

Abstract
Yes-associated protein (YAP) plays an important role in hepatocarcinogenesis, although the potential role of YAP in non-neoplastic liver diseases remains largely unknown. We report herein that YAP in Kupffer cells (KCs) enhances the production of pro-inflammatory cytokines and promotes the development of non-alcoholic steatohepatitis. Our data show that the expression of YAP is significantly increased in KCs of wild type mice fed HFD. We then generated mice with macrophage/monocyte-specific deletion of YAP (YAPϕKO) or TLR4 (TLR4ϕKO) and the animals were fed high fat diet (HFD) or treated with LPS. Our data showed that YAPϕKO mice fed HFD exhibited lower serum ALT/AST levels and less hepatic inflammation when compared to their littermate controls. LPS treatment induced the accumulation of YAP in KCs in vitro and in mice which was prevented by macrophage/monocyte-specific deletion of TLR4 (TLR4ϕKO). LPS transcriptionally activates YAP via AP-1 in macrophages/KCs. The LPS-induced YAP further enhances the expression of pro-inflammatory cytokines (including MCP-1, TNF-α and IL-6) through YAP association with the TEAD-binding motif in the promoter region of inflammatory cytokines. Forced overexpression of active YAP (YAP5SA) in KCs enhanced the production of pro-inflammatory cytokines. Treatment of HFD-fed mice with verteporfin inhibited Kupffer cell activation, reduced liver inflammation and decreased serum ALT/AST levels. Analyses of liver tissues from NASH patients reveal that YAP is increased in KCs and that the level of YAP in human liver tissues is positively correlated with the expression of pro-inflammatory cytokines.
Conclusions:
This study describes an important role of YAP in Kupffer cells for regulation of liver inflammation in NASH. Our findings suggest that inhibition of YAP may represent a novel and effective therapeutic strategy for NASH treatment.
Keywords: Non-alcoholic fatty liver disease, Inflammation, Lipopolysaccharide
INTRODUCTION
Non-Alcoholic Fatty Liver Disease (NAFLD) is a major cause of chronic liver disease which frequently leads to liver cirrhosis and the development of hepatocellular carcinoma (1). To date, multiple factors including lipotoxicity, endoplasmic reticulum (ER) stress, oxidative stress, gut microbiota, and epigenetic factors have been implicated in the pathogenesis of NAFLD (2–4), although the precise mechanisms underlying NAFLD progression from non-alcoholic fatty liver (NAFL) to non-alcoholic steatohepatitis (NASH) remain to be further defined.
The Hippo pathway and its effector protein Yes-associated protein (YAP), has been implicated in liver regeneration and carcinogenesis (5–8). Notably, mice with liver specific knockout of the Hippo pathway kinase MST 1/2 (which leads to YAP activation) and mice with tetracycline-induced expression of active YAP (YAP S127A) both show liver enlargement and cancer development (9–13). Mechanistically, activated YAP translocates into the nucleus where it binds to the transcription factor TEA domain (TEAD) and promotes the transcription of target genes regulating cell survival, proliferation and stemness (11). Accordingly, proliferation of hepatocytes and expansion of liver progenitor cells have been documented in the livers of MST 1/2-double knockout mice (13). While a proliferative and oncogenic role of YAP is well established, it remains unclear whether YAP is involved in the pathogenesis of non-neoplastic liver diseases. In this context, increased expression of YAP has been reported in hepatic stellate cells (HSCs) (14) and in reactive bile ductules (15) in patients with liver fibrosis, although the functional impact and mechanism of YAP signaling in hepatic cells and diseases remain largely unexplored.
Kupffer cells (KCs) play a critical role in the progression of NAFLD by secreting pro-inflammatory cytokines, including TNF-α among others (16). Gut microbiota-derived endotoxin/lipopolysaccharide (LPS) has been linked to KCs activation and progression of NAFLD (17, 18). Mechanistically, LPS is known to activate KCs toward M1 pro-inflammatory phenotypes through Toll-like receptor 4 (TLR4) (19). Studies have shown that LPS/TLR4 signaling promotes the activation of pro-inflammatory cascades, including the nuclear factor-kB (NF-kB), AP-1 and interferon regulatory factor 3 (IRF3) pathways (18, 20). However, it remains unknown whether LPS/TLR4 signaling in Kupffer cells may influence other signaling molecules leading to NAFLD progression.
In the current study, we report for the first time that YAP signaling in Kupffer cells induces hepatic inflammation through production of pro-inflammatory cytokines (including MCP-1, TNF-α and IL-6) in the setting of NAFLD. We show that YAP is significantly increased in the livers of HFD-fed mice and that Kupffer cells are the predominant liver cells with high level of YAP expression in NAFLD. We then generated mice with macrophage/monocyte-specific deletion of YAP and observed that these mice, when fed HFD, exhibited reduced serum transaminases (AST/ALT) with decreased cytokine production/liver inflammation, in comparison to HFD-fed WT mice. Our further studies demonstrate a novel interplay between LPS and YAP signaling pathways in Kupffer cells which are implicated in NASH progression. We observed that treatment of HFD-fed mice with the YAP inhibitor verteporfin inhibited Kupffer cell activation, reduced liver inflammation, and decreased the levels of AST and ALT. Our results presented in this study suggest that inhibition of YAP may represent an effective therapeutic strategy in NASH treatment.
MATERIAL AND METHODS
Animal experiments.
The mice used in this study (YAPflox/flox, TLR4flox/flox, LysM-Cre, and C57BL/6) mice were obtained from Jackson Laboratory (Bar Harbor, ME). To generate macrophage/monocyte-specific YAP or TLR4 knockout mice, YAPflox/flox or TLR4flox/flox were crossed with LysM-Cre mice, producing LysM Cre+ YAPf/+ or LysM Cre+ TLR4f/+. These mice were subsequently crossed with TLR4f/f or YAP f/f to generate LysM Cre+ TLR4f/f (TLR4ϕKO) or LysM Cre+ YAPf/f (YAPϕKO) mice. The genotype of mice was determined by polymerase chain reaction (PCR) using tail tissues. The primer sequences used for genotyping are listed in Supplementary Table 1. The macrophage/monocyte-specific knockout mice (YAPϕKO or TLR4ϕKO) and age-matched wild type littermate (Cre negative) were fed with normal chow diet (NCD) or high fat diet (60% kal fat) for 12 weeks. For LPS experiment, six week old male C57BL/6 mice were intraperitoneally injected with either LPS (0.25 mg/Kg, daily) or PBS for 3 weeks. All animal studies were performed with the Tulane University IACUC approval.
Reagents and Antibodies.
Mouse diets were obtained from Research Diet, Inc. (New Brunswick, NJ). Verteporfin, SR11302, SP600125, and Bay11–7082 were purchased from Cayman chemical (Ann Arbor, MI). LPS and anti-β-actin antibody were purchased from Sigma (St Louis, MO). The antibodies against YAP, c-jun, c-fos, MST1, MST2, and LATS were purchased from Cell signaling Technology (Beverly, CA). The anti-F4/80, anti-CD14, and anti-rabbit Alexa 594 were purchased from Abcam (Cambridge, MA). pYAP5SA, pmIL-6FL, pMCP-Luc plasmids were obtained from Addgene, and pmTNF-α-GLuc was obtained from Genecopoeia (Rockville, MD).
Imagestream analysis.
To obtain non-parenchymal cells from liver tissues, mouse livers were perfused and digested with collagenase solution. After filtering liver cell suspension, hepatocytes were removed by centrifugation (50g for 2 min), and the non-parenchymal cells were collected. For F4/80 staining, the cells were stained with anti-F4/80 antibody conjugated with FITC. For YAP staining, the cells were stained with rabbit anti-YAP antibody, followed by anti-rabbit Alexa594 secondary antibody. The cells were stained with DAPI for nuclear counterstaining. The stained single cells were gated and analyzed by Amnis Imagestream X (Millipore).
Kupffer cell isolation and transfection.
Mouse primary Kupffer cells were separated by gradient centrifugation as previously described (21). Briefly, the mouse liver was perfused and digested with collagenase solution. The liver was further digested with DMEM containing 1% collagenase in a 37°C water bath for 30 min in vitro. After filtering the liver cells suspension, the cells were centrifuged twice at 50 g for 2 min to remove hepatocytes. The hepatic non-parachymal cells were further centrifuged at 400 g for 10 min. The cell pellets were resuspended with percoll solution and centrifuged at 1600 g for 17 min without brake. The Kupffer cell layers (between 25% and 50% percoll gradient) were collected, washed with PBS, and resuspended in DMEM containing 10% FBS and 100 U/ml Penicillin/Streptomycin. The cells were cultured in 6-well plate at 37°C. After 2 hours, non-adherent cells were removed by aspiration and adherent cells (Kupffer cells) were washed with PBS three times. The isolated Kupffer cells were identified by immunofluorescence staining with anti-F4/80 (Abcam). For transfection, the cells were seeded in 6-well plates and 2 μg of YAP5SA plasmid or control plasmid (pcDNA) per well was transfected using Targefect-Raw according to the manufacturer’s instruction (Targeting system).
RNA isolation and Quantitative RT-PCR.
Total RNA was isolated from KCs or liver tissues using Trizol (Invitrogen) following manufacturer’s instructions, and 1 μg of RNA was reverse transcribed into cDNA using iScript Reverse Transcription Supermix (Bio-Rad). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed with an SsoAdvanced SYBR Green Supermix (Bio-Rad) using Bio-Rad CFX96 Real-time PCR detection system. The primer sequences used in this study are listed in Supplementary Table 1.
Immunohistochemistry.
Liver tissues were fixed with 10% formalin and embedded in paraffin. Parffin-embedded sections (4μm) were stained with hematoxylin and eosin using standard protocol. Immunohistochemistry was performed as we previously described (21). Primary antibodies against YAP (Cell Signaling Technology) and F4/80 (Abcam) were used for immunohistochemical staining.
Immunofluorescence staining.
Primary Kupffer cells were fixed with 4% paraformaldehyde for 15 min at room temperature and washed with PBS three times. After blocking, the cells were incubated with the anti-YAP antibody (Cell signaling) for overnight at 4°C. The cells were then rinsed three times in PBS and incubated with the secondary antibody (Alexa Fluor® 594-conjugated anti-rabbit IgG) for 1–2 hours at room temperature. After washing with PBS, the cells were counterstained with DAPI (Roche) for 5 min and visualized using fluorescent microscopy.
Chromatin Immunoprecipitation (ChIP) assay.
The ChIP assay was performed using the Simplechip Enzymatic chromatin IP kit (Cell signaling Technology, Billerica, MA) according to the manufacturer’s instruction. Briefly, the cells were cross-linked with 37% formaldehyde and the nuclei were subsequently extracted. After fragmentation by sonication and enzymatic digestion, the DNA-protein complex was subjected to immunoprecipitation with 8 μg of anti c-jun, anti-YAP antibody (Cell signaling Technology) or rabbit IgG as control. Purified ChIP DNA was amplified by real-time quantitative PCR using specific primers. The primer sequences are listed in Supplementary Table 1.
Promoter luciferase reporter activity assay.
The cells were transfected with 1 μg of promoter-Luciferase vectors (pmIL-6FL, pMCP-Luc and pmTNF-α-GLuc) using Targefect Raw reagents in 6-well plates. Luciferase activity assays were performed 48 hours after transfection using the Luciferase Reporter Assay System (Promega, Madison, WI) or Secrete-Pair Gaussia Luciferase assay kit (GeneCopoeia, Rockville, MD). Luciferase activities were measured using a Centro XS3 LB 960 microplate fluorescence reader (Berthold Technologies, Ontario, Canada). The luciferase activity values were normalized to protein concentrations.
Western blotting.
Cells were lysed with RIPA buffer containing protease inhibitors cocktail (Roche). After sonication on ice, the samples were centrifuged at 12,000g for 15 min at 4°C and the supernatants were collected. Cellular proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes (BioRad). The membranes were then blocked with 5% nonfat milk in PBS-T for 1 hr, followed by incubation at 4°C overnight with antibodies against YAP, c-Jun, c-fos, MST-1, MST-2, LATS1 or β-actin at appropriate dilutions. After washing three times with PBS/T, the membranes were incubated with secondary antibody (IRDye conjugated IgG, LI-COR) in PBS-T containing 5% nonfat milk for 1 hr. The signals were detected by using the Odyssey Imaging System (LI-COR).
Electrophoretic mobility shift assay (EMSA).
Nuclear extracts were isolated from Raw 264.7 cells treated with or without LPS. EMSA was performed using Odyssey Infrared EMSA kit (LI-COR). Briefly, 1 μg of nuclear protein was analyzed for DNA binding to IRDye 800 labeled, double stranded oligonucleotides corresponding to the YAP promoter region containing AP-1 motif. The DNA sequences are listed in Supplementary Table 1. The labeled oligonucleotides were incubated with 1 μg of nuclear proteins for 20 minutes at room temperature in binding buffer containing 25 mM DTT, 2.5% Tween 20, 1 μg Poly dI-dC, 1% NP-40, 100 mM MgCl2. Binding was competed by 200X unlabeled oligonucleotides. The binding complexes were resolved by electrophoresis using 5% TBE gels (Bio-Rad), and the signals were detected by using the Odyssey Imaging System (LI-COR).
Human NASH tissue gene expression data set analysis.
We searched the Gene Expression Omnibus (GEO) datasets of human NAFLD patients from NCBI database. Datasets GSE48452 and GSE61260 containing detailed patient’s history and key pathological parameters of NASH (fat, inflammation, and fibrosis) were analyzed. Included in the analysis were patients with age of < 70 years old. Patients receiving bariatric surgery were excluded for analysis. Control liver tissue samples showing inflammation or fibrosis were excluded.
Immunoflourescence analysis for YAP and CD14 in human NASH tissues.
Human NASH tissue microarrays were obtained from Sekisui XenoTech, LLC (Kansas City, KS) (containing five normal liver and twelve NASH cases). Immunofluorescence was performed as we previously described (21). Briefly, the tissue slide was deparaffinized, rehydrated, and antigen retrieved using 10 mM citrate buffer (pH 6.0). After blocking with 5% normal goat serum, the slide was stained with primary antibody against YAP (Cell Signaling Technology) and CD14 (Abcam). For autofluorescence quenching, the slides were incubated with quenching solutions (Vector, Burlingame, CA) according to the manufacturer’s instruction. The nuclei were stained with DAPI. The colocalization analysis was performed using the ImageJ software.
Statistical analysis.
Quantitative data are expressed as Mean ± SD or SEM from at least three replicates. Statistical significance between two groups was analyzed using a two-tailed t test. A P value <0.05 was considered statistically significant.
RESULTS
YAP is upregulated in Kupffer cells in the setting of NAFLD
We examined the expression of YAP in the livers of normal chow diet (NCD; 5% fat) and high fat diet (HFD; 60% fat)-fed mice (experimental protocol outlined in Figure 1A). Our data showed that the livers from HFD-fed mice showed increased expression of YAP when compared to the livers from NCD-fed mice (Figure 1B). Immunohistochemistry (IHC) and immunofluorescence (IF) analyses show that HFD-induced YAP is predominantly localized in Kupffer cells (Figure 1C and 1D), while the hepatocytes from HFD-fed mice exhibit only trace staining of YAP. Under immunofluorescence, YAP is co-localized with F4/80 (a marker for macrophages/Kupffer cells) in the livers of HFD-fed mice (Figure 1D) (the levels of YAP and F4/80 are low in the livers of NCD-fed mice).
Figure 1. Increased expression of YAP in Kupffer cells of HFD-fed mice.

(A) Schematic outline of experimental approaches and analyses. (B) C57BL/6 wild type mice were fed normal chow diet (NCD) or high fat diet (HFD) for 12 weeks (n=6–8 per group). (left panel) Body weight; (mid panel) Liver-to-Body weight ratio; (right panel) H&E staining and YAP Western blotting of liver tissues. (C) Immunohistochemical staining for YAP of liver sections from mice fed NCD or HFD. (D) Immunofluorescence analysis for YAP and F4/80 of liver sections from mice fed NCD or HFD. (E) Expression of YAP in F4/80+ Kupffer cells was measured by ImageStream X in mice fed NCD or HFD (n=4 per group). (F) qRT-PCR analysis for YAP mRNA expression in primary Kupffer cells and hepatocytes obtained from NCD- or HFD-fed mice (n=4 per group). The data are expressed as mean±S.D. ***P<0.001.
To further characterize the expression and intracellular localization of YAP, we analyzed YAP expression by using imaging flow cytometry. As shown Figure 1E, increased fluorescence intensity of YAP in the nucleus was observed in F4/80-positive Kupffer cells from HFD-fed mice compared with NCD-fed mice. We then isolated primary Kupffer cells as well as hepatocytes from HFD-fed mice and NCD-fed mice to measure YAP mRNA levels by qRT-PCR. As shown in Figure 1F, YAP mRNA expression is notably upregulated in primary Kupffer cells from HFD-fed mice in comparison to NCD-fed mice. We observed that the level of YAP in primary hepatocytes from HFD-fed mice was slightly higher than in hepatocytes from NCD-fed mice, although the degree of YAP increase in hepatocytes was to a lesser extent in comparison to Kupffer cells. Taken together, our findings indicate that YAP is upregulated predominantly in Kupffer cells in the setting of NAFLD.
Deletion of YAP in Kupffer cells reduces inflammation but not steatosis in the setting of NAFLD
To assess the role of YAP in Kupffer cells, we generated macrophage/monocyte-specific YAP knockout mice (YAPϕKO) by crossing YAPfl/fl mice to mice bearing a LysM-Cre transgene. Successful deletion of YAP in YAPϕKO mice was confirmed by qRT-PCR analysis using isolated primary Kupffer cells from the livers (Figure 2A). The YAPϕKO mice and their littermate wild type controls (LysM Cre−; YAPfl/fl or YAPfl/+) were then subjected to HFD feeding protocol (outlined in Figure 2B). As shown in Figure 2C, YAPϕKO mice exhibited decreased AST and ALT levels under HFD feeding when compared to the WT mice. The YAPϕKO mice showed reduced lobular inflammation when compared to the WT mice, although the degree of steatosis was comparable between the two groups (Figure 2D). The YAPϕKO mice did not show significant differences in the expression of lipogenesis genes when compared to WT mice (Supplementary Figure S1A). Also, we did not observe significant differences in fibrosis and the expression of fibrogenesis genes between WT and YAPϕKO mice (Supplementary Figure S1B and S1C). Next, we analyzed the expression of pro-inflammatory cytokines in the liver tissues from HFD-fed YAPϕKO mice. We observed that the YAPϕKO mice showed reduced expression of several pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6 and MCP-1, in liver tissues (Figure 2E) and in isolated Kupffer cells (Figure 3A). Furthermore, our data showed that the numbers of F4/80-positive Kupffer cells were decreased in the livers of YAPϕKO mice in comparison to WT mice (Figure 2F). These findings suggest an important role of YAP in Kupffer cells for regulation of cytokine production and liver inflammation in NAFLD.
Figure 2. Deletion of YAP in Kupffer cells/macrophages reduces hepatic inflammation.

(A) Mice with macrophage/monocyte-specific deletion of YAP (YAPϕKO mice) were generated by crossing YAPfl/fl mice with LysM Cre mice. The expression of YAP in Kupffer cells isolated from YAPϕKO and their matched littermate control mice (LysM Cre−; YAPfl/fl or YAPfl/+) were determined by qRT-PCR analysis (Bottom panel). (B) Schematic outline of experimental approaches and analyses. (C) The levels of serum transaminases (AST and ALT) measured from YAPϕKO (n=5) and WT mice (n=11) fed HFD (mean±S.E.M.). (D) H&E staining and lobular inflammatory score of liver tissues (mean±S.E.M.). (E) qRT-PCR analysis for inflammatory cytokines in the liver tissues from HFD-fed WT (n=4) and YAPϕKO mice (n=6). (F) Immunohistochemical staining for F4/80 in liver tissues. The numbers of F4/80-positive Kupffer cells per 200X field were counted (mean±S.E.M.). The qRT-PCR data are expressed as mean±S.D. ***P<0.001.
Figure 3. Macrophage/monocyte-specific YAP deletion inhibits M1 phenotype.

(A) qRT-PCR analysis for pro-inflammatory cytokines in Kupffer cells isolated from HFD-fed WT (n=4) and YAPϕKO mice (n=6). (B-C) qRT-PCR analysis for specific M1 and M2 markers in Kupffer cells isolated from HFD-fed WT (n=4) and YAPϕKO (n=6). The data are expressed as mean±S.D. ***P<0.001.
Given that M1/M2 polarization of Kupffer cells is importantly associated with NAFLD progression (23, 24), we further examined the expression of M1 and M2 markers in Kupffer cells from HFD-fed YAPϕKO mice. As shown in Figure 3B and 3C, the Kupffer cells isolated from HFD-fed YAPϕKO mice showed reduced expression of M1 markers (CD80 and CD86), but increased expression of M2 marker (Mgl2 and Retlna), when compared to HFD-fed WT mice. These results suggest that YAP activation in Kupffer cells promotes M1 polarization which may contribute to liver inflammation in the setting of NAFLD.
LPS/TLR4 signaling regulates YAP expression in Kupffer cells
It is well known that gut-derived endotoxin/lipopolysaccharide (LPS)/Toll-like receptor 4 (TLR4) signaling-mediated Kupffer cell activation plays an important role in the development of NASH(17, 20). In our system, we sought to further determine whether LPS/TLR4 signaling might influence YAP activation in Kupffer cells. By incubating primary Kupffer cells with LPS, we observed that LPS treatment increased YAP protein and mRNA, as documented by immunofluorescence and qRT-PCR analyses (Figure 4A). In parallel, we analyzed the effect of LPS on Kupffer cell YAP expression in vivo. For the latter purpose, we administered LPS to wild type C57BL/6 mice via daily intraperitoneal (i.p.) injection (0.25 mg/kg for 3 weeks) and the mice were subsequently sacrificed to document the expression of YAP in Kupffer cells. Imaging flowcytometry analysis showed that LPS treatment increased YAP protein in F4/80-positive KCs (Figure 4B). qRT-PCR analysis showed that LPS treatment increased the expression of YAP (in addition to TNF-α) in Kupffer cells (Figure 4B).
Figure 4. LPS/TLR4 signaling induces YAP transcription in Kupffer cells in vitro and in vivo.


(A) Primary Kupffer cells isolated from C57/BL6 WT mice were treated with LPS (1 μg/ml) for 24 hrs in 2% FBS containing DMEM medium. The expression of YAP was determined by immunofluorescence and qRT-PCR. (B) WT mice (C57/BL6) were treated with LPS (0.25mg/kg) for 3 weeks (daily i.p. injection) (n=4 per group). The expression of YAP was analyzed by imagestream X (representative images are shown in the left panel). The bar graphs (right panel) represent YAP and TNF-α mRNA levels in primary Kupffer cells isolated from LPS-treated mice. (C) Generation of TLR4ϕKO mice. Deletion of TLR4 was verified by measuring mRNA in Kupffer cells isolated from WT and TLR4ϕKO mice (RT-PCR and qRT-PCR). (D) Effects of LPS on YAP and TNF-α expression in Kupffer cells isolated from WT and TLR4ϕKO mice. The isolated primary Kupffer cells were treated with LPS for 6 hrs. The expression of YAP and TNF-α was determined by qRT-PCR. The data are expressed as mean±S.D. ***P<0.001.
To further investigate the role of TLR4 for YAP regulation in KCs, we generated macrophage/monocyte-specific TLR4 null mice (TLR4ϕKO) by crossing Tlr4fl/fl mice to LysM-Cre mice (Figure 4C). Successful deletion of TLR4 in the produced TLR4ϕKO mice was confirmed by depletion of KC TLR4 mRNA as determined by qRT-PCR analysis. We then isolated KCs from TLR4ϕKO mice and the corresponding littermate controls; the obtained KCs were subsequently treated with LPS to determine YAP expression (Figure 4D). Our data showed that LPS-induced YAP expression in KCs from TLR4ϕKO mice was significantly lower when compared to KCs from control mice. As expected, the mRNA level of TNF-α in KCs from TLR4ϕKO mice was also lower when compared to KCs from the control mice (Figure 4D).
As NF-kB is well-documented in LPS/TLR4 signaling, we performed further studies to assess the relative contribution of NF-kB and YAP signaling pathways to the inflammatory response in Kupffer cells (KCs). Specifically, we compared the inhibitory effects of the NF-kB inhibitor Bay11–7082 versus the YAP inhibitor verteporfin on LPS-induced inflammatory cytokine production in primary KCs. We observed that LPS-induced inflammatory cytokine production was inhibited to a similar extent by either NF-kB or YAP inhibition (Supplementary Figure S2). Therefore, both NF-kB and YAP signaling pathways are implicated in LPS-mediated inflammation in KCs. Taken together, our findings reveal an important role of LPS/TLR4 signaling for regulation of YAP in KCs.
YAP activation increases the production of pro-inflammatory cytokines in Kupffer cells
To delineate the effect of YAP on pro-inflammatory cytokine expression, we isolated mouse primary Kupffer cells, and the cells were transfected with constitutively active form of YAP (YAP5SA) in which the five Ser residues were replaced with Ala. Successful transfection of YAP was verified by detection of YAP immunoflorescence in trasnsfected cells (Figure 5A). We observed that overexpression of active YAP (YAP5SA) led to significant increase in the production of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6 and MCP-1, in primary KCs (Figure 5B). Consistent with the latter observations, YAP5SA also increased the production of TNF-α, IL-1β, IL-6 and MCP-1 in Raw 264.7 cells (murine macrophage cell line) (Figure 5C).
Figure 5. YAP activation enhances the expression of pro-inflammatory cytokines in murine Kupffer cells/macrophages.
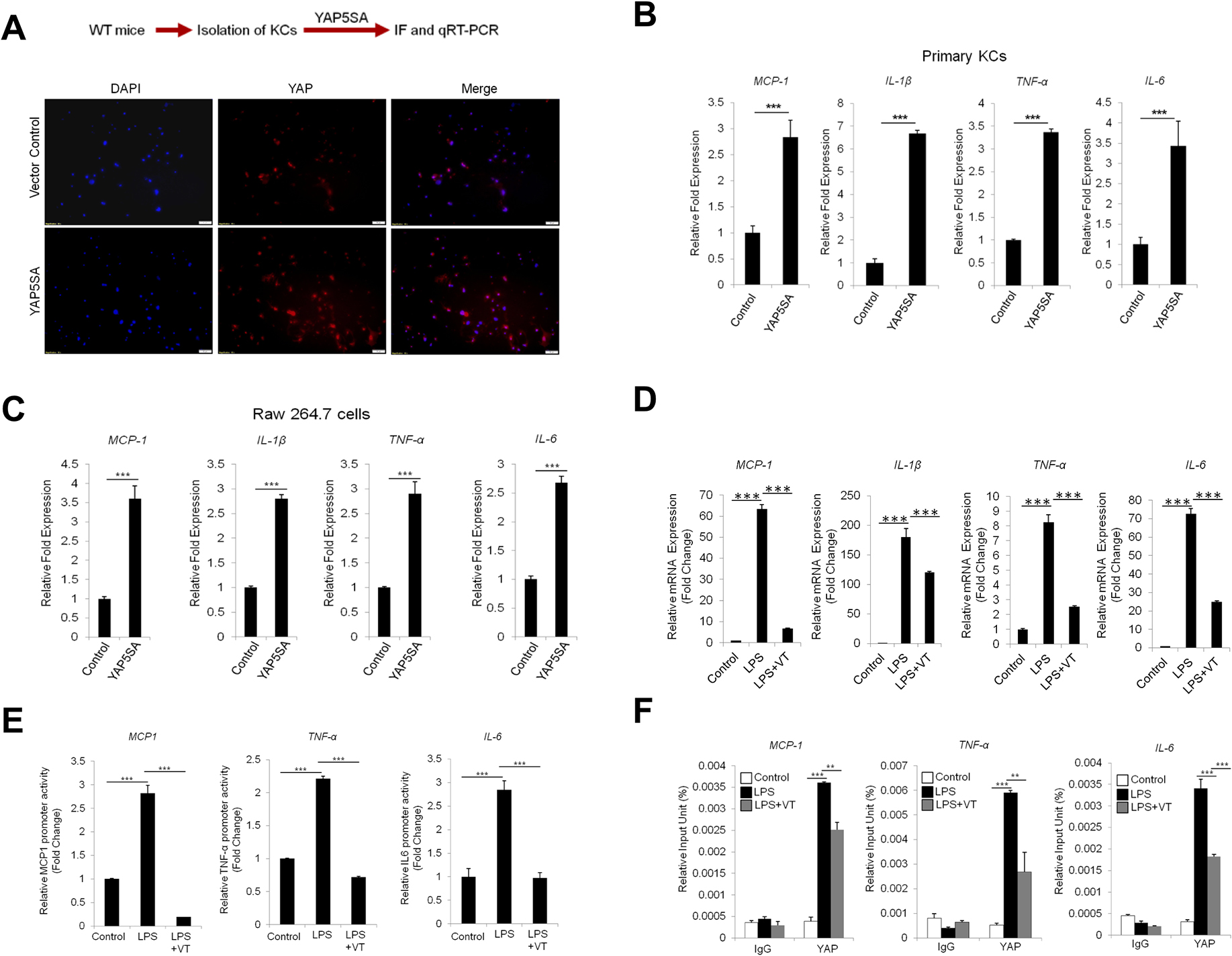
(A) Schematic outline of experimental analysis (top). Primary Kupffer cells isolated from C57BL/6 mice were transfected with 2μg of YAP5SA or control vector using Targefect-Raw agents. The expression of YAP is analyzed by immunofluorescence. (B) The expression of pro-inflammatory cytokines in Kupffer cells was measured by qRT-PCR (48 hr post-transfection). (C) The expression of pro-inflammatory cytokines in Raw 264.7 cells (48 hr post-transfection). (D) Raw264.7 cells were pre-treated with verteporfin (1 μM) for 1 hour followed by LPS (1 μg/ml) treatment for 24 hours. The expression of pro-inflammatory cytokines was determined by qRT-PCR. (E) Raw264.7 cells were transfected with the promoter reporter constructs for specific pro-inflammatory cytokines (pmIL-6FL, pMCP-Luc or pTNF-α-GLuc). The cells were then treated with verteporfin (1 μM) for 1 hour followed by LPS treatment (1 μg/ml) for 6 hrs. The luciferase activities were measured as described in the Material and Methods. (F) Raw264.7 cells were treated with veteporfin for 1 hour followed by LPS treatment for 24 hours. DNA-protein complexes were obtained and incubated with anti-IgG or anti-YAP antibodies. The enriched DNAs were then amplified by qRT-PCR using primers covering the TEAD biding site at the promoter regions of inflammatory cytokine genes (MCP-1, TNF-α, and IL-6). The data are expressed as mean±S.D. ***P<0.001.
We next utilized verteporfin, a pharmacological inhibitor of YAP, to assess its effect on cytokine production. Our data showed that verteporfin treatment markedly inhibited LPS-induced expression of pro-inflammatory cytokines including TNF-α, IL-1β, IL-6 and MCP-1 (Figure 5D). Accordingly, verteporfin treatment also inhibited LPS induced promoter-luciferase activities of pro-inflammatory cytokines (Figure 5E). The latter findings suggest that YAP activation up-regulates the transcription of pro-inflammatory cytokines.
YAP is known to regulate gene transcription by interacting with TEAD transcriptional factors (transcriptional enhancer associated domain). By performing sequence alignment analysis, we had identified the presence of TEAD binding motif in the promoter regions of the above-indicated inflammatory cytokines. Based on this information, we then performed ChIP assay to determine whether LPS treatment might influence YAP binding to the promoter region of the cytokine genes which contain the TEAD motif. Our data showed that LPS treatment enhanced YAP binding to the promoter regions of TNF-α, IL-6 and MCP-1 and their associations were reduced by the YAP inhibitor verteporfin (Figure 5F). These data indicate that YAP enhances the transcription of pro-inflammatory cytokines in Kupffer cells/macrophages.
LPS induces YAP gene expression through activation of AP-1 in macrophages
It is well established that LPS-mediated TLR4 signaling activates gene expression through key transcription factors including AP-1 (17, 20). In our system, we observed that LPS-induced YAP expression was inhibited by pre-treatment with the AP-1 inhibitor (SR11302) as well as by its up-stream JNK inhibitor (SP600125) in Kupffer cells and in Raw 264.7 cells (Figure 6A). We had performed sequence alignment analysis for the YAP gene and identified AP-1 binding site between −2072 and −2078 in the YAP promoter region (illustrated in Figure 6B). Based on this information, we performed DNA pull down assay to determine whether LPS treatment might alter AP-1 association with the YAP promoter. We observed that LPS treatment increased the binding of c-jun and c-fos to the AP-1 binding site in the YAP promoter region (Figure 6B). Moreover, ChIP-qPCR assay revealed the enrichment of c-jun at the YAP promoter region in response to LPS treatment (Figure 6C). We further performed electrophoretic mobility shift assay (EMSA) by using fluorescent labeled, AP1 motif-containing YAP promoter oligonucleotide and nuclear extracts from Raw264.7 cells (with or without LPS treatment). As shown in Figure 6D, LPS treatment increased the binding to the AP-1 motif in YAP promoter. These findings demonstrate an important role of AP-1 in LPS-induced YAP transcription in macrophages.
Figure 6. LPS induces YAP expression through activation of AP1.

(A) Primary Kupffer cells and Raw264.7 cells were pre-treated with AP-1 inhibitor (SR11302) or JNK inhibitor (SP600125) for 1 hour followed by LPS (1 μg/ml) treatment for 24 hours. The expression of YAP was determined by qRT-PCR. (B) DNA pull down assay was performed using biotinylated probe containing AP-1 motif at the YAP promoter region. Mutant probe with indicated AP1 site mutation was used as negative control. LPS induces endogenous c-jun and c-fos bindings to AP-1 binding sites in mouse YAP promoter region. (C) Chromatin immunoprecipitation (ChIP) assay. Raw264.7 cells were treated with LPS for 24 hrs. DNA-protein complexes were obtained and incubated with anti-IgG or anti-c-jun antibody. The enriched DNAs were then amplified by qRT-PCR using primers covering the AP-1 biding site at the YAP promoter region. (D) Electrophoretic mobility shift assay (EMSA) was performed by using fluorescent labeled, AP1 motif-containing YAP promoter oligonucleotide and nuclear extracts from Raw264.7 cells (with or without LPS treatment). LPS treatment increased the binding to the AP-1 motif in YAP promoter (the binding was eliminated by cold competition with unlabeled oligonucleotides). The quantitative data are expressed as mean±S.D. ***P<0.001.
Given that YAP activity is largely regulated by Hippo pathway kinases including LATS and MST1/2, we performed further studies to determine whether LPS might regulate the Hippo pathway kinases. Our data showed that LPS treatment led to reduction of MST1 protein, but not MST2 and LATS proteins (Supplementary Figure S3A). On the other hand, the mRNA of MST1 was not altered by LPS treatment (Supplementary Figure S3B). The latter observation suggests that LPS signaling may reduce MST1 protein through a post-transcriptional regulatory mechanism. In this context, a previous study has reported that histone deacetylases (HDAC)-mediated deacetylation of MST1 leads to its degradation through lysosome-dependent manner (25). Therefore, we performed further studies to examine whether HDAC might be implicated in LPS-mediated reduction of MST1 protein. Indeed, our data showed that LPS-induced reduction of MST1 protein was reversed by pre-treatment with the HDAC inhibitor, Trichostatin A (TSA) (Supplementary Figure S3C). In parallel, we observed that LPS-induced YAP accumulation and nuclear localization was also reversed by pre-treatment with TSA (Supplementary Figure S3D). These findings suggest that LPS signaling activates YAP through HDAC-mediated MST1 degradation.
The YAP inhibitor, Verteporfin, attenuates liver inflammation in NAFLD
To assess the potential therapeutic impact of YAP inhibition for NASH treatment, we administered the YAP inhibitor verteporfin to HFD-fed mice. Specifically, C57BL/6 mice were fed HFD for 12 weeks, followed by treatment with verteporfin for 3 weeks (100mg/kg, i.p. at 3-day intervals, with continued HFD feeding) (procedures outlined in Figure 7A). Under this protocol, we observed that verteporfin treatment reduced the levels of liver transaminases (AST and ALT) (Figure 7A). Verteporfin treatment also significantly inhibited the production of pro-inflammatory cytokines in Kupffer cells (Figure 7B). The mice treated with verteporfin exhibited less lobular inflammation when compared to the non-treatment group, albeit both groups show a similar degree of hepatosteatosis (Figure 7C). We then measured the liver triglyceride content in verteporfin-treated mice and observed that the triglyceride levels in the liver tissues were not significantly altered by verteporfin treatment (Figure 7D). Also, verteporfin treatment did not significantly alter the body weight (Figure 7E). On the other hand, we observed that the verteporfin-treated group had reduced F4/80-positive Kupffer cells in the liver tissues when compared to the non-treatment group (Figure 7F). These findings provide important pre-clinical evidence for utilization of YAP inhibitor as a novel therapy for NASH treatment.
Figure 7. The YAP inhibitor, Verteporfin, attenuates hepatic inflammation in NAFLD.

(A) C57BL/6 WT mice were fed HFD for 12 weeks, followed by treatment with Verteporfin every other day for 3 weeks. The levels of liver enzymes (AST/ALT) were determined upon sacrifice (the data are expressed as mean±S.E.M.; n=5/group). (B) Kupffer cells were isolated from mice with indicated treatments, and the expression of inflammatory cytokines in isolated KCs was measured by qRT-PCR. (C) H&E staining and lobular inflammation score in liver tissues. The quantitative data are expressed as mean±S.E.M.. (D) Quantification of liver triglyceride levels per tissue weight (g) in NCD-fed mice (n=3) or HFD-fed mice treated with or without verteporfin (n=5 per groups). (E) Body weight of HFD-fed mice with or without verteporfin treatment (n=5 per groups). (F) Immunohistochemical staining for F4/80. The numbers of F4/80-positive Kupffer cells per 200 X field were counted. (mean±S.E.M.). The qRT-PCR data are expressed as mean±S.D. *P<0.05, ***P<0.001.
YAP is increased and correlated with pro-inflammatory cytokines in patients with NASH
To assess the clinical relevance of our findings, we performed gene expression analysis in NASH patients using Gene Expression Omnibus (GEO) datasets (GSE48452 and GSE61260) obtained from NCBI database. As shown Figure 8A, YAP expression was significantly increased in liver tissues of patients with NASH compared to normal livers. The level of YAP was positively correlated with the expression of pro-inflammatory cytokines (IL1β, MCP1 and TNF) (Figure 8B). Moreover, the level of YAP was positively correlated with the M1 markers (CD80 and CD86), but negatively correlated with the M2 marker (Arginase1) (Figure 8C). Furthermore, we observed that the level of YAP was positively correlated with the stage of liver fibrosis and the expression of fibrogenesis genes (Collagen 1 alpha1 and α-SMA) (Figure 8D). Under immunofluorescence analysis, we observed that the liver tissues from NASH patients showed increased expression of YAP which is co-localized with the activated human Kupffer cell marker, CD14(26, 27) (Figure 8E). Together, our results demonstrate a pro-inflammatory role of YAP signaling in Kupffer cells and its crosstalk with LPS/TLR4 signaling in the pathogenesis of NASH (illustrated in Figure 8F).
Figure 8. YAP expression in patients with NASH.

(A) Gene expression analysis in liver tissues from NASH patients using Gene Expression Omnibus (GEO) datasets (GSE48452 and GSE61260) from NCBI database. ***P<0.001. n=28 (normal) and n=41 (NASH). (B) Correlation test between YAP and pro-inflammatory cytokines expression. Correlation coefficient (r2) was calculated by Pearson correlation test. (C) Correlation test between YAP and M1/M2 markers expression. Both normal and NASH tissues were included in the correlation tests. (D) YAP expression analysis in liver tissues from NASH patients with different stages of liver fibrosis (top panel). The bottom panel shows correlation test between YAP and fibrosis genes (Col1a1: collagene type1a, and Acta2: α-smooth muscle actin). (E) Immunofluorescence analysis for YAP and CD14 in liver tissues from NASH patients. The images are representative stains from 5 normal and 12 NASH patients. Colocalization test (Mander’s coefficients) was performed by using imageJ. (F) Illustration of mechanisms by which YAP in Kupffer cells enhances the production of pro-inflammatory cytokines and promotes the development of NASH.
DISCUSSION
Despite the well-recognized role of Hippo/YAP signaling in hepatic carcinogenesis, the effect and mechanism of Hippo/YAP signaling in non-neoplastic liver diseases are largely unknown. In the current study, we provide the first evidence that the Hippo signaling effector YAP is notably increased in the liver tissues in NAFLD and that Kupffer cells represent the predominant liver cells with high level of YAP expression induced by HFD feeding. We show that YAP in Kupffer cells promotes hepatic inflammation by up-regulating the expression of pro-inflammatory cytokines. Our data indicate that deletion of YAP in Kupffer cells or inhibition of YAP by pharmacological inhibitor attenuated liver inflammation in association with NAFLD in mice. Further mechanistic studies demonstrate that LPS transcriptionally activates YAP in Kupffer cells which further induces the expression of pro-inflammatory cytokines (including MCP-1, TNF-α and IL-6) through YAP association with the TEAD-binding motif in the promoter region of inflammatory cytokines. Our findings provide novel evidence for a pivotal role of YAP in Kupffer cells which importantly contributes to liver inflammation and NASH progression.
The Hippo/YAP signaling is recognized as a critical regulator of cell proliferation and survival. In the current study, we provide novel evidence that activation of YAP in Kupffer cells plays a critical role in inflammation during NASH progression. Indeed, Kupffer cell activation is known to contribute to liver inflammation in NASH by recruiting other immune cells and promoting hepatocyte injury (16). Studies have shown that clodronate-mediated depletion of Kupffer cells attenuated liver inflammation and prevented liver injury in mouse models of NASH (28, 29). Our data presented in this study show that Kupffer cell-specific depletion of YAP or administration of the YAP inhibitor verteporfin effectively decreases hepatic inflammation and reduces serum transaminases in HFD-fed mice. These findings suggest that inhibiting YAP activation in Kupffer cells may represent a new therapeutic strategy for effective treatment of NASH.
In NASH, LPS derived from gut microbial activates Kupffer cells which in turn leads to secretion of pro-inflammatory cytokines (18). Accordingly, suppression of LPS by modulation of gut-microbial through probiotic or antibiotic treatment diminished NASH in leptin-deficient mice (30, 31). For NASH induced by MCD or fructose diet, mice with defective TLR4 signaling exhibit lower level of liver inflammation and reduced tissue injury when compared to the wild type mice (29, 32). In the current study, we describe a novel crosstalk between the LPS/TLR4 signaling and the Hippo/YAP pathway in Kupffer cells. Our data showed that YAP expression is increased by LPS treatment in vitro and in vivo. We further demonstrated that AP-1 is involved in transcriptional regulation of YAP in KCs/macrophages. Consistent with these results, we observed that deletion of TLR4 in Kupffer cells attenuated LPS-induced YAP expression. Furthermore, we observed that inhibition of YAP by verteporfin reduced LPS-induced pro-inflammatory cytokine expressions.
Macrophage M1/M2 polarization is closely associated with the progression of NAFLD (29). Specifically, M1 polarized Kupffer cells possess pro-inflammatory phenotypes that secreting pro-inflammatory cytokines, while M2 polarized Kupffer cells exhibit anti-inflammatory phenotypes. Accordingly, increased M1 Kupffer cells have been reported in NASH (23, 30). To date, several transcription factors including peroxisome proliferator-activated receptors (PPARs) and the Kruppel-like factor4 (KLF4) are known to affect macrophage M1/M2 polarization (33–36). In the current study, we observed that HFD-fed YAPϕKO mice exhibited reduced M1 markers but increased M2 markers in Kupffer cells. Our findings provide novel evidence that YAP importantly regulates Kupffer cell M1/M2 polarization.
By analyzing gene expression profiles in human liver tissues from NASH patients, we observed that the level of YAP was positively correlated with the stage of liver fibrosis and the expression of fibrogenesis genes (Figure 8D). This observation supports the notion that Kupffer cell-mediated inflammation contributes to stellate cell activation and fibrogenesis in NASH. However, in mouse model of NAFLD induced by high fat diet feeding, we observed that deletion of YAP in Kupffer cells/macrophages did not significantly alter the degree of liver fibrosis or the expression of fibrogenesis genes (Supplementary Figure S1). A possible explanation for the divergence phenomenon may be the limitation of the high fat diet feeding protocol in mice which induces relatively low level of liver fibrosis.
Verteporfin has been used in photodynamic therapy (PDT) for patients with subfoveal choroidal neovascularization (CNV), which has been recently identified as a YAP inhibitor (37, 38). In our study, administration of verteporfin to mice significantly decreased HFD-induced liver inflammation and production of pro-inflammatory cytokines, although it did not significantly alter the degree of steatosis. This observation is corroborated by the fact that KC-specific YAP deletion reduced liver inflammation but not steatosis in the setting of HFD feeding. Therefore, our findings suggest that YAP activation in KCs may only regulate inflammatory responses, rather than lipid metabolism. Given the critical role of inflammation in NASH progression, our data provide novel insight for use of YAP inhibitor as a new therapeutic strategy for the treatment of NASH in patients.
In conclusion, our findings demonstrate that YAP-mediated Kupffer cell activation critically contributes to NASH progression through production of pro-inflammatory cytokines and perpetuation of liver inflammation. Further, our data reveal that LPS/TLR4 signaling transcriptionally actives YAP in Kupffer cells, which provides novel mechanistic insight for LPS-mediated inflammation in the progression of NAFLD. Therefore, inhibition of YAP may represent a new therapeutic strategy for the treatment of NASH.
Supplementary Material
Grant support:
The work in the authors’ laboratory is supported by NIH R01 grants (CA226281, CA219541 and CA102325).
Abbreviations:
- YAP
Yes-associated protein
- NASH
Non-alcoholic steatohepatitis
- NAFLD
Non-alcoholic fatty liver disease
- KCs
Kupffer cells
- HFD
High fat diet
- NCD
Normal chow diet
- LPS
lipopolysaccharide
- TLR4
Toll-like receptor 4
- MCP-1
monocyte chemoattractant protein 1
- TNF-α
Tumor necrosis factor alpha
- IL-6
Interleukin 6
- IL-1β
Interleukin 1 beta
- MST-1/2
mammalian Ste20-like kinase 1/2
- LATS
Large tumor suppressor kinase
- TEAD
TEA domain
- AST
Aspartate aminotransferase
- ALT
Alanine aminotransferase
- AP-1
Activator protein 1
- VT
Verteporfin
- ChIP
Chromatin immunoprecipitation
- IHC
Immunohistochemistry
- IF
Immunofluorescence
- qRT-PCR
quantitative reverse transcription polymerase chain reacton
- WT
wild-type
Footnotes
Disclosures: The authors have declared that no conflict of interest exists
REFERENCE
- 1.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11–20. [DOI] [PubMed] [Google Scholar]
- 2.Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arab JP, Arrese M, Trauner M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu Rev Pathol 2018;13:321–350. [DOI] [PubMed] [Google Scholar]
- 4.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 2010;24:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol 2011;13:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh SH, Swiderska-Syn M, Jewell ML, Premont RT, Diehl AM. Liver regeneration requires Yap1-TGFbeta-dependent epithelial-mesenchymal transition in hepatocytes. J Hepatol 2018;69:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moya IM, Halder G. The Hippo pathway in cellular reprogramming and regeneration of different organs. Curr Opin Cell Biol 2016;43:62–68. [DOI] [PubMed] [Google Scholar]
- 9.Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci U S A 2010;107:1431–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009;16:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007;130:1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 2007;17:2054–2060. [DOI] [PubMed] [Google Scholar]
- 13.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A 2010;107:1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, Hoorens A, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 2015;63:679–688. [DOI] [PubMed] [Google Scholar]
- 15.Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, Diehl AM. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J Hepatol 2015;63:962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol 2009;51:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aron-Wisnewsky J, Gaborit B, Dutour A, Clement K. Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin Microbiol Infect 2013;19:338–348. [DOI] [PubMed] [Google Scholar]
- 18.Abu-Shanab A, Quigley EM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2010;7:691–701. [DOI] [PubMed] [Google Scholar]
- 19.Miura K, Ohnishi H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol 2014;20:7381–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Machado MV, Cortez-Pinto H. Gut microbiota and nonalcoholic fatty liver disease. Ann Hepatol 2012;11:440–449. [PubMed] [Google Scholar]
- 21.Kwon H, Song K, Han C, Chen W, Wang Y, Dash S, Lim K, et al. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2016;63:1155–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song K, Han C, Zhang J, Lu D, Dash S, Feitelson M, Lim K, et al. Epigenetic regulation of MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology 2013;58:1681–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014;59:130–142. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W, Zhang X, et al. Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene 2016;35:4048–4057. [DOI] [PubMed] [Google Scholar]
- 26.Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H, Nishiyama K, et al. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. J Leukoc Biol 2013;94:1325–1336. [DOI] [PubMed] [Google Scholar]
- 27.Su GL, Goyert SM, Fan MH, Aminlari A, Gong KQ, Klein RD, Myc A, et al. Activation of human and mouse Kupffer cells by lipopolysaccharide is mediated by CD14. Am J Physiol Gastrointest Liver Physiol 2002;283:G640–645. [DOI] [PubMed] [Google Scholar]
- 28.Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem 2012;287:40161–40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 2007;47:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003;37:343–350. [DOI] [PubMed] [Google Scholar]
- 31.Solga SF, Diehl AM. Non-alcoholic fatty liver disease: lumen-liver interactions and possible role for probiotics. J Hepatol 2003;38:681–687. [DOI] [PubMed] [Google Scholar]
- 32.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009;50:1094–1104. [DOI] [PubMed] [Google Scholar]
- 33.Yao Q, Liu J, Zhang Z, Li F, Zhang C, Lai B, Xiao L, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) induces the gene expression of integrin alphaVbeta5 to promote macrophage M2 polarization. J Biol Chem 2018;293:16572–16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong X, Liu H. Honokiol attenuates diet-induced non-alcoholic steatohepatitis by regulating macrophage polarization through activating peroxisome proliferator-activated receptor gamma. J Gastroenterol Hepatol 2018;33:524–532. [DOI] [PubMed] [Google Scholar]
- 35.Kapoor N, Niu J, Saad Y, Kumar S, Sirakova T, Becerra E, Li X, et al. Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J Immunol 2015;194:6011–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS, Koo SH, et al. RORalpha Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep 2017;20:124–135. [DOI] [PubMed] [Google Scholar]
- 37.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26:1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt-Erfurth U, Hasan T. Mechanisms of action of photodynamic therapy with verteporfin for the treatment of age-related macular degeneration. Surv Ophthalmol 2000;45:195–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.