

Abstract
The basidiomycete Cryptococcus neoformans is not only a clinically important pathogen, but also a model organism for studying microbial pathogenesis and eukaryotic biology. One key factor behind its rise as a model organism is its genetic amenability. The widely used methods for transforming the C. neoformans species complex are Agrobacterium-mediated transformation (AMT) for random insertional mutagenesis and biolistic transformation for targeted mutagenesis. Electroporation was introduced to C. neoformans in early 1990s. Although electroporation is economic and yields a large number of transformants, introduced DNA rarely integrates into cryptococcal genome, which limits its use. Biolistic transformation, although costly and inefficient, has been the only method used in targeted mutagenesis in the past two decades. Several modifications, including the use of a donor DNA with split markers, a drug-resistant selection marker, and a recipient strain deficient in non-homologous end joining (NHEJ), have since modestly increased the frequency of genome integration and the rate of homologous replacement of the DNA introduced by electroporation. However, electroporation was not the method of choice for transformation until the recent adoption of CRISPR-Cas9 systems. We have developed a Transient CRISPR-Cas9 coupled with Electroporation System (TRACE), which dramatically facilitates targeted mutagenesis in the Cryptococcus species complex. TRACE combines the high transformation efficiency of electroporation with the high rates of DNA integration due to the transiently expressed CRISPR-Cas9. Here, we briefly discussed the history of electroporation for Cryptococcus transformation and provided detailed procedures for electroporation and the cassettes construction of the TRACE system for various genetic manipulations.
Keywords: Electroporation, TRACE, CRISPR-Cas9, Gene deletion, Safe haven, Complementation, Overexpression, Protein tagging, Allele Swap
Introduction
Electroporation is a commonly used method for transformation in microbiology and for transfection in mammalian cells. It uses a high electrical pulse to create temporary pores in cell membranes through which foreign molecules like nucleic acids can pass into cells (Rivera et al., 2014; Ruiz-Díez, 2002). Electroporation was first used to transform a fungal species, the model yeast Saccharomyces cerevisiae, by Hashimoto et al in 1985 (Hashimoto et al., 1985). Electroporation was later adopted to transform other fungal species, such as Neurospora crassa (Chakraborty et al., 1991), Aspergillus niger (Ozeki et al., 1994), and Candida albicans (Samaranayake and Hanes, 2011). Electroporation was the first reported approach to transform the basidiomyceteous fungal pathogen Cryptococcus neoformans (Edman and Kwon-Chung, 1990).
Unlike what has been observed in S. cerevisiae, genome integration of the introduced DNA is rare in C. neoformans. Homologous recombination is even rarer (Edman and Kwon-Chung, 1990; Varma et al., 1992). In the initial electroporation experiments, C. neoformans serotype D auxotrophic strains were transformed with a nutrient selection marker (Chang and Kwon-Chung, 1994; Chang and Kwon-Chung, 1998; Chang et al., 1996; Davidson et al., 2000). The homologous recombination rates among transformants range from 1/100,000 to 1/1000, as most introduced DNA fragments were episomally maintained in this fungus. Consequently, the vast majority of transformants are unstable and DNA introduced by electroporation often gets lost in the absence of selection. Later attempts in serotype A strains for targeted gene deletion using a dominant drug-selection marker modestly increased efficiency in genome integration (Lin et al., 2015). However, even among the few stable transformants, the homologous replacement rates were low (typically 2% –50% in serotype A strains and 1% –4% in serotype D strains) (Lin et al., 2015). The low homologous replacement rates are likely due to a preference for non-homologous end joining (NHEJ) over homologous recombination (HR) in DNA repair during non-division phase in this organism (Arras and Fraser, 2016; Sonoda et al., 2006). It is important to point out that the predominance of episomal maintenance of electroporation-introduced DNA in multiple copies is useful for generating multi-copy genomic libraries (Mondon et al., 2000). Such libraries constructed in the episomally maintained pPM8 shuttle vector, for example, were used successfully to identify multi-copy suppressors (Fox et al., 2003).
Given the high cost of biolistic transformation, the simplicity in electroporation procedure, the low operation cost, and the high transformation efficiency of electroporation in C. neoformans make it imperative to overcome two major problems: non-genome integration and the predominance of NHEJ. To tip the balance towards HR, a recipient strain lacking the CKU80 gene, which encodes a key component in the DNA binding process during NHEJ, was used (Critchlow and Jackson, 1998; Goins et al., 2006; Valencia et al., 2001; Walker et al., 2001). However, Cku80 may have a potential role in stress adaptation and virulence (Liu et al., 2008) and the reliance on a cku80Δ mutant background would restrict isolates that can be genetically manipulated.
A breakthrough in efficient and accurate editing of cryptococcal genome came with the advent of engineering genomes with CRISPR-Cas9. It solves both issues facing cryptococcal transformation by electroporation: a double-stranded break (DSB) created by CRISPR-Cas9 at the designated location promotes integration of donor DNA at that specific locus and the presence of homologous arms in the donor DNA facilitates HR repair at the DSB (Fan and Lin, 2018; Lin et al., 2015; Wang et al., 2016). We developed a Transient CRISPR-Cas9 coupled with Electroporation (TRACE) system to avoid the issue of unwanted Cas9 integration in the genomes of transformants. With TRACE, we can obtain transformants with high frequency of homologous replacement in both serotype A and serotype D strains (Fan and Lin, 2018). In this system, three linear DNA fragments, carrying a single guide RNA (sgRNA) expression cassette, an endonuclease Cas9 expression cassette, and a donor DNA (such as a gene deletion construct), are mixed and co-transformed into Cryptococcus cells by electroporation. In a single gene deletion experiment, the vast majority of the hundreds or thousands of transformants obtained via TRACE have the target gene disrupted (up to 90% in serotype A strain H99 and 70% in serotype D strains XL280 and JEC21) (Fan and Lin, 2018). Lowering the dose of the sgRNA and the Cas9 cassettes, which carry no selection marker, lowers their chance of being integrated into the genome. Besides single gene deletion, we use TRACE for one-step deletion of multiple genes by a single gRNA (Fan and Lin, 2018) or by multiple gRNAs (Hu et al., 2019), gene complementation (Fan and Lin, 2018; Lin et al., 2019), gene overexpression (Lin et al., 2019), promoter swapping, and gene knock-in (e.g. for protein tagging at the native locus). Thus, the high transformation efficiency of electroporation coupled with a high HR rate caused by CRISPR-Cas9 make TRACE an efficient and cost-effective method to manipulate cryptococcal genomes.
Materials
Equipment and Supplies:
Eppendorf Multiporator (the 4308 Electroporation System for eukaryotic cells, bacteria, and yeast cells) or Bio-Rad Gene Pulser Xcell™, 2 mm gap electroporation cuvettes (VWR Signature™ Disposable Electroporation Cuvettes, #89047–208), refrigerated centrifuge (Eppendorf centrifuge 4524R), shaker incubator, spectrophotometer, 50 mL culture tubes, 15 mL culture tubes, 5 mm disruption glass beads (RPI, #9831), disruptor genie (VWR #101454–126), PureLink PCR purification kit (Invitrogen, #K310001), and sterile toothpicks.
Media and Chemicals:
Lysis buffer (100 mM Tris-HCl pH 8.0, 50 mM EDTA, 1% SDS); 7 M ammonium acetate; chloroform; isopropanol; ethanol; Phusion Flash High-Fidelity PCR Master Mix (ThermoFisher, #F548L); GoTaq Green Master Mix (Promega, #M7123); 3 M sodium acetate; 1 M dithiothreitol (DTT) solution; Electroporation Buffer or EB buffer (10 mM Tris-HCl pH 7.5, 1 mM MgCl2, 270 mM Sucrose; filter sterilized using 0.22 μm filter).
YPD medium (2% w/v peptone, 1% yeast extract, 2% glucose, 2% agar) with the selective drug (G418: 100 μg/mL, clonNAT or NAT for simplicity: 100 μg/mL, or hygromycin: 200–250 μg/mL) for the selection of drug resistant transformants; YNB minimal medium (6.7 g/L yeast nitrogen base w/o amino acids, 2% w/v glucose, 2% w/v agar) for the selection of prototrophic transformants.
Methods/Protocols
1. Construction of DNA fragments for the TRACE system
Unlike Aspergillus, Candida, or Saccharomyces species, Cryptococcus species cannot efficiently undergo HR with the donor DNA carrying microhomologous arms (30–60 bp). It requires longer homologous arms (0.5–1 kb). To generate a gene deletion construct, the 5’ and the 3’ homologous arms of the deletion region and a drug selection marker are first amplified separately by PCR using primers that produce overlapping ends. Then, the 5’ homologous arm, the drug marker, and the 3’ homologous arm are fused into a single molecule through a double joint PCR (overlap extension PCR) to generate the donor DNA (Kim et al., 2012; Kim et al., 2009; Yu et al., 2004) (Figure 1C). For gene complementation and gene overexpression, integration of the construct into an identified gene-free “safe-haven” region is recommended to avoid position effects and to minimize the risk of unintended disruption of the genome. Such safe havens are identified in both serotype A and D strains (Arras et al., 2015; Upadhya et al., 2017)(Fan and Lin, in preparation). TRACE can also be easily adopted for knocking in a tag to the 3’ end of the gene of interest at its native locus. In this method, sgRNA is preferentially designed to target a region near the 3’ end of the coding sequence. The donor DNA carrying the tag sequence is designed with homologous arms flanking the tag. To avoid the donor DNA being cleaved by CRISPR-Cas9, a carefully designed sgRNA is required. Sometimes donor DNA with synonymous substitution which removes the PAM site (NGG) is required.
Figure 1. Diagrams of the cassettes used in a TRACE system.
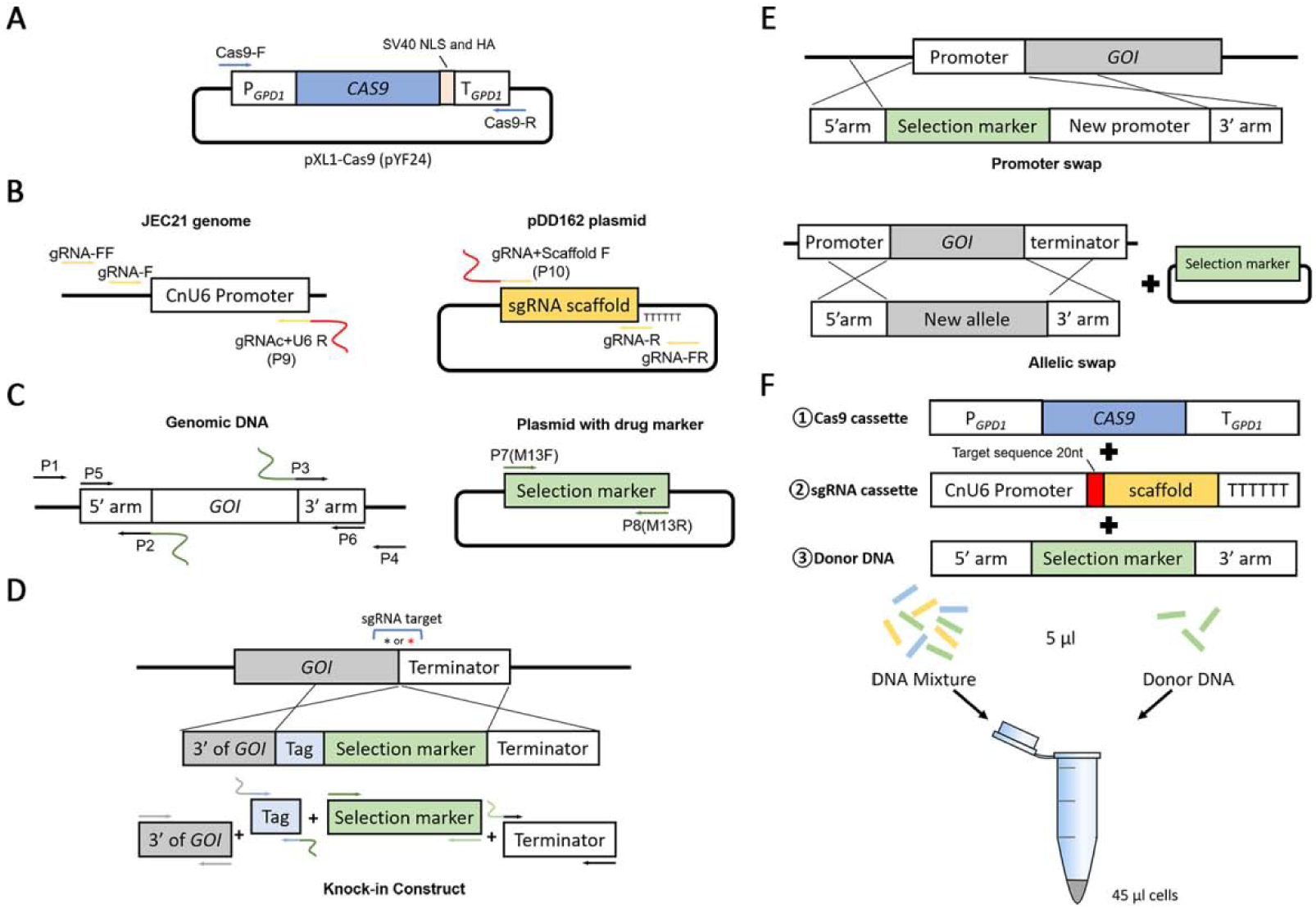
A. The simplified layout of the pXL1-Cas9 (pYF24) plasmid. The Cas9 construct (~7kb) for TRACE is amplified from this vector with the Cas9-F/Cas9-R primer set. B. The gRNA construct is amplified by fusing the U6 promoter and the gRNA scaffold in an overlap extension PCR with primers gRNA-F/gRNA-R. The U6 promoter and the gRNA scaffold are amplified from the genomic DNA of strain JEC21 and the vector pDD162 respectively. C. The gene deletion construct is amplified with the primer set P5/P6 by fusing the 5’ arm, the selection marker, and the 3’ arm in a double-joint PCR. The homologous arms are amplified from the genomic DNA of the parent strain and the drug selection marker is amplified from a vector with the M13F/M13R primer set. D. The native-tagging knock-in construct is generated by fusing 4 fragments: the 3’ end of GOI, the tag cassette, the selection marker, and the downstream of GOI (terminator). Asterisks indicate the locations of the gRNA target sequences. E. Both promoter swap and ORF allelic swap constructs contain two homologous arms in the donor construct. However, in allelic swap, the selection marker needs to be co-transformed into cell separately. F. The linear Cas9, gRNA, and donor DNA are co-transformed into Cryptococcus cells in the TRACE system.
In brief, a TRACE transformation requires Cas9, sgRNA, and a donor DNA construct. Depending on the purpose, the donor DNA could be a gene deletion construct (1.2), a gene complementation or gene overexpression construct (1.3), a knock-in construct for tagging (1.4), or an allele-swapping construct (either coding or non-coding region, 1.5).
1.1. Cas9 and gRNA constructs for TRACE
1.1.1. Preparation of the Cas9 expression cassette
The Cas9 construct with the constitutive promoter PGPD1 can be readily amplified by PCR from the vector pXL1-Cas9 (pYF24) using Cas9-F/Cas9-R primer pair as illustrated in Figure 1A and listed in Table 1 (Fan and Lin, 2018). This construct will be used for every TRACE transformation.
Table 1.
Primers for the CRISPR-Cas9 constructs and screening.
| Cas9-F | GGTGACGCTGTGAGAGTGG | For Cas9 construct |
| Cas9-R | GGGCCCCTCTTCACGTGG | amplification from pXL1- Cas9 (pYF24) plasmid (~7kb product) |
| gRNA-FF | GGCTCAAAGAGCAGATCAATG | For U6 promoter amplification from JEC21 genomicDNA (~700bp product) |
| gRNAcomplementary (Indicated as letter P)-P9 | PPPPPPPPPPPPPPPPPPPPAACAGTATACCCTGCCGGTG | |
| gRNA(indicated as letter N)-P10 | NNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTT | For gRNA scaffold amplification from plasmid pDD 162 (~400bp product) |
| gRNA-FR | CCTCTGACACATGCAGCTCC | |
| gRNA-F | CCATCGATTTGCATTAGAACTAAA AACAAAGCA | For amplification of final gRNA construct (~400bp product) |
| gRNA-R | CCGCTCGAGTAAAACAAAAAAGC ACCGAC | |
| gRNAcomplementary (SH2)-U6R | AGACTCCACAGCCTAAGATCAACAGTATACCCTGCCGGTG | The primers for SH2 gRNA amplification. SH2 is the safe haven locus in H99 strain. The gRNA sequences are in bold. |
| gRNA(SH2)-ScaffoldF | GATCTTAGGCTGTGGAGTCTGTTTTAGAGCTAGAAATAGCAAGTT | |
| Cas9-screen-F | CCATGGACAAAAAATACAGC | For screening the presence of Cas9 construct (~1.7kb product) |
| Cas9-screen-R | CTTGAAGTAGTCCTCCTTGAG |
1.1.2. Preparation of single guide RNA expression cassette
To make the sgRNA construct, the U6 promoter is amplified with primers gRNA-FF/P9 from the genomic DNA of strain JEC21 (Figure 1B). The scaffold/terminator is amplified with primers gRNA-FR/P10 from the vector pDD162 (Figure 1B). The two PCR products are purified, mixed at 1:1 molar ratio, and fused together with a joint PCR to generate the gRNA expression cassette. Sufficient amount of the full sgRNA construct is amplified by nested primer set gRNA-F/gRNA-R using the joint PCR product as the template. Clean the PCR products for transformation.
The gRNA target sequence (upstream of PAM and exclude PAM) can be designed using sgRNAcas9_V3.0_GUI software as described in previous works (Fan and Lin, 2018) with following parameter (sgRNA length: 20 nt; GC%: 40~60%; offset distance of paired-gRNAs: 22~32; and maximum number of mismatches:5). The gRNA target sequence can also be designed using the Eukaryotic Pathogen CRISPR guide RNA/DNA Design Tool (http://grna.ctegd.uga.edu/) embedded in FungiDB. Both tools can provide a list of gRNA target sequences after selecting the type of RNA guided nuclease (SpCas9), the fungal genome (e.g. C. neoformans H99), and target DNA region. Both programs can predict potential off-target sites. In general, gRNA target sequences which start with G and have single specific site in the genome are preferred. Target sequence with the least number of mismatches outside of seed sequence (12–15 nucleotides upstream of PAM sequence) can also be used. Additional G can be added to the 5’ of the designed 20nt target sequence to facilitate gRNA transcription.
Although higher concentrations of the sgRNA and the Cas9 DNA constructs increase the transformation efficiency, too much sgRNA and Cas9 increases their chance of being ectopically integrated into the genome, which should be avoided. We recommend 70–100 ng of the gRNA construct and 100–250 ng of the Cas9 construct for a TRACE experiment.
1.2. Gene deletion construct
1.2.1. Design primers for amplifying the homologous arms.
Six primers (P1 to P6) are designed to make the gene deletion construct as illustrated in Figure 1C and in Table 2. The use of the nested primer set (P5/P6) is not required, but it increases the efficiency and specificity in the fusion or double-joint PCR reactions. P1 and P4 can also be used later for screening transformants by diagnostic PCRs or sequencing. In addition to the sequence designed for amplification of the desired genomic DNA region, P2 and P3 also carry overhang sequences (red in Table 2, green in Figure 1C) which complement primers that are used to amplify the selection marker (M13F and M13R for our drug selection markers).
To make a clean gene deletion mutant, deletion of the whole open reading frame is preferred. However, some genes may overlap with each other. In that case, avoid the overlapped sequences and minimize the risk of disrupting neighbor gene’s ORF or promoter region.
Table 2.
Primer design (deletion of the ADE2 gene in H99)
| Gene locus | Primer | Sequence (5’ to 3’) | Purpose |
|---|---|---|---|
| CNAG_02294 ADE2 | P1 | CTCCGTGTACCACGCTGC | 1st Round of PCR for 5’arm. (pair with P2) |
| P2 | CTGGCCGTCGTTTTAC-CCACCGCCTGAGGATG | 1st Round of PCR for 5’arm. (pair with PI) Red sequence is overhang complement P7 |
|
| P3 | GTCATAGCTGTTTCCTG-GTCGACATTCTGGAGGAGG | 1st Round of PCR for 3’arm. (pair with P4) Red sequence is overhang complement P8 |
|
| P4 | GAAGATGAGGATATTGCCGAAG | 1st Round of PCR for 3’arm. (pair with P3) | |
| P5 | GTGTCGATGGCAGATCTCC | 3rd Round of PCR for full fragment (pair with P6) | |
| P6 | GTCGGTCAAAGGACATCTG | 3rd Round of PCR for full fragment (Pair with P5) | |
| P7 | GTAAAACGACGGCCAG (M13F) | Amplify drug marker, (pair with P8) | |
| P8 | CAGGAAACAGCTATGAC (M13R) | Amplify drug marker, (pair with P7) |
1.2.2. 1st round of PCR: Amplification of the 5’ homologous arm, the selection marker, and the 3’ homologous arm.
The 5’ arm and the 3’ arm are amplified with P1/P2 and P3/P4 from the wild-type genomic DNA. The marker gene that confers resistance to NAT, G418, or hygromycin is amplified from the vector pPZP-NATcc, pPZP-NEO1, or pPZP-HYG2 (Walton et al., 2005). All these drug selection markers can be amplified using the universal M13F and M13R primers from these plasmids. Other plasmids carrying the correct drug selection marker can also be used. In that case, primers other than M13F/M13R and different complementing sequences in primers P2/P3 should be used. Check the quality of the PCR products by electrophoresis. Purify and concentrate the PCR products using a PCR purification kit or other purification methods such as salt precipitation. Purified PCR products can be stored at −20°C for future use.
We use the high fidelity Phusion DNA polymerase to amplify the fragments to minimize the chances of mutations. The extension time depends on the fragment length & efficiency of the polymerase.
The drug selection markers amplified with M13F/M13R primers are universally used for making deletion constructs here.
1.2.3. 2nd round of PCR: Fusion of the selection marker with the 5’ and the 3’ arms.
Prepare a 25 μL Phusion polymerase PCR reaction with a molar ratio of 1:2:1 for the 5’ arm, the selection marker, and the 3’ arm. Do not add any primer. These three DNA
Calculate the volume of each fragment by the following equation:
[e.g. if the 5’ arm is 1kb and the purified PCR conc. is 82.9 ng/μL, then Vol= 1×200/82.9=2.41 μL]
1.2.4. 3rd round of PCR: Amplification of the full-length deletion construct.
Use 1 μL of the above PCR product as the template and amplify the full-length deletion construct with the nested primers P5 and P6 in a 50 μL Phusion polymerase PCR. Check the quality of the products by gel electrophoresis, purify the PCR products, and store the purified DNA for transformation.
1.3. Complementation/Overexpression constructs
Gene complementation or overexpression constructs are often integrated into the genome randomly, which can cause insertions in hot spots and unexpected gene disruption. With the CRISPR-Cas9 system, it becomes relatively easy to integrate such a construct at a specific genetic locus such as a safe-haven locus (Arras et al., 2015; Upadhya et al., 2017). It is found that linear plasmids or PCR constructs, with or without homologous arms, can integrate into the designated DSB site created by CRISPR-Cas9 with TRACE at a relatively high frequency. Thus, gene complementation/overexpression constructs lacking the homologous arms can integrate at the safe haven site with TRACE.
1.3.1. Clone the ORF into a vector for gene complementation or gene overexpression.
Gene of interest can be cloned into a vector carrying a fungal section marker and a proper fungal promoter (e.g. the TEF1 or the GPD1 promoter for gene overexpression, the native promoter for complementation).
1.3.2. PCR amplify the construct or linearize the plasmid.
Design primers and amplify the whole construct (including fungal selection marker) with a high fidelity Phusion polymerase. A linearized plasmid rather than a PCR product can also be used. DNA constructs can be used in TRACE after purification.
The PCR products are preferred over the linearized plasmids as fewer unnecessary DNA sequences will integrate into the Cryptococcus genome. Using a high fidelity polymerase reduces the chance of mutation in PCR products.
1.4. Knock-in constructs
Tagging proteins are used widely in cell biological and biochemical studies to investigate gene functions. For instance, fluorescent protein tagged proteins provide information regarding their subcellular localization. Proteins with fluorescence tags (e.g. GFP, RFP) or epitope tags (e.g. Myc-tag, HA-tag, His-tag, or FLAG-tag) also enable biochemical and microscopic analyses such as western, co-immunoprecipitation, ChIP-DNA seq, and immune-fluorescence tools etc. In addition, tagging the gene of interest at its native locus is often preferred over ectopic expression. Knocking-in a sequence encoding a tag to the native locus of GOI can be easily achieved using CRISPR-Cas9. Here we briefly describe the design of a knock-in construct for TRACE.
1.4.1. Design and construct a knock-in cassette.
The knock-in construct is composed of four parts: two homologous arms, the tag construct and the selection marker (Figure 1D). To knock in the tagging sequence at the 3’ end of the ORF, the 5’ homologous arm is immediately upstream of the stop codon of the gene of interest, not including the stop codon (3’ of GOI in Figure 1D). The 3’ arm is homologous to the downstream sequence of gRNA cutting site. The gRNA recognition site should be designed close to the stop codon. The terminator region is preferred over the ORF region (see 1.4.2 for detail).
Firstly, design primers with overhang for fusion PCR. Make sure that the overhang is complementary to the next fragment specifically (Figure 1D). Secondly, amplify each fragment individually and then use the overlap extension PCR to fuse the 4 fragments into a single knock-in piece using similar procedures as described in step 1.2.3 and 1.2.4. The assembled knock-in construct can be cloned into a vector or used directly for transformation. Sequence the construct to rule out any mutation. Make sure that the tag is in frame with the ORF.
Sometimes, the fusion of 4 fragments may not be efficient. An alternative approach is to first fuse the 5’ arm with tag, and the selection marker with the 3’ arm to generate two bigger fragments. Then fuse the two bigger fragments into one knock-in construct. Other gene assembly methods instead of fusion PCR, such as Gibson assembly, NEBuilder HiFi DNA assembly and Golden Gate assembly can also be used to make the construct.
1.4.2. Site-directed mutagenesis on the knock-in construct.
Because the knock-in construct uses the 3’ sequence of GOI as one homologous arm, the knock-in construct will also have the Cas9 cutting site if sgRNA is designed to target the region before the stop codon (black asterisk in Figure 1D). Cas9 will recognize both the donor knock-in construct and the genome DNA and then generate double strand breaks. In this case, make a point mutation in knock-in construct to remove the PAM sequence.
An alternative approach is to design the gRNA in the terminator of GOI (red asterisk in Figure 1D) and to have the 3’ homologous arm after the PAM. In this way, there is no recognition site of the designed gRNA in the knock-in construct.
1.5. Allele-swapping constructs
Allele exchange/swapping is to replace a region with a similar but different sequences. This is mostly used to study the effect of polymorphisms in the ORF region or the regulatory region of a gene. For allele-swapping, we adopt the same homologous recombination strategy for the donor DNA construction as for gene knockout or gene knock-in. One major difference is the position of the selection marker in the donor DNA. The donor DNA for gene deletion is composed of 3 parts: the 5’ homologous arm, the selection marker, and the 3’ homologous arm in that order (③in Figure 1F). The tagging knock-in construct includes 4 components: the 5’ homologous arm, the tag cassette, the selection marker, and the 3’ homologous arm (Figure 1D). The promoter swapping construct contains 4 parts: the 5’ homologous arm, the selection marker, the desired promoter, and the 3’ homologous arm. If the ORF of gene of interest is swapped with an another allele, then the donor DNA includes the 5’ homologous arm, the allelic ORF, and the 3’ homologous arm. The selection marker in this case should be co-transformed in a separate construct (Figure. 1E). The procedures to construct the donor DNAs is similar to the steps described in sections 1.2 and 1.4.
2. Transformation by Electroporation
2.1. Inoculate 3 mL liquid YPD medium with the recipient cryptococcal strain in a 15 mL culture tube. Incubate the culture overnight (13–16h) at 30°C with shaking at 250 rpm. The initial OD600 of the culture should be around 0.2 based on a spectrophotometer reading.
Use cells freshly grown from −80°C glycerol stocks for transformation. Do not use cells that have been growing on a plate for 3 days or more for transformation.
2.2. Transfer the overnight culture to a 250 mL glass flask containing 100 mL YPD liquid medium and adjust the inoculum to OD600 = 0.2. Incubate at 30 °C with shaking at 250 rpm for 4 hours or longer until the culture reaches the mid-log phase (OD600 = 0.6~1.0). Monitor OD600 of the culture every half an hour after about 3 hours of incubation.
Growth and competency of C. neoformans is highly impaired by Lysol or bleach. Clean the glass flasks thoroughly and avoid using these agents in cleaning the flasks used for electroporation.
2.3. Collect the cells in a 50 mL falcon tube by centrifugation at 4000 rpm at 4°C for 5 min. Remove as much YPD medium as possible.
Pour the supernatant and let the tube with cell pellets stand on ice for 30 seconds. Remove the residual liquid by pipette tips.
2.4. Wash the cell pellet twice with ice-cold sterile ddH2O.
Use at least 30 mL of ice-cold water to thoroughly wash the cell pellet each time. Remove as much of the residual liquid as possible after the last wash.
2.5. Resuspend the cell pellet thoroughly in 10 mL ice-cold EB buffer on ice by pipetting, then add 10 μL of 1M DTT. Gently swirl or invert the cell suspension. Incubate the cell suspension on ice for 30 min to 1 hour.
Make a bulk of EB buffer for long term use. Aliquot a small volume for each experiment. Keep the aliquot EB buffer on ice before experiment.
2.6. Prepare 2 mm gap electroporation cuvettes during the incubation time. Chill cuvettes on ice.
2.7. Collect cells by centrifugation at 4000 rpm at 4°C for 5 min.
Remove as much buffer as possible using pipette tips.
2.8. Gently resuspend cells in 220–250 μL ice-cold EB buffer without DTT by pipetting.
Keep cells on ice during sample preparation. These cells should be enough for 5 TRACE transformation reactions.
2.9. Transfer 45 μL of cells into a pre-chilled 1.7 mL microcentrifuge tube. Add 5 μL of DNA mix to 45 μL of cells and mix them gently (keep cuvettes cold). Transfer the DNA-cell mix to a pre-chilled electroporation cuvette. Make sure that cells sit at the bottom of the cuvette and fill the gap evenly.
For electroporation with a single DNA fragment, use up to 2 μg of DNA.
In the CRISPR-Cas9 system developed by the Zhu’s lab (Wang et al., 2016), 3 μg of linearized plasmids or purified PCR products were used.
In the TRACE system, the mixture of three fragments is used: the sgRNA cassette (~100 ng), the Cas9 cassette (~100 ng), and the donor construct (~2 μg) (Fan and Lin, 2018).
2.10. Electroporation can be done using an Eppendorf multiporator or a BioRad gene pulser.
- Eppendorf multiporator
- Use the Bacterial mode (symbol: an empty circle)
- V=2kv with τ optimized for 5ms
- The time constant value after electroporation should be 3.6–4.1 ms.
- BioRad gene pulser
- 0.45 kV, 125 μF, 400–600 Ω
- (or V=0.48 kv, R=400 ohm, C=250 uFD)
- The time constant value after electroporation should be 30–37 ms.
A range of parameters for a BioRad gene pulser should be tested for an optimal performance in a new setting.
2.11. Add 1 mL of liquid YPD into the cuvette after electroporation and transfer cells to a new 1.7 mL microcentrifuge tube. Incubate cells at 30 °C with shaking at 220 rpm for two hours and then plate cells onto selective medium plates.
The recovering step allows time for the cells to express the selection marker. It is not necessary to have the recovering time if auxotrophic selection markers are used.
Because of high transformation rates, typically only 1/5–1/3 of the cells from the transformation are plated (~200–300 μl out of the 1 mL of the recovered cell suspension), which usually yields ~30–600 colonies. Efficiency may vary in different settings. Figure 2A shows a transformation plate from an ADE2 deletion TRACE experiment.
Figure 2. The stability test of transformants.

A. A picture of an ADE2 gene deletion transformation plate using TRACE. Transformants were grown on YPD plate supplemented with the selection drug. The ade2Δ colonies appear red once the adenine in the media is depleted after several days of growth. B. A diagram shows the replica plating test to screen for stable transformants. Transformants are inoculated onto the master plate and the plate is replicated onto nonselective media for 4 consecutive times. On the fifth passage (P5), cells were replicated onto a selective and a nonselective medium plate. C. The pictures of transformants on a nonselective plate (left) and a selective plate (plate) on passage 5. Transformants that grow robustly on both plates (indicated with black triangles) are considered stable transformants. Those with spotty growth on selective medium are considered non-stable transformants.
2.12. Incubate the plates at 30°C. Colonies should show up in 2–3 days.
3. Stability test and PCR confirmation
3.1. Pick colonies to a new selective medium plate. Incubate them at 30°C for one day. This plate will serve as the master plate for stability test and for further screening of the transformants. The master plate can be stored at 4°C for up to 10 days.
3.2. To test the stability of the transformants, replicate cells onto a fresh non-selective YPD plate each day. On the fifth passage, replicate cells onto a non-selective YPD medium plate and a selective medium plate supplemented with NAT, G418, or hygromycin as shown in Figure 2B. Stable and nonstable transformants can be distinguished visually as shown in Figure 2C.
Colonies that show robust growth on both the non-selective and the selective plates are stable transformants. Spotty growth on the selective medium compared to that on the non-selective medium indicates instability. Colonies of unstable transformants will show cell imprints in some areas on the selective medium but with no or little growth. The majority of the transformants generated from TRACE are stable.
3.3. Pick 8–20 of the stable transformants from the master plate and patch them onto a fresh YPD plate.
3.4. Extract the genomic DNA of the picked transformants for diagnostic PCR screens. (see below for the mini-genomic DNA extraction procedures).
3.5. Verify the transformants by diagnostic PCRs.
3.5.1 For gene deletion, the primer pair P1/P6 can be used for a diagnostic PCR. Both the mutants and the WT strain will yield a positive band, but the products are most likely of different sizes. If the drug selection marker is of the same size as the DNA region being replaced, then a restriction enzyme digestion of the PCR amplicons should be able to distinguish the deletion mutant from the wild type. In addition, verify the lack of the ORF and correct integration of the selection marker at the targeted locus in the candidate transformants by PCRs.
3.5.2 For gene complementation/overexpression at the safe haven site, homologous arms are not required. Any linear construct, including overexpression cassette, the gRNA construct, or the Cas9 construct, has a chance to integrate at the SH site. Use one primer close to the terminus of the overexpression/complementation construct to pair with a primer in the flanking sequence of the safe haven site to verify correct integration. Without homologous arms, the construct can integrate at either forward or reverse direction.
3.5.3 For mutants generated via TRACE, candidates with unwanted Cas9 integration as reflected by positive PCR amplification of the full or a part of the Cas9 construct from the genome should be excluded.
3.6. Save the confirmed candidates in glycerol stocks (15% glycerol) at −80°C.
If the transformation plates contain a lot of transformants, the chance of one visible colony is derived from more than one transformants is increased. In that case, streak the correct candidates on a selective medium plate for single colonies. Confirm these candidates again with PCR.
Make a glycerol stock for the mutant as soon as the genotype is confirmed to minimize the risk of accumulating other mutations or epigenetic changes. If the confirmation process is expected to be long, store the candidates as glycerol stocks first.
3.8. Test the phenotypes of the selected candidates. Use a genetic linkage assay or gene complementation to validate the causative effect of the genetic manipulation and the observed phenotype (Lin et al., 2019; Lin et al., 2010).
4. Cryptococcus genomic DNA mini-preparation
4.1. Grow the selected candidates on YPD agar plate (8–16 candidates/plate). Collect and suspend cells in 1.7 mL microcentrifuge tubes with 500 μL of lysis buffer and add approximate 200 μL of 0.5 mm glass beads.
4.2. Break cells in the disruptor genie for 4 minutes. Centrifuge the tubes at 6000 ×g for 1 minute and transfer the supernatants into new 1.7 mL microcentrifuge tubes with 275 μL of 7 M ammonium acetate. Incubate the lysates at 65°C for 5 minutes and then chill the tubes on ice for 3–5 minutes.
4.3. Add 500 μL of chloroform into each tube and vigorously invert the tubes multiple times before centrifugation at 15000 ×g for 5–10 minutes. Transfer the supernatant into a new microcentrifuge tube and mix with 1 volume of cold isopropanol.
4.4. Precipitate the genomic DNA by centrifugation at 15000 ×g for 10 minutes and then wash DNA once with 700 μL of 70% ethanol. Air dry. Dissolve DNA in 50–100 μL of water.
Discussion
The development of CRISPR-Cas9 systems for genome editing has greatly advanced genetic manipulations in a wide spectrum of species. Several CRISPR-Cas9 transformation systems have been developed for the Cryptococcus species complex (Arras et al., 2016; Fan and Lin, 2018; Wang, 2018). Here we detailed the experimental design and procedures of the transient CRISPR-Cas9 coupled with electroporation system (TRACE) (Fan and Lin, 2018). Some of the procedures that we discussed here could be adapted for other fungal species.
Here we provide examples for single gene deletion, and gene complementation/overexpression, native-locus tagging, and allele swapping. In addition to these applications, TRACE transformation can be used in multi-gene deletion (Fan and Lin, 2018), promoter swapping, and many more (Hu et al., 2019). TRACE has also been successfully used to knockout multiple unrelated genes in one experiment, with a mixture of multiple gRNAs, deletion constructs (with the same drug selection marker), and the Cas9 construct (Hu et al., 2019). TRACE is tremendously useful for deleting multiple genes with only a single selection marker. This is valuable particularly in species where the number of dominant selection markers is limited.
The position of gRNA target sites in Cryptococcus species seems to have little effect on the efficiency of gene deletions, probably due to the long homologous arms present in the donor DNAs. Using gRNAs at the 5’ end, the 3’ end, or in the middle of a 6.4kb gene (ORF is 5.6kb) worked well in gene deletion in Cryptococcus species (Lin et al., 2019).
In addition to the DNA expression cassettes described here, the CRISPR-Cas9 ribonucleoprotein complex has been used in transformation of multiple fungi as well, including Aspergillus fumigatus, Magnaporthe oryzae, and Cryptococcus species (Al Abdallah et al., 2017; Wang, 2018). Using the commercial CRISPR-Cas9 ribonucleoprotein complex will cost more, but it eliminates any potential issues caused by integration of the Cas9 and gRNA construct into the genome.
Table 3:
Fusion of three fragments
| PCR mixture | PCR program | |||
|---|---|---|---|---|
| Fragment 1 | 5’arm (from 1st PCR) | A μL | 95°C, 30s | |
| Fragment 2 | NAT, NEO or HYG(from 1st PCR) | B μL | 95°C, 15s | 20 cycles |
| Fragment 3 | 3’arm (from 1st PCR) | C μL | 50°C, 4 minutes | |
| Enzyme | 2X Phusion Flash PCR Master Mix | 12.5 μL | 72°C, X s | |
| H2O | 12.5-A-B-C μL | 72°C, 5 minutes | ||
| TOTAL | 25 μL | 20°C, ∞ | ||
Funding
This work was supported by National Institutes of Health (http://www.niaid.nih.gov/Pages/default.aspx) (R01AI140719 to XL). Dr. Xiaorong Lin holds an Investigator Award in the Pathogenesis of Infectious Disease from the Burroughs Wellcome Fund (http://www.bwfund.org/)(1012445 to XL). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al Abdallah Q, et al. , 2017. A Simple and Universal System for Gene Manipulation in Aspergillus fumigatus: In Vitro-Assembled Cas9-Guide RNA Ribonucleoproteins Coupled with Microhomology Repair Templates. mSphere. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arras SD, et al. , 2015. A genomic safe haven for mutant complementation in Cryptococcus neoformans. PLoS One. 10, e0122916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arras SD, et al. , 2016. Targeted Genome Editing via CRISPR in the Pathogen Cryptococcus neoformans. PLoS One. 11, e0164322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arras SD, Fraser JA, 2016. Chemical Inhibitors of Non-Homologous End Joining Increase Targeted Construct Integration in Cryptococcus neoformans. PLoS One. 11, e0163049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty BN, et al. , 1991. An electroporation-based system for high-efficiency transformation of germinated conidia of filamentous fungi. Canadian Journal of Microbiology. 37, 858–863. [DOI] [PubMed] [Google Scholar]
- Chang YC, Kwon-Chung KJ, 1994. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol Cell Biol. 14, 4912–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Kwon-Chung KJ, 1998. Isolation of the third capsule-associated gene, CAP60, required for virulence in Cryptococcus neoformans. Infect Immun. 66, 2230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, et al. , 1996. The second capsule gene of Cryptococcus neoformans, CAP64, is essential for virulence. Infect Immun. 64, 1977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow SE, Jackson SP, 1998. DNA end-joining: from yeast to man. Trends in Biochemical Sciences. 23, 394–398. [DOI] [PubMed] [Google Scholar]
- Davidson RC, et al. , 2000. Gene disruption by biolistic transformation in serotype D strains of Cryptococcus neoformans. Fungal Genet Biol. 29, 38–48. [DOI] [PubMed] [Google Scholar]
- Edman JC, Kwon-Chung KJ, 1990. Isolation of the URA5 gene from Cryptococcus neoformans var. neoformans and its use as a selective marker for transformation. Mol Cell Biol. 10, 4538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Lin X, 2018. Multiple Applications of a Transient CRISPR-Cas9 Coupled with Electroporation (TRACE) System in the Cryptococcus neoformans Species Complex. Genetics. 208, 1357–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DS, et al. , 2003. Phospholipid-binding protein Cts1 controls septation and functions coordinately with calcineurin in Cryptococcus neoformans. Eukaryot Cell. 2, 1025–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goins CL, et al. , 2006. Improvements to gene deletion in the fungal pathogen Cryptococcus neoformans: absence of Ku proteins increases homologous recombination, and co-transformation of independent DNA molecules allows rapid complementation of deletion phenotypes. Fungal Genet Biol. 43, 531–44. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, et al. , 1985. A Novel Method for Transformation of Intact Yeast-Cells by Electroinjection of Plasmid DNA. Applied Microbiology and Biotechnology. 21, 336–339. [Google Scholar]
- Hu P, et al. , 2019. Elucidation of the determinant for orchestration of solo unisexual cycle in an important human fungal pathogen. bioRxiv. 867408. [Google Scholar]
- Kim MS, et al. , 2012. Targeted gene disruption in Cryptococcus neoformans using double-joint PCR with split dominant selectable markers. Methods Mol Biol. 845, 67–84. [DOI] [PubMed] [Google Scholar]
- Kim MS, et al. , 2009. An efficient gene-disruption method in Cryptococcus neoformans by double-joint PCR with NAT-split markers. Biochem Biophys Res Commun. 390, 983–8. [DOI] [PubMed] [Google Scholar]
- Lin J, et al. , 2019. Transcription factor Znf2 coordinates with the chromatin remodeling SWI/SNF complex to regulate cryptococcal cellular differentiation. Commun Biol. 2, 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, et al. , 2015. Generation of stable mutants and targeted gene deletion strains in Cryptococcus neoformans through electroporation. Med Mycol. 53, 225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, et al. , 2010. Transcription factors Mat2 and Znf2 operate cellular circuits orchestrating opposite- and same-sex mating in Cryptococcus neoformans. PLoS Genet. 6, e1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu OW, et al. , 2008. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 135, 174–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondon P, et al. , 2000. A novel episomal shuttle vector for transformation of Cryptococcus neoformans with the ccdB gene as a positive selection marker in bacteria. FEMS Microbiol Lett. 187, 41–5. [DOI] [PubMed] [Google Scholar]
- Ozeki K, et al. , 1994. Transformation of Intact Aspergillus niger by Electroporation. Bioscience, Biotechnology, and Biochemistry. 58, 2224–2227. [DOI] [PubMed] [Google Scholar]
- Rivera AL, et al. , 2014. Physical methods for genetic transformation of fungi and yeast. Physics of Life Reviews. 11, 184–203. [DOI] [PubMed] [Google Scholar]
- Ruiz-Díez B, 2002. Strategies for the transformation of filamentous fungi. Journal of Applied Microbiology. 92, 189–195. [DOI] [PubMed] [Google Scholar]
- Samaranayake DP, Hanes SD, 2011. Milestones in Candida albicans gene manipulation. Fungal Genetics and Biology. 48, 858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, et al. , 2006. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst). 5, 1021–9. [DOI] [PubMed] [Google Scholar]
- Upadhya R, et al. , 2017. A fluorogenic C. neoformans reporter strain with a robust expression of m-cherry expressed from a safe haven site in the genome. Fungal Genet Biol. 108, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia M, et al. , 2001. NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature. 414, 666. [DOI] [PubMed] [Google Scholar]
- Varma A, et al. , 1992. Molecular and genetic analysis of URA5 transformants of Cryptococcus neoformans. Infect Immun. 60, 1101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JR, et al. , 2001. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 412, 607–14. [DOI] [PubMed] [Google Scholar]
- Walton FJ, et al. , 2005. Novel gene functions required for melanization of the human pathogen Cryptococcus neoformans. Mol Microbiol. 57, 1381–96. [DOI] [PubMed] [Google Scholar]
- Wang P, 2018. Two Distinct Approaches for CRISPR-Cas9-Mediated Gene Editing in Cryptococcus neoformans and Related Species. mSphere. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. , 2016. A ‘suicide’ CRISPR-Cas9 system to promote gene deletion and restoration by electroporation in Cryptococcus neoformans. Sci Rep. 6, 31145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, et al. , 2004. Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol. 41, 973–81. [DOI] [PubMed] [Google Scholar]