

ABSTRACT
The efficacy of immune checkpoint inhibitors has been shown to depend on preexisting antitumor immunity; thus, their combination with cancer vaccines is an attractive therapeutic approach. Plasmacytoid dendritic cells (PDC) are strong inducers of antitumor responses and represent promising vaccine candidates. We developed a cancer vaccine approach based on an allogeneic PDC line that functioned as a very potent antigen-presenting cell in pre-clinical studies. In this phase Ib clinical trial, nine patients with metastatic stage IV melanoma received up to 60 million irradiated PDC line cells loaded with 4 melanoma antigens, injected subcutaneously at weekly intervals. The primary endpoints were safety and tolerability. The vaccine was well tolerated and no serious vaccine-induced side effects were recorded. Strikingly, there was no allogeneic response toward the vaccine, but a significant increase in the frequency of circulating anti-tumor specific T lymphocytes was observed in two patients, accompanied by a switch from a naïve to memory phenotype, thus demonstrating priming of antigen-specific T-cells. Signs of clinical activity were observed, including four stable diseases according to IrRC and vitiligoïd lesions. Four patients were still alive at week 48. We also demonstrate the in vitro enhancement of specific T cell expansion induced by the synergistic combination of peptide-loaded PDC line with anti-PD-1, as compared to peptide-loaded PDC line alone. Taken together, these clinical observations demonstrate the ability of the PDC line based-vaccine to prime and expand antitumor CD8+ responses in cancer patients. Further trials should test the combination of this vaccine with immune checkpoint inhibitors.
KEYWORDS: Plasmacytoid dendritic cells, vaccination, peptide-vaccines, melanoma, clinical study, immunotherapy, immune checkpoint blockers
Introduction
The clinical benefit of immune checkpoint inhibitors (ICI) in the treatment of cancer patients stands as proof that the immune system can help control tumor growth. Anti-tumor cytotoxic CD8 + T-cells are suggested to be the key effectors in the ICI mechanisms of action because they can recognize tumor-associated-antigens, and lyse antigenic tumor cells. Indeed, other than the expression of PD-L1 in the tumor microenvironment, both the expression of immunogenic tumor antigens by cancer cells, and the presence of tumor-infiltrating CD8 + T-cells have been associated with the clinical efficacy of ICI in numerous cancer indications.1-3
ICI is now the gold standard treatment for metastatic melanoma, 4 accounting for the substantial increase of 5-year overall survival rates from less than 10% to up to 40-50%.5 Melanoma cells express multiple antigens, many of them as the result of the high number of mutations displayed by malignant cells, 6 while others are shared tumor-associated antigens, tissue-specific (e.g. Melan-A/MLANA, Tyrosinase/TYR, gp100/PMEL) or belonging to the MAGE-type antigens family encoded by cancer-germline genes (e.g. MAGEA, CTAG1B).7 Nevertheless, spontaneous antitumor T cell responses are often unable to control melanoma growth. Resistance can arise from the exclusion of T cells from the tumor, or from T cell dysfunction induced by the tumor microenvironment.8 This latter could be related to dendritic cell (DC) dysfunction, and we have described the features of melanoma-infiltrating plasmacytoid dendritic cells (PDCs), which skew T cell differentiation toward Th2 and Treg profiles through OX40L and ICOSL expression.9
In this context, efficient activation of functional antitumor T cells is crucial to improve patient outcomes, and vaccination represents a promising therapeutic option. Several vaccination approaches have been tested, especially in melanoma, including peptide-, DNA-, or vector-based vaccines, 10-12 however their efficacy may be affected when the functionality of the patient’s DC is compromised. Vaccines may also rely on antigen-loaded dendritic cell injection, thus bypassing in situ DC dysfunction. Among the DC populations, plasmacytoid dendritic cells (PDC) are of great interest, 13 as they are potent type 1 IFN producers and can induce strong CTL responses.14 Only one clinical trial was performed using autologous PDC, in which favorable observations were made: systemic type I interferon signature after each vaccination, vaccine-induced expansion of high-affinity T cell clones and increased overall survival.15 In addition, the activation of PDC by intratumoral injection of TLR ligands demonstrated a clinical benefit in cancer patients.16
We developed an original therapeutic vaccine approach based on a proprietary allogeneic plasmacytoid dendritic cell line (‘PDC line’). PDC line displays a professional antigen-presenting cell activity and can prime naïve CD8+ cells derived from cord blood (Plumas, unpublished data). In preclinical models PDC line loaded with viral or melanoma-associated antigens led to highly efficient expansion of antigen-specific T cells.17-19 We showed recently that PDC line loaded with neoantigens was able to prime naïve CD8+ T cells from healthy donors and efficiently expand neoantigen-specific T cells.20 The resulting T cells were highly functional in terms of IFN-γ secretion and cytotoxic activity. Their antitumor activity was evaluated in a humanized mouse model in which vaccinations with peptide-loaded PDC line led to tumor growth inhibition, with the recruitment of anti-vaccine T cells to the tumor site.17 Moreover, the stimulation of specific T cells was demonstrated ex vivo with lymphocytes from melanoma patients, and the primed T cells displayed cytolytic activity that was specific for the autologous tumor cells.17,21 Based on this proof of concept, we conducted a phase I clinical trial (GeniusVac-Mel4), to test the safety of the allogeneic PDC line loaded with four melanoma antigens in monotherapy, and its ability to elicit antitumor immune responses in metastatic melanoma patients.
Material and methods
Study design
This open-label, non-randomized, Phase Ib study was conducted at 3 clinical centers in France (Grenoble University Hospital, Center Léon Bérard (Lyon) and Nantes University Hospital). The protocol was approved by the CPP Sud Est V (ethical committee) and the national competent authorities for the safety of medicine and health products (ANSM). All patients gave written informed consent after being explained the whole study by the investigator. Patients were split into three groups according to the dose (4, 20 or 60 × 106 cells/injection) and received a total of three weekly injections of the vaccine. The primary endpoints were safety and tolerability evaluation. Secondary endpoints were immunological responses against melanoma antigens and clinical activity. The study was conducted in accordance with the ethical principles of the Helsinki declaration. The study was registered with the Eudract number 2012-003124-20 and the clinicaltrials.gov number NCT01863108. The start time of the study treatment (first administration of the investigational product) was considered as the starting point of follow-up. The duration of follow-up for each patient for this analysis was 48 weeks (± 1 week).
Patients
Eligibility criteria included American Joint Committee on Cancer (AJCC) stage IIIC or IV confirmed unresectable metastatic melanoma. Other eligibility criteria included HLA-A*0201 positivity, OMS performance score <3 and failure of at least one line of systemic treatment. Exclusion criteria included primary ocular melanoma, chemotherapy, immunotherapy or radiotherapy within 4 weeks preceding inclusion, treatment with drugs under development within 4 weeks or cerebral metastasis (with some exceptions).
Additional in vitro experiments were performed to evaluate the synergy between GeniusVac and the immune checkpoint blocker anti-PD-1, with peripheral blood mononuclear cells from 12 additional metastatic melanoma patients. These cells came from heparinized blood samples collected in the department of dermatology in Grenoble-Alpes University Hospital at the time of cancer diagnosis, and included in the biological sample collection DC-2008-787. As controls, blood samples were obtained from 14 healthy donors (HD) following“Etablissement Français du Sang” (EFS, Grenoble) approved protocols.
Vaccine preparation
PDC line was generated from the leukemic cells of a patient with PDC leukemia. For this clinical trial, two clinical batches of PDC line were generated from the primary cell bank fully certified in terms of identity, purity, and viral security. The PDC line was amplified in CellSTACK Chambers (CellBIND Surface, Corning Life Sciences), starting from an aliquot of the frozen primary cell bank. The Drug Product (DP) was composed of four cellular preparations, each of them loaded separately with one melanoma antigen: MLANA26-35L (ELAGIGILTV), MAGEA3271-279 (YMDGTMSQV), PMEL209-217 (FLWGPRALV) and TYR369-377 (IMDQVPFSV), (Symprosis, Marseille, France)). The cells were then irradiated (60 Gy, 137Cs γ-ray irradiation in a Biobeam GM device, Eckert and Ziegler, Leipzig, Germany), mixed, washed, and stored in liquid nitrogen in Cryomacs freezing bags (Miltenyi) (Figure 1(a)). Irradiation was used to block cell proliferation and avoid any risk of tumorigenesis, as confirmed by tumorigenic assays in mice (not shown), while preserving the immunostimulatory activity of the PDC line, as demonstrated in pre-clinical experiments.22 Cell phenotyping confirmed the expression of CD123, HLA-A2, CD4, and HLA-DR molecules and the absence of CD11 c (see supplementary table 1 for the list of antibodies).
Figure 1.
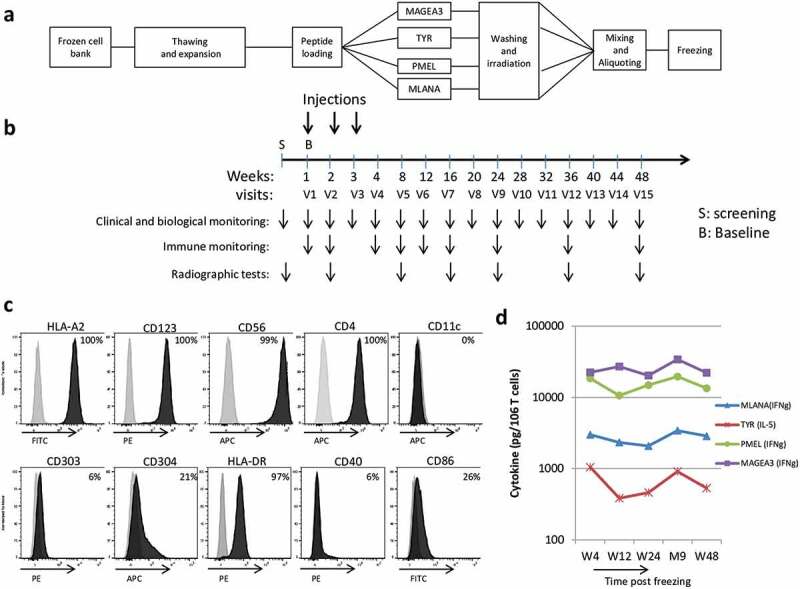
GeniusVac-Mel4 vaccine production, an overview of trial design, and drug product characterization.
(a) GeniusVacMel4 was produced as a cell therapy product, by amplification of the PDC line. The cells were then split and loaded with the four tumor-associated peptides, washed, irradiated and put back together before freezing in ready-to-use aliquots. (b) After screening, patients received 3 weekly intradermal injections of GeniusVacMel4, and their follow-up consisted in clinical and biological monitoring, with blood samples collected for immunomonitoring. (c) The DP was characterized by flow cytometry and displayed a PDC-like expression profile as well as CD56, the hallmark of leukemic PDC. Isotype matched controls (gray histograms) and the test markers (dark histograms) are shown. (d) The stability of the frozen drug product was evaluated with a potency assay consisting in assessing its T cell stimulatory capacity by measuring the cytokine secretion (IL-5 or IFNγ, cytometric beads array method) by Ag-specific T cell clones cocultured 18 h with the DP. ‘W’ stands for ‘week’ and ‘M’ for ‘month.’
The potency of the DP was verified by measuring its capacity to stimulate cytokine secretion after coculture of the DP with Ag-specific T cell clones directed against melanoma antigens. Four HLA-A2 restricted T cell clones were used: WOLF-IVS-B (anti-TYR 369–377), DYMA 811-327/4(anti-PMEL209-217) and PENA-297/22 (anti-MAGEA3271-279) obtained from P. Coulie, and 24-B7 (anti-MLANA26-35L) obtained from B. Dreno. T cell clones were thawed, and seeded in culture (25,000 T cells) with the DP (10,000 cells) in 200 µL (round-bottomed 96-well plates), in RPMI complete medium, supplemented with IL-2 (10 U/mL, Peprotech) and 10% fetal calf serum. After 20 h, supernatants were harvested, frozen, and their cytokine content was determined by cytometric bead array (CBA, BD Bioscience, IFN-γ and IL-5, depending on the clones). Positive controls were PMA+ionomycin-activated cells, negative controls were cells incubated in the medium.
Vaccine administration
DP was thawed and reconditioned in syringes of 1.75 mL ready for injection. The cell concentration was adapted in order to obtain the final doses: 4, 20, or 60 × 106 cells in the injected volume of 1.5 mL. For patients recruited at the University hospital of Nantes, DP was shipped in nitrogen gas (Dryshipper) to the Nantes cell therapy unit to be thawed and processed. The injection was performed within 6 h after syringe preparation.
Each weekly vaccination was performed by three neighboring subcutaneous injections in the patient’s thigh.
Evaluations
The study evaluation was mainly focused on safety and tolerability (based on clinical laboratory tests, vital signs, full clinical examination and occurrence of (S)AEs). Brain, lung, abdomen, and pelvis CT scans were performed at weeks 8, 16, 24, 36 and 48, completed by a tumor evaluation using a scanner and clinical evaluation at weeks 2, 8, 16, 24, 36 and 48.
Clinical responses were evaluated during and at the end of follow-up according to guidelines for evaluation criteria for solid tumors (RECIST 1.1) and using the immune-related response criteria (irRC). Immune responses were also assessed, according to the trial schedule shown in Figure 1(b).
Immunomonitoring
Blood samples were obtained at different time points (W1, W2, W4, W8, W12, W16, W24, W36, and W48). Blood without anticoagulant was processed for Anti-HLA antibodies detection and heparinized blood was used for immunomonitoring and mixed lymphocyte reaction (MLR) assays.
Circulating peptide-specific T lymphocytes percentages were determined by flow cytometry by dextramer labeling on fresh peripheral blood mononuclear cells (PBMC obtained by density gradient centrifugation) (supplementary table 1). Percentages of antiviral T cells (influenza and Epstein-Barr Virus) were measured as a control. A threefold increase in the proportion of dextramer positive T cells was considered positive. The proportions of naïve, effector memory (EM and EMRA), and central memory (CM) T lymphocytes among these specific T lymphocytes were measured (Flow cytometry, CD45RA/CCR7 differential expression on dextramer positive cells (supplementary table 1)). A 1.5 fold increase in specific Effector Memory T cell frequency was considered positive.
Allogeneic humoral and cellular responses were evaluated by measuring anti-HLA antibodies in the sera of patients, and by measuring patients’ lymphocyte proliferation in MLR. MLR was performed by co-culturing the patient’s PBMC with the irradiated PDC line in a 1:1 ratio (50,000 cells), in 200 µL in 96-well round-bottom plates. Proliferation was measured at day 6 after 18-h incubation with 3 H-thymidine. The HLA typing of patients and the PDC line cells is given in Supplementary Table 2. The proliferation of PBMC with irradiated autologous PBMC was the negative control, and proliferation against irradiated PBMC from an unrelated healthy donor (cell collection EFS DC-2008-787) was used as a positive control. IgG antibodies to HLA class I and class II antigens were evaluated by Luminex technology (kit LM-HLA class I/II deluxe screening, Gen-Probe) in the Histocompatibility laboratory of EFS, Grenoble, France).
The frequency of circulating immune cell subsets was measured by flow cytometry, with the antibodies listed in supplementary table 1. Whole blood was used for sub-population determination (T,B NK DC, or Treg cells) with the appropriate staining (supplementary table 1). PBMCs were used directly for dextramer staining (supplementary table 1). Samples were analyzed with a BD FACSCanto III instrument and the DIVA software, and data analysis was performed using the FlowJo software (Supplementary fig 1 depicts the gating strategy to identify the various cell subsets).
Patient 0101 required the surgical excision of ulcerative and necrotic budding lesions on the right lower limb (AE noted grade 3) and one of these lesions were analyzed by immunohistochemistry and flow cytometry to document the T cell infiltrate and the expression of PD-1 and PD-L1. One part of the tumor was paraffin embedded, while the other was mechanically and enzymatically dissociated to recover a single cell suspension. Dextramer staining (HLA-A2:MLANA, HLA-A2:PMEL, HLA-A2:TYR and HLA-A2:MAGEA3) was performed on fresh cells as described previously for PBMC. PD-1 and PD-L1 expression were analyzed on thawed cells, with the antibodies listed in supplementary table 1. Immunohistochemistry staining was performed on 3 µm fixed paraffin-embedded tissue sections using a Benchmark XT autostainer (Roche/Ventana Medical Systems, Tucson AZ, USA). After deparaffinization, antigen retrieving was carried out with Cell Conditioning 1 (CC1) standard antigen retrieval buffer (Tris/Borate/EDTA pH 8.4) for either 30 min (CD163), 60 min (CD3, CD8, HLA-ABC, PD1) or 90 min at 98°C (PDL1). After incubation at 37°C with primary antibodies (supplementary table 1), the revelation was achieved by using UltraView Universal DAB detection kit (Ventana Medical System – Roche – Ref. 05269806001) with amplification for PDL1 (Amplification Kit – Roche – Ref. 05266114001).
Ex vivo evaluation of peptide-loaded PDC line and anti-PD-1 combination
Thawed PBMC from 12 metastatic melanoma patients and 14 healthy volunteers (sample collection DC-2008-787) were used to analyze by flow cytometry the expression of PD-1 on MLANA-specific T cells (identified by CD3, CD8,and dextramer labeling, supplementary Table 1). To study the synergy between anti-PD-1 and peptide-loaded PDC line, PBMC (2x106 cells) from melanoma patients were co-cultured with or without the irradiated PDC line loaded with MLANA and PMEL peptides (pep-PDC line, 0.2 × 106 cells) in the presence of recombinant human IL-2 (Proleukin, Novartis Pharma, 200 UI/ml) with or without clinical-grade anti-PD1 (Pembrolizumab, 10 µg/ml, Merk) for 1 week in complete medium supplemented with 10% FCS. The cells were then harvested and stained with FITC- HLA-A2:MLANA and PE-HLA-A2:PMEL dextramers, together with anti-CD3 and -CD8. The fold increase of specific T cells following culture with pep-PDC line (with or without anti-PD-1) was calculated with the following formula: .
Statistical analysis
Statistical analyses are not relevant for such a clinical trial with only nine patients split into three treatment groups. Qualitative and quantitative data are described for each patient, or each group of patients. Data from all patients recruited in the study were collected and used for analysis.
In the in vitro experiments analyzing the synergy between anti-PD-1 and pep-PDC line, non-parametric Kruskal–Wallis or Friedman (with Dunn’s posttest), Wilcoxon signed-rank test or Mann–Whitney tests were used (GraphPad Prism software).
Results
Vaccine production and characterization
Two clinical batches of PDC line were produced. The PDC line expressed high levels (more than 97% of positive cells) of CD123, CD4, HLA-A2 and HLA-DR, while being negative for CD11 c. Intermediate levels of CD304 (21%) and CD86 (26%) were detected, and the expression of CD40 and CD303 was barely detectable (Figure 1(c)). The stability of the stored drug product (DP) was checked, and tests demonstrated that the Ag-loaded, y-irradiated PDC line retained for at least 12 months its ability to present the 4 antigenic peptides to specific T cell clones, triggering their cytokine secretion (Figure 1(d)). The viability of the DP was always over 70% before injection to the patients (not shown).
Demographics and trial completion
Ten patients were screened, 9 of whom (four men and five women) were recruited between June 2013 and March 2016 in Grenoble and Nantes Hospitals (no patient was recruited in Center Léon Bérard, Lyon) (see Table 1 for patients’ characteristics). Patients were aged between 35.4 and 84.9 (mean = 71.8). All patients had stage IIIC/IV melanoma at study initiation and had already received several treatments. Three patients received 4 × 106 cells per administration in group 1, 3 patients received 20 × 106 cells in group 2 and 3 patients received 60 × 106 cells in group 3. No patient stopped the treatment because of side effects. Four patients with progressive disease were withdrawn from the study in order to start a new treatment (patients 0301 (fostemustin), 0501 (dabrafenib), 0103 (ipilimumab) and 0403 (nivolumab)). Three of them (0301, 0401, and 0203) died very soon after treatment switch, but patient 0501 was still alive at week 48. In addition, three other patients (0101, 0403, and 0601) were also alive at week 48.
Table 1.
Overall demographics and activity.
| ID | Age (years) | sex | TNM | Stage (AJCC 2009) | Nb of prior systemic treatments | Prior immunotherapy | Cohort | Dose (cells) | Clinical response RECIST♣ | Clinical response IrRC♣ | Melan A immune response (fold increase) | Clinical outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0101 | 75 | F | T4N3M1 c | IV | 3 | - | 1 | 4x106 | PD | PD | 1.17 ♦ | alive |
| 0201 | 80 | F | T3N2bM1 c | IV | 1 | - | 1 | 4x106 | PD | PD | 30* | D (32) |
| 0301 | 35 | M | T4bN1bM1 c | IV | 2 | ICI(2) | 1 | 4x106 | PD | PD | 1.1 | D (9) |
| 0401 | 61 | M | T4bN3M1 c | IV | 1 | - | 2 | 20x106 | PD | PD | 5.4* | D (26) |
| 0501 | 84 | F | T2xN3M1a | IV | 2 | IFN AD | 2 | 20x106 | PD | PD | NI | alive |
| 0103 | 72 | M | T3bN2bM1a | IV | 1 | IFN AD, VACCIN | 2 | 20x106 | PD | - | NI | D (20) |
| 0203 | 76 | F | T3aN1bM1a | IV | 6 | VACCIN(2), TIL, ICI(2) | 3 | 60x106 | PD | PD | 1 | D (18) |
| 0403 | 79 | M | T4bN2cM1b | IV | 1 | - | 3 | 60x106 | SD | - | NI | alive |
| 0601 | 79 | F | T1xN3M1a | IV | 2 | - | 3 | 60x106 | SD | SD | 1.8 | alive |
ICI: immune checkpoint inhibitor
IFN AD: interferon alpha adjuvant
TIL: tumor-infiltrating lymphocytes
D (): death (weeks post first vaccine injection)
NI: not interpretable
*: significant increase associated with naive/memory T cell switch
♦: Fold increase calculated from W1, since W0 data were missing
♣: RECIST and IrRC results at the end of patient follow-up
Product safety
All nine patients reported adverse events (AEs) during the study. As shown in supplementary figure 2, the most frequently reported adverse events were general disorders and administration site events, i.e. asthenia and pain. Other frequent AEs were nausea, abdominal pain, ascites, gastroeosophageal reflux disease, chest pain, bronchitis, decreased appetite, hemoptysis, dyspnea, and vertigo. AEs were distributed roughly evenly among all three groups of patients with 20 (33%), 14 (23%) and 26 (43%) events for Groups 1, 2 and 3, respectively (Figure 2(a)).
Figure 2.
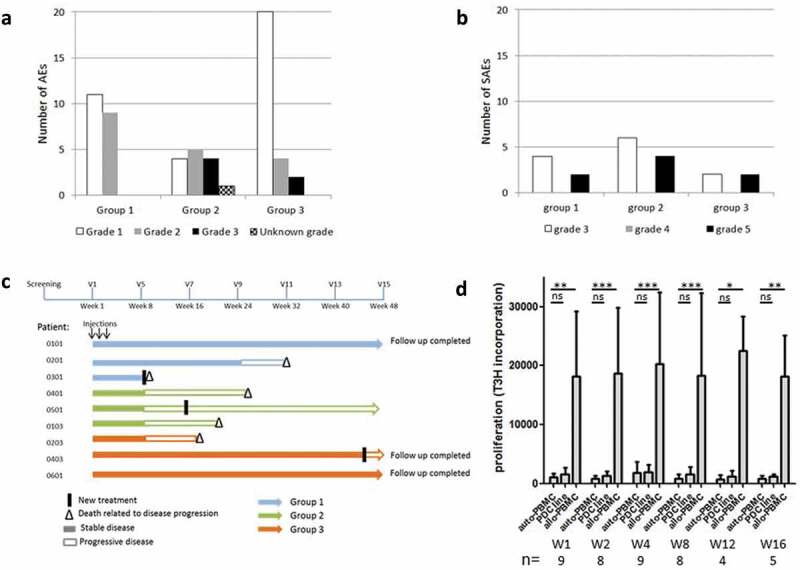
Toxicity profile, patients follow-up and alloimmunization.
The total number of adverse events (a) and severe adverse events (b) in all areas is shown, according to the group of patients. Patients in Group 1, 2 and 3 received three injections of 4, 20 and 60 × 106 cells of the vaccine, respectively. (c) Swimmer plot displaying overall survival, clinical response (IrRC), treatment dose and treatment switch in patients, over the 48 weeks of follow-up. Each bar represents one patient (n = 9). (d) Allo-immunization in patients was assessed by mixed lymphocyte cultures, analyzing the proliferation (3H-Thymidine incorporation) of patients’ PBMC after a 6-day coculture with the irradiated PDC line (1:1 ratio). The proliferation of PBMC with irradiated autologous PBMC was the negative control (medium), and proliferation against allogeneic irradiated PBMC from an unrelated healthy volunteer (allo-PBMC) was used as a positive control. The significance was evaluated by the Friedman test, followed by Dunn’s multiple comparison post-test.
Twenty serious adverse events in 8 patients were reported during the study. They did not appear to be linked to the administered dose of the DP (Figure 2(b)). The relationship to study treatment was assessed for all SAEs. In patient 0103, two SAEs were considered related to study treatment: pain (W7, grade 3), lymphadenitis (W7, grade 3); the two other SAEs were probably not caused by the treatment: pulmonary embolism (W14, grade 5), and superficial thrombophlebitis (W14, grade 5). Three other SAE had an unknown relationship to study treatment (skin necrosis leading to lesion excision (patient 0101, W20, grade 3) and nervous system disorder (patient 0201, W26, grade 3)). All other SAEs were considered unrelated. Among the 20 SAEs, 12 were rated severe (grade 3) and 8 were associated with the death of 5 patients (grade 5).
Clinical activity
Five out of the nine patients had been previously treated by two or more lines of treatment, and four had already received immunotherapies (anti-CTLA-4, anti-PD-1, type I IFN, or vaccine). Taking into account the poor prognosis of these patients with stage IV melanoma, it is not possible to evaluate whether the GeniusVac-Mel4 vaccine provided a clinical benefit. Five patients out of nine died of disease progression (2, 2 and 1 patient in group 1, 2 and 3, respectively). These deaths were linked to disease progression and not to the study treatment (Figure 2(c)). One death occurred early (W9) for patient 0301 who had pulmonary metastases at inclusion, and relapsed after ICI treatment, the other deaths occurred between week 18 and week 32. According to Recist 1.1 criteria, two patients (0403 and 0601) from group 3 (60x106 cells) had a stable disease throughout the study. For these patients, the disease progressed at week 44 and 48, respectively, and they were treated with ICI. According to IrRC criteria, three patients had stable disease for 16 to 24 weeks, and one patient remained with the stable disease until week 48. For patient 0101 (group 1), the progression of some lesions was observed at week 16, while others regressed (see one example in Figure 3(a)), and no new cancer treatment was initiated. Patient 0101’s vitiligoid lesions on cutaneous metastases worsened, while new vitiligoid lesions appeared on a non-tumor area in patient 0501 at W3, suggestive of an anti-melanocyte immune response. These two patients were still alive 48 weeks after treatment initiation.
Figure 3.
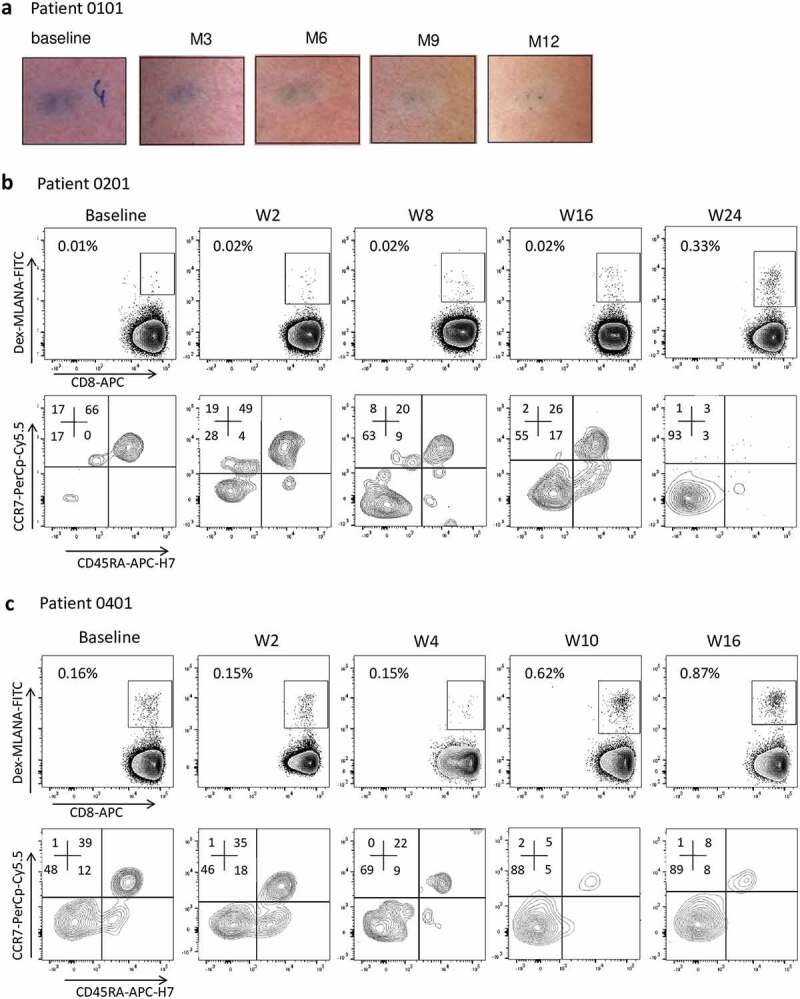
Clinical and immunological responses in patients treated with GeniusVac-Mel4.
(a) Example of a skin metastasis regression in patient 0101. A zone of depigmentation around the metastasis can be observed, characteristic of vitiligo development. (b and c) Immunomonitoring on fresh PBMC showing the evolution of anti-MLANA T cell percentages among CD45+CD3+CD8+ T cells in patients 0201 (group 1) and 0401 (group 2). Specific T cells were analyzed by flow cytometry, with dextramer labeling. Specific T cells were further characterized in terms of naïve/memory profile, according to their differential expression of CCR7 and CD45RA.
Measurement of anti-vaccine immune response
The presence of an allogeneic response directed toward the vaccine was evaluated. Specific anti-HLA class I and class II antibodies were measured with the Luminex technology and no antibody directed against HLA molecules expressed by PDC line could be detected (not shown). In other experiments, cellular immune responses directed against PDC line were measured by MLR. The low proliferation observed when patients’ PBMC were cultured with PDC line was not different from the negative control condition (autologous irradiated PBMC), and a strong cell proliferation was observed against allogeneic PBMC from unrelated donors (positive control) (Figure 2(d)).
We conclude that patients did not mount any detectable alloresponse to the vaccine even at the highest dose used (60x106 of cells) and with repeated injections (3 times in 3 weeks).
Analysis of cell subset variations was performed by flow cytometry immunophenotyping on fresh blood samples, and the percentages of T cells (CD4+, CD8+, and regulatory T cells), B cells, dendritic cells (myeloid and plasmacytoid), natural killer cells, and monocytes were measured. These cell subsets remained mostly stable all along with the trial follow-up (supplementary figure 1).
We then evaluated the specific responses against the four antigenic peptides presented by PDC line by flow cytometry, using multimer staining of peripheral blood, without any prior in vitro activation or expansion step. The limit of detection of the peptide-specific T-cell frequency was equal to 0.01% of the CD8+ T cells. Due to this limit, anti-MAGEA3 and anti-PMEL T cells were not detectable, and anti-TYR T cells were detected in only one patient (0601, Group 3). In this patient, the vaccine-induced a 1.9 fold increase of the Ag-specific T cells when compared to baseline, at two time points (W8 and W36, not shown).
“As expected, T cells directed toward MLANA peptides were the easiest to detect, with 6 out of 9 patients with detectable anti-MLANA T cells (Table 1, supplementary fig 3). Among these six patients, 2 (0401 and 0201) had a significant increase of anti-MLANA T cells compared to baseline: x5.4 and x30, respectively (Figure 3(b,c)). Interestingly, as shown in Figure 3(b,c), these cells switched from naïve to memory phenotype at two time points at least (patient 0201 from 17% memory at baseline to 63%, 55%, and 93% at weeks 8, 16 and 24, respectively; and patient 0401 from 48% to 69%, 88%, and 89% at weeks 4, 10 and 16, respectively; see gating strategy supplementary figure 1).
For patient 0101, a progressive ulcerative cutaneous metastasis was resected at week 20 and analyzed by histochemistry and flow cytometry. Immunohistological examination (Figure 4(a)) shows a dermal metastasis surrounded by fibrotic tissue and displaying a pattern of “non-infiltrated melanoma.” Despite the presence of necrosis, only a few CD163+ macrophages and CD3 + T lymphocytes were observed in the tumor bed. Immune cells were more numerous at the periphery of the tumor, in the fibrotic zone surrounding the tumor. A small proportion of T lymphocytes were CD8 + T cells, and HLA-class I molecules were expressed by the tumor cells. Interestingly, melanoma cells and macrophages in the center of the metastasis expressed lower levels of PD-L1 than the tumor cells and macrophages in the invasive margin. PD-1 was mostly negative, with however rare clusters of positive lymphocytes in the invasive margin (see arrows).
Figure 4.
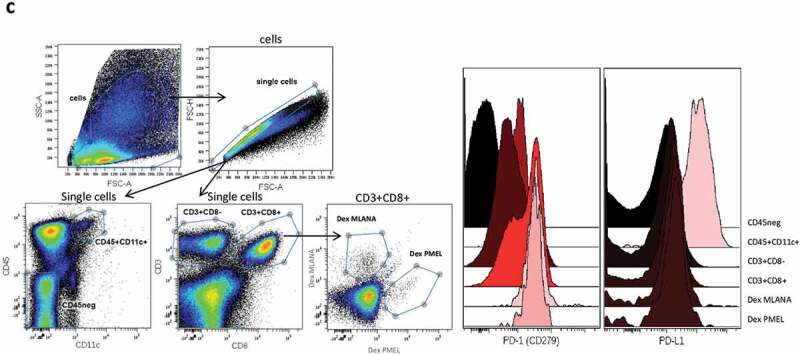
Continued.
Figure 4.
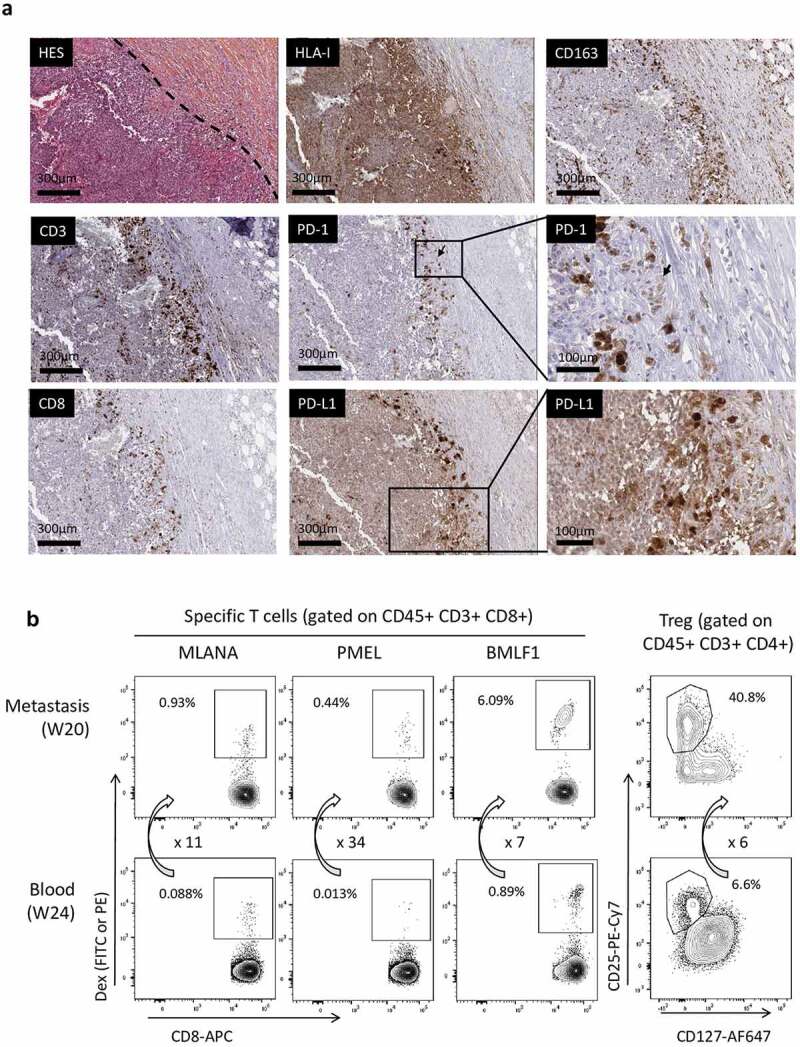
On-treatment metastasis (week 20) analysis in patient 0101.
(a) Immunohistochemical analysis of the paraffin-embedded tissue showing the low intratumoral infiltration by lymphocytes, which are more numerous in the fibrotic area surrounding the tumor. PD-1 was expressed at a low level on rare and small clusters of lymphocytes, while PD-L1 was found expressed at high levels by cells located at the tumor margin. (b) Comparison of specific T cell proportions in a cutaneous metastasis (Week 20) and in the blood (week 24) of patient 0101. The tumor was mechanically and enzymatically dissociated and immunophenotyping was performed on the freshly obtained cell suspension. Specific T cells (directed against MLANA, PMEL, or the BMLF1 EBV antigen) percentages were measured on gated CD45+CD3+CD8+ T lymphocytes. Arrows and associated numbers represent the fold increase of specific T cells or Treg cells in the metastasis as compared to blood. (c) Frozen cells from this metastasis were analyzed by flow cytometry for PD-1 and PD-L1 expression, focusing on anti-MLANA and anti-PMEL T cells and on CD11 c+ myeloid cells. Left panels show the gating strategy and right histograms show the differential expression of PD-1 and PD-L1 by the various cell types.
The analysis of the metastasis by flow cytometry indicated that anti-MLANA, -PMEL, and -BMLF1 T cells were highly enriched as compared to the blood collected at week 24 (fold increases of 10.6, 33.8, and 6.8, respectively, Figure 4(b)) or at week 16 (not shown). The enrichment appeared to be higher for the anti-tumor than the antiviral T cells, suggesting a preferential recruitment of the anti-vaccine T cells into the tumor bed. In accordance with immunohistochemistry, CD8+ T cells represented 40% of T cells in the metastasis (not shown) and a high proportion of T cells were Treg cells (41% of CD4+ T cells in the tumor as compared to 6.6% in the blood). An evaluation of PD-1 and PD-L1 expression was also performed on thawed cells from this metastasis, focusing on anti-MLANA and anti-PMEL T cells and on myeloid cells (CD11 c+ CD45+). As shown in Figure 4(c), PD-1 was highly expressed by the anti-vaccine T cells as compared to the complete CD3+CD8+ population. PD-1 expression was lower on CD11 c+ myeloid cells and on CD3+CD8− cells than on CD3+CD8+ cells. PD-L1 was highly expressed on myeloid CD11 c+ cells. Altogether, these observations suggest that the anti-vaccine T cells that were recruited from the blood into the metastasis could not enter into the tumor bed, and that the PD-1/PD-L1 axis may block the antitumoral activity of the T cells primed or restimulated by GeniusVac-Mel4.
Ex vivo evaluation of peptide-loaded PDC line and anti-PD-1 combination
We used HLA-A*02:01 BMC from healthy volunteers (n = 14) and melanoma patients (n = 12) to analyze PD-1 expression on anti-MLANA T cells. PD-1 was expressed on a significantly higher proportion of anti-MLANA T cells of melanoma patients (mean = 37.3%) than on bulk CD8+ T cells (21.3%, p = .046) or anti-MLANA T cells of healthy volunteers (14.4%, p = .014) (Figure 5(a)).
Figure 5.
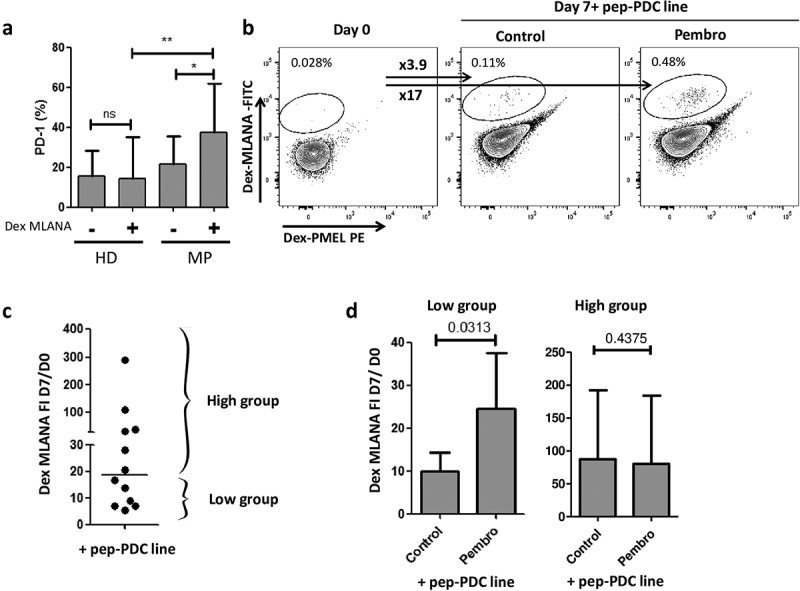
Toward a combination of GeniusVac with immune checkpoint inhibitors.
(a) Basal PD-1 expression (percentage of cells expressing PD-1) was measured by flow cytometry on whole CD8+ T cells and on anti-MLANA T cells of healthy donors (HD, n = 14) and melanoma patients (MP, n = 12). Statistical significance was evaluated by Wilcoxon signed-rank test for comparison of PD-1 expression between anti-MLANA and non-MLANA-specific T cells, and by Kruskal–Wallis test with Dunn’s Multiple Comparison test for comparison of PD-1 expression between melanoma and HD cells. (b) Example of anti-MLANA T cell amplification after a 7-day coculture of PBMC from one melanoma patient with the peptide-loaded irradiated PDC line (10:1 ratio), in the presence of IL-2, with or without blocking anti-PD-1 (pembrolizumab, rightmost plot). FI is the calculated fold increase of specific T cells between day 0 and day 7 and is labeled as ‘xFI’ (arrows). (c) Each dot represents the FI for one coculture, bar is set at the median (= 17), and is used to separate the cultures that displayed low vs high amplification of specific T cells when PBMC from melanoma patients (n = 12) were stimulated with the peptide-loaded PDC line alone. (d) Histograms display the mean with a standard deviation of the fold increase FI, for the two groups of patients defined in (c), upon stimulation by peptide-loaded PDC line without (control) or with Pembrolizumab (Pembro). The significance was evaluated by the Mann–Whitney test, indicating that there is a benefit of combining the PDC line with anti-PD-1 when the proliferation induced by the PDC line alone is lower.
The benefit of combining peptide-loaded PDC line with anti-PD-1 (Pembrolizumab) was evaluated by measuring the amplification of anti-MLANA T cells of 12 melanoma patients after a 7-day coculture (in the presence of IL-2). Of note, the culture duration (7 days) was not sufficient to detect anti-PMEL T cells (limit of detection = 0.01%). As shown in an example (Figure 5(b)), we observed a higher amplification of anti-MLANA T cells with than without PD-1 blockade. The analysis of all cocultures demonstrated that the benefit of PD-1 blockade was higher when the proliferation induced by PDC line alone was low (below the median FI = 17) (Figure 5(b,c)).
Discussion
With the advent of antibodies against immune checkpoints, cancer treatment entered in a new era. However, despite the clinical benefit observed in a large series of cancer indications, few patients respond to monotherapy. Thus, to increase the proportion of patients that could benefit from ICI, many clinical trials are conducted that combine these drugs with other treatments such as chemotherapy, targeted therapy or radiotherapy.23 Since ICI efficacy has been shown to depend on the presence of preexisting antitumor T cells, its combination with antitumor vaccination represents an attractive therapeutic approach.10 Vaccination with tumor-specific antigens can prime antitumor T cells whose restimulation can then be boosted by ICI. Moreover, it ought to increase the ratio between antitumor T cells and antimicrobial or antiself T cells, decreasing the autoimmune and inflammatory side effects of ICI. Diverse vaccine platforms can be used for therapeutic vaccination including proteins/peptides, RNA or DNA, viral-vector-based, tumor cells or dendritic cells-based vaccines.24 Among dendritic cells, plasmacytoid dendritic cells are of great interest.13 In murine models, PDC that were properly loaded with tumor antigens and activated induced strong CTL responses and tumor regression.17,25
To overcome the difficulty of obtaining PDC in autologous settings, we have developed an innovative, off-the-shelf cell product for vaccination, based on an allogeneic HLA-A*02:01 peptide-loaded PDC line. We demonstrated in pre-clinical studies that it acts as a potent professional antigen-presenting cell to prime and expand naïve and memory antigen-specific T-cells.17-19,21 Its efficacy as a therapeutic vaccine for melanoma was evaluated in vivo in humanized mice and ex vivo with cells from melanoma patients.17,21 These results led us to design a first-in-human vaccine clinical trial based on this allogeneic PDC line, irradiated and loaded with tumor-specific and tumor-associated antigenic peptides (GeniusVac-Mel4 clinical trial). As the cell line expresses HLA-A*02:01, the study treatment was restricted to HLA-A*02:01 patients (this allele is present in about 50% of the Caucasian population).
The GeniusVac-Mel4 clinical trial assessed the feasibility, safety, and tolerance of multiple subcutaneous injections of the PDC line vaccine in patients with metastatic melanoma. These objectives were met, as the trial demonstrated that three weekly injections of up to 60 × 106 cells were safe and well tolerated for a long period of time, as some patients were followed for 1 year. This result is in line with those of previously used allogeneic antitumor vaccine modalities.26-29 Interestingly, PDC line induced neither humoral nor cellular allogeneic responses, which opens the possibility to administer higher doses of PDC line or increase the number of injections. The mechanisms responsible for this absence of detectable alloreaction are currently under investigation.
Firm conclusions about clinical responses cannot be drawn from this small first-in-man study but some signs of clinical activity are worth mentioning. Two patients from cohort 3 at the highest dose (60x106 cells) displayed a stable disease (RECIST criteria) up to week 48. Four out of the nine treated patients were still alive after 48 weeks and 2 of them did not need any other anti-cancer treatment. Given that these four patients had metastatic disease, GeniusVac-Mel4 might have slowed tumor progression. In one of these patients (0101), the blood anti-MLANA T cells became progressively enriched in memory cells and high frequencies of anti-MLANA and anti-PMEL T cells were present in the metastasis resected at week 20. As the vitiligoid lesions of this patient concomitantly worsened, the vaccine may have contributed to the favorable clinical course.
The induction of a specific immune response was directly detected ex vivo in 2 out of 6 evaluable patients, associated with a switch from naïve to memory Ag-specific T cells, confirming the immunogenic potential of GeniusVac-Mel4, even with three administrations only. Most therapeutic anticancer vaccine trials comprise many more injections of the drug product15,30-34 over several months.17,30-34 Very few clinical trials have documented ex vivo detection of anti-vaccine T cells, 35-37 as in most cases anti-vaccine T cells were detected after several rounds of restimulation in vitro. In the trial performed by Tel et al. with autologous PDC loaded with PMEL and TYR peptides, the ex vivo detection of anti-vaccine T cells proved negative while 7 of 15 vaccinated patients showed increased frequencies of blood anti-PMEL CD8 + T cells detected after a 2-week in vitro restimulation.15 Here, the direct ex vivo detection of anti-MLANA T cells expansion illustrates the immunogenicity of GeniusVac-Mel4. Admittedly, the frequency of naive CD8+ T cells to the MLANA26-35 peptide presented by HLA-A*02:01 molecules is known to be about 100-fold higher than that of CD8+ T cells to other HLA-peptide combinations, 38 which lowers the threshold of detectability of anti-MLANA responses. Higher sensitivities of T cell response detection might be obtained through TCR repertoire sequencing and analyses of T cells located in tissues. Indeed, we have observed higher percentages of anti-MLANA and anti-PMEL T cells in the metastasis of a patient 0101 than in his blood, indicating recruitment to the tumor or local proliferation.
Despite the demonstrated potency of the PDC line vaccine, it is likely that local or systemic immunosuppression prevents full antitumor T cell activity. Thus, in the metastasis resected from patient 0101, there was a huge proportion of regulatory T cells, and T cells were located at the periphery of the tumor, a high PD-1 expression was detected on the antitumor T cells and PD-L1 was present on myeloid cells. In this context, priming or restimulating antitumor T cells with the vaccine may synergize with ICI.1-3,39,40 In a first evaluation of combined treatment, we explored the synergy between the PDC vaccine and PD-1 blockade on the priming of anti-vaccine T cells. Our results obtained ex vivo with cells from melanoma patients clearly show a higher proliferation of anti-vaccine T cells induced by peptide-loaded PDC line with anti-PD-1 as compared to without anti-PD-1.
The results of this phase I clinical study show that this off-the-shelf PDC line-based vaccine is safe, not rejected and can prime and expand tumor-specific T cells in patients with advanced cancer. While improvements in the drug product and its administration schedule can be envisaged, it will be of great interest to evaluate this vaccine in combination with immune checkpoint inhibitors. Even if this clinical trial addressed only HLA-A*02:01 patients, it is interesting to note that the cell line expresses other HLA molecules like HLA-B*07:02, and could be engineered to express functionally other HLA molecules of interest (manuscript in preparation) to enlarge the target patient population. The numerous advantages of this vaccine approach (easy scaling-up, low cost, ready-to-use) make it suitable for large-scale deployment in several kinds of cancer, in particularly in lung cancer (NCT03970746).
Funding Statement
Proof-of-concept CLARA (GeniusVacMel4), EFS APR 2010-2011, Ligue contre le Cancer: Comité de la Savoie, EFS-RA, Fondation SILAB (TherapDC).
Acknowledgments
We are grateful to the melanoma patients and healthy volunteers who participated in this study. We also thank members from PDC*line Pharma (Isabelle Tabah-Fish, Béatrice Devos, Karine Laulagnier, Clarisse Maurin, Marie Genin, Pedro Alves, Estelle Leplus); from Grenoble-Alpes University Hospital (CHUGA) Dermatology, Biological Resource and Immunology departments (Laure Bondier, Catherine Portal, Pascal Mossuz, Philippe Lorimier, and Marie-Christine Jacob); from the Saint-Ismier Cell Therapy Unit (Marie-Jeanne Richard, Marie Favrot, Virginie Persoons, Harald Hegelhofer); from Geneva Solid Drug Development (JP Combette, C Deloche); from the Lyon Centre Leon Bérard and Cancer Research Center (P. Casier, C. Caux); and from Grenoble EFS Research and Development Laboratory (Jean-Paul Molens, Alexandre Walencik, Tania Dufeu-Duchesne, David Laurin and all the team).
Disclosure of Potential Conflicts of Interest
Joel Plumas is Chief Scientific Officer since 2014 of PDC*line Pharma. Pierre Coulie is a member of the Scientific Advisory Board of PDC*line Pharma.
Supplementary material
The supplementary data for this article can be accessed on the Publisher’s website.
References
- 1.Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(7):1019–13. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6(8):827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A, Ugurel S.. Melanoma. Lancet. 2018;392(10151):971–984. doi: 10.1016/S0140-6736(18)31559-9. [DOI] [PubMed] [Google Scholar]
- 5.Ugurel S, Rohmel J, Ascierto PA, Flaherty KT, Grob JJ, Hauschild A, Larkin J, Long GV, Lorigan P, McArthur GA, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies-update 2017. Eur J Cancer. 2017;83:247–257. doi: 10.1016/j.ejca.2017.06.028. [DOI] [PubMed] [Google Scholar]
- 6.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faramarzi S, Ghafouri-Fard S.. Melanoma: a prototype of cancer-testis antigen-expressing malignancies. Immunotherapy. 2017;9(13):1103–1113. doi: 10.2217/imt-2017-0091. [DOI] [PubMed] [Google Scholar]
- 8.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aspord C, Leccia MT, Charles J, Plumas J. Plasmacytoid dendritic cells support melanoma progression by promoting Th2 and regulatory immunity through OX40L and ICOSL. Cancer Immunol Res. 2013;1(6):402–415. doi: 10.1158/2326-6066.CIR-13-0114-T. [DOI] [PubMed] [Google Scholar]
- 10.Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11(1):8. doi: 10.1186/s13045-017-0552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parmiani G, Russo V, Maccalli C, Parolini D, Rizzo N, Maio M. Peptide-based vaccines for cancer therapy. Hum Vaccin Immunother. 2014;10(11):3175–3178. doi: 10.4161/hv.29418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-Cerdeira C, Carnero Gregorio M, Lopez-Barcenas A, Sanchez-Blanco E, Sanchez-Blanco B, Fabbrocini G, Bardhi B, Sinani A, Guzman RA. Advances in immunotherapy for melanoma: a comprehensive review. Mediators Inflamm. 2017;2017:3264217. doi: 10.1155/2017/3264217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schettini J, Mukherjee P. Physiological role of plasmacytoid dendritic cells and their potential use in cancer immunity. Clin Dev Immunol. 2008;2008:106321. doi: 10.1155/2008/106321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, Bhardwaj N. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 2003;101(9):3520–3526. doi: 10.1182/blood-2002-10-3063. [DOI] [PubMed] [Google Scholar]
- 15.Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, Boerman OC, Croockewit S, Oyen WJG, van Rossum M, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013;73(3):1063–1075. doi: 10.1158/0008-5472.CAN-12-2583. [DOI] [PubMed] [Google Scholar]
- 16.Iribarren K, Bloy N, Buque A, Cremer I, Eggermont A, Fridman WH, Fucikova J, Galon J, Špíšek R, Zitvogel L, et al. Trial Watch: immunostimulation with Toll-like receptor agonists in cancer therapy. Oncoimmunology. 2016;5(3):e1088631. doi: 10.1080/2162402X.2015.1088631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aspord C, Charles J, Leccia M-T, Laurin D, Richard M-J, Chaperot L, Plumas J. A novel cancer vaccine strategy based on HLA-A*0201 matched allogeneic plasmacytoid dendritic cells. PLoS One. 2010;5(5):e10458. doi: 10.1371/journal.pone.0010458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aspord C, Laurin D, Richard MJ, Vie H, Chaperot L, Plumas J. Induction of antiviral cytotoxic T cells by plasmacytoid dendritic cells for adoptive immunotherapy of posttransplant diseases. Am J Transplant. 2011;11(12):2613–2626. doi: 10.1111/j.1600-6143.2011.03722.x. [DOI] [PubMed] [Google Scholar]
- 19.Martinet J, Leroy V, Dufeu-Duchesne T, Larrat S, Richard MJ, Zoulim F, Plumas J, Aspord C. Plasmacytoid dendritic cells induce efficient stimulation of antiviral immunity in the context of chronic hepatitis B virus infection. Hepatology. 2012;56(5):1706–1718. doi: 10.1002/hep.25879. [DOI] [PubMed] [Google Scholar]
- 20.Plumas J, Alve sP, Chaperot L, Hannani D. Priming and expansion of neo-antigen specific-T cells thanks to an off-the-self cell-based drug product. Ann Oncol. 2017;28:xi6–xi29. In: Oncology Ao, editor. ESMO Immuno-Oncology Congress 2017. [Google Scholar]
- 21.Aspord C, Leccia MT, Salameire D, Laurin D, Chaperot L, Charles J, Plumas J. HLA-A(*)0201(+) plasmacytoid dendritic cells provide a cell-based immunotherapy for melanoma patients. J Invest Dermatol. 2012;132(10):2395–2406. doi:S0022-202X(15)35489-0[pii]10.1038/jid.2012.152. [DOI] [PubMed] [Google Scholar]
- 22.Aspord C, Charles J, Leccia MT, Laurin D, Richard MJ, Chaperot L, Plumas J. GENiusVac, a novel antitumor vaccine strategy based on allogeneic plasmacytoid dendritic cells. Rev Med Int. 2011;32(5):329–332. doi: 10.1016/j.revmed.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 23.Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, Dong H. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Front Immunol. 2018;9:1739. doi: 10.3389/fimmu.2018.01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013;119:421–475. doi: 10.1016/B978-0-12-407190-2.00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Domizio J, Demaria O, Gilliet M. Plasmacytoid dendritic cells in melanoma: can we revert bad into good? J Invest Dermatol. 2014;134(7):1797–1800. doi: 10.1038/jid.2014.155. [DOI] [PubMed] [Google Scholar]
- 26.Srivatsan S, Patel JM, Bozeman EN, Imasuen IE, He S, Daniels D, Selvaraj P. Allogeneic tumor cell vaccines: the promise and limitations in clinical trials. Hum Vaccin Immunother. 2014;10(1):52–63. doi: 10.4161/hv.26568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anguille S, Willemen Y, Lion E, Smits EL, Berneman ZN. Dendritic cell vaccination in acute myeloid leukemia. Cytotherapy. 2012;14(6):647–656. doi: 10.3109/14653249.2012.693744. [DOI] [PubMed] [Google Scholar]
- 28.Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15(7):e257–67. doi: 10.1016/S1470-2045(13)70585-0. [DOI] [PubMed] [Google Scholar]
- 29.Schmitt A, Hus I, Schmitt M. Dendritic cell vaccines for leukemia patients. Expert Rev Anticancer Ther. 2007;7(3):275–283. doi: 10.1586/14737140.7.3.275. [DOI] [PubMed] [Google Scholar]
- 30.Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 31.Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, Jassem J, Yoshimura M, Dahabreh J, Nakayama H, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(6):822–835. doi: 10.1016/S1470-2045(16)00099-1. [DOI] [PubMed] [Google Scholar]
- 32.Quoix E, Ramlau R, Westeel V, Papai Z, Madroszyk A, Riviere A, Koralewski P, Breton J-L, Stoelben E, Braun D, et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol. 2011;12(12):1125–1133. doi: 10.1016/S1470-2045(11)70259-5. [DOI] [PubMed] [Google Scholar]
- 33.Andtbacka RH, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, Amatruda T, Zager JS, Cranmer L, Hsueh E, et al. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM Phase III clinical trial. Ann Surg Oncol. 2016;23(13):4169–4177. doi: 10.1245/s10434-016-5286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel PM, Ottensmeier CH, Mulatero C, Lorigan P, Plummer R, Pandha H, Elsheikh S, Hadjimichael E, Villasanti N, Adams SE, et al. Targeting gp100 and TRP-2 with a DNA vaccine: incorporating T cell epitopes with a human IgG1 antibody induces potent T cell responses that are associated with favourable clinical outcome in a phase I/II trial. Oncoimmunology. 2018;7(6):e1433516. doi: 10.1080/2162402x.2018.1433516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini J-C, Romero P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 2005;115(3):739–746. doi: 10.1172/JCI23373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith JW, Walker EB, Fox BA, Haley D, Wisner KP, Doran T, Fisher B, Justice L, Wood W, Vetto J, et al. Adjuvant immunization of HLA-A2-positive melanoma patients with a modified gp100 peptide induces peptide-specific CD8+ T-cell responses. J Clin Oncol. 2003;21(8):1562–1573. doi: 10.1200/JCO.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Zippelius A, Pittet MJ, Batard P, Rufer N, de Smedt M, Guillaume P, Ellefsen K, Valmori D, Liénard D, Plum J, et al. Thymic selection generates a large T cell pool recognizing a self-peptide in humans. J Exp Med. 2002;195(4):485–494. doi: 10.1084/jem.20011658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dammeijer F, Lau SP, van Eijck CHJ, van der Burg SH, Aerts J. Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev. 2017;36:5–15. doi: 10.1016/j.cytogfr.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 40.Collins JM, Redman JM, Gulley JL. Combining vaccines and immune checkpoint inhibitors to prime, expand, and facilitate effective tumor immunotherapy. Expert Rev Vaccines. 2018;17(8):697–705. doi: 10.1080/14760584.2018.1506332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.