, Hodoniczky et al.37 described a positive impact of terminal galactose of IgG only on CDC activity, but not ADCC and FcγRIIIa binding. Likewise, an exciting aspect of Fc-glycans also concerns the type of sialic acid bond. It is known that commercial cell lines such as Chinese hamster ovary produces only glycans with α-2,3 sialic acid linkages, whereas human endogenous Fc-glycans contain exclusively α-2,6- sialic acid residues. Consistent with our results, Lin et al.7 observed a stronger interaction for a 2,6-sialylated type than did the other glycoforms when investigating commercial mAbs versus their glycoengineered sialic acids variants.
The individual contribution of each glycofrom may also be evaluated by thermal stability studies, where the protein’s unfolding temperatures are determined using differential scanning calorimetry, circular dichroism or fluorescence-based activity assays (e.g., nanoDSF). Herein, we demonstrated that terminal glycan engineering has no significant influence on the mAb stability itself. Similar to prior literature, where Tm of CH2 domain was significantly reduced only in case of partially deglycosylated acceptors and terminally mannosylated mAb.38 However, when glycovariants are in complex with CD16a the changes in Tm are clearly observed. By coupling a temperature controlling unit to a standard nESI source, we were able to monitor the release of free mAb in a relatively simple manner and deduce that terminal galactose and α-2,6-sialylic acid with additional H-bounds show a stabilizing effect, opposite to a truncated and α-2,3-sialylic variant. Moreover, it was previously demonstrated that the IgG1 – FcγRIIIa interaction is driven by a favorable binding enthalpy (ΔH) where, for instance, fucose depletion contributes favorably to a ΔH and thus increases the binding constant.17 Yet, an intriguing question to be answered is if α-2,6-sialylation affects the binding affinity more strongly than galactosylation itself. In both cases, data obtained from native MS and SPR experiments suggest a rather similar binding behavior, which should, however, be interpreted with caution, as for ST6 only 54% of G2S2 F constituted a homogenous fraction. Furthermore, from MST experiments we learned that binding of the mAbs must occur close to the label site, as the crystal structure of CD16a shows several lysine residues being close to or even involved in the binding interface.19,27 Therefore, the interaction seems to be sufficiently affected, letting us conclude that label-free strategies resolve mAb:CD16a interaction more adequately.
The label-free approach of native MS makes it a powerful technique that allows one to study protein interactions without compromising their natural folded state, e.g., by covalent attachment of a detection tag. Under non-denaturing conditions, proteins and their complexes are transferred from the liquid to the gas phase in a very soft ionization process, followed by MS detection. Information on conformation, protein homogeneity and stoichiometry of the binding can be obtained, where the latter two cannot be determined with more commonly used SPR technique. Nonetheless, the qTOF mass analyzers we used are characterized by low resolution and sensitivity, especially when dealing with heterogeneous biomolecules like CD16a. Thus, being aware that recorded spectra may pose a certain limitation with regard to data analysis, we developed a data evaluation approach to fit the complex peak area equally for all investigated glycovariants. Based on this, we were able to distinguish a binding pattern and found Kds in the low µM range. An alternative solution, though with higher instrument cost, is a new Orbitrap mass spectrometer.39 With modifications to boost the performance in the high m/z range and with improved resolution and sensitivity, the full glycoproteoform of proteins such as human fetuin have been detected.40
In conclusion, we describe the development of a native ESI-MS system as a new way for assessing the bioactivity of glycoengineered antibodies. We report, to the best of our knowledge, for the first time direct binding of mAbs to a heterogenous CD16a receptor in the gas phase. By simple titration in native conditions, we were able to both monitor the antibody-receptor interaction and further analyze the influence of Fc N-glycosylation on binding equilibria. We showed that galactose and α-2,6-sialic acid improve the binding affinity. Additionally, the use of temperature control nESI source revealed information about the thermal stability of the whole mAb-Cd16a complex, indicating that differences in enthalpy contribute to the differences in binding between different glycoforms. Our system fully reflects the results obtained with other standard biophysical method such as SPR and immunofluorescence assay with engineered NK cells. Thus, it delivers new insight into the mechanism of glycoprotein-glycoprotein interaction.
Materials and methods
mAb glycovariants production
An in-house WT mAb with a heterogeneous glycosylation pattern (c = 65 mg/ml) was used as the starting material. For enzymatic preparation of the hypo-galactosylated (G0 F) variant, 200 µl of the β1,4-galactosidase (Prozyme, GKX-5014) was added to WT (40 mg) and incubated for 24 hours at 37°C in a reaction buffer (Prozyme, GKX5014). For enzymatic preparation of the hyper-galactosylated (G2 F) variant, WT (250 mg) was added into a reaction buffer containing 10 mM of UDP-Gal, 5 mM MnCl2, 100 mM MES at pH 6.5. Next, 230 µl of the β1,4-galactosyltransferase (Roche, cat. no. 08098182103) was added to the sample and incubated for 24 hours at 37°C. For enzymatic ST3 variant preparation, the α2,3-sialyltransferase (Roche, cat. no. 07429916103) was incubated with a G2 F in a 1:10 ratio with addition of CMP-NANA (c = 10 mg/ml in 20 mM His/HCl buffer). Furthermore, alkaline phosphatase (AP, Roche, cat. no. 03359123001) and ZnCl2 were added to the mixture with a final concentration of 200 nM AP and 0.1 mM ZnCl2. Analogously, the ST6 variant was prepared by incubation of the α2,6-sialyltransferase (Roche, cat. no. 08098174103) with G2 F substrate in a 1:10 ratio in a presence of CMP-NANA (c = 10 mg/ml in 20 mM His/HCl buffer), AP and ZnCl2. Both ST3 and ST6 reaction mixtures were incubated for 23 hours at 37°C. Lastly, WT and all glycovariants were purified with Protein A chromatography and formulated into a 20 mM His/HCl buffer.
The N-linked glycosylation pattern was determined using the 2AB-HILIC chromatography. For the enzymatic reaction, PNGaseF from NEB (PNGaseF 500.000 U/mL, NEB, P0705 L) was used. 2 µl of the enzyme was applied for deglycosylation of 200 µg antibody with 1 h incubation time at 45°C. Subsequently, the 2AB labeled N-glycans were determined using a UPLC system with a HILIC column (ACQUITY UPLC BEH Glycan 1,7 μm, 2.1 × 150 mm).
Sample preparation
Prior to native MS analysis, all mAbs and recombinant Human Fc gamma RIIIa/CD16a (V176) protein (ACROBiosystems, Newark, USA) were extensively buffer-exchanged against ammonium acetate buffer (150 mM, pH 7.4) using 10-kDa-MWCO centrifugal filters (Amicon Ultra Centrifugal Filter, Merck) and pre-equilibrated with the same buffer. The concentration of the proteins was determined by measuring the UV absorbance at 280 nm with a Nanodrop 2000 spectrophotometer (Thermo Scientific, Madison, WI, USA). The ammonium acetate buffer was used as a blank. The monomer concentration of the mAb and CD16a was calculated according to the Lambert-Beer law using a percent extinction coefficient ϵ280 nm = 1.885 and ϵ280 nm = 1.65 predicted from the sequence of amino acids, respectively.
Native mass spectrometry
Native ESI-MS experiments were performed using a commercial hybrid quadrupole time-of-flight mass spectrometer (Q-TOF Ultima API; Micromass, Manchester, UK) with a modified 32 k quadrupole and a high-pressure collision cell. Up to 5 μL of sample was loaded into a borosilicate glass emitter with an inner diameter of 0.75 mm and an outer diameter of 1.0 mm. The spray was obtained with a commercial nano-ESI source at ambient temperature by applying 2.0–3.0 kV and a backing pressure of 0.5 bar. Gentle transmission of the ions from atmospheric pressure to vacuum was supported by increasing the pressure in the first pumping compartment to 3.5 mbar. The pressure inside the collision cell was adjusted to 5.5–6.0x10–3 mbar, whereas in the TOF it was 4.0 × 10–7 mbar. The cone voltage, rf lens 1, and the collision energy were optimized to obtain efficient ion transfer and good signal intensity. All spectra were recorded in positive ion mode in the 1000–15000 m/z mass range, with a scan time of 1 s. Approximately 100 individual scans were summed to produce a mass spectrum. Data were recorded and processed with the MassLynx 4.0 software (Waters, Manchester, UK).
Routinely, the mass spectrometer was calibrated with 25 mg/mL aqueous of cesium iodide (CsI). The CsI spectra of were averaged, smoothed with a Savitzky-Golay algorithm, and centroided with 80% peak height. The m/z axis was calibrated after applying a polynomial fit.
TCnESI
MS melting experiments were conducted by using a TCnESI source coupled to the q-TOF mass spectrometer. The details about construction and performance of the TCnESI source are described by Marchand et al.26 Briefly, the capillary emitter is embedded between two copper blocks, which are either heated or cooled by a Peltier element (Adaptive ET-127-10-13-RS, 15.7 V, 37.9 W) powered by a Kikusui PMX18-5A supply (Kikusui Electonics Corp., Japan) and remotely controlled by a LabView software (National Instruments, USA). The temperature of the copper block was monitored using a bolt-mounted thermistor sensor (5000 Ω, ON-950-44005, Omega, standard accuracy of 0.2°C from 0 to 70°C) connected to a Keithley 2701E multimeter (Equipements Scientifiques, Garchies, France) and analyzed with LabView software. The LabView program consists of a loop adjusting the temperature. A ramp was programmed to scan from 20 to 50°C at 2°C/min, with a single nanospray emitter lasting for 30 to 60 min.
Data processing
Multiple ESI spectra were processed using R software (version 3.4.2) with the MALDIquant, MALDIquantForeign and hyperSpec packages. First, the mAb and CD16a area under a curve were calculated with the trapezoid function that approximate value of the definite integral a and b of a continuous function according to the following equation:
| (1) |
Subsequently, a linear regression was introduced to separate the overlapping peaks derived from the monomer mAb charge state distribution. The peaks detected above the regression line were included in the total mAb area and thus subtracted from the total complex area. Subsequently, the bound fraction of mAb was calculated from the ratio of the integrated complex [CD16a:mAb] to the total area of mAb conserved as [mAb]+[CD16a:mAb] = [mAbT]. For unbound mAb, indicated as [mAb], a sum of all charge states was considered. The Kds were determined using the binding model (Equation (2)) describing the equilibrium of a single ligand species to a single uniform population of receptor binding sites.41
Native mass spectra were deconvoluted for MW determination using UniDec software.42
nanoDSF
The nanoDSF experiment was performed on a Tycho NT.6 instrument (NanoTemper Technologies, Munich, Germany). Each antibody was analyzed in a concentration of 6 µM in 10 mM PBS, pH 7.4. The temperature gradient ranged from 35 to 95°C, and the heating rate was 30°C/min. Inflection temperatures were calculated from the first derivative of the spectra obtained at the ratio of the signals at 350 nm to those at 330 nm with Excel 2016 software for Macintosh (Microsoft Corp., Redmond, USA). The data were plotted with Prism version 8.2.1 software (GraphPad Software, San Diego, USA).
MST
Lyophilized CD16a was reconstituted with 500 µl sterile deionized H2O to a final concentration of 200 µg/ml (≈ 4 µM). CD16a labeling was performed by amine coupling, i.e. coupling of the dye to free amines of CD16a. 100 µl of a 4 µM CD16a solution were mixed with 100 µl of a 10 µM solution of 2nd Generation RED-NHS dye (NanoTemper Technologies, Munich, Germany) and incubated for 30 minutes. The dye was subsequently removed by gravitational flow chromatography and the protein concentration determined by UV-spectroscopy (BioSpec nano, Shimadzu, Kyoto, Japan) . Optimal assay conditions for the protein were determined prior to any test with a pretest on a Monolith NT.115 Pico system (NanoTemper Technologies, Munich, Germany). Therefore, 15 µl of 10 nM labeled CD16a was mixed with 15 µl buffer and measured in premium capillaries to check for adsorption to the capillary walls and aggregation of the protein upon application of a temperature gradient of 4 K. 10 mM PBS with 0.025% Tween was the buffer system used, resulting in no observable adsorption to the capillary walls and no aggregation of the labeled CD16a at 5 nM final concentration. The antibodies were diluted 1:1 (v/v), where 10 µl of each concentration were incubated with 10 µl of 10 nM labeled CD16a for 30 minutes at room temperature and then measured at 25°C on a Monolith NT.115 Pico. All measurements were performed in “premium” capillaries (NanoTemper Technologies, Munich, Germany) at 20% excitation power, an MST-power of 40% (equal to 4 K in temperature shift) and with 20 seconds of laser-on time. The ligand-induced fluorescence change was observed in all measurements. Subsequently, a denaturing test to check for unspecific binding was performed (SD-test). The three highest and three lowest concentrations (with regard to the antibody concentration) of antibody+CD16a mixtures were denatured by incubating 10 µl of the antibody+CD16a mixture with 10 µl of 80 mM DTT and 8% SDS at 95°C for 5 minutes. Afterward, the fluorescence before heat application (initial fluorescence) was remeasured. In order to support a ligand-induced fluorescence change, the initial fluorescence had to be below 10% between the three highest and three lowest concentrations of antibody. This condition was fulfilled by all tested antibodies concluding ligand-induced fluorescence change. The initial fluorescence signal data were analyzed with MO.Affinity version 2.3 software (NanoTemper Technologies, Munich, Germany). Additionally, the MST traces were analyzed in the intervals between four and five seconds after the laser had been turned on. The data were plotted with Prism version 8.2.1 software (GraphPad Software, San Diego, USA).
SPR
The SPR experiments were performed on a Bruker SPR-32 instrument (Bruker Daltonics SPR, Hamburg, Germany). A His-tag-capturing antibody (ThermoFisher, Watham, USA, Cat. No. MA1-21315) was immobilized on a High Capacity Sensor (Bruker Daltonics SPR, Hamburg, Germany) by amine coupling. The sensor gold surface was activated with a mixture of 100 mM N-hydroxysuccinimide and 25 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide injected with a flow rate of 5 µl/min for 8 minutes of contact time. The antibody was immobilized in 10 mM PBS at pH 7.4 and at a protein concentration of 20 µg/ml with 8 minutes of contact time to reach a final immobilization level of at least 1500 RU. A 1 M ethanolamine solution was injected at a flow rate of 5 µl/min to inactivate the surface with 8 minutes of contact time. All experiments were performed in 10 mM PBS at pH 7.4 and 25°C. CD16a was captured at 200 nM with 10 minutes of contact time at a 5 µl/min flow rate. A buffer injection was followed to let the signal stabilize for 5 minutes. The antibodies were injected in triplicates both in concentrations of 0.75 µM and 1.5 µM, with a 25 µl/min flow rate, a contact time of 2 minutes, and 3 minutes of dissociation time. Each antibody injection was preceded and followed by a buffer injection. Sensor surface regeneration was performed with 10 mM glycine at pH 1.5, with 2 minutes of contact time and a flow rate of 25 µl/min. The data were analyzed with the Sierra Analyzer software (version 3.1.14, Bruker Daltonics SPR, Hamburg, Germany) by “double subtraction” (i.e., subtraction of the reference surface and buffer injections from the raw data). The antibody binding capacity was determined from the equilibrium signal, which was normalized by the MW and immobilization level of CD16a according to Equation (3). The relative binding capacity was then calculated with respect to the binding capacity of WT antibody.
Cell culture
CD16+ NK-92 cells were purchased from American Type Culture Collection (PTA-6967). The cells were cultivated in an alpha minimal essential medium (Sigma Aldrich) according to a previously published protocol.43 The medium contained 10% heat inactivated defined fetal bovine serum (HyClone Laboratories, Logan), 10% heat inactivated horse serum (Gibco), 2 mM L-glutamine (Gibco), 100 µM 2-mercaptoethanol (Gibco), 2 mM folic acid (Sigma Aldrich), 1 mM sodium pyruvate (Gibco) and 100 µg/mL streptomycin/penicillin (Gibco). Additionally, after sterile filtration medium was supplemented with recombinant interleukin-2 (PeproTech) at a concentration 200 IU/mL for the first culture days and then, titrated down to 100 IU/mL. The cells were passed at a 1:5 ratio into fresh medium every 3 days and incubated at 6% of CO2 at 37°C. To keep the cells consistent and to avoid an overgrowth, the culture was maintained for a maximum 4 weeks.
Fluorescence-activated cell sorting analysis
Prior to surface staining, cells were washed and suspended in PBS buffer at a pH 7.4 containing 1% of bovine serum albumin and 2 mM EDTA solution (FACS buffer). Subsequently, the NK cells expressing a high affinity variant of CD16 were counted and split to obtain equal numbers of approximately 250,000 cells per well. For immunofluorescence staining, we used an indirect protocol, where the cells were first incubated for 30 min on ice with a serial dilution of primary antibody, here WT and glycovariants. Six concentration points in a range of 0 to 125 µg/mL were investigated. Afterward, the cells were washed and incubated with a PE-conjugated anti-human F(ab’)2 fragment (dilution ratio 1:100, Jackson Immuno-Research, Cat. No. 109-116-097) for another 30 min on ice and washed with a FACS buffer. All flow cytometry data where measured on a BD FACSAria III (BD Biosciences) instrument and analyzed using FlowJo software.
Funding Statement
This work was supported by the ETH Zurich Postdoctoral Fellowship Program [17-2 FEL-22], cofunded by the Marie Curie Actions for People COFUND Program..
Acknowledgments
J.H. would like to acknowledge the support received from the ETH Zurich Postdoctoral Fellowship Program, cofunded by Marie Curie Actions for People COFUND Program. The authors gratefully acknowledge Dr. Adrien Marchand for his valuable advice and discussion on TCnESI source application.
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Data availability statement
The original data used in this publication are made available in a curated data archive at ETH Zurich (https://www.research-collection.ethz.ch) under the DOI 10.3929/ethz-b-000388768.
References
- 1.Reusch D, Tejada ML.. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25:1325–11. doi: 10.1093/glycob/cwv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nimmerjahn F, Ravetch JV.. Fcγ receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 3.Zahavi D, AlDeghaither D, O’Connell A, Weiner LM. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antib Ther. 2018;1:7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flynn GC, Chen X, Liu YD, Shah B, Zhang Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol Immunol. 2010;47:2074–82. doi: 10.1016/j.molimm.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Ruhaak LR, Uh HW, Deelder AM, Dolhain REJM, Wuhrer M. Total plasma N-glycome changes during pregnancy. J Proteome Res. 2014;13:1657–68. doi: 10.1021/pr401128j. [DOI] [PubMed] [Google Scholar]
- 6.Chen CL, Hsu JC, Lin CW, Wang CH, Tsai MH, Wu CY, Wong CH, Ma C. Crystal structure of a homogeneous IgG-Fc glycoform with the N-Glycan designed to maximize the antibody dependent cellular cytotoxicity. ACS Chem Biol. 2017;12:1335–45. doi: 10.1021/acschembio.7b00140. [DOI] [PubMed] [Google Scholar]
- 7.Lin C-W, Tsai M-H, Li S-T, Tsai T-I, Chu K-C, Liu Y-C, Lai M-Y, Wu C-Y, Tseng Y-C, Shivatare SS, et al. A common glycan structure on immunoglobulin G for enhancement of effector functions. Proc Natl Acad Sci. 2015;112:10611–16. doi: 10.1073/pnas.1513456112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Gloria Meng Y, Weikert SHA, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–40. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 9.Hajduk J, Wolf M, Steinhoff R, Karst D, Souquet J, Broly H, Morbidelli M, Zenobi R. Monitoring of antibody glycosylation pattern based on microarray MALDI-TOF mass spectrometry. J Biotechnol. 2019;302:77–84. doi: 10.1016/j.jbiotec.2019.06.306. [DOI] [PubMed] [Google Scholar]
- 10.Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, Wakitani M, Niwa R, Sakurada M, Uchida K, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng. 2004;87:614–22. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- 11.Markham A. Obinutuzumab: first global approval. Drugs. 2016;76:397–403. doi: 10.1007/s40265-016-0540-0. [DOI] [PubMed] [Google Scholar]
- 12.Karst DJ, Scibona E, Serra E, Bielser J-M, Souquet J, Stettler M, Broly H, Soos M, Morbidelli M, Villiger TK. Modulation and modeling of monoclonal antibody N-linked glycosylation in mammalian cell perfusion reactors. Biotechnol Bioeng. 2017;114:1978–90. doi: 10.1002/bit.26315. [DOI] [PubMed] [Google Scholar]
- 13.Manabe S, Yamaguchi Y, Matsumoto K, Fuchigami H, Kawase T, Hirose K, Mitani A, Sumiyoshi W, Kinoshita T, Abe J, et al. Characterization of antibody products obtained through enzymatic and nonenzymatic glycosylation reactions with a glycan oxazoline and preparation of a homogeneous antibody-drug conjugate via Fc N-Glycan. Bioconjug Chem. 2019;30:1343–55. doi: 10.1021/acs.bioconjchem.9b00132. [DOI] [PubMed] [Google Scholar]
- 14.Thomann M, Schlothauer T, Dashivets T, Malik S, Avenal C, Bulau P, Rüger P, Reusch D. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS One. 2015;10:1–16. doi: 10.1371/journal.pone.0134949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mastrangeli R, Palinsky W, Bierau H. Glycoengineered antibodies: towards the next-generation of immunotherapeutics. Glycobiology. 2018;29:199–210. doi: 10.1093/glycob/cwy092. [DOI] [PubMed] [Google Scholar]
- 16.Maenaka K, Van der Merwe PA, Stuart DI, Jones EY, Sondermann P. The human low affinity Fcγ Receptors IIa, IIb, and III Bind IgG with fast kinetics and distinct thermodynamic properties. J Biol Chem. 2001;276:44898–904. doi: 10.1074/jbc.M106819200. [DOI] [PubMed] [Google Scholar]
- 17.Okazaki A, Shoji-Hosaka E, Nakamura K, Wakitani M, Uchida K, Kakita S, Tsumoto K, Kumagai I, Shitara K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcγRIIIa. J Mol Biol. 2004;336:1239–49. doi: 10.1016/j.jmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Kato K, Sautès-Fridman C, Yamada W, Kobayashi K, Uchiyama S, Kim H, Enokizono J, Galinha A, Kobayashi Y, Fridman WH, et al. Structural basis of the interaction between IgG and Fcγ receptors. J Mol Biol. 2000;295:213–24. doi: 10.1006/jmbi.1999.3351. [DOI] [PubMed] [Google Scholar]
- 19.Roberts JT, Barb AW. A single amino acid distorts the Fc γ receptor IIIb/CD16b structure upon binding immunoglobulin G1 and reduces affinity relative to CD16a. J Biol Chem. 2018;293:19899–908. doi: 10.1074/jbc.RA118.005273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leney AC, Heck AJR. Native mass spectrometry: what is in the name? J Am Soc Mass Spectrom. 2017;28:5–13. doi: 10.1007/s13361-016-1545-3. [DOI] [PubMed] [Google Scholar]
- 21.Heck AJR. Native mass spectrometry: A bridge between interactomics and structural biology. Nat Methods. 2008;5:927–33. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 22.Silveira JA, Fort KL, Kim D, Servage KA, Pierson NA, Clemmer DE, Russell DH. From solution to the gas phase: stepwise dehydration and kinetic trapping of substance p reveals the origin of peptide conformations. J Am Chem Soc. 2013;135:19147–53. doi: 10.1021/ja4114193. [DOI] [PubMed] [Google Scholar]
- 23.Susa AC, Xia Z, Tang HYH, Tainer JA, Williams ER. Charging of proteins in native mass spectrometry. J Am Soc Mass Spectrom. 2017;28:332–40. doi: 10.1007/s13361-016-1517-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tong W, Wang G. How can native mass spectrometry contribute to characterization of biomacromolecular higher-order structure and interactions? Methods. 2018;144:3–13. doi: 10.1016/j.ymeth.2018.04.025. [DOI] [PubMed] [Google Scholar]
- 25.Wang G, De Jong RN, Van Den Bremer ETJ, Parren PWHI, Heck AJR. Enhancing accuracy in molecular weight determination of highly heterogeneously glycosylated proteins by native tandem mass spectrometry. Anal Chem. 2017;89:4793–97. doi: 10.1021/acs.analchem.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marchand A, Rosu F, Zenobi R, Gabelica V. Thermal denaturation of DNA G-quadruplexes and their complexes with ligands: thermodynamic analysis of the multiple states revealed by mass spectrometry. J Am Chem Soc. 2018;140:12553–65. doi: 10.1021/jacs.8b07302. [DOI] [PubMed] [Google Scholar]
- 27.Sondermann P, Huber R, Oosthuizen V, The JU. 3.2-Å crystal structure of the human IgG1 Fc fragment–FcγRIII complex. Nature. 2000;406:267–73. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 28.Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 2011;3:568–76. doi: 10.4161/mabs.3.6.17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamaguchi Y, Nishimura M, Nagano M, Yagi H, Sasakawa H, Uchida K, Shitara K, Kato K. Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim Biophys Acta - Gen Subj. 2006;1760:693–700. doi: 10.1016/j.bbagen.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Subedi GP, Barb AW. The immunoglobulin G1 N-glycan composition affects binding to each low affinity Fc γ receptor. MAbs. 2016;8:1512–24. doi: 10.1080/19420862.2016.1218586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vermeer AWP, Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brader ML, Estey T, Bai S, Alston RW, Lucas KK, Lantz S, Landsman P, Maloney KM. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm. 2015;12:1005–17. doi: 10.1021/mp400666b. [DOI] [PubMed] [Google Scholar]
- 33.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci. 2008;97:1414–26. doi: 10.1002/jps.21104. [DOI] [PubMed] [Google Scholar]
- 34.Jerabek-Willemsen M, André T, Wanner R, Roth HM, Duhr S, Baaske P, Breitsprecher D. MicroScale thermophoresis: interaction analysis and beyond. J Mol Struct. 2014;1077:101–13. doi: 10.1016/j.molstruc.2014.03.009. [DOI] [Google Scholar]
- 35.Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics. 2010;9:1716–28. doi: 10.1074/mcp.M900540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol. 2008;20:471–78. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Hodoniczky J, Yuan ZZ, James DC. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol Prog. 2005;21:1644–52. doi: 10.1021/bp050228w. [DOI] [PubMed] [Google Scholar]
- 38.Wada R, Matsui M, Kawasaki N. Influence of N-glycosylation on effector functions and thermal stability of glycoengineered IgG1 monoclonal antibody with homogeneous glycoforms. MAbs. 2019;11:350–72. doi: 10.1080/19420862.2018.1551044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fort KL, Van De Waterbeemd M, Boll D, Reinhardt-Szyba M, Belov ME, Sasaki E, Zschoche R, Hilvert D, Makarov AA, Heck AJR. Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst. 2018;143:100–05. doi: 10.1039/C7AN01629H. [DOI] [PubMed] [Google Scholar]
- 40.Lin YH, Franc V, Heck AJR. Similar Albeit Not the same: in-depth analysis of proteoforms of human serum, bovine serum, and recombinant human fetuin. J Proteome Res. 2018;17:2861–69. doi: 10.1021/acs.jproteome.8b00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hulme EC, Trevethick MA. Ligand binding assays at equilibrium : validation and interpretation. Br J Pharmacol. 2010;161:1219–37. doi: 10.1111/j.1476-5381.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP, Robinson CV. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem. 2015;87:4370–76. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miah SMS, Campbell KS. Expression of cDNAs in human natural killer cell lines by retroviral transduction. Methods Mol Biol. 2010; 612:447–63. doi: 10.1007/978-1-60761-362-6_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original data used in this publication are made available in a curated data archive at ETH Zurich (https://www.research-collection.ethz.ch) under the DOI 10.3929/ethz-b-000388768.