

Abstract
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease. The disease is characterized by marked variability in morphological expression and natural history, ranging from asymptomatic to heart failure or sudden cardiac death. Left ventricular hypertrophy and abnormal ventricular configuration result in dynamic left ventricular outflow obstruction in most patients. The goal of pharmacological therapy in HCM is to alleviate the symptoms, and it includes pharmacotherapies and septal reduction therapies. In this review, we summarize the relevant clinical issues and treatment options of HCM.
Abbreviations: CMR, cardiac magnetic resonance; HCM, hypertrophic cardiomyopathy; HF, heart failure; ISH, interventricular septal hypertrophy; LVH, left ventricular hypertrophy; LVOT, left ventricular outflow tract; SCD, sudden cardiac death; SAM, systolic anterior motion
1. Introduction
The first modern description of HCM was provided in 1958 by Robert Teare who published a series of cases of asymmetrical hypertrophy or muscular hamartoma of the heart in young people with sudden death. However, in the early 1960 s, Dr. Eugene Braunwald and colleagues were responsible for describing the histological aspects, the clinical presentation, and the treatment of this disease [1], [2], [3]. HCM is the most common genetic cardiomyopathy; it is present in a variety of races and ethnic groups [4]. The majority of individuals with HCM have normal or near-normal life expectancy [5]. The disease is characterized by marked variability in morphological expression and natural history. Depending on the mutation, there is variability in family transmission, degree of hypertrophy, evolution, and prognosis of the disease.
1.1. Definition and prevalence
The diagnosis of HCM is based on the presence of nondilated left ventricular hypertrophy (LVH), identified by echocardiography or magnetic resonance imaging, which occurs in the absence of another cardiac, systemic, metabolic, or syndromic disease [5], [6], [7]. HCM typically manifests as asymmetric septal hypertrophy, although other patterns, such as apical, concentric, lateral wall, and right ventricular forms, can occur [8].
Epidemiological studies estimate a prevalence of 1 in 500 persons in the general population, and the disease has been identified in 122 countries (representing 90% of the world population) [9], [10], [11]. Recent studies suggest that approximately 20 million people worldwide are affected by HCM, well beyond the population initially thought, although it is estimated that only 10% of cases are clinically identified and only 6% symptomatic [4], [11].
1.2. Etiology
HCM is associated with mutations in one of the genes encoding proteins of the cardiac sarcomere, Z-disc, and calcium-controlling proteins (Table 1) [12], [13], [14]. Twenty genes have been related to the disease, and over 2000 different mutations have been identified in disease-affected individuals, but the most common genes involved with HCM are β-myosin heavy chain (MYH7) and myosin binding protein C (MYBPC3) [10], [11], [12], [13]. Genetic phenocopies, such as Fabry's disease, amyloidosis, Danone's disease, and Friederich's ataxia, may be associated with HCM [7].
Table 1.
Genes implicated in HCM.
| Gene | Protein | Chromosome | Frequency (%) |
|---|---|---|---|
| Myofilament genes | |||
| TTN | Titin | 2 | 1 |
| MYH7 | β-Myosin heavy chain | 14 | 15–25 |
| MYH6 | α-Myosin heavy chain | 14 | 1 |
| MYL2 | Regulatory myosin light chain | 12 | 2 |
| MYL3 | Essential myosin light chain | 3 | 1 |
| MYBPC3 | Myosin-binding protein C | 11 | 15–25 |
| TNNT2 | Cardiac troponin T | 1 | 5 |
| TNNI3 | Cardiac troponin I | 19 | 5 |
| TPM1 | α-Tropomyosin | 15 | 5 |
| ACTC | Cardiac α-actin | 15 | 1 |
| TNNC1 | Cardiac troponin C | 3 | 1 |
| Genes of Z disk | |||
| LBD3 | Lim domain binding 3 | 10 | 1–5 |
| CSRP3 | Cysteine- and glycine-rich protein 3 | 17 | 1 |
| TCAP | Tcap (telethonin) | 17 | 1 |
| VCL | Vinculin/Metavinculin | 10 | 1 |
| ACTN2 | Actinin, α2 | 1 | 1 |
| MYOZ2 | Myozenin 2 (calsarcin 1) | 4 | 1 |
| NEXN | Nexilin | 1 | <1 |
| Calcium controlling genes | |||
| JPH2 | Junctophilin-2 | 20 | 1 |
| PLN | Phospholamban | 6 | 1 |
More recently, sarcomere mutations have also been recognized as the cause of dilated cardiomyopathy, idiopathic restrictive cardiomyopathy, and LV noncompaction [15].
The SHaRe registry showed that HCM patients with sarcomere mutations have a 2-folder greater risk for adverse outcomes, highest for ventricular arrhythmias, compared to those without mutations [16].
Up to 5% of patients may have two distinct pathogenic mutations, and a minority (<1%), three pathogenic mutations. Patients who manifest earlier and have more severe phenotypes often have multiple mutations [17], [18], [19].
1.3. Pathophysiology
The association between LVH and histopathological changes characteristic of the disease, such as myofibril disruption and increased interstitial space with fibrosis formation, results in the occurrence of diastolic dysfunction, frequently found in patients with HCM [20], [21].
Septal hypertrophy favors the occurrence of high left ventricular outflow tract (LVOT), resulting in a “suction” effect (Venturi effect), which leads to the traction of the anterior mitral valve leaflet towards the interventricular septum with consequent emergence of subaortic systolic gradient and LVOT obstruction. The obstructive form is defined as LVOT gradient ≥30 mmHg. Approximately 70% of HCM patients have LVOT obstruction at rest or with provocative maneuvers [22].
Nonobstructive HCM generally carries a favorable prognosis. The exception is “burned-out” or end-stage HCM, wherein the phenotype transitions to a dilated cardiomyopathy (wall thinning, cavity dilation, systolic dysfunction, and secondary pulmonary hypertension), with a poor prognosis.
Myocardial ischemia is common and of multifactorial origin. It is related to several pathophysiological mechanisms that act alone or synergistically and are responsible for the angina symptom found in 50% of the population with the disease. The mechanisms are (1) excessive increase in muscle mass, with consequent increase in oxygen demand by the myocardium; (2) insufficient coronary microvascular structure in relation to increased ventricular mass; (3) increased ventricular diastolic pressures and microvascular compression, decreasing blood flow mainly in the subendocardial region; and (4) thickening of the coronary artery walls with decreased arteriolar lumen [23].
Moreover, the left atrium is usually dilated as a result of the high resistance to ventricular filling caused by diastolic dysfunction and reflux effect through the mitral valve.
1.4. Morphologic patterns of HCM
The morphologic expression of HCM is widely variable and heterogeneous because HCM may affect any portion of the LV. Asymmetric septal involvement is the most common form of the disease, and other variants include midventricular, apical and concentric HCM, as shown in the figure below (Fig. 1).
Fig. 2.

(A) ECG showing LVH in a patient with HCM; (B) ECG of a patient with apical cardiac hypertrophy (Yamaguchi syndrome).
Fig. 1.

HCM phenotypes. Diagrams show focal basal septum HCM (A), diffuse septum (B), concentric and diffuse HCM (C), midventricular HCM (D) and apical HCM (E). Adapted from Baxi et al. [24].
2. Clinical presentation and diagnosis
2.1. Natural history
HCM is one of the few cardiovascular diseases that can manifest at any phase of life, from infancy to the seventh decade of life. Most patients have normal life expectancy without the need for major therapeutic interventions [5], [6], [7]. Mortality rates have been substantially revised over the years. Population studies performed on highly selected tertiary center patients after the initial description of the disease showed an annual mortality of around 4% to 6%. However, the selection bias probably overestimated the risk of the disease by creating a concept that HCM would be a “malignant” heart disease with a poor prognosis and reduced life expectancy [5]. Recent studies with large populations and longer follow-up have shown annual mortality rates from 0.9% to 1.5%, being reduced to 0.5% per year after the development of new therapeutic interventions, such as implantable cardioverter defibrillator (ICD) and heart transplantation [5], [7], [11], [25], [26], [27].
A small proportion of patients may develop complications like sudden cardiac death (SCD) that usually affects younger (<30 years) and asymptomatic or oligosymptomatic individuals [25], [26], [27], and progressive heart failure (HF) with preserved systolic function. Occasionally (<5% of cases), the disease may develop with left ventricular dysfunction (“end stage” disease) [28] and atrial fibrillation (AF), with significant risk of stroke [5], [7], [29], [30].
However, data from the SHaRe registry demonstrated that mortality in HCM patients is significantly higher than in the US general population. Although SHaRe data confirmed that absolute mortality in young patients with HCM is low, it was 4-fold higher than expected for 20- to 29-year-old, and the rate of SCD, which is the leading cause of death HCM, was much lower in this registry (16%) than that shown by previous studies (around 40%) [16].
2.2. Physical examination
Physical examination is usually normal in asymptomatic patients without LVOT obstruction. The ictus is broad, strong, and shifted to the left, and a systolic thrill can be palpated at the tip level or at the lower left sternal border. An inverted paradoxical pulse may occur. The jugular venous pulse may have an elevated “a” wave due to marked atrial contraction resulting from decreased ventricular distensibility.
The carotid pulse increases rapidly and then decreases in mesosystole as gradient formation occurs followed by a secondary increase, giving the appearance of bisferiens.
In cardiac auscultation, the most common finding is the presence of a B4, which is explained by a vigorous atrial contraction. B2 is unfolded and, patients with marked obstruction of the left ventricular outflow tract may experience a paradoxical unfolding. In the majority of patients with the obstructive form, a rough “crescent-decreasing” systolic murmur is heard, which begins shortly after B1, audible along the low left sternal border resulting from turbulent flow in the narrow LV outflow tract. When mitral regurgitation is present, a soft, holosystolic murmur in the apex radiates into the armpit.
Some maneuvers modify the intensity of the murmur and assist in the clinical diagnosis of the disease (Table 2).
Table 2.
Effects of interventions on the LVOT gradient and murmur in patients with hypertrophic cardiomyopathy.
| Intervention | Gradient and cardiac murmur |
|---|---|
| Valsalva maneuver | Increase |
| Orthostatic position | Increase |
| Post extrasystole | Increase |
| Squatting position | Decrease |
| Handgrip maneuver | Decrease |
3. Electrocardiogram (ECG)
ECG is altered in up to 95% of cases, and the disease has no characteristic pattern. The most common abnormalities are ST-segment and T-wave changes, followed by evidence of LVH. Other electrocardiographic alterations may be present in HCM as shown in Table 3.
Table 3.
ECG features of HCM.
| ECG abnormalities in HCM |
|---|
|
|
|
|
|
| Apical HCM |
|
3.1. Diagnosis
The diagnosis of HCM rests on the detection of increased LV wall thickness by any imaging modality in the absence of another cardiac or systemic disease that itself would be capable of producing the magnitude of hypertrophy [5], [7].
In an adult, HCM is usually recognized by maximal LV wall thickness ≥ 15 mm, with wall thickness of 13 to 14 mm considered borderline, as measured by any imaging technique (echocardiography or cardiac magnetic resonance imaging), particularly in the presence of other compelling information (e.g., family history of HCM) [5], [7].
In the case of children, increased LV wall thickness is defined as wall thickness ≥2 standard deviations above the mean (z score ≥2) for age, sex, or body size [5], [7].
Table 4 summarizes the recommendations of the main imaging techniques in patients with suspected HCM.
Table 4.
Imaging techniques in patients with suspected HCM.
| Imaging techniques | Recommendations |
|---|---|
| Genetic testing |
|
| Eletrocardiogram (ECG) |
|
| Exercise testing |
|
| Ambulatory ECG monitoring |
|
| Transthoracic echocardiography |
|
| Cardiac magnetic resonance |
|
Transthoracic echocardiography has been the mainstay for imaging the HCM phenotype, and it remains the initial test for patients due to its portability, widespread access, and reliability in quantifying dynamic outflow tract gradients (Fig. 3). It can also determine changes in the mitral valve, as well as the follow-up of the disease [31]. For the diagnosis, the only important parameter is the maximum ventricular wall thickness in any LV segment, and careful evaluation of all LV segments during the examination is essential because the anterolateral wall and apex hypertrophies can be difficult to see. Diagnostic criteria by echocardiography are ventricular wall thickness ≥15 mm in at least one LV myocardial segment or ≥13 mm in patients with a first-degree relative with confirmed HCM. The presence of the LVOT gradient is defined as nonobstructive when the LVOT gradient is <30 mmHg at rest, obstructive when the LVOT gradient is ≥30 mmHg at rest, and latent obstructive form when the LVOT gradient is <30 mmHg at rest, and on exertion when ≥30 mmHg [5], [7], [32], [33].
Fig. 3.
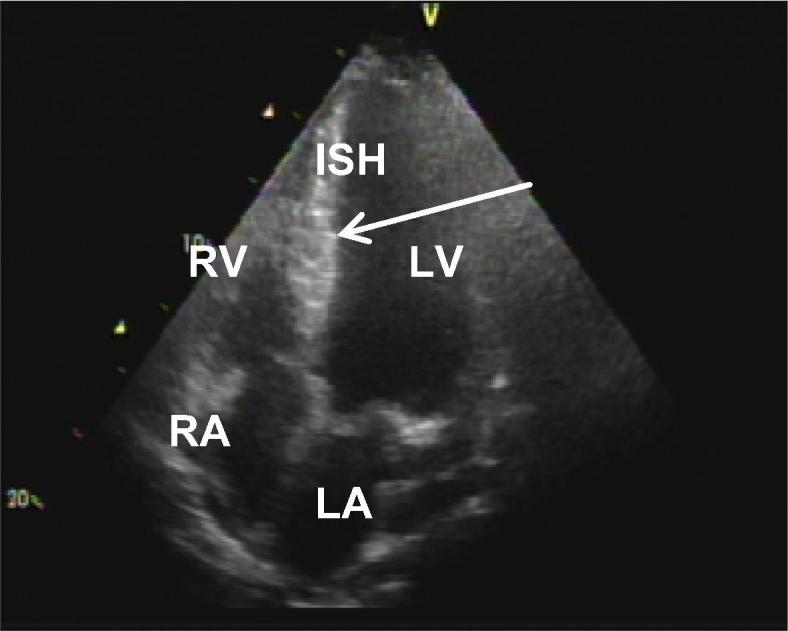
Two-dimensional echocardiogram. Left ventricular longitudinal long-axis 2-chamber in tele-diastole showing a mid-apical hypertrophy (arrow) and highlighting interventricular septal hypertrophy (ISH = 36 mm).
Echocardiography is currently the most practical technique for screening for HCM and is recommended at 12-month intervals during adolescence and every 5 years in adults [39], [40], as well as at the onset of symptoms suggestive of HCM [5], [7]. In preclinical phases, reduced mitral tissue Doppler velocities may precede the development of LVH, and reduction in strain rates may also provide an incremental value in identifying subclinical forms [34], [35].
Cardiac magnetic resonance (CMR) should be ordered to establish the diagnosis of HCM in cases where areas of hypertrophy are technically difficult to assess by conventional echocardiography [36] (Fig. 4, Fig. 5). It can also be performed for the differential diagnosis of myocardial hypertrophies found in other situations, such as athlete's heart, hypertensive heart disease, and deposition diseases (Fabry's disease and amyloidosis) [37]. Through infusion of paramagnetic contrast, CMR identifies myocardial fibrosis, found in up to 65% of HCM patients, with often multifocal, irregular distribution, and not respecting coronary anatomy [37], [38], [39], [40], [41], [42]. The presence of extensive myocardial fibrosis is associated with a higher risk of LV dilation and systolic dysfunction. A recent multicenter study showed that patients with fibrosis extension ≥15% of LV mass have twice the risk for SCD compared with patients without fibrosis regardless of other risk factors [40].
Fig. 4.

An asymptomatic 36-year-old man with HCM. CMR images demonstrate mild asymmetric septal hypertrophy (A and B) and late gadolinium enhancement (arrows in C) in mid-septal wall and located near the right ventricular insertion points.
Fig. 5.

A 17-year-old male patient with NYHA class II and massive LVH. (A) Electrocardiogram with LVH; (B) Transthoracic echocardiogram parasternal short-axis view shows severe asymmetric septal hypertrophy involving the anterior septum (wall thickness of 65 mm) and posterior LV (wall thickness of 24 mm); (C) M-mode echocardiogram recording of anterior systolic movement (ASM) and mitral leaflet septal contact (arrows); (D) CMR demonstrating the massive LVH.
Nuclear imaging can provide measurements of LV volumes and ejection function, RV volumes and ejection function, and identify thickened myocardium without a definable cause and myocardial ischemia (Table 5) [34].
Table 5.
Nuclear imaging evaluation of patients with HCM.
| 1. Myocardial perfusion |
| 2. LV volumes and ejection function by radionuclide and gated SPECT |
| 3. Coronary flow reserve by PET |
| 4. Cardiac metabolism by PET (research application) |
Myocardial ischemia in the absence of epicardial coronary artery disease is a feature of HCM and is shown to play a significant role in the clinical course of these patients [43].
Although myocardial ischemia is by no means the sole etiology of SCD in HCM, several lines of investigation provide evidence for its role as a major contributor to its risk. First, extensive remodeling of the intramural coronary arteries is seen in young individuals who had died suddenly as the first manifestation of HCM [44]. Second, inducible myocardial ischemia has been frequently found among young survivors of cardiac arrest [45]. Third, myocardial ischemia is often more commonly seen in HCM patients with massive left ventricular hypertrophy, dynamic left ventricular outflow tract obstruction, or hypotensive response to exercise [46], [47]. Finally, high prevalence of late gadolinium enhancement in areas of hypertrophy has been correlated with the presence of non-sustained ventricular tachycardia and cardiac mortality. Despite this evidence, assessment of myocardial ischemia has not been considered part of the evaluation and clinical management of patients with HCM.
In addition, the degree of microvascular dysfunction in patients with HCM assessed by Positron Emission Tomography (PET), has already been shown to be an independent predictor of clinical deterioration and death [48].
However, the new guidelines do not routinely recommend performing nuclear imaging in the prognostic assessment of HCM [5] due to the potential risks of radiation exposure and that the benefits of the information gained sufficiently balance those risks [49]. This concept may be particularly important in patients with HCM, who in general will be younger compared with other subgroups of patients being evaluated for heart disease.
DNA analysis is the definitive method for the diagnosis of HCM. It should be requested whenever possible in the index case for the possibility of family screening and genetic counseling. In approximately 40–60% of cases, the mutation responsible for the disease is identified [50]. Despite initial optimism that “malignant” or “benign” mutations would be identifiable among patient populations, the most recent clinical genetic studies have not been able to demonstrate the association of genetic mutations with prognostic evolution or SCD. Thus, the test result does not influence treatment strategies and does not identify high-risk SCD patients who may benefit from ICD therapy [51], [52], [53].
HCM is usually diagnosed using non-invasive methods, without the need for endomyocardial biopsy (EMB). However, histopathological findings can be useful for the differential diagnosis of other cardiomyopathies, especially Fabry's disease and amiloidosis [54].
4. Environmental phenotype modulators of HCM
Recently, there has been a great interest in environmental phenotype modulators of HCM. Fumagalli et al showed that obesity is highly prevalent among patients with HCM and is associated with an increased likelihood of LVOT obstruction and atrial fibrillation [55].
Sedentary lifestyle has been associated with increased cardiovascular morbidity and mortality in the general population. However, competitive sports have been discouraged by international consensus [56], [57], although there is a growing understanding that exercise, even at high intensity, can benefit HCM patients [58]. Only 1 published randomized controlled trial has examined the effect of exercise training in patients with HCM. Saberi et al. showed that moderate-intensity physical activity improves exercise capacity in patients with HCM [59]. High-quality evidence from multiple prospective randomized controlled trials regarding the efficacy and safety of high-intensity exercise compared with moderate-intensity exercise in patients with HCM is lacking. Although current guidelines recommend only low-intensity physical activity, both moderate- and high-intensity exercise may be safe and effective in selected groups of patients with HCM. The benefits of regular physical activity can outweigh the negative effects of a sedentary lifestyle [60].
Studies to better define the risk of high-intensity exercise in HCM are ongoing (LIVE-HCM/LQT and HIIT-HCM). The LIVE-HCM/LQT trial aims to evaluate how lifestyle and exercise impact the well-being of individuals with hypertrophic cardiomyopathy (HCM) and long QT syndrome (LQTS). The HIIT-HCM trial aims to examine the efficacy and safety of supervised high-intensity exercise in adults with HCM.
Recently, the Sport Cardiology Section of the European Association of Preventive Cardiology (EAPC) updated the recommendations for practicing cardiologists and sport physicians managing athletes with cardiomyopathies. The document states that exercise participation should be considered on an individual basis. Findings that might represent absolute contraindications for sports participation include history of aborted SCD, unexplained syncope, exercise-induced ventricular tachycardia (VT), high ESC 5-year risk score, significant increase in left ventricular (LV) outflow gradient (>50 mmHg), and abnormal blood pressure response to exercise. Adult athletes with mild clinical expressions of HCM and low ESC risk score, may selectively be allowed to participate in competitive sports, with the exception of those sports with intrinsic risk of harm with syncope (eg, scuba diving). Genotype positive – phenotype negative status should not equate to having HCM and thus should not preclude participation in sports [61].
4.1. Management
HCM treatment is indicated in the presence of symptoms. Treatment of patients with HCM requires a thorough understanding of the natural history and must be individualized to the patient. The general approach is outlined in Fig. 6.
Fig. 6.

Prognostic pathways and primary treatment strategies within the broad clinical spectrum of HCM. ICD = implantable cardioverter defibrillator; RF = radiofrequency.
4.2. Pharmacotherapy
The goal of pharmacological therapy in HCM is to alleviate the symptoms of exertional dyspnea, palpitations, and angina and thereby improve patients' quality of life. However, previous studies have not been able to demonstrate benefits in the use of medications to prevent disease progression [5], [7].
Beta-blockers (BB) and calcium channel blockers (CCB) have been used since the 1960 s and should be used in progressive doses until a resting heart rate of 60 bpm is reached. These drugs improve exertional dyspnea and chest pain by inhibiting sympathetic heart stimulation; decreasing oxygen consumption by reducing heart rate, contractility, and myocardial stress during systole; and increasing the diastolic filling period. Decreased LVOT flow obstruction during exercise would lead to improvement of dyspnea and syncope. Despite these therapeutic effects, it is now known that these drugs do not decrease the incidence of ventricular arrhythmias or sudden death [5], [7], [62], [63]. CCB should be avoided in patients with the obstructive form because its vasodilatory effect may worsen obstruction in LVOT [64]. Disopyramide, an antiarrhythmic drug, has an important negative inotropic effect that, in obstructive forms, leads to a decrease or even disappearance of the LVOT gradient, with improvement of symptoms [65]. Diuretics may be considered in patients with signs of pulmonary congestion [66].
The potential beneficial effects of inhibitors of the renin-angiotensin-aldosterone system as anti-hypertrophic and anti-fibrotic agents were tested in animal models of HCM. Treatment with an angiotensin II receptor blocker (losartan) has shown that transforming growth factor (TGF)-β is an important mediator of profibrotic signals and that early inhibition of TGF-β diminishes the development of fibrosis [67].
The INHERIT (INHibition of the renin angiotensin system in hypertrophic cardiomyopathy and the Effect on hypertrophy - a Randomised Intervention Trial with losartan) trial was designed to assess the effect of 100 mg of losartan in promoting the regression of LV hypertrophy in HCM. This study, however, failed to show any significant reduction in the fibrotic mass proportion (evaluated using late gadolinium enhancement in CMR) with the losartan [68].
The currently ongoing VANISH (Valsartan for Attenuating Disease Evolution In Early Sarcomeric HCM) trial is a multicenter, double-blind, randomized controlled trial designed to determine whether treatment with valsartan will have a beneficial effect in attenuating disease evolution in early HCM. The study also will measure the impact of valsartan treatment on disease pathology, clinical outcomes and assessment of symptom burden, and incidence of adverse drug reactions [69].
Atrial fibrillation (AF) is a common complication and occurs in up to 20% of individuals with HCM. This is likely due to raised left ventricular end diastolic pressure related to ventricular dysfunction caused by LVOT obstruction. AF is associated with clinical deterioration due to HF. Therefore, restoration of sinus rhythm is desirable [70], [71]. CHA2DS2-VASc score is not recommended for use in patients with HCM. For rhythm maintenance, amiodarone (sotalol alternative), catheter ablation, and the MAZE surgical technique (in patients undergoing surgery) should be considered [5], [6], [7], [8], [72], [73], [74]. Oral anticoagulation is recommended in all patients with AF where possible; thromboembolism is a common consequence of AF (up to 30%) [5], [7], [11], [75].
Chronic anticoagulation with vitamin K antagonists (VKAs) is recommended in patients with HCM and AF.
There are currently no data available from randomized controlled trials on the efficacy of direct oral anticoagulants (DOACs) in reducing thromboembolic risk in this population. Two retrospective trials [76], [77] in patients with HCM undergoing oral anticoagulation did not find any significant difference between DOACs and vitamin K antagonists (VKA) in ischemic or bleeding events. Current guidelines [5], [7] recommend the use of DOACs in significant difficulties with INR monitoring, side effects or patients are unable to perform INR monitoring. However, the most recent ESC Guidelines for the Management of AF (2016) have recommended that there is no preference between VKA or DOACs in these patients [78].
5. Invasive therapy
Invasive treatment is indicated in symptomatic patients (NYHA III-IV) despite optimal medical therapy with an LVOT gradient ≥50 mmHg (at rest or provoked) [5], [6], [7], [11]. Table 6 summarizes indications of septal reduction therapies.
Table 6.
Eligible patients for invasive therapy.
| Clinical: NYHA functional classes III or IV, syncope or other symptoms that interfere with quality of life despite optimal medical therapy. |
| Hemodynamic: LVOT gradient ≥50 mmHg (at rest or provoked) associated with septal hypertrophy and systolic anterior motion of the mitral valve. |
| Anatomic: Targeted anterior septal thickness sufficient to perform the procedure safely and effectively in the judgment of the individual operator. |
5.1. Surgical septal myectomy
Surgical septal myectomy has been the gold standard for septal reduction. The procedure removes a small portion of muscle from the basal ventricular septum to reduce the LVOT gradient (Morrow's surgery) [2], [79], [80].
Long-term studies with>50 years of follow-up have shown that surgical myectomy reliably reverses heart failure symptoms, permanently abolishing obstruction (90% of cases), restoring normal LV filling pressure and reducing or abolishing mitral regurgitation [81], [82]. Operative mortality for septal myectomy ranges from <1% at experienced centers to up to 3–4% when associated with mitral valve repair. The main perioperative complications are complete atrioventricular (AV) block, aortic valve insufficiency, and interventricular communication [82], [83], [84]. The use of intraoperative transesophageal echocardiography has assisted surgeons in septal resection and correction of concomitant structural abnormalities, with improved surgical outcomes and reduced complication rates [5], [7]. Male patients <50 years old with left atrium size <46 mm without atrial fibrillation have the best surgical results [82], [83].
Another important data is the association of the patient sex with outcomes after surgical myectomy. It has been shown that women with HCM are diagnosed at an older age, tend to be more symptomatic at presentation, have more obstructive form and have reduced survival compared with men. Meghji et al. have showed that women undergoing septal myectomy were older, more symptomatic at presentation and had more severe obstructive form, in addition to being more likely to have moderate to severe mitral regurgitation and higher right ventricular systolic pressure than men. However, survival following septal myectomy was independent of female sex after adjustment for important baseline prognostic factors. Improved care of women with obstructive HCM should focus on early identification of disease and prompt surgical referral of appropriate patients who do not respond to medical treatment [85].
5.2. Alcohol septal ablation
Alcoholic septal ablation was first successfully performed in 1994 by Sigwart in three patients with the obstructive form of HCM who had not improved with clinical treatment. After this procedure, patients had a significant decrease in LVOT gradient, with improvement in symptoms and functional class [86]. The procedure consists of occlusion of the major septal branch of the anterior descending artery and injection of absolute alcohol (1.5 to 2.5 mL) through the cardiac catheterization technique, which causes a septal infarction (10% of the LV wall, 30% of the septum, on average), with the formation of scarring and septal reduction [87]. Currently, there are questions as to whether the scar that formed on the septum could contribute as an arrhythmogenic focus at risk for SCD [88].
Alcohol septal ablation has become an alternative to surgical myectomy in select patients, such as those with advanced age, significant comorbidities, or who oppose open chest surgery. The procedure should be avoided in patients with septal thickness <16 mm or >30 mm [5], [7]. Results of alcohol septal ablation are similar to those of surgical myectomy in terms of gradient reduction, improvement of symptoms, and exercise capacity, with mortality similar to that of isolated myectomy. Complete atrioventricular (AV) block is the main complication (7–20% of patients), and 12% of patients need to repeat the procedure due to residual obstruction [89], [90], [91], [92], [93].
Fig. 7 shows a comparison between two strategies for septal reduction in patients with HCM.
Fig. 7.

Comparison between septal reduction therapies.
On the other hand, Kim et al analyzed patients who were hospitalized for septal myectomy or alcohol septal ablation in the US between 2003 and 2011. The authors showed that most centers that provide septal reduction therapy performed few procedures, below the threshold recommended by the American College of Cardiology Foundation/American Heart Association and concluded that hospitals with a low septal myectomy volume are associated with worse in-hospital outcomes, including higher mortality [94].
5.3. Pacing therapy
The rationale for pacing in patients with HCM include: (1) negative inotropic effect and reduced hypercontractility of the LV; (2) asynchronous septal activation and delayed septal thickening; (3) limitation of abnormal mitral valve motion; (4) interaction with LV filling, and (5) ventricular remodeling [95].
The presence of the electrode at the RV tip determines a change in contractile activation of the myocardium, which changes from bottom to top and from right to left, with paradoxical movement of the interventricular septum, which moves away from the posterior wall of the LV during systole, increasing the ventricular chamber and reducing the LVOT gradient.
However, Maron et al. in a double-blind trial have demonstrated the placebo effect as responsible for the improvement of symptoms with device implantation [96].
Therefore, the definitive indication of atrioventricular pacing in HCM is reserved only for very symptomatic patients, refractory to pharmacological treatment, not candidates for septal myectomy or alcoholic septal ablation [5], [7].
5.4. Heart transplantation
In the US, HCM accounts for 1–5% of heart transplants with survival rates similar to the transplant rate in patients with other cardiomyopathies, including those with ischemic etiology [97], [98]. Heart transplantation is indicated in patients with end-stage disease and left ventricular dysfunction or in those with nonobstructive HCM with preserved systolic function (restrictive form) who develop refractory heart failure symptoms due to diastolic dysfunction [5], [6], [7], [11], [99].
5.5. Novel therapeutic strategies
In severely symptomatic patients with HCM, small LV cavity, and nonobstructive physiology, apical myectomy to enlarge the LV improves symptoms for patients who would otherwise be relegated to cardiac transplantation. Percutaneous mitral valve repair to limit systolic anterior motion (SAM) is a novel approach to treating LVOT obstruction in drug-refractory patients with obstructive HCM who are not candidates for septal reduction therapy [100]. However, there is controversy about whether mitral valve abnormalities should be surgically repaired at the time of myectomy [96]. Currently, it is considered that the surgical mitral valve repair in association with septal myectomy is the preferred approach [101]. Surgical transapical myectomy can be performed in patients in whom the hypertrophic myocardium is confined to mid-apical segments [102], [103].
Perhexiline acts by reducing fatty-acid oxidation through inhibition of the enzymes carnitine palmitoyl transferase-1 and 2, which are crucial transporters of long chain fatty acids into the mitochondria. This inhibition thus induces a change in cellular metabolism, reducing the consumption of fatty acids and favoring the use of carbohydrates as a metabolic substrate, which is associated with greater efficiency in the use of oxygen for the production of ATP in the myocardium [104]. Abozguia et al showed that perhexiline ameliorates cardiac energetic impairment, corrects diastolic dysfunction, and increases exercise capacity in patients with symptomatic HCM [105].
Trials investigating the effect of cardiac myosin inhibitors such as mavacamten (EXPLORER-HCM and MAVERICK-HCM) and CK-274 (REDWOOD-HCM) on LVOTO and HCM associated diastolic heart failure are underway.
Mavacamten (MYK-461) is a small molecule that reduces contractility of cardiac myocytes by inhibiting ATPase activity of myosin. Heitner et al have showed that mavacamten reduced mean postexercise LVOT gradient from 103 mmHg at baseline to 19 mmHg and improve exercise capacity and symptoms at 12 weeks in patients with obstructive HCM [106]. The ongoing PIONEER-HCM study has evaluated the efficacy, safety, and tolerability of mavacamten in subjects with symptomatic HCM and LVOT obstruction aged 18–70 years.
Treatment with N-acetylcysteine (NAC), a precursor to the most abundant intracellular non-protein thiol pool against oxidative stress, reversed cardiac hypertrophy and interstitial fibrosis in a rabbit HCM model [107] However, the HALT-HCM study performed in 42 humans showed that treatment with NAC for 12 months had small effect sizes on indices of cardiac hypertrophy or fibrosis [108].
Finally, gene-silencing with CRISPR/Cas9 gene-editing technology may someday play a role in the prevention of disease development before the appearance of clinical manifestations [109].
6. Management of patients at risk of SCD
SCD is a feared and often unpredictable complication of HCM [5], [6], [7], [11] that usually affects asymptomatic patients under 35 years of age, resulting from ventricular arrhythmias caused by autonomic hyperactivity secondary to LVOT obstruction, microvascular ischemia, myocardial fibrosis, and myocyte derangement [110], [111].
In patients with a personal history of cardiac arrest, ventricular fibrillation, or sustained ventricular tachycardia, SCD risk rises to about 10% per year, and in this setting, ICD is the only effective therapy for preventing SCD [5], [6], [7], [112].
For primary prevention, the great challenge is to identify the candidates for the device (Fig. 6) due to the low incidence of SCD, the clinical individuality of each patient, the wide variation in the literature to define risk factors, and the morbidity attributed to ICD [113], [114], [115], [116], [117], [118]. It is recommended that all HCM patients undergo SCD risk stratification to identify those who will benefit from ICD implantation.
In 2014, the European Society of Cardiology developed a new risk stratification score that defines the 5-year SCD risk - HCM-Risk-SCD Calculator [http://www.doc2do.com/hcm/webHCM.html] [119]. This score takes into account age (inversely proportional to risk), LV wall thickness, left atrial diameter, LVOT gradient, family history of SCD, nonsustained ventricular tachycardia (NSVT), and unexplained syncope. ICD may be considered in patients with a risk of 4–6%, and in patients with a 5-year risk ≥ 6%, an ICD should be considered [7].
The other model, advocated by the American College of Cardiology/American Heart Association (Table 7), states that ICD can be considered in the presence of one or more of the following risk factors: NSVT; family history of SCD; LV wall thickness ≥30 mm; unexplained syncope; and abnormal blood pressure response during exercise (defined as a decrease in a blood pressure or inability to increase blood pressure by ≥20 mmHg during exercise).
Table 7.
Major and possible risk factors for SCD in patients with HCM. Adapted from Elliot PM et al. [25].
| Major risk factors | |
|---|---|
| 1. Family history of sudden death (<50 years). | |
| 2. Unexplained and recurrent syncope. | |
| 3. Ventricular wall thickness ≥30 mm. | |
| 4. Documented nonsustained ventricular tachycardia (NSVT) (>three beats with heart rate >120 bpm)* | |
| 5. Systolic blood pressure drop ≥20 mmHg on exertion* | |
| Number of major risk factors | Estimated 6-year survival from SCD (%) |
|
95 |
|
93 |
|
82 |
Possible risk factors: LVOT gradient ≥30 mmHg; high-risk genetic mutation; myocardial fibrosis (>15% LV mass) on CMR, LV apical aneurysm.
Low risk (SCD incidence 0.2–0.4% per year): Absence of established risk factors; absent or mild symptoms; left atrium ≤45 mm; ventricular wall thickness ≤20 mm; LVOT gradient <50 mmHg.
Only when associated with other possible risk factors.
ICD indication for primary prevention can also be defined by the presence of established risk factors, and the sum of these factors may further refine the identification of patients at high risk for SCD, as shown in Table 7 [5], [120], [121], [122], [123].
Although most guidelines suggest that a single risk factor may justify ICD, the decision should always be individualized taking into account the patient's age and desire, the positive predictive value of the present risk factor, as well as the technical and financial conditions of health care and the possible complications of device implantation [5], [7].
7. General considerations
Every individual with a family history of HCM should be followed for the risk of developing the disease (Fig. 8). For first-degree relatives, ideally the echocardiogram should be performed annually from 12 to 20 years of age, and before this period only in patients who have symptoms or who wish to participate in high-intensity sports competitions. After a person is 20 years old, echocardiography assessments are recommended every 3 to 5 years [5], [7]. The practice of intense physical exercise and competitive sports is contraindicated for patients with HCM. Low-intensity activities with maximum O2 consumption <40% may be released. For patients with positive genotype and negative phenotype, there is no restriction on physical exercise [5], [7], [124], [125]. Prophylaxis for infectious endocarditis should be considered for patients with the obstructive form when undergoing dental treatment [126]. Pregnancy is often well tolerated in asymptomatic patients with little risk of prematurity and maternal mortality, but the high potential for genetic transmission of the disease indicates genetic counseling before planning a pregnancy.
Fig. 8.

Follow-up of patients with HCM. SCD = sudden cardiac death; AF = atrial fibrillation; HCM = hypertrophic cardiomyopathy.
Goland et al. report maternal and foetal outcomes from an international cohort of women with HCM who entered the ROPAC database. Sixty pregnant women with HCM (41.7% obstructive) were included. No maternal mortality occurred in this cohort. However, a major adverse cardiovascular event (MACE) defined as death, heart failure, arrhythmia, or thrombo-embolic event occurred in ~23%. Although most women with HCM tolerated pregnancy well, cardiovascular complications were not uncommon and predicted by pre-pregnancy status facilitating pre-pregnancy counselling and targeted antenatal care [127].
In the presence of LVOT gradient >50 mmHg or symptoms refractory to drug treatment, pregnancy is considered high risk [128], [129].
References
- 1.Teare D. Asymetrical hypertrophy of the heart in young adults. Br. Heart J. 1958;20:1–8. doi: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrow A.G., Lambrew C.T., Braunwald E. Idiopathic hypertrophic subaortic stenosis: II, operative treatment and the results of pre- and postoperative hemodynamic evaluations. Circulation. 1964;30(suppl 4):120–151. [PubMed] [Google Scholar]
- 3.Braunwald E, Lambrew CT, Rockoff SD, Ross J Jr, Morrow AC. Idiopathic hypertrophic subaortic stenosis. A description of the disease based upon an analysis of 64 patients. Circulation. 1964;30(Suppl. IV):IV3-IV119. [DOI] [PubMed]
- 4.Maron B.J., Rowin E.J., Maron M.S. Global burden of hypertrophic cardiomyopathy. JACC Heart Fail. 2018;6:376–378. doi: 10.1016/j.jchf.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Gersh B.J., Maron B.J., Bonow R.O. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic: a report of the american college of cardiology foundation/american heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2011;58:212–260. doi: 10.1016/j.jacc.2011.10.825. [DOI] [PubMed] [Google Scholar]
- 6.Maron B.J., Ommen S.R., Semsarian C., Spirito P., Olivotto I., Maron M.S. Hypertrophic cardiomyopathy present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 2014;64:83–99. doi: 10.1016/j.jacc.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Elliot P.M., Anastasakis A., Borger M.A. ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC) Eur Heart J. 2014;284:1–55. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 8.Syed I.S., Ommen S.R., Breen J.F., Tajik A.J. Hypertrophic cardiomyopathy: identification of morphological subtypes by echocardiography and cardiac magnetic resonance imaging. J. Am. Coll. Cardiol. Img. 2008;1:377–379. doi: 10.1016/j.jcmg.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Maron B.J., Gardin J.M., Flack J.M., Gidding S.S., Kurosaki T.T., Bild D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. coronary artery risk development in (Young) adults. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 10.Arteaga E., Ianni B.M., Fernandes F., Mady C. Benign outcome in a long-term follow-up of patients with hypertrophic cardiomyopathy in Brazil. Am. Heart J. 2005;149:1099–1105. doi: 10.1016/j.ahj.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 11.Maron B.J. Clinical course and management of hypertrophic cardiomyopathy. N. Engl. J. Med. 2018;379:655–668. doi: 10.1056/NEJMra1710575. [DOI] [PubMed] [Google Scholar]
- 12.Marston S., Copeland O., Jacques A. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 13.Bos J.M., Towbin J.A., Ackerman M.J. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009;54:201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- 14.Richard Pascale, Villard Eric, Charron Philippe, Isnard Richard. The genetic bases of cardiomyopathies. J. Am. Coll. Cardiol. 2006;48(9):A79–A89. [Google Scholar]
- 15.Ho C.Y., Charron P., Richard P. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc. Res. 2015;105(4):397–408. doi: 10.1093/cvr/cvv025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho C.Y., Day S.M., Ashley E.A. Genotype and lifetime burden of disease in hypertrophic cardiomiopathy. Circulation. 2018;138:1387–1398. doi: 10.1161/CIRCULATIONAHA.117.033200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingles J., Doolan A., Chiu C., Seidman J., Seidman C., Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J. Med. Genet. 2005;42 doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Girolami F., Ho C.Y., Semsarian C. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010;55:1444–1453. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- 19.Marian A.J. Modifier genes for hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2002;17:242–252. doi: 10.1097/01.HCO.0000013803.40803.6A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies M.J., McKenna W.J. Hypertrophic cardiomyopathy-pathology and pathogenesis. Histopathology. 1995;26:493–500. doi: 10.1111/j.1365-2559.1995.tb00267.x. [DOI] [PubMed] [Google Scholar]
- 21.Hughes S.E. The pathology of hypertrophic cardiomyopathy. Histopathology. 2004;44:412–427. doi: 10.1111/j.1365-2559.2004.01835.x. [DOI] [PubMed] [Google Scholar]
- 22.Maron M.S., Olivotto I., Zenovich A.G. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114:2232–2239. doi: 10.1161/CIRCULATIONAHA.106.644682. [DOI] [PubMed] [Google Scholar]
- 23.Maron B.J., Wolfson J.K., Epstein S.E., Roberts W.C. Intramural (‘small vessel’) coronary artery disease in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1986;8:545–557. doi: 10.1016/s0735-1097(86)80181-4. [DOI] [PubMed] [Google Scholar]
- 24.Baxi A.J., Restrepo C.S., Vargas D. Hypertrophic cardiomyopathy from A to Z: genetics, pathophysiology, imaging, and management. Radiographics. 2016;36(2):335–354. doi: 10.1148/rg.2016150137. [DOI] [PubMed] [Google Scholar]
- 25.Elliot P.M., Poloniecki J., Dickie S. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J. Am. Coll. Cardiol. 2000;36:2212–2218. doi: 10.1016/s0735-1097(00)01003-2. [DOI] [PubMed] [Google Scholar]
- 26.Bos J.M., Maron B.J., Ackerman M.J. Role of family history of sudden death in risk stratification and prevention of sudden death with implantable defibrillators in hypertrophic cardiomyopathy. Am. J. Cardiol. 2010;106:1481–1486. doi: 10.1016/j.amjcard.2010.06.077. [DOI] [PubMed] [Google Scholar]
- 27.Spirito P., Autore C., Rapezzi C. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119:1703–1710. doi: 10.1161/CIRCULATIONAHA.108.798314. [DOI] [PubMed] [Google Scholar]
- 28.Melacini P., Basso C., Angelini A. Clinicopathological profiles of progressive heart failure in hypertrophic cardiomyopathy. Eur. Heart J. 2010;31:2111–2123. doi: 10.1093/eurheartj/ehq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olivotto I., Cecchi F., Casey S.A., Dolara A., Traverse J.H., Maron B.J. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517–2524. doi: 10.1161/hc4601.097997. [DOI] [PubMed] [Google Scholar]
- 30.Guttmann O.P., Rahman M.S., O’Mahony C., Anastasakis A., Elliott P.M. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100:465–472. doi: 10.1136/heartjnl-2013-304276. [DOI] [PubMed] [Google Scholar]
- 31.Wigle E.D. The diagnosis of hypertrophic cardiomyopathy. Heart. 2001;86:709–714. doi: 10.1136/heart.86.6.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maron B.J., McKenna W.J., Danielson G.K. American college of cardiology; committee for practice guidelines. european society of cardiology. J. Am. Coll. Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 33.Elliott P.M., Gimeno J.R., Tome M.T. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2006;27:1933–1941. doi: 10.1093/eurheartj/ehl041. [DOI] [PubMed] [Google Scholar]
- 34.Nagueh S.F., Bierig S.M., Budoff M.J. American Society of Echocardiography; American Society of Nuclear Cardiology; Society for Cardiovascular Magnetic Resonance; Society of Cardiovascular Computed Tomography. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: Endorsed by the American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. J. Am. Soc. Echocardiogr. 2011;24(5):473–498. doi: 10.1016/j.echo.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Nagueh S.F., Bachinski L.L., Meyer D. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128–130. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rickers C., Wilke N.M., Jerosch-Herold M., Casey S.A., Panse P., Panse N. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation. 2005;112:855–861. doi: 10.1161/CIRCULATIONAHA.104.507723. [DOI] [PubMed] [Google Scholar]
- 37.Moon J.C., McKenna W.J., McCrohon J.A., Elliott P.M., Smith G.C., Pennell D.J. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J. Am. Coll. Cardiol. 2003;41:1561–1567. doi: 10.1016/s0735-1097(03)00189-x. [DOI] [PubMed] [Google Scholar]
- 38.Choudhury L., Mahrholdt H., Wagner A., Choi K.M., Elliot M.D., Klocke F.J. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2002;40:2156–2164. doi: 10.1016/s0735-1097(02)02602-5. [DOI] [PubMed] [Google Scholar]
- 39.Shiozaki A.A., Senra T., Arteaga E. Myocardial fibrosis detected by cardiac CT predicts ventricular fibrillation/ventricular tachycardia events in patients with hypertrophic cardiomyopathy. J. Cardiovasc. Comput. Tomogr. 2013;7:173–181. doi: 10.1016/j.jcct.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan R.H., Maron B.J., Olivotto I. Prognostic value of quantitative contrastenhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130:484–495. doi: 10.1161/CIRCULATIONAHA.113.007094. [DOI] [PubMed] [Google Scholar]
- 41.Weng Z., Yao J., Chan R.H. Prognostic value of LGE-CMR in HCM: a metaanalysis. JACC Cardiovasc Imaging. 2016;9:1392–1402. doi: 10.1016/j.jcmg.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 42.Shiozaki A.A., Senra T., Arteaga E. Correlation between myocardial fibrosis and cardiac sudden death in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2008;51(10 suppl A) A152-3. [Google Scholar]
- 43.Shirani J., Dilsizian V. Nuclear cardiac imaging in hypertrophic cardiomyopathy. J. Nucl. Cardiol. 2011;18(1):123–134. doi: 10.1007/s12350-010-9279-2. [DOI] [PubMed] [Google Scholar]
- 44.Shirani J., Pick R., Roberts W.C., Maron B.J. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J. Am. Coll. Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 45.Dilsizian V., Bonow R.O., Epstein S.E., Fananapazir L. Myocardial ischemia detected by thallium scintigraphy is frequently related to cardiac arrest and syncope in young patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1993;22:796–804. doi: 10.1016/0735-1097(93)90193-5. [DOI] [PubMed] [Google Scholar]
- 46.Cannon R.O., 3rd, McIntosh C.L., Schenke W.H., Maron B.J., Bonow R.O., Epstein S.E. Effect of surgical reduction of left ventricular outflow obstruction on hemodynamics, coronary flow, and myocardial metabolism in hypertrophic cardiomyopathy. Circulation. 1989;79:766–775. doi: 10.1161/01.cir.79.4.766. [DOI] [PubMed] [Google Scholar]
- 47.Cannon R.O., 3rd, Schenke W.H., Maron B.J. Differences in coronary flow and myocardial metabolism at rest and during pacing between patients with obstructive and patients with nonobstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 1987;10:53–62. doi: 10.1016/s0735-1097(87)80159-6. [DOI] [PubMed] [Google Scholar]
- 48.Cecchi F., Olivotto I., Gistri R. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomiopathy. N Eng J Med. 2003;349:1027–1035. doi: 10.1056/NEJMoa025050. [DOI] [PubMed] [Google Scholar]
- 49.Cerqueira M.D., Allman K.C., Ficaro E.P. Recommendations for reducing radiation exposure in myocardial perfusion imaging. J. Nucl. Cardiol. 2010;17:709–718. doi: 10.1007/s12350-010-9244-0. [DOI] [PubMed] [Google Scholar]
- 50.Marsiglia J.D., Credidio F.L., de Oliveira T.G. Screening of MYH7, MYBPC3, and TNNT2 genes in Brazilian patients with hypertrophic cardiomyopathy. Am. Heart J. 2013;166:775–782. doi: 10.1016/j.ahj.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 51.Ho C.Y. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–2440. doi: 10.1161/CIRCULATIONAHA.110.978924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andersen P.S., Havndrup O., Hougs L. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum. Mutat. 2009;30:363–370. doi: 10.1002/humu.20862. [DOI] [PubMed] [Google Scholar]
- 53.Ingles J., Burns C., Bagnall R.D. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ. Cardiovasc. Genet. 2017;10 doi: 10.1161/CIRCGENETICS.116.001620. [DOI] [PubMed] [Google Scholar]
- 54.Leone O., Veinot J.P., Angelini A. 2011 consensus statement on endomyocardial biopsy from the Association for European Cardiovascular Pathology and the Society for Cardiovascular Pathology. Cardiovasc. Pathol. 2012;21:245–274. doi: 10.1016/j.carpath.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 55.Fumagalli C., Maurizi N., Day S.M. Association of obesity with adverse long-term outcomes in hypertrophic cardiomyopathy. JAMA Cardiol. 2019:1–8. doi: 10.1001/jamacardio.2019.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pelliccia A., Corrado D., Bjørnstad H.H. Recommendations for participation in competitive sport and leisure-time physical activity in individuals with cardiomyopathies, myocarditis and pericarditis. Eur. J. Cardiovasc. Prev. Rehabil. 2006;13(6):876–885. doi: 10.1097/01.hjr.0000238393.96975.32. [DOI] [PubMed] [Google Scholar]
- 57.Maron B.J., Udelson J.E., Bonow R.O. American Heart Association Electrocardiography and Arrhythmias Committee of Council on Clinical Cardiology, Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and American College of Cardiology. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 3: hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132(22):e273–e280. doi: 10.1161/CIR.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 58.Papoutsidakis N., Heitner S., Ingles J. Participation in thrill-seeking activities by patients with hypertrophic cardiomyopathy: individual preferences, adverse events and physician attitude. Am. Heart J. 2019;214:28–35. doi: 10.1016/j.ahj.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 59.Saberi S., Wheeler M., Bragg-Gresham J. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy. JAMA. 2017;317(13):1349–1357. doi: 10.1001/jama.2017.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dias K.A., Link M.S., Levine B.D. Exercise training for patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2018;72(10):1157–1165. doi: 10.1016/j.jacc.2018.06.054. [DOI] [PubMed] [Google Scholar]
- 61.Pelliccia A., Solberg E.E., Papadakis M. Recommendations for participation in competitive and leisure time sport in athletes with cardiomyopathies, myocarditis, and pericarditis: position statement of the Sport Cardiology Section of the European Association of Preventive Cardiology (EAPC) Eur. Heart J. 2019;40(1):19–33. doi: 10.1093/eurheartj/ehy730. [DOI] [PubMed] [Google Scholar]
- 62.Sherrid M.V., Shetty A., Winson G. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with β-blockade or verapamil. Circ Heart Fail. 2013;6:694–702. doi: 10.1161/CIRCHEARTFAILURE.112.000122. [DOI] [PubMed] [Google Scholar]
- 63.Nistri S., Olivotto I., Maron M.S. Beta-blockers for prevention of exercise-induced left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Am. J. Cardiol. 2012;110:715–719. doi: 10.1016/j.amjcard.2012.04.051. [DOI] [PubMed] [Google Scholar]
- 64.Toshima H., Koga Y., Nagata H., Toyomasu K., Itaya K., Matoba T. Comparable effects of oral diltiazem and verapamil in the treatment of hypertrophic cardiomyopathy. Double-blind Crossover Study. Jpn Heart. 1986;27:701–715. doi: 10.1536/ihj.27.701. [DOI] [PubMed] [Google Scholar]
- 65.Sherrid M.V., Barac I., McKenna W.J. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2005;45:1251–1258. doi: 10.1016/j.jacc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 66.Maron B.J., Rowin E.J., Udelson J.E., Maron M.S. Clinical spectrum and management of heart failure in hypertrophic cardiomyopathy. JACC Heart Fail. 2018;6:353–363. doi: 10.1016/j.jchf.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 67.Teekakirikul P., Eminaga S., Toka O. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by nonmyocyte proliferation and requires TGF-β. J. Clin. Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015;3:123-31. [DOI] [PubMed]
- 69.The Design of the Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) Trial. Am Heart J. 2017;187:145-55. [DOI] [PMC free article] [PubMed]
- 70.Rowin E.J., Hausvater A., Link M.S. Clinical profile and consequences of atrial fibrillation in hypertrophic cardiomyopathy. Circulation. 2017;136:2420–2436. doi: 10.1161/CIRCULATIONAHA.117.029267. [DOI] [PubMed] [Google Scholar]
- 71.Siontis K.C., Geske J.B., Ong K., Nishimura R.A., Ommen S.R., Gersh B.J. Atrial fibrillation in hypertrophic cardiomyopathy: prevalence, clinical correlations, and mortality in a large high-risk population. J. Am. Heart Assoc. 2014;3(3) doi: 10.1161/JAHA.114.001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roy D., Talajic M., Dorian P. Amiodarone to prevent recurrence of atrial fibrillation. Canadian trial of atrial Fibrillation Investigators. N. Engl. J. Med. 2000;342:913–920. doi: 10.1056/NEJM200003303421302. [DOI] [PubMed] [Google Scholar]
- 73.Di Donna P., Olivotto I., Delcrè S.D. Efficacy of catheter ablation for atrial fibrillation in hypertrophic cardiomyopathy: impact of age, atrial remodelling, and disease progression. Europace. 2010;12:347–355. doi: 10.1093/europace/euq013. [DOI] [PubMed] [Google Scholar]
- 74.Santangeli P., Di Biase L., Themistoclakis S. Catheter ablation of atrial fibrillation in hypertrophic cardiomyopathy: long-term outcomes and mechanisms of arrhythmia recurrence. Circ. Arrhythm. Electrophysiol. 2013;6:1089–1094. doi: 10.1161/CIRCEP.113.000339. [DOI] [PubMed] [Google Scholar]
- 75.Guttmann O.P., Rahman M.S., O'Mahony C., Anastasakis A., Elliott P.M. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100(6):465–472. doi: 10.1136/heartjnl-2013-304276. [DOI] [PubMed] [Google Scholar]
- 76.Noseworthy P.A., Yao X., Shah N.D., Gersh B.J. Stroke and bleeding risks in NOAC- and Warfarin-treated patients with hypertrophic cardiomyopathy and atrial fibrillation. J. Am. Coll. Cardiol. 2016;67:3020–3021. doi: 10.1016/j.jacc.2016.04.026. [DOI] [PubMed] [Google Scholar]
- 77.Dominguez F., Climent V., Zorio E. Direct oral anticoagulants in patients with hypertrophic cardiomyopathy and atrial fibrillation. Int. J. Cardiol. 2017;248:232–238. doi: 10.1016/j.ijcard.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 78.Kirchhof P., Benussi S., Kotecha D. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. Heart J. 2016;37:2893–2962. doi: 10.1093/eurheartj/ehw210. [DOI] [PubMed] [Google Scholar]
- 79.Dearani J.A., Ommen S.R., Gersh B.J., Schaff H.V., Danielson G.K. Surgery insight: septal myectomy for obstructive hypertrophic cardiomyopathy-the Mayo Clinic experience. Nat. Clin. Pract. Cardiovasc. Med. 2007;4:503–512. doi: 10.1038/ncpcardio0965. [DOI] [PubMed] [Google Scholar]
- 80.Morrow A.G., Brockenbrough E.C. Surgical treatment of idiopathic hypertrophic subaortic stenosis. Am. Surg. 1961;154:181–189. doi: 10.1097/00000658-196108000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maron B.J. Surgical myectomy remains the primary treatment option for severely symptomatic patients with obstructive hypertrophic cardiomyopathy. Circulation. 2007;116:196–206. doi: 10.1161/CIRCULATIONAHA.107.691378. [DOI] [PubMed] [Google Scholar]
- 82.Ommen S.R., Maron B.J., Olivotto I. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2005;46:470–476. doi: 10.1016/j.jacc.2005.02.090. [DOI] [PubMed] [Google Scholar]
- 83.Kotkar K.D., Said S.M., Dearani J.A., Schaff H.V. Hypertrophic obstructive cardiomyopathy: the Mayo Clinic experience. Ann. Cardiothorac. Surg. 2017;6:329–336. doi: 10.21037/acs.2017.07.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rastegar H., Boll G., Rowin E.J. Results of surgical septal myectomy for obstructive hypertrophic cardiomyopathy: the Tufts experience. Ann. Cardiothorac. Surg. 2017;6:353–363. doi: 10.21037/acs.2017.07.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meghji Z., Nguyen A., Fatima B. Survival differences in women and men after septal myectomy for obstructive hypertrophic cardiomyopathy. JAMA Cardiol. 2019;4(3):237–245. doi: 10.1001/jamacardio.2019.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sigwart U. Non-surgical myocardial reduction of hypertrophic obstructive cardiomyopathy. Lancet. 1995;346:211–214. doi: 10.1016/s0140-6736(95)91267-3. [DOI] [PubMed] [Google Scholar]
- 87.Seggewis H., Faber L., Gluchmam V. Percutaneous transluminal septal ablation in hypertrophic obstructive cardiomyopathy. Thorac. Cardiovasc. Surg. 1999;47:94–100. doi: 10.1055/s-2007-1013118. [DOI] [PubMed] [Google Scholar]
- 88.Ten Cate F.J., Soliman O.I., Michels M. Long-term outcome of alcohol septal ablation in patients with obstructive hypertrophic cardiomyopathy: a word of caution. Circ. Heart Fail. 2010;3:362–369. doi: 10.1161/CIRCHEARTFAILURE.109.862359. [DOI] [PubMed] [Google Scholar]
- 89.Zeng Z., Wang F., Dou X., Zhang S., Pu J. Comparison of percutaneous transluminal septal myocardial ablation versus septal myectomy for the treatment of patients with hypertrophic obstructive cardiomyopathy: a meta-analysis. Int. J. Cardiol. 2006;112:80–84. doi: 10.1016/j.ijcard.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 90.Leonardi R.A., Kransdorf E.P., Simel D.L., Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ. Cardiovasc. Interv. 2010;3:97–104. doi: 10.1161/CIRCINTERVENTIONS.109.916676. [DOI] [PubMed] [Google Scholar]
- 91.Geitzen F.H., Leuner Ch.J., Raute-Kreinsen U. Acute and long-term results after transcoronary ablation of septal hypertrophy (TASH) Eur. Heart J. 1999;20:1342–1354. doi: 10.1053/euhj.1999.1520. [DOI] [PubMed] [Google Scholar]
- 92.Fifer M.A., Sigwart U. Hypertrophic obstructive cardiomyopathy: alcohol septal ablation. Eur. Heart J. 2011;32:1059–1064. doi: 10.1093/eurheartj/ehr013. [DOI] [PubMed] [Google Scholar]
- 93.Nagueh S.F., Groves B.M., Schwartz L. Alcohol septal ablation for the treatment of hypertrophic obstructive cardiomyopathy: a multicenter North American registry. J. Am. Coll. Cardiol. 2011;58:2322–2328. doi: 10.1016/j.jacc.2011.06.073. [DOI] [PubMed] [Google Scholar]
- 94.Kim L.K., Swaminathan R.V., Looser P. Hospital volume outcomes after septal myectomy and alcohol septal ablation for treatment of obstructive hypertrophic cardiomyopathy: US nationwide inpatient database, 2003–2011. JAMA Cardiol. 2016;1(3):324–332. doi: 10.1001/jamacardio.2016.0252. [DOI] [PubMed] [Google Scholar]
- 95.Daubert C., Gadler F., Mabo P., Linde C. Pacing for hypertrophic obstructive cardiomyopathy: an update and future directions. Europace. 2018;20(6):908–920. doi: 10.1093/europace/eux131. [DOI] [PubMed] [Google Scholar]
- 96.Maron B.J., Nishimura R.A., McKenna W.J. Assessment of permanent dual-chamber pacing as a treatment for drug-refractory symptomatic patients with obstructive hypertrophic cardiomyopathy. a randomized, double-blind, crossover study (M-Pathy) Circulation. 1999;99:2927–2933. doi: 10.1161/01.cir.99.22.2927. [DOI] [PubMed] [Google Scholar]
- 97.Maron M.S., Kalsmith B.M., Udelson J.E., Li W., DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ. Heart Fail. 2010;3:574–579. doi: 10.1161/CIRCHEARTFAILURE.109.922872. [DOI] [PubMed] [Google Scholar]
- 98.Rowin E.J., Maron B.J., Abt P. Impact of advanced therapies in improving survival to heart transplant in patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 2018;121:986–996. doi: 10.1016/j.amjcard.2017.12.044. [DOI] [PubMed] [Google Scholar]
- 99.Rowin E.J., Maron B.J., Kiernan M.S. Advanced heart failure with preserved systolic function in nonobstructive hypertrophic cardiomyopathy: under-recognized subset of candidates for heart transplant. Circ. Heart Fail. 2014;7:967–975. doi: 10.1161/CIRCHEARTFAILURE.114.001435. [DOI] [PubMed] [Google Scholar]
- 100.Sorajja P., Pedersen W.A., Bae R. First experience with percutaneous mitral valve plication as primary therapy for symptomatic obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2016;67:2811–2818. doi: 10.1016/j.jacc.2016.03.587. [DOI] [PubMed] [Google Scholar]
- 101.Sherrid M.V., Balaram S., Kim B. The mitral valve in obstructive hypertrophic cardiomyopathy: a test in context. J. Am. Coll. Cardiol. 2016;67(15):1846–1858. doi: 10.1016/j.jacc.2016.01.071. [DOI] [PubMed] [Google Scholar]
- 102.Said S.M., Schaff H.V., Abel M.D., Dearani J.A. Transapical approach for apical myectomy and relief of midventricular obstruction in hypertrophic cardiomyopathy. J. Card. Surg. 2012;27:443–448. doi: 10.1111/j.1540-8191.2012.01475.x. [DOI] [PubMed] [Google Scholar]
- 103.Scudeler T.L., Rezende P.C., Oikawa F.T., da Costa L.M., Hueb A.C., Hueb W. A case of mid-apical obstructive hypertrophic cardiomyopathy treated with a transapical myectomy approach: a case report. J. Med. Case Rep. 2014;8:364. doi: 10.1186/1752-1947-8-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Direct evidence that perhexiline modifies myocardial substrate utilization from fatty acids to lactate. J Cardiovasc Pharmacol. 1995;25:469-72. [DOI] [PubMed]
- 105.Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122:1562-9. [DOI] [PubMed]
- 106.Heitner S.B., Jacoby D., Lester S.J. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann. Intern. Med. 2019;170(11):741–748. doi: 10.7326/M18-3016. [DOI] [PubMed] [Google Scholar]
- 107.Lombardi R., Rodriguez G., Chen S.N. Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation. 2009;119:1398–1407. doi: 10.1161/CIRCULATIONAHA.108.790501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Marian A.J., Tan Y., Nagueh S.F. Hypertrophy regression with N-Acetylcysteine in hypertrophic cardiomyopathy (HALT-HCM): a randomized placebo controlled double blind pilot study. Circ. Res. 2018;122:1109–1118. doi: 10.1161/CIRCRESAHA.117.312647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Repetti G.G., Toepfer C.N., Seidman J.G., Seidman C.E. Novel therapies for prevention and early treatment of cardiomyopathies. Circ. Res. 2019;124:1536–1550. doi: 10.1161/CIRCRESAHA.119.313569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McKenna W.J., Deanfield J.E. Hypertrophic cardiomyopathy: an important cause of sudden death. Arch. Dis. Child. 1984;59:971–975. doi: 10.1136/adc.59.10.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arteaga E., Araújo A.Q., Bernstein M. Prognostic value of the collagen volume fraction in hypertrophic cardiomyopathy. Arq. Bras. Cardiol. 2009;92:216–220. doi: 10.1590/s0066-782x2009000300010. [DOI] [PubMed] [Google Scholar]
- 112.Mirowski M., Reid P.R., Mower M.M. Termination of malignant ventricular arrhythmias with an implanted automatic defibrillator in human beings. N. Engl. J. Med. 1980;303:22–24. doi: 10.1056/NEJM198008073030607. [DOI] [PubMed] [Google Scholar]
- 113.Maron B.J., Shen W.K., Link M.S. Efficacy of implantable cardioverter-defibrillator for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N. Engl. J. Med. 2000;342:365–374. doi: 10.1056/NEJM200002103420601. [DOI] [PubMed] [Google Scholar]
- 114.Begley D.A., Mohiddin S.A., Tripodi D., Winckler J.B., Fananapazir L. Efficacy of implantable cardioverter defibrillator therapy for primary and secondary prevention of sudden death in hypertrophic cardiomyopathy. PACE. 2003;26:1887–1896. doi: 10.1046/j.1460-9592.2003.00285.x. [DOI] [PubMed] [Google Scholar]
- 115.Maron B.J., Spirito P., Shen W.K. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405–412. doi: 10.1001/jama.298.4.405. [DOI] [PubMed] [Google Scholar]
- 116.Syska P., Przybylski A., Chojnowska L. Implantable cardioverter-defibrillator in patients with hypertrophic cardiomyopathy: efficacy and complications of the therapy in long-term follow-up. J. Cardiovasc. Eletrophysiol. 2010;21:883–889. doi: 10.1111/j.1540-8167.2009.01716.x. [DOI] [PubMed] [Google Scholar]
- 117.Lin G., Nishimura R.A., Gersh B.J., Ommen S., Ackerman M., Brady P.A. Device complications and inappropriate implantable cardioverter-defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709–714. doi: 10.1136/hrt.2008.150656. [DOI] [PubMed] [Google Scholar]
- 118.Maron B.J., Spirito P., Ackerman M.J. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2013;61:1527–1535. doi: 10.1016/j.jacc.2013.01.037. [DOI] [PubMed] [Google Scholar]
- 119.O’Mahony C., Jichi F., Pavlou M. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD) Eur. Heart J. 2014;35:2010–2020. doi: 10.1093/eurheartj/eht439. [DOI] [PubMed] [Google Scholar]
- 120.Oka K., Tsujino T., Nakao S. Symptomatic ventricular tachyarrhythmia is associated with delayed gadolinium enhancement in cardiac magnetic resonance imaging and with elevated plasma brain natriuretic peptide level in hypertrophic cardiomyopathy. J. Cardiol. 2008;52:146–153. doi: 10.1016/j.jjcc.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 121.Monserrat L., Elliott P.M., Gimeno J.R., Sharma S. Penas Lado-M, McKenna WJ. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J. Am. Coll. Cardiol. 2003;42:873–879. doi: 10.1016/s0735-1097(03)00827-1. [DOI] [PubMed] [Google Scholar]
- 122.Spirito P., Bellone P., Harris K.M., Bernabo P., Bruzzi P., Maron B.J. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med. 2000;342:1778–1785. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 123.Maki S., Ikeda H., Muro A. Predictors of sudden cardiac death in hypertrophic cardiomyopathy. Am. J. Cardiol. 1998;82:774–778. doi: 10.1016/s0002-9149(98)00455-x. [DOI] [PubMed] [Google Scholar]
- 124.Pelliccia A., Zipes D.P., Bethesda Maron BJ. Conference #36 and the European Society of Cardiology Consensus Recommendations revisited a comparison of U.S. and European criteria for eligibility and disqualification of competitive athletes with cardiovascular abnormalities. J. Am. Coll. Cardiol. 2008;52:1990–1996. doi: 10.1016/j.jacc.2008.08.055. [DOI] [PubMed] [Google Scholar]
- 125.Sharma S., Elliot P., Whyte G., Mahon N., Whipp B., McKenna W.J. Utility of cardiopulmonary exercise in the assessment of clinical determinants of functional capacity in hypertrophic cardiomyopathy. Am. J. Cardiol. 2000;86:162–168. doi: 10.1016/s0002-9149(00)00854-7. [DOI] [PubMed] [Google Scholar]
- 126.Spirito P., Rapezzi C., Bellone P. Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99:2132–2137. doi: 10.1161/01.cir.99.16.2132. [DOI] [PubMed] [Google Scholar]
- 127.Goland S., van Hagen I.M., Elbaz-Greener G. Pregnancy in women with hypertrophic cardiomyopathy: data from the European Society of Cardiology initiated Registry of Pregnancy and Cardiac disease (ROPAC) Eur. Heart J. 2017;38:2683–2690. doi: 10.1093/eurheartj/ehx189. [DOI] [PubMed] [Google Scholar]
- 128.Avila W.S., Amaral F.M., Ramires J.A. Influence of pregnancy on clinical course and fetal outcome of women with hypertrophic cardiomyopathy. Arq. Bras. Cardiol. 2007;88:480–485. doi: 10.1590/s0066-782x2007000400019. [DOI] [PubMed] [Google Scholar]
- 129.Autore C., Conte M.R., Piccininno M. Risk associated with pregnancy in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2002;40:1864–1869. doi: 10.1016/s0735-1097(02)02495-6. [DOI] [PubMed] [Google Scholar]