

Abstract
Background
Although aortic regurgitation (AR) is a clinically important condition that is becoming increasingly common, few relevant murine models and mechanistic studies exist for this condition. In this study, we attempted to delineate the pathological and molecular changes and address the roles of some potentially relevant molecules in an animal model of surgically induced AR.
Methods
AR was induced by puncturing the aortic valve leaflets in C57BL/6J mice under echocardiographic guidance.
Results
As early as 1 week following AR, the left ventricles (LV) displayed marked impairments in diastolic function and coronary flow reserve (CFR), as well as cardiac hypertrophy and chamber dilatation at both end-systole and end-diastole. LV free wall thickening and cardiomyocyte hypertrophy in LV were observed 2 weeks following of AR while a decline in ejection fraction was not seen until after 4 weeks. Nppa (natriuretic peptide A) and Nppb (natriuretic peptide B) increased over time, in conjunction with prominent Akt activation as well as slight CaMKII (Ca2+/calmodulin-dependent protein kinase II) activation and biphasic changes in β-arrestin-2 expression. Treatment of AR mice with Akt inhibition exacerbated the eccentric hypertrophy, while neither inhibition of CaMKII nor β-arrestin-2 overexpression influenced the response to AR.
Conclusions
Our structural, functional, molecular and therapeutic analyses reveal that Akt, but not CaMKII or β-arrestin-2, plays a regulatory role in the development of LV remodeling after AR in Mice. These results may shed important light on therapeutic targets for volume overloaded cardiomyopathy.
Keywords: Aortic regurgitation, eccentric hypertrophy, volume overload, signaling effector, mouse
Introduction
Aortic regurgitation (AR) is characterized by volume overload-induced eccentric hypertrophy due to diastolic reflux of blood from the aorta into the left ventricle (LV) (1,2). The Framingham Heart Study reported that the overall prevalence of AR was 13% in men and 8.5% in women (1,3). It’s anticipated that the prevalence of AR will increase even further due to the rapidly growing aged population worldwide (3,4). Although laudable progress has been made in the management of AR, the pathophysiological mechanisms involved in chronic AR remain not fully elucidated (5). Moreover, although mouse models of cardiac disease are common, in part because of abundant knowledge of their genome, appropriate and clinically relevant mouse models of AR for investigation of its impact on cardiac remodeling are still not fully ascertained (6).
Volume overload often occurs clinically as a result of AR (7). Most animal models of volume overload have used arteriovenous fistulae (aortocaval shunts) (8,9) which, however, rarely occur in humans (10). Moreover, arteriovenous fistulae result in mixing of arterial and venous blood and do not recapitulate the increases in diastolic blood flow in the ventricles observed with valve disorders, thereby potentially limiting the relevance and applicability of these animal models (11,12). Taking these issues into consideration, we have previously established an AR model to induce volume overload in mice, by disrupting the aortic valve cusps. In this context, we focused initially on macrovascular hemodynamic profiles (13,14). Compared with arteriovenous fistulae models, this novel volume overload model induced by AR has clinically distinct stress on the heart characterized by back flow versus pure forward flow, and shows less stress on veins. In a more recent study, we measured changes of several load-related signaling effectors in response to 2 weeks of AR, and found that Akt and Ca2+/calmodulin-dependent kinase II (CaMKII) were activated in conjunction with reduced β-arrestin-2 levels (15). Given these previous data, we have designed the present study, which aimed at extending our knowledge on the effects of AR over a more extended period (from adaptive hypertrophy to heart failure), with a particular focus on the potential regulatory roles of Akt, β-arrestin-2, and CaMKII on the overall cardiac remodeling process.
Methods
Mice and experimental protocol
Animal experiments followed the recommendations of Guide for the Care and Use of Laboratory Animals (No. 85-23, revised 1996; National Institutes of Health, Bethesda, MD, USA). The experimental protocol (No. 2016-048, approved February 18, 2016) was approved by the Animal Care and Use Committee of Zhongshan Hospital, Fudan University. The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. A total of 161 male C57BL/6J mice (including 53 mice for the survival study), 11–14 weeks old, weighing 22.0–28.0 g, purchased from the Shanghai Branch of the National Rodent Laboratory Animal Resources of China, were enrolled to serially investigate the LV response after the AR operation. The animals were housed at 24±2 °C under 12-h light/12-h dark cycles, with free access to tap water and mouse chow. Echocardiographic evaluation was performed 1 day (0W) before the AR operation, as well as 1 week (1W), 2W, 4W and 8W after the AR operation. At each time point, 8 mice for each group were used for hemodynamic studies followed by histological (n=4 for each group) and molecular (n=8 for each group) assessments.
Creation of the AR model
AR surgery was performed under the guidance of ultrasound imaging, according to the methods we have previously described (13,14). Briefly, after the mice were anesthetized with 2% isoflurane, a plastic catheter containing a metal wire was inserted into the right common carotid artery, then the metal wire was advanced through the catheter to puncture the aortic valve until the Doppler ultrasound showed significant diastolic retrograde flow (>200 mm/s) in the aortic arch (Figure 1). Finally, the catheter together with the metal wire was withdrawn, and the right common carotid artery was enclosed. Meloxicam (0.13 mg each) was administered subcutaneously for analgesia. The corresponding sham-operated mice underwent the same procedure without the puncturing of the aortic valve.
Figure 1.

Creation and evaluation of aortic regurgitation (AR) in mice. (A) The plastic catheter and the metal wire used in AR surgery. Scale bar =1 cm; (B) echocardiographic imaging of the region of ascending aorta and aortic valves which contains the plastic catheter and the metal wire, as labeled, just prior to aortic valve disruption; (C) schematic of the surgery procedure. From left to right, before, during and after aortic valves puncture; (D) color Doppler images of aortic arch flow during cardiac systole and diastole from sham-operated and AR mice 2 weeks after surgery. Red indicates flow toward, and blue indicates flow away from the transducer; (E) pulsed wave Doppler recordings of aortic arch flow during cardiac systole and diastole from sham-operated and AR mice. In this view, the systolic flow is downwards, and the diastolic flow is upwards (regurgitant). The scale in the panel is in units of meters/second (m/sec).
Echocardiography
Cardiac structure and function were assessed by M-mode and Doppler echocardiography at the aforementioned time points, using high-frequency ultrasound systems Vevo770 and Vevo2100 (VisualSonics, Toronto, ON, Canada). The center frequencies of the transducers were 30-MHz, providing resolutions of 115 µm (lateral) × 55 µm (axial). The echocardiographic assessment had good intra- and inter-observer agreement, as reported in our previous studies (13,14,16,17). During the echocardiographic assessment, the mouse was positioned on a heating pad to maintain normothermia and anesthetized with 1% isoflurane. Heart rate was maintained at around 500 bpm, and the intercept angle between the Doppler beam line and flow direction was controlled within 60 degrees (18). The structure or hemodynamics of the LV and aortic arch, and basal left coronary artery (LCA) flow were measured during 1% isoflurane, and hyperemic LCA flow was induced by 140 µg·kg−1·min−1 adenosine infusion via tail vein during 1% isoflurane, according to the methods we described previously (13,14,16,17). Assessment parameters were listed as follows.
LV structure and function
The M-mode recording was made in a left parasternal long-axis view (19). LV end-diastolic (LVEDD) and end-systolic (LVESD) dimensions, LV posterior wall end-diastolic (LVPWTd) and end-systolic (LVPWTs) thickness, were measured, and LV ejection fraction (LVEF) and fractional shortening (LVFS), stroke volume (SV), and cardiac output (CO), were calculated from the following formulas, as we described previously (15,16):
| %EF = 100×[7.0÷(2.4+LVEDD)×LVEDD3-7.0÷(2.4+LVESD)×LVESD3]÷[7.0÷(2.4+LVEDD)×LVEDD3], |
| %FS = 100×[(LVEDD-LVESD)÷LVEDD], |
| SV = LVEDV-LVESV, |
| CO = SV×heart rate. |
LCA flow
The LCA was imaged from a modified left parasternal long axis view (19). With the Doppler sample volume placed in the proximal LCA, the flow spectrum was recorded. Coronary flow reserve (CFR) was calculated as the ratio of hyperemic (induced by adenosine infusion) to basal (no adenosine) diastolic area (velocity-time integral) of the LCA flow.
Aortic arch flow
The aortic arch flow was estimated using pulse-wave Doppler recordings (Figure 1D). Aortic regurgitation was identified by positive (reverse) flow profiles. The aortic arch flow during systole is shown in Figure 1D as negative going signals while regurgitant flow during diastole is identified as positive flow profiles and was only observed after aortic valve puncture. Mice were only included in our studies when the positive (reverse) flow profiles were above 200 mm/sec. The systolic and diastolic aortic flows were quantified using the velocity-time integral which was calculated by measuring the area of the negative and positive flow profiles in order to estimate the systole (SVTIa) and diastole (DVTIa) of aortic arch flow.
Mitral flow
The mitral Doppler flow waveform was acquired in the apical four-chamber view. The E/A ratio was calculated by the division of the peak velocity of the early ventricular filling wave (E wave) by the late ventricular filling wave caused by atrial contraction (A wave). The isovolumic relaxation time (IVRT) was measured from the closure point of the aortic valve to the onset of filling by the opening of the mitral valve, while the isovolumic contraction time (IVCT) was from the closure point of the mitral valve to the start of ejection by the opening of the aortic valve. The ventricular ejection time (ET) was from the start to the end of the aortic flow. The cardiac performance (Tei) index was calculated by the formula (IVCT+IVRT)/ET (20).
In principle, the diastolic function can be measured via E/A ratio or via systolic and diastolic time intervals obtained from the mitral flow waveforms (21,22). However, as reported in murine hearts with myocardial infarction (23), we found that the E and A waves were difficult to separate in AR mice, making it difficult to calculate the of E/A ratio. Thus, we used the diastolic function using the IVRT, IVCT), ventricular ET, and cardiac performance (Tei) index, all of which could be reliability estimated from our Doppler flow recordings, to assess diastolic function, as described below.
Invasive hemodynamic study
In vivo cardiac performance was evaluated by invasive hemodynamic measurement via a carotid artery catheter as we have previously described (18). Briefly, a micromanometer (Millar 1.4F, SPR 835; Millar Instruments, Houston, TX, USA) connected to a Power Laboratory system (AD Instruments, Castle Hill, Australia), was inserted into the left common carotid artery (for the dextral one was permanently occluded during the AR surgery) and carefully advanced into the LV. Heart rate, left ventricular end-systolic (LVESP) and end-diastolic (LVEDP) pressure, and maximal contraction and relaxation velocity (+dp/dt and −dp/dt) were recorded.
Histological analysis
After mice were euthanized, the hearts were excised, rinsed in saline, and heart weights and heart weight-to-body weight ratio (HW/BW) were measured. The hearts were then fixed in 10% formalin, embedded in paraffin, sectioned at a 4-µm thickness in the short axis at the papillary muscle level, and then stained with hematoxylin and eosin (H&E) for cardiomyocyte size, or with Masson trichrome for cardiac fibrosis. Digital photographs were taken using an image analysis system (Qwin V3, Leica, Wetzlar, Germany), at a magnification of 400 times for myocyte size evaluation, and of 200 times for fibrosis evaluation. With an image analysis system (Image-Pro Plus 5.0, Media Cybernetics, Inc. Bethesda, MD, USA), two or three sections from each heart were measured, and three high-power fields from each section were randomly chosen. Cross-sectional areas of left ventricular cardiomyocytes were measured. The extent of fibrosis was expressed as the ratio of Masson trichrome-stained area to total left ventricular area.
Quantitative real-time PCR
Total RNA was extracted from LV samples using Trizol reagent (catalog 15596026, Invitrogen, Carlsbad, CA, USA). The reverse transcription of PCR was performed using TOYOBO ReverTra Ace-α-RT-PCR kit (catalog FSQ-101, TOYOBO CO., Ltd., Osaka, Japan) according to the manufacturer’s instruction. The real-time PCR was performed in a volume of 25 µL in a 96-well plate using a CFX96 real-time PCR detection system (CFX96, Bio-Rad, Hercules, CA, USA). The targeted fetal genes (natriuretic peptide A, Nppa; and natriuretic peptide B, Nppb) and β-actin were amplified using their specific primers. The primers used for PCR analysis were: (I) Nppa gene, 5’-GGTGTCCAACACAGATCTGA-3’, and antisense: 5’-CCACTAGACCACTCATCTAC-3’; (II) Nppb gene, sense: 5’-GCCATTTCCTCCGACTTTTCTC-3’ and antisense: 5’-GAGGTCACTCCTATCCTCTGG-3’; (III) β-actin gene, sense: 5’-CCAGTTGGTAACAATGCCATGT-3’, and antisense: 5’-GGCTGTATTCCCCTCCATCG-3’. Quantification was performed according to a comparative Ct method (∆∆ CT).
Drug/vector treatments
We have previously shown that AR induces changes in Akt and CaMKII activity and in β-arrestin-2 expression (15). We in this study further investigated the role of these molecular factors in AR induced cardiac remodeling by inhibition of CaMKII and Akt, as well as by over-expression of β-arrestin-2. For CaMKII blockade, mice (n=8 for AR, n=4 for Sham) were treated with 0.1 mL of KN-93 (3 mg·kg−1·d−1, MedChem Express, Princeton, NJ, USA) administered by peritoneal injection each day for 28 days post-AR. For Akt blockade, mice (n=8 for AR, n=4 for Sham) were treated with 0.1 mL of Wortmannin (30 µg·kg−1·d−1, MedChem Express, Princeton, NJ, USA) administered by peritoneal injection each day for 28 days post AR. We chose Wortmannin as a PI3K/Akt inhibitor for it is normally used to inhibit Akt in heart (24,25). Another group of mice (n=8 for AR, n=4 for Sham) was administered with the same volume of saline. To over-express β-arrestin-2, mice (n=8 for AR, n=4 for Sham) received 0.1 mL (5E+11 v.g./mouse) of an adeno-associated virus (β-arrestin-2-AAV9, Obio Technology, Shanghai, China) administered by tail vein injection 4 weeks before the AR operation. An additional group of mice (n=8 for AR, n=4 for Sham) was administered with the same volume of scrambled AAV9 (Obio Technology, Shanghai, China). Considering no significant difference in cardiac morphology and function was found between saline and scrambled AAV9 treated mice (data not shown), these mice served as control mice (n=16 for AR, n=8 for Sham).
Western blotting
Total protein extracted from homogenized LV tissues was electrophoresed in 12% polyacrylamide gel and transferred to PVDF membrane (Millipore, Billerica, MA, USA). The blotted membranes were incubated with antibodies against Akt (1:1,000, catalog #9272, Cell Signaling Technology, Danvers, MA, USA), p-Akt (1:1,000, catalog #9271, Cell Signaling Technology, Danvers, MA, USA), β-arrestin-2 (1:1,000; catalogue #3857, Cell Signaling Technology, Danvers, MA, USA), CaMKII (1:1,000, catalogue #3362, Cell Signaling Technology, Danvers, MA, USA), p-CaMKII (1:1,000, catalog #12716, Cell Signaling Technology, Danvers, MA, USA). After three washes, the blotted membranes were then incubated with horseradish peroxidase-conjugated rabbit secondary antibody (1:5,000, catalog 31460, Thermo Scientific, Waltham, MA, USA). The antigen-antibody complexes were detected with ECL chemiluminescence reagents (catalog RPN2106, GE Healthcare, Piscataway, NJ, USA) and the densitometry was visualized using a LAS-3000 imaging system (FUJIFILM, Kanagawa, Japan).
Statistics
We used the Kurtosis and Skewness test to assess the normality of the data. Mean ± SEM was used to express normal distributed quantitative data. Multiple comparisons were conducted by one-way ANOVA with Student-Newman-Keuls (S-N-K) test. P<0.05 was considered statistically significant. Statistical evaluations were performed using the software SPSS 15.0 (SPSS Inc., Chicago, IL, USA).
Results
Structural and hemodynamic remodeling induced by AR
After the AR surgery, the integrated area of flow profile during both systole (SVTIa) and diastole (DVTIa) increased significantly (P<0.05) when compared to that of the sham operated mice, and these parameters did not change noticeably over the subsequent 8 weeks (Figure 2, Table 1). Moreover, AR led to a near doubling of the total (integrated) blood flow during systole with the extra systolic flow matching the estimated integrated flow during diastole at each time point (Table 1). These results establish that we were able to reproducibly generate AR in mice, by the aforementioned surgical procedure.
Figure 2.

Pulse wave Doppler flow spectra and M-mode echocardiography in mice before aortic regurgitation AR (0W), and either 1, 2, 4, or 8 weeks after AR. (A) Doppler aortic arch flow velocity for mice at the indicated time points. The systolic flow is downwards, and the diastolic flow is upwards (regurgitant). Notice the appearance of backward flow following AR. Scale bar =1 m/s; (B) M-mode images of the left ventricle for the indicated time points demonstrating progressive LV dilatation following AR. Scale bar =2 mm; (C) mitral Doppler spectra. As discussed in the Methods Section, E waves (i.e., Doppler flow signals associated with early ventricular filling) and A waves (flow signals associated with atrial contraction) could be consistently identified in normal mice and 1 week after AR, but these signals merged at later stages after AR. Scale bar =1 m/s; (D) basal (without adenosine) coronary flow velocity waveforms. Scale bar =200 mm/s; (E) D/S VTIa, diastolic/systolic velocity-time integral of aortic arch flow; (F) LVEDD, left ventricular end-diastolic dimension measured using M-mode; (G) LVESD, left ventricular end-systolic dimension; (H) SV, stroke volume; (I) LVPWTd, left ventricular posterior wall end-diastolic thickness; (J) LVPWTs, left ventricular posterior wall end-systolic thickness; (K) LVEF, left ventricular ejection fraction; (L) LVFS, left ventricular fractional shortening; (M) ET, ejection time; (N) Tei index; (O) CFR, coronary flow reserve. *, P<0.05 vs. sham; †, P<0.05 vs. 0W; ‡, P<0.05 vs. 1W; §, P<0.05 vs. 2W. LVEDD, left ventricular end-diastolic dimension measured using M-mode; LVESD, left ventricular end-systolic dimension; SV, stroke volume; LVPWTd, left ventricular posterior wall end-diastolic thickness; LVPWTs, left ventricular posterior wall end-systolic thickness; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; ET, ejection time; CFR, coronary flow reserve.
Table 1. General information and echocardiographic characteristics of mice subjected to AR or sham operation.
| Parameter | 0W | 1W | 2W | 4W | 8W | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sham | AR | Sham | AR | Sham | AR | Sham | AR | Sham | AR | |||||
| Heart weight (g) | 0.11±0.01 | 0.11±0.02 | 0.11±0.01 | 0.16±0.01*† | 0.12±0.01 | 0.18±0.01*† | 0.12±0.01 | 0.20±0.01*†‡ | 0.13±0.01†‡§ | 0.22±0.01*†‡§ | ||||
| Body weight (g) | 24.69±0.33 | 24.31±0.42 | 25.33±0.62 | 24.43±0.44 | 26.52±0.44 | 26.03±0.50 | 26.87±0.75†‡ | 26.19±0.59 | 28.43±0.77†‡ | 27.64±0.67†‡ | ||||
| HW/BW (mg/g) | 4.49±0.14 | 4.25±0.14 | 4.38±0.06 | 5.93±0.20*† | 4.37±0.06 | 6.54±0.15*† | 4.44±0.08 | 7.40±0.23*†‡§ | 4.48±0.08 | 7.75±0.19*†‡§ | ||||
| HR (bpm) | 505±17 | 517±12 | 497±15 | 517±8 | 480±7 | 503±7 | 507±11 | 519±7 | 513±12 | 517±8 | ||||
| SVTIa (cm) | 3.30±0.13 | 2.94±0.12 | 3.06±0.11 | 5.78±0.15*† | 3.10±0.08 | 6.15±0.22*† | 3.18±0.11 | 6.44±0.34*† | 3.13±0.11 | 6.66±0.35*† | ||||
| DVTIa (cm) | 0 | 0 | 0 | 2.57±0.16*† | 0 | 2.76±0.13*† | 0 | 2.73±0.16*† | 0 | 2.63±0.11*† | ||||
| D/S VTIa | 0 | 0 | 0 | 0.46±0.04*† | 0 | 0.46±0.03*† | 0 | 0.43±0.03*† | 0 | 0.42±0.03*† | ||||
| LVEDD (mm) | 3.72±0.06 | 3.69±0.05 | 3.80±0.08 | 4.66±0.05*† | 3.85±0.05 | 4.83±0.06*† | 3.93±0.07 | 5.32±0.12*†‡§ | 3.98±0.08 | 5.85±0.13*†‡§|| | ||||
| LVESD (mm) | 2.43±0.08 | 2.40±0.07 | 2.58±0.11 | 3.22±0.05*† | 2.59±0.07 | 3.31±0.07*† | 2.67±0.09 | 3.98±0.12*†‡§ | 2.68±0.09 | 4.77±0.17*†‡§|| | ||||
| SV (μL) | 38.0±1.4 | 37.3±1.1 | 37.4±1.5 | 58.9±1.7*† | 39.5±1.1 | 64.5±1.7*† | 40.5±1.2 | 67.0±2.7*† | 43.5±1.6†‡ | 62.5±2.5*† | ||||
| CO (mL/min) | 16.7±1.8 | 19.2±0.5 | 18.5±1.3 | 28.2±1.5*† | 17.0±1.7 | 32.2±1.2*† | 19.7±1.2 | 33.9±1.9*† | 21.8±1.2†‡§ | 33.7±1.6*† | ||||
| LVEF (%) | 63.11±1.67 | 63.72±1.62 | 60.89±2.60 | 58.39±1.18 | 61.99±1.51 | 59.58±1.02 | 60.81±1.81 | 48.94±1.41*†‡§ | 62.35±1.67 | 36.49±2.04*†‡§|| | ||||
| LVFS (%) | 33.78±1.22 | 34.62±1.18 | 32.88±1.70 | 30.90±0.77 | 33.17±1.03 | 31.79±0.72 | 32.38±1.25 | 25.10±0.86*†‡§ | 33.42±1.20 | 17.97±1.11*†‡§|| | ||||
| LVPWTd (mm) | 0.75±0.02 | 0.78±0.03 | 0.73±0.02 | 0.74±0.02 | 0.78±0.02 | 0.86±0.02*†‡ | 0.78±0.02 | 0.91±0.02*†‡ | 0.77±0.02 | 0.93±0.02*†‡ | ||||
| LVPWTs (mm) | 1.11±0.02 | 1.13±0.02 | 1.08±0.02 | 1.10±0.02 | 1.12±0.02 | 1.23±0.02*†‡ | 1.11±0.03 | 1.22±0.03*†‡ | 1.13±0.03 | 1.19±0.03‡ | ||||
| IVCT (ms) | 11.86±0.79 | 11.82±0.68 | 12.29±1.42 | 10.65±0.61 | 12.36±0.59 | 13.28±1.11 | 13.26±1.24 | 11.10±1.49 | 11.98±0.86 | 11.64±0.55 | ||||
| ET (ms) | 47.17±1.13 | 47.38±1.37 | 46.87±0.63 | 58.06±1.29*† | 45.97±1.19 | 60.77±0.91*† | 49.88±2.00 | 62.37±2.86*† | 46.61±1.51 | 58.56±1.32*† | ||||
| IVRT (ms) | 15.97±0.52 | 15.87±0.42 | 17.09±0.87 | 15.74±0.62 | 16.39±0.35 | 17.30±0.66 | 17.98±1.24 | 17.72±1.16 | 16.67±0.79 | 18.26±0.59 | ||||
| Tei index | 0.59±0.03 | 0.58±0.02 | 0.63±0.05 | 0.46±0.02*† | 0.63±0.02 | 0.50±0.02*† | 0.63±0.04 | 0.47±0.03*† | 0.62±0.02 | 0.51±0.01*† | ||||
| Basal DVTIc (cm) | 1.28±0.07 | 1.35±0.07 | 1.32±0.04 | 2.15±0.10*† | 1.46±0.08 | 2.37±0.09*† | 1.46±0.07 | 2.35±0.21*† | 1.38±0.11 | 2.45±0.12*† | ||||
| Hyperemic DVTIc (cm) | 4.08±0.19 | 4.02±0.36 | 4.01±0.23 | 3.88±0.40 | 4.00±0.33 | 3.84±0.27 | 4.15±0.21 | 3.81±0.52 | 4.18±0.31 | 3.73±0.14 | ||||
| CFR | 3.19±0.29 | 2.97±0.21 | 3.03±0.13 | 1.80±0.13*† | 2.72±0.21 | 1.65±0.15*† | 2.86±0.12 | 1.59±0.14*† | 3.04±0.08 | 1.54±0.12*† | ||||
*, P<0.05 vs. the corresponding values of sham-operated mice; †, P<0.05 vs. the corresponding values of 0 week pre-operation; ‡, P<0.05 vs. the corresponding values of 1 week post-operation; §, P<0.05 vs. the corresponding values of 2 weeks post-operation; ||, P<0.05 vs. the corresponding values of 4 weeks post-operation. AR, aortic regurgitation; HW/BW, ratio of heart weight to body weight; HR, heart rate; SVTIa, systolic velocity-time integral of aortic arch flow; DVTIa, diastolic velocity-time integral of aortic arch flow; LVPWTd, left ventricular posterior wall end-diastolic thickness; LVPWTs, left ventricular posterior wall end-systolic thickness; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; SV, stroke volume; CO: cardiac output; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; DVTIc, diastolic velocity-time integral of left coronary artery flow; CFR, coronary flow reserve; IVCT, isovolumic contraction time; ET, ejection time; IVRT, isovolumic relaxation time.
AR also induced marked elevations (~2-fold) in the estimated SV along with increases in both LVEDD and LVESD within the first week post-surgery (Figure 2). Thereafter, the hearts underwent progressive dilatation over the subsequent 8 weeks with a relatively larger increase in LVESD than LVEDD, when compared to baseline sham-operated mice. Despite this progressive ventricular dilatation, stroke volume (SV) did not increase further with time. By contrast, although estimated ejection fraction (LVEF) and fraction shortening (LVFS) was not altered initially following AR, these parameters did decline by the 4th week following AR with a further decline thereafter (Figure 2, Table 1), suggesting systolic dysfunction at later time points.
These changes observed, with echocardiography, in cardiac structure and function, were associated with corresponding hemodynamic changes. For example, the LVESP was significantly elevated beginning as early as 1 week post-surgery and with further increases at later time points, consistent with the progressive elevations in SV. These elevations in LVESP were associated, as expected, with prolongation of the ejections times (ETs) as well as progressive suppression of both the Tei index (Figure 2, Table 1) and maximal dp/dt (dp/dtmax), which was in agreement with progressive reductions in LVEF, albeit with an earlier onset (Figure 3). Nevertheless, the isovolumic contraction times (i.e., IVCT) were unaffected by AR. More interesting perhaps is the early decline in the magnitude of the minimum dp/dt (-dp/dtmin) which was seen at 1W post-surgery (Figure 3), suggesting possible early impairment of diastolic function. Indeed, LVEDP was progressively elevated over the 8-week period (Figure 3). We attempted to assess diastolic function further by estimating the E/A ratio, but we found that the E and A waves were not readily separated in most mice 1W post-surgery. However, despite the elevations in LVEDP, the isovolumic relaxation (i.e., IVRT) remained normal in the AR mice, as compared with the sham operated mice, regardless of the time point, suggesting that active relaxation is not markedly affected by severe prolonged volume overload (Figure 3D, Table 1).
Figure 3.

Invasive left ventricular hemodynamics in mice before aortic regurgitation AR (0W), and at 1, 2, 4, and 8 weeks after AR. (A) Left ventricular pressure (unit: mmHg); (B) left ventricular contraction velocity and relaxation velocity (unit: mmHg/s); (C) LVESP; (D) LVEDP; (E) maximal +dp/dt; (F) minimal −dp/dt. *, P<0.05 vs. sham; †, P<0.05 vs. 0W; ‡, P<0.05 vs. 1W; §, P<0.05 vs. 2W. LVESP, left ventricular end-systolic pressure; LVEDP, left ventricular end-diastolic pressure.
The left ventricular dilatation induced by AR was associated with cardiac hypertrophy as indicated by progressive elevations in gross appearance (Figure 4), heart-weight to body-weight ratios beginning after the 1st week following AR (Table 1), while LV cardiomyocyte cross-sectional areas (Figure 4), as well as LVPWTd and LVPWTs (Figure 2), were measurably increased beginning 2 weeks following AR. Another notable finding was the presence of mild interstitial fibrosis in LV wall at all time points (Figure 4), with no evidence for differences in the amount of perivascular connective tissue between AR and sham-operated mice (Figure 4, Figure S1).
Figure 4.

Gross samples and histological analyses of mouse hearts before aortic regurgitation AR (0W), and at 1, 2, 4, and 8 weeks after AR. (A) Representative pictures of whole heart (n=15–28 per group). Scale bar =1 mm; (B) HE-stained heart sections (n=8–14 per group). Scale bar =1 mm; (C) HE-stained cardiomyocytes (n=200–350 cells per group). Scale bar =20 µm; (D) Masson-stained interstitial fibrosis (n=32–56 slides per group). Scale bar =50 µm; (E) Masson-stained perivascular fibrosis (n=8–14 slides per group). Scale bar =50 µm; (F) quantification of CSA of cardiomyocytes; (G) quantification of interstitial fibrosis. *, P<0.05 vs. sham; †, P<0.05 vs. 0W; ‡, P<0.05 vs. 1W, §, P<0.05 vs. 2W; ||, P<0.05 vs. 4W.
Figure S1.

Representative images of Masson-stained perivascular fibrosis in mice subjected to 4-week AR, TAC, or corresponding sham operations (scale bar, 50 µm). Perivascular fibrosis is more conspicuous in TAC mice than in AR mice. TAC, transverse aortic constriction. AR, aortic regurgitation.
Consistent with the elevated SV and LVESP, we found that left coronary blood flow, as measured by the basal velocity-time integral of left coronary artery flow (basal DVTIc), was augmented (P<0.05) by similar amounts at all time points, as might be expected from the increases in cardiac output needed with the flow regurgitation associated with valve disruption. By contrast, the hyperemic DVTIc induced by adenosine infusion (see Methods) was unaffected by AR. Resultantly, the CFR, estimated using the ratio of hyperemic to basal DVTIc, was compromised (Figure 2, Table 1).
Changes in gene expression and signaling pathways induced by AR
In association with the LV hypertrophy and reduced contractility, both Nppa and Nppb expression increased (P<0.05) progressively with time in AR mice, whilst remaining constant in sham-operated mice (Figure 5). We then focused on Akt, CaMKII and β-arrestin-2 which may be involved in the structural and functional changes induced by AR. As shown in Figure 6, we confirmed that CaMKII phosphorylation (Figure 6A) and Akt phosphorylation (Figure 6C) are both increased in the early period following AR. But interestingly, while CaMKII activity (Figure 6B) continued to increase progressively with time with a ~2-fold increase after 8 weeks following AR, Akt phosphorylation peaked at 1 week after AR with a ~6-fold increase and gradually waned thereafter to ~2-fold after 8 weeks following AR. By contrast, β-arrestin-2 expression was suppressed in the first 2 weeks following AR, consistent with our previous results (15), but then gradually increased thereafter, being ~30% higher than sham-operated controls at the 8-week time point.
Figure 5.
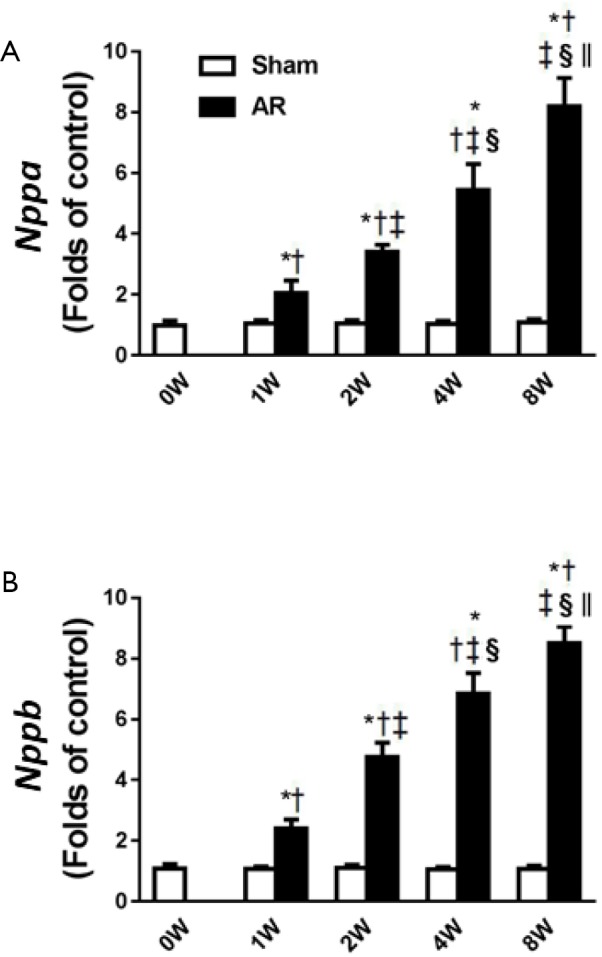
The sequential changes in Nppa and Nppb before aortic regurgitation AR (0W), and at 1, 2, 4, and 8 weeks after AR. (A) Nppa; (B) Nppb. †, P<0.05 vs. 0W; ‡, P<0.05 vs. 1W; §, P<0.05 vs. 2W; ||, P<0.05 vs. 4W.
Figure 6.

Time course changes in load-related signaling pathways of CaMKII (A), β-arrestin-2 (B), and Akt (C) before aortic regurgitation AR (0W), and at 1, 2, 4, and 8 weeks after AR. Original immunoblots are shown below the graphs. p, phosphorylated protein; t, total protein; p/r, phosphorylation ratio; †, P<0.05 vs. 0W; ‡, P<0.05 vs. 1W; §, P<0.05 vs. 2W; ||, P<0.05 vs. 4W.
The results above (Figures 2-6) establish a temporal correlation following AR between the level of CaMKII activation and the hypertrophy (HW-to-BW ratios and cardiomyocyte CSA), the expression of hypertrophic markers and the impaired contractility, consistent with CaMKII being a major hypertrophic signaling effector linked to adverse remodeling and impaired heart function (26). To explore this possible connection further, mice were treated with 3 mg·kg−1·d−1 KN-93 or given the same volume of saline administered by peritoneal injection each day for 28 days post AR. As shown in Figure S2, KN-93 caused a ~75% reduction in CaMKII activity at day 28 after AR. However, despite this reduction in CaMKII activity, KN-93 had minimal effects on the degree of hypertrophy or changes in heart function (Figure 7), when compared to controls, supporting the possibility that the adverse effects induced by AR occur independent of changes in CaMKII activity. Similarly, although a two-fold over-expression of β-arrestin-2 was observed in the heart following tail vein injection of β-arrestin-2-AAV9 (5E+11 v.g./mouse) 4 weeks before the AR operation, the pattern of changes in hypertrophy and heart function was minimally affected by the increases in the β-arrestin-2 expression, when compared with mice injected with AAV expressing scrambled peptide, which did not affect LV structure or function (see Methods).
Figure S2.

The intervention of Akt, β-arrestin-2, and CaMKII in the hearts of AR mice. (A) The protein level of CaMKII after KN-93 treatment; (B) the protein level of β-arrestin-2 after β-arrestin-2 AAV9 treatment; (C) the protein level of Akt after wortmannin treatment. *, P<0.05 vs. Sham; †, P<0.05 vs. control.
Figure 7.

Effects of the intervention of Akt, β-arrestin-2, and CaMKII on cardiac structure and function at 4 weeks after AR. (A) The mRNA levels of Nppa after wortmannin, KN-93, and β-arrestin-2 AAV9 treatment, respectively; (B) the mRNA levels of Nppb after wortmannin, KN-93, and β-arrestin-2 AAV9 treatment, respectively; (C) M-mode images of the left ventricle (unit: mm). Scale bar =2 mm; (D) left ventricular pressure (unit: mmHg); (E) ventricular contraction velocity and relaxation velocity (unit: mmHg/s); (F) HW/BW, heart weight-to-body weight ratio; (G) LVPWTd; (H) LVPWTs; (I) LVEDD; (J) LVESD; (K) LVEF; (L) LVFS; (M) LVESP; (N) LVEDP; (O) maximal +dp/dt. (P) Minimal −dp/dt. *, P<0.05 vs. Sham; †, P<0.05 vs. control; ‡, P<0.05 vs. KN93; §, P<0.05 vs. β-arrestin-2. LVPWTd, left ventricular posterior wall end-diastolic thickness; LVPWT, left ventricular posterior wall end-systolic thickness; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; LVESP, left ventricular end-systolic pressure; LVEDP, left ventricular end-diastolic pressure.
On the other hand, treatment of mice with the PI3K/Akt inhibitor Wortmannin (30 µg·kg−1·d−1) by peritoneal injection post AR caused a ~70% reduction in Akt activity at day 28 after AR which clearly aggravated eccentric cardiac hypertrophy and heart function, indicative of a critical protective role of Akt in volume overloaded hearts (Figure 7) when compared to control saline-injected mice (see Methods).
Discussion
This study has been undertaken to (I) delineate, to our knowledge, for the first time, the structural, functional and molecular progression changes of the LV in response to volume overload over an 8-week period following AR in mice; (II) discern load-dependent alterations in gene expression and signaling pathways through intervention of relevant signaling effectors in this volume overload murine model. The level of volume overload in our model is quite severe as characterized by a ~50% increase in SV along with a value of 0.4–0.5 for the DVTIa/SVTIa ratio (14), values that were more-or-less maintained over the entire 8-week period following induction of AR. This AR clearly induced a large increase in workload on the heart as documented by the over 60% increase in coronary blood flow, which led to progressive LV eccentric hypertrophy and dilatation (in remarkable contrast to the already well documented pressure overload cardiac remodeling), characterized by mild increases in free wall thickness and marked elevations in heart weight as well as increased cross-sectional area of LV cardiomyocytes. This progressive eccentric hypertrophy in turn correlated with LV dysfunction, as measured by reductions in ejection fraction and rates of pressure development and relaxation as well as moderate interstitial fibrosis in the absence of perivascular fibrosis. These structural and functional changes in the LV were strongly affected by Akt blockers but not CaMKII blockers or β-arrestin-2 expression levels (Figure 7).
Mechanical overload is a prominent trigger to induce remodeling in the heart, which is not only an adaptive state before the development of overt heart failure, but also an independent risk factor of major cardiac events (27-30). Although the regurgitant flow was similar during the observation period, three stages of cardiac remodeling could be identified following AR. Within the first week, we observed abrupt cardiac dilatation with preserved contractile function (as measured by EF and dP/dt) and increased work (measured by coronary flow), accompanied by relatively small changes in LV hypertrophy as characterized by HW/BW ratios, CM cross-sectional areas and posterior LV wall thickness. Between 1 and 2 weeks after AR, no further LV dilatation or elevations in coronary flow occurred despite clear evidence of cardiac hypertrophy without measurable depression in hemodynamic function (except for a slight decline in –dpdtmin). Thereafter, the LV underwent progressive deterioration characterized by prominent cardiac dilatation and dysfunction. The phenotypic evolution observed in our studies is somewhat similar to some clinical observations, suggesting that our model might have translational relevance for studying volume overload conditions.
CFR represents the maximal oxygen delivery capacity during increasing oxygen demand, therefore also reflecting cardiac reserve (31). In the current study, the AR mice showed an eccentrically hypertrophic heart, suggesting a state of high-energy consumption, as indicated in other studies in rats (32). In parallel, the CFR were impaired as early as 1 week after AR operation, albeit the LV systolic function was still largely preserved (Figure 2). Coincidently, in other animal models such as pressure overload, we and others also found CFR reduction at early adaptive phases of cardiac remodeling (13,16,33). Thereby, the CFR showed a predictive role in cardiac function in AR induced cardiomyopathy. It is suggested that the reduced CFR is attributed to the robust increase of basal coronary flow. Since cardiomyocytes already extract oxygen with great efficiency at rest, it’s not surprising that the only way of meeting pronounced demand is to increase coronary blood flow, as indicated elsewhere (31).
Since merged E and A waves were presented in most cases of AR mice, it is difficult to calculate the E/A ratio, as similarly reported in other studies evaluating diseased mouse hearts (23,34). Tei index is a noninvasive and reliable method of combined systolic and diastolic myocardial function (35). In this study we used the Tei index, which was derived from systolic/diastolic time intervals, to evaluate diastolic function. The Tei index was significantly decreased during the observation period, which was mainly attributed to ET, because ET was significantly elongated, whereas IVRT and IVCT were of normal value. The increase in ET may be explained as being a result of increased SV in this study, implicating the heart produces more cardiac stroke work against vascular resistance (9), as similarly shown in patients with AR (35).
Fibrosis is a key feature of various cardiomyopathies and compromises cardiac performance (36). It is well documented that cardiac fibrosis is remarkable in other mechanical overload scenarios such as press overload cardiac hypertrophy (15). However, cardiac fibrosis in volume overload remains controversial. Previous literature indicates remarkable (10,37) or unaltered cardiac fibrosis (8,9,38) in the volume overload hearts of mice with aortocaval fistula. In this study using the novel model of AR, mild interstitial fibrosis in LV wall at all time points was presented. The variations in fibrosis among different studies may depend on the type of volume load (aortocaval fistula vs. aortic regurgitation) and the timing of assessment. We also found that the elevated fibrosis was mainly attributed to an increase in interstitial fibrosis rather than in perivascular fibrosis, for no evidence was found for differences in the amount of perivascular connective tissue between AR and sham-operated mice. We and others have well documented that aortic structure underwent considerable remodeling under pressure overload (39). Unlike pressure overload in that study, the AR model in the current study was characterized by volume overload, suggesting that systolic pressure, rather than diastolic pressure, is a robust driver for perivascular fibrosis. We also surprisingly found that fibrosis and cardiac dilatation/function could be separate during this disease course. The reason may be in volume overload, fibrosis was not as marked as that in pressure overload (8,9,15), and up to 8 weeks of volume overload in this study did not show more conspicuous fibrosis. Of note, previous studies also show that cardiac fibrosis could remain similar while cardiac function improves or worsens (36,38).
Akt regulates multiple aspects of cellular effects on myocyte growth, survival and metabolism (40). Akt activation promotes the growth of the heart while preserving cardiac function (physiological hypertrophy), suggesting a beneficial role for Akt activation in cardiac remodeling (26). Interestingly, the chamber phenotype in physiological hypertrophy involves eccentric LV remodeling just as in volume overload (41,42). However, it is controversial whether Akt activation is elevated or unaltered in chronic volume overload (8,38). In the current study, Akt was markedly activated early after AR but gradually waned thereafter (Figure 6). The dynamic changes in Akt activation may explain the discrepancy of the role of Akt. Our study corroborated a previous study in which Akt was activated at 1 week after aortocaval fistula operation (9). Considering it’s well documented that Akt can be activated by multifarious extracellular stimuli (43) and is cardioprotective (44,45), it is not surprising that Akt was activated early after AR. The gradual decreased Akt phosphorylation status seems to contribute to the progression of heart failure. To reinforce this notion, we used Wortmanin which was widely used to suppress the Akt activity well (24,25). Correspondingly, administration of Wortmannin aggravates eccentric cardiac hypertrophy in this study, strengthening the potential protective role of Akt in volume overloaded hearts.
CaMKII is an important Ca2+-dependent hypertrophic signaling effector, induceing phosphorylation and nuclear export of class II histone deacetylases (HDACs), leading to the relief of HDAC-mediated transcriptional repression of myocyte enhancer factor 2 (MEF2) and MEF2-dependent transcription of hypertrophic genes (46). However, in the current study, although we found CaMKII was slightly or modestly activated during 8 weeks of AR, KN-93 was ineffective in attenuating AR induced eccentric hypertrophy. Our findings were also previously suggested by several other studies using a dihydropyridine calcium channel blocker (nifedipine), in humans or rats with aortic regurgitation. Those studies found that nifedipine was not effective at slowing the development of LV remodeling (7,47). Of note, a recent study reported that genetic deletion of CaMKII delta did not mitigate adverse myocardial remodeling in aortocaval shunt mice (48). Moreover, we recently also found that calcineurin (another important Ca2+-dependent hypertrophic signaling regulator) was prominently activated in TAC but marginally activated in AR 2 weeks after AR (15). Collectively, it suggests Ca2+-dependent hypertrophic signaling pathways are not significantly involved in volume overloaded cardiac hypertrophy. Correspondingly, as to mechanical overload induced cardiac hypertrophy differential pharmaceutical considerations should be made, depending on the elevated load occurs in systole, or in diastole.
A recent breakthrough concept in G protein-coupled receptor (GPCR) signaling is arrestin-biased agonism. β-arrestin-2 specifically initiates GPCR conformational changes, linking activated GPCRs to downstream signaling events in a G protein-independent, arrestin-biased manner (49,50). Although the critical roles of β-arrestin-2 in pressure overload and myocardial infarction are well documented (51-53), its role in volume overload remains largely unknown. We found β-arrestin-2 was suppressed at 2 weeks after AR operation, but then gradually increased hereafter. We then over-expressed, via AAV gene transfer, β-arrestin-2 protein in the hearts of mice before AR operation. Surprisingly, β-arrestin-2 overexpression failed to improve cardiac remodeling and function in AR mice, suggesting a marginal role of β-arrestin-2 in eccentric hypertrophy induced by chronic volume overload. It is noteworthy that this newly identified effect of β-arrestin-2 is different from a previously recognized notion that β-arrestin-2 is cardioprotective, as shown in pressure overloaded mice (52,54). Intriguingly, it has been reported that β-arrestin-2 can induce G protein-independent activation of ERK1/2 (51,52) and CaMKII (55), both of which are much less activated in volume overload versus pressure overload (15). Moreover, a recent study elegantly showed β-arrestin-2 was a pathogenic factor in cardiac ischemia-reperfusion injury by inhibiting GPCR-independent Akt cell survival signaling pathway (56). This suggests that different stimuli may trigger distinct β-arrestin-2 conformational states and activation, causing different, or even opposite effects in the heart. Our findings may also help broaden the concept of biased agonism and individualized therapeutics. Previous studies showed that some AT1-R (a classic GPCR) blockers (ARBs) suppressed both AngII and mechanical stress (arrestin biased) mediated cardiac hypertrophy, functioning as inverse agonists, whereas other ARBs prohibited only AngII induced hypertrophy, without the function of inverse agonism (57). Considering that β-arrestin-2 does not show a regulatory role in the LV response to AR, it is reasonable to assume that inverse agonist may not demonstrate efficacious superiority in volume overload cardiac hypertrophy.
Study limitations
The signaling effectors (such as Akt) examined in this study have multiple downstream targets, which were not fully investigated in this study. Evaluation of several other parameters and potential targets such as mammalian target of rapamycin, (glycogen synthase kinase-3β) and myosin light-chain kinase, could assist in a more detailed analysis of the pathways involved in the progression of eccentric hypertrophy (40).
We did not perform comparative experiments at an in vitro cellular level, as we aimed to characterize the progressive evolution in a murine model of eccentric hypertrophy. Moreover, the heart is an organ, not isolated cardiomyocytes only. Therefore it could be viewed as an advantage to delineate the progression in an in vivo state, rather than in an in vitro one, which is restricted to cardiomyocytes.
Conclusions
We characterized the progressive evolution of LV eccentric hypertrophy and failure in a novel mouse model of volume overload surgically induced by puncture of aortic valves. The process of the LV remodeling was structurally, functionally and molecularly different from that well documented in pressure overload models such as transverse aortic constriction. We further found that Akt, rather than β-arrestin-2 or calcium handling proteins, may be a useful therapeutic target for this kind of volume overload. Although the specific molecular drivers of eccentric hypertrophy remain to be further elucidated, our present work may shed important light on eccentric hypertrophy mechanisms and have the potential to personalize pharmaceutical options for different types of cardiac hypertrophy.
Acknowledgments
The authors are grateful to Prof. Weili Shen and Ms. Chenglin Huang, from the Department of Hypertension, Ruijin Hospital, for their experimental assistance and helpful comments on this manuscript. We are grateful to Mr. Jianguo Jia, Mr. Chunjie Yang, Mrs. Bingyu Li, Mrs. Sanli Qian, Mrs. Weiwei Zhang, and Mr. Zhenzhong Zhang, from Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, for their kindness with technical expertise. We also would like to thank Mr. Guoping Zhang, for his assistance in lab management.
Funding: This work was supported by the National Natural Science Foundation of China (31430039, 81670228, 81770274, 31672376, and 81500191), Laboratory Animal Science Foundation of Science and Technology Commission of Shanghai Municipality (16140901100), Scientific Research Foundation of Shanghai Municipal Commission of Health (201640044), Science and Technology Planning Project of Guangdong Province (2016A020216019 and 2015A030302035).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The experimental protocol (No. 2016-048, approved February 18, 2016) was approved by the Animal Care and Use Committee of Zhongshan Hospital, Fudan University.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Singh JP, Evans JC, Levy D, et al. Prevalence and clinical determinants of mitral, tricuspid, and aortic regurgitation (the Framingham Heart Study). Am J Cardiol 1999;83:897-902. 10.1016/S0002-9149(98)01064-9 [DOI] [PubMed] [Google Scholar]
- 2.Baumgartner H, Falk V, Bax JJ, et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur Heart J 2017;38:2739-91. 10.1093/eurheartj/ehx391 [DOI] [PubMed] [Google Scholar]
- 3.Coffey S, Cairns BJ, Iung B. The modern epidemiology of heart valve disease. Heart 2016;102:75-85. 10.1136/heartjnl-2014-307020 [DOI] [PubMed] [Google Scholar]
- 4.Kodali SK, Velagapudi P, Hahn RT, et al. Valvular Heart Disease in Patients >/=80 Years of Age. J Am Coll Cardiol 2018;71:2058-72. 10.1016/j.jacc.2018.03.459 [DOI] [PubMed] [Google Scholar]
- 5.Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016;37:2129-200. 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 6.Houser SR, Margulies KB, Murphy AM, et al. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 2012;111:131-50. 10.1161/RES.0b013e3182582523 [DOI] [PubMed] [Google Scholar]
- 7.Plante E, Lachance D, Beaudoin J, et al. Comparative study of vasodilators in an animal model of chronic volume overload caused by severe aortic regurgitation. Circ Heart Fail 2009;2:25-32. 10.1161/CIRCHEARTFAILURE.108.801548 [DOI] [PubMed] [Google Scholar]
- 8.Mohamed BA, Schnelle M, Khadjeh S, et al. Molecular and structural transition mechanisms in long-term volume overload. Eur J Heart Fail 2016;18:362-71. 10.1002/ejhf.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toischer K, Rokita AG, Unsold B, et al. Differential cardiac remodeling in preload versus afterload. Circulation 2010;122:993-1003. 10.1161/CIRCULATIONAHA.110.943431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Champetier S, Bojmehrani A, Beaudoin J, et al. Gene profiling of left ventricle eccentric hypertrophy in aortic regurgitation in rats: rationale for targeting the beta-adrenergic and renin-angiotensin systems. Am J Physiol Heart Circ Physiol 2009;296:H669-77. 10.1152/ajpheart.01046.2008 [DOI] [PubMed] [Google Scholar]
- 11.Zhou YQ, Zhu SN, Foster FS, et al. Aortic regurgitation dramatically alters the distribution of atherosclerotic lesions and enhances atherogenesis in mice. Arterioscler Thromb Vasc Biol 2010;30:1181-8. 10.1161/ATVBAHA.110.204198 [DOI] [PubMed] [Google Scholar]
- 12.Shimoni S, Zilberman L, Edri O, et al. Thoracic aortic atherosclerosis in patients with aortic regurgitation. Atherosclerosis 2011;218:107-9. 10.1016/j.atherosclerosis.2011.05.026 [DOI] [PubMed] [Google Scholar]
- 13.You J, Wu J, Ge J, et al. Comparison between adenosine and isoflurane for assessing the coronary flow reserve in mouse models of left ventricular pressure and volume overload. Am J Physiol Heart Circ Physiol 2012;303:H1199-207. 10.1152/ajpheart.00612.2012 [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Wu J, Zhu D, et al. Characterization of coronary flow reserve and left ventricular remodeling in a mouse model of chronic aortic regurgitation with carvedilol intervention. J Ultrasound Med 2015;34:483-93. 10.7863/ultra.34.3.483 [DOI] [PubMed] [Google Scholar]
- 15.You J, Wu J, Zhang Q, et al. Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. Am J Physiol Heart Circ Physiol 2018;314:H552-62. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Zhou YQ, Zou Y, et al. Evaluation of Bi-ventricular Coronary Flow Patterns Using High-Frequency Ultrasound in Mice with Transverse Aortic Constriction. Ultrasound Med Biol 2013;39:2053-65. 10.1016/j.ultrasmedbio.2013.04.022 [DOI] [PubMed] [Google Scholar]
- 17.Wu J, You J, Jiang G, et al. Noninvasive estimation of infarct size in a mouse model of myocardial infarction by echocardiographic coronary perfusion. J Ultrasound Med 2012;31:1111-21. 10.7863/jum.2012.31.7.1111 [DOI] [PubMed] [Google Scholar]
- 18.Wu J, You J, Li L, et al. Early estimation of left ventricular systolic pressure and prediction of successful aortic constriction in a mouse model of pressure overload by ultrasound biomicroscopy. Ultrasound Med Biol 2012;38:1030-9. 10.1016/j.ultrasmedbio.2012.01.018 [DOI] [PubMed] [Google Scholar]
- 19.You J, Wu J, Jiang G, et al. Olmesartan Attenuates Cardiac Remodeling Through DLL4/Notch1 Pathway Activation in Pressure Overload Mice. J Cardiovasc Pharmacol 2013;61:142-51. 10.1097/FJC.0b013e31827a0278 [DOI] [PubMed] [Google Scholar]
- 20.Shingu Y, Amorim P, Nguyen TD, et al. Myocardial performance (Tei) index is normal in diastolic and systolic heart failure induced by pressure overload in rats. Eur J Echocardiogr 2010;11:829-33. 10.1093/ejechocard/jeq077 [DOI] [PubMed] [Google Scholar]
- 21.Zhou YQ, Zhu Y, Bishop J, et al. Abnormal cardiac inflow patterns during postnatal development in a mouse model of Holt-Oram syndrome. Am J Physiol Heart Circ Physiol 2005;289:H992-1001. 10.1152/ajpheart.00027.2005 [DOI] [PubMed] [Google Scholar]
- 22.Gao S, Ho D, Vatner DE, et al. Echocardiography in Mice. Curr Protoc Mouse Biol 2011;1:71-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan LJ, Wang T, Kahn ML, et al. High-resolution echocardiographic assessment of infarct size and cardiac function in mice with myocardial infarction. J Am Soc Echocardiogr 2011;24:219-26. 10.1016/j.echo.2010.11.001 [DOI] [PubMed] [Google Scholar]
- 24.Ng SS, Tsao MS, Nicklee T, et al. Wortmannin inhibits pkb/akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. Clin Cancer Res 2001;7:3269-75. [PubMed] [Google Scholar]
- 25.Kanashiro-Takeuchi RM, Takeuchi LM, Rick FG, et al. Activation of growth hormone releasing hormone (GHRH) receptor stimulates cardiac reverse remodeling after myocardial infarction (MI). Proc Natl Acad Sci U S A 2012;109:559-63. 10.1073/pnas.1119203109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 2016;97:245-62. 10.1016/j.yjmcc.2016.06.001 [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther 2016;161:52-66. 10.1016/j.pharmthera.2016.03.005 [DOI] [PubMed] [Google Scholar]
- 28.Wu J, You J, Wang S, et al. Insights into the activation and inhibition of angiotensin II type 1 receptor in the mechanically loaded heart. Circ J 2014;78:1283-9. 10.1253/circj.CJ-14-0470 [DOI] [PubMed] [Google Scholar]
- 29.Bernardo BC, Weeks KL, Pretorius L, et al. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 2010;128:191-227. 10.1016/j.pharmthera.2010.04.005 [DOI] [PubMed] [Google Scholar]
- 30.Gao RR, Wu XD, Jiang HM, et al. Traditional Chinese medicine Qiliqiangxin attenuates phenylephrine-induced cardiac hypertrophy via upregulating PPARgamma and PGC-1alpha. Ann Transl Med 2018;6:153. 10.21037/atm.2018.04.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gan LM, Wikstrom J, Fritsche-Danielson R. Coronary Flow Reserve from Mouse to Man-from Mechanistic Understanding to Future Interventions. J Cardiovasc Transl Res 2013;6:715-28. 10.1007/s12265-013-9497-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roussel E, Drolet MC, Walsh-Wilkinson E, et al. Transcriptional Changes Associated with Long-Term Left Ventricle Volume Overload in Rats: Impact on Enzymes Related to Myocardial Energy Metabolism. Biomed Res Int 2015;2015:949624. 10.1155/2015/949624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartley CJ, Reddy AK, Madala S, et al. Doppler estimation of reduced coronary flow reserve in mice with pressure overload cardiac hypertrophy. Ultrasound Med Biol 2008;34:892-901. 10.1016/j.ultrasmedbio.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnelle M, Catibog N, Zhang M, et al. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J Mol Cell Cardiol 2018;114:20-8. 10.1016/j.yjmcc.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haque A, Otsuji Y, Yoshifuku S, et al. Effects of valve dysfunction on Doppler Tei index. J Am Soc Echocardiogr 2002;15:877-83. 10.1067/mje.2002.121198 [DOI] [PubMed] [Google Scholar]
- 36.Byrne NJ, Levasseur J, Sung MM, et al. Normalization of cardiac substrate utilization and left ventricular hypertrophy precede functional recovery in heart failure regression. Cardiovasc Res 2016;110:249-57. 10.1093/cvr/cvw051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lachance D, Plante E, Bouchard-Thomassin AA, et al. Moderate exercise training improves survival and ventricular remodeling in an animal model of left ventricular volume overload. Circ Heart Fail 2009;2:437-45. 10.1161/CIRCHEARTFAILURE.108.845487 [DOI] [PubMed] [Google Scholar]
- 38.Olsen NT, Dimaano VL, Fritz-Hansen T, et al. Hypertrophy signaling pathways in experimental chronic aortic regurgitation. J Cardiovasc Transl Res 2013;6:852-60. 10.1007/s12265-013-9503-y [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Wu J, Li L, et al. Effect of an acute mechanical stimulus on aortic structure in the transverse aortic constriction mouse model. Clin Exp Pharmacol Physiol 2011;38:570-6. 10.1111/j.1440-1681.2011.05544.x [DOI] [PubMed] [Google Scholar]
- 40.Sussman MA, Volkers M, Fischer K, et al. Myocardial AKT: the omnipresent nexus. Physiol Rev 2011;91:1023-70. 10.1152/physrev.00024.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moc C, Taylor AE, Chesini GP, et al. Physiological activation of Akt by PHLPP1 deletion protects against pathological hypertrophy. Cardiovasc Res 2015;105:160-70. 10.1093/cvr/cvu243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schnelle M, Sawyer I, Anilkumar N, et al. NADPH oxidase-4 promotes eccentric cardiac hypertrophy in response to volume overload. Cardiovasc Res 2019. [Epub ahead of print]. 10.1093/cvr/cvz331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev 2006;20:3347-65. 10.1101/gad.1492806 [DOI] [PubMed] [Google Scholar]
- 44.Li GH, Shi Y, Chen Y, et al. Gelsolin regulates cardiac remodeling after myocardial infarction through DNase I-mediated apoptosis. Circ Res 2009;104:896-904. 10.1161/CIRCRESAHA.108.172882 [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Xia Z, La Cour KH, et al. Activation of Akt rescues endoplasmic reticulum stress-impaired murine cardiac contractile function via glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeation pore opening. Antioxid Redox Signal 2011;15:2407-24. 10.1089/ars.2010.3751 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest 2013;123:37-45. 10.1172/JCI62839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evangelista A, Tornos P, Sambola A, et al. Long-term vasodilator therapy in patients with severe aortic regurgitation. N Engl J Med 2005;353:1342-9. 10.1056/NEJMoa050666 [DOI] [PubMed] [Google Scholar]
- 48.Mohamed BA, Elkenani M, Jakubiczka-Smorag J, et al. Genetic deletion of calcium/calmodulin-dependent protein kinase type II delta does not mitigate adverse myocardial remodeling in volume-overloaded hearts. Sci Rep 2019;9:9889. 10.1038/s41598-019-46332-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nuber S, Zabel U, Lorenz K, et al. beta-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 2016;531:661-4. 10.1038/nature17198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhabyeyev P, Zhang H, Oudit GY. Is beta-Arrestin 2 a Magic Bullet for Heart Failure Treatment? Hypertension 2017;70:887-9. 10.1161/HYPERTENSIONAHA.117.09943 [DOI] [PubMed] [Google Scholar]
- 51.Rakesh K, Yoo B, Kim IM, et al. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal 2010;3:ra46. 10.1126/scisignal.2000769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang S, Gong H, Jiang G, et al. Src is required for mechanical stretch-induced cardiomyocyte hypertrophy through angiotensin II type 1 receptor-dependent beta-arrestin2 pathways. PLoS One 2014;9:e92926. 10.1371/journal.pone.0092926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCrink KA, Maning J, Vu A, et al. beta-Arrestin2 Improves Post-Myocardial Infarction Heart Failure via Sarco(endo)plasmic Reticulum Ca2+-ATPase-Dependent Positive Inotropy in Cardiomyocytes. Hypertension 2017;70:972-81. 10.1161/HYPERTENSIONAHA.117.09817 [DOI] [PubMed] [Google Scholar]
- 54.Kim KS, Abraham D, Williams B, et al. beta-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am J Physiol Heart Circ Physiol 2012;303:H1001-10. 10.1152/ajpheart.00475.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mangmool S, Shukla AK, Rockman HA. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol 2010;189:573-87. 10.1083/jcb.200911047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Jin L, Song Y, et al. beta-arrestin 2 mediates cardiac ischemia-reperfusion injury via inhibiting GPCR-independent cell survival signalling. Cardiovasc Res 2017;113:1615-26. 10.1093/cvr/cvx147 [DOI] [PubMed] [Google Scholar]
- 57.Wang X, Ye Y, Gong H, et al. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1-7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J Mol Cell Cardiol 2016;97:180-90. 10.1016/j.yjmcc.2016.05.012 [DOI] [PubMed] [Google Scholar]