

Abstract
Emery‐Dreifuss muscular dystrophy (EDMD) is a rare muscular dystrophy, but is particularly important to diagnose due to frequent life‐threatening cardiac complications. EDMD classically presents with muscle weakness, early contractures, cardiac conduction abnormalities and cardiomyopathy, although the presence and severity of these manifestations vary by subtype and individual. Associated genes include EMD, LMNA, SYNE1, SYNE2, FHL1, TMEM43, SUN1, SUN2, and TTN, encoding emerin, lamin A/C, nesprin‐1, nesprin‐2, FHL1, LUMA, SUN1, SUN2, and titin, respectively. The Online Mendelian Inheritance in Man database recognizes subtypes 1 through 7, which captures most but not all of the associated genes. Genetic diagnosis is essential whenever available, but traditional diagnostic tools can help steer the evaluation toward EDMD and assist with interpretation of equivocal genetic test results. Management is primarily supportive, but it is important to monitor patients closely, especially for potential cardiac complications. There is a high potential for progress in the treatment of EDMD in the coming years.
Keywords: cardiomyopathy, contractures, emerin, Emery‐Dreifuss, laminopathy, muscular dystrophy
Abbreviations
- AAV
adeno‐associated virus
- ACE
angiotensin‐converting enzyme
- AV
atrioventricular
- CK
creatine kinase
- CMT
Charcot–Marie–Tooth disease
- CRT
cardiac resynchronization therapy
- DCM
dilated cardiomyopathy
- ECG
electrocardiogram
- EDMD
Emery‐Dreifuss muscular dystrophy
- EF
ejection fraction
- EMG
electromyography
- ERK
extracellular‐signal regulated kinase
- FHL1
four‐and‐a‐half LIM domains 1
- FPLD
familial partial lipodystrophy
- ICD
implantable cardioverter defibrillator
- LAP2
lamina‐associated polypeptide 2
- LGMD
limb‐girdle muscular dystrophy
- LINC
linker of nucleoskeleton‐and‐cytoskeleton
- LMNA
lamin A/C
- MAPK
mitogen‐activated protein kinase
- mTOR
mammalian target of rapamycin
- NAD+
nicotinamide adenine dinucleotide
- Nup
nucleoporin
- OMIM
Online Mendelian Inheritance in Man
- SCD
sudden cardiac death
- SUN1
SAD1 and UNC84 domain containing protein 1
- SUN2
SAD1 and UNC84 domain containing protein 2
- TMEM43
transmembrane protein 43
1. HISTORY
Emery‐Dreifuss muscular dystrophy (EDMD) has a distinct clinical presentation that led to its description as a classic clinical entity many years before the genetic etiologies were identified. An early description of a muscular dystrophy with early contractures was made by Cestan and Lejonne in 1902.1 In 1955, Becker and Kiener described a slowly progressive X‐linked muscular dystrophy with a later onset than Duchenne muscular dystrophy and a slightly reduced average lifespan.2 It was not until 1966, however, when Emery and Dreifuss provided a more detailed description of the clinical features and typical progression of the disease that would later assume their names.3 In 1979, this disease became officially known as Emery‐Dreifuss muscular dystrophy (EDMD).4
2. EPIDEMIOLOGY
A meta‐analysis estimated that the pooled prevalence of EDMD in all age groups was 0.39 per 100,000.5 However, this study noted significant heterogeneity of results in the four primary articles from which this estimate was derived. This is not surprising, as the studies examined four diverse populations representing Northern Ireland,6 Northern England,7 Assuit, Egypt,8 and Hong Kong.9 Sporadic mutations are believed to be infrequent for EMD, but are becoming increasingly recognized for LMNA.10 In the autosomal dominant and recessive forms of EDMD, males and females are equally affected, while the X‐linked form primarily affects males, with some disease manifestations among female carriers.11
3. CLINICAL PRESENTATION
Clinically, the classic form of EDMD can be thought of as a triad within a triad. The classic overall triad consists of early contractures, progressive muscle weakness and atrophy, and cardiac abnormalities. The second triad captures the pattern of contractures, most prominently involving neck extension, elbow flexion, and heel cord tightening.
The contractures frequently emerge in the first decade of life, but become more evident and bothersome during the growth spurt that often occurs in adolescence. They can affect the paraspinal ligaments and posterior cervical musculature so significantly that the patient's neck may become fixed in an extended position. The cervical spine rigidity may become prominent enough to alter neck anatomy, thereby leading to dysphagia.12 It is important to remember that early contractures, including congenital contractures, may be associated with other diseases. Collagenopathies such as Ullrich congenital muscular dystrophy and Bethlem myopathy may present with early contractures, as may SEPN1‐related myopathy.13 Numerous underlying diseases have been found to cause arthrogryposis multiplex congenita, a pleiomorphic syndrome of congenital contractures.14
A common early motor symptom is difficulty with walking or running. Muscle weakness and atrophy become evident by the second or third decade. The weakness commonly presents in a “humeroperoneal” pattern, affecting the proximal arms (specifically the biceps and triceps, with relative sparing of the deltoids and infraspinatus) and distal legs (predominantly the peroneal muscles, with sparing of the thighs and intrinsic foot musculature). Neck weakness is almost universal and scapular winging is common, with sparing of facial muscles.10
Cardiac complications are found in the majority of EDMD patients. Atrial tachyarrhythmias, atrial standstill, ventricular tachyarrhythmias and cardiomyopathy are the most common manifestations. Symptoms usually start after the second decade of life with palpitations, presyncope and syncope, exercise intolerance, and heart failure symptoms. Frequently, cardiac manifestations precede the onset of significant skeletal muscle weakness. Compared with the background population, EDMD1 female carriers have an increased risk of developing cardiac complications, such as conduction abnormalities and sudden death, often in the absence of significant neuromuscular symptoms.11. The incidence of heart failure can exceed 60% in patients older than 50 years with LMNA mutations, including those with EDMD.15
4. GENETICS
Several genes have been implicated in the pathogenesis of EDMD. Among them, EMD, LMNA, SYNE1, SYNE2, FHL1, and TMEM43 have all been assigned to specific EDMD subtypes (EDMD1, EDMD2 and EDMD3, EDMD4, EDMD5, EDMD6, and EDMD7, respectively) (Table 1). These subtypes are recognized in the Online Mendelian Inheritance in Man (OMIM) database.16 Other genes that have been associated with this disease are SUN1 and SUN2, as well as TTN.17–23 There are still more causative genes yet to be discovered, as over 60% of patients with EDMD do not have detectable mutations in EMD or LMNA, the two most common genes.24
Table 1.
EDMD subtypes
| Subtype | Gene | Protein | Inheritance | Age of onset | Muscle weakness | Contractures | Cardiac Involvement |
|---|---|---|---|---|---|---|---|
| 1 | EMD | Emerin | X‐linked recessive | 4–5 years |
‐Develops early in course ‐Usually slowly progressive ‐Often humeroperoneal distribution in early stages |
‐Typically the initial symptom ‐Most often involving the elbows, Achilles tendons, cervical spinal muscles |
‐Typically emerges after skeletal muscle weakness and contractures ‐Includes conduction defects, arrhythmias, hypertrophic cardiomyopathy |
| 2 | LMNA | LMNA | Autosomal Dominant | Usually 3–6 years, but rarely before age 3 |
‐May be the initial symptom ‐Unpredictable severity, but frequently severe enough to result in loss of ambulation ‐Preferential involvement of biceps brachii may be a feature |
Develop after muscle weakness |
‐Often the initial manifestation of disease ‐Includes conduction defects, arrhythmias, dilated cardiomyopathy |
| 3 | LMNA | LMNA | Autosomal Recessive |
‐Variable ‐Can range from 14 months to 24 years |
‐Variable pattern, including limb‐girdle or diffuse muscle involvement ‐Variable severity, but can be severe enough to lead to immobilization |
Present, with variable involvement of Achilles tendons, elbows, neck |
‐Variable ‐If present, can include supraventricular and/or ventricular arrhythmias |
| 4 | * SYNE1 | Nesprin‐1 | Autosomal Dominant | 11 years old |
Gradually progressive |
Present | Variable |
| 5 | SYNE2 | Nesprin‐2 | Autosomal Dominant | Childhood | Proximal muscle weakness | Usually not present |
‐Usually present ‐Includes arrhythmias, dilated cardiomyopathy, heart failure |
| 6 | FHL1 | FHL1 | X‐linked recessive | Most range from 4 to 14 years, rarely in adulthood |
‐Variable pattern ‐Typically involves some combination of scapular, humeral, pelvic, peroneal, and/or axial regions ‐May have facial, bulbar, or respiratory involvement |
‐Usually present ‐May include rigid spine |
‐Usually present ‐Occurs after skeletal muscle manifestations ‐Includes conduction defects, arrhythmias, hypertrophic cardiomyopathy |
| 7 | TMEM43 | LUMA | Autosomal Dominant | Adulthood |
Proximal muscle weakness and atrophy |
Not present in reported cases | Cardiac conduction defects |
| N/A | * SUN1 | SUN domain‐containing protein 1 | Autosomal Recessive | 10 years old | Mild |
Spine rigidity |
None |
| N/A | ** SUN2 | SUN domain‐containing protein 2 | NA | NA | NA | NA | NA |
| N/A | TTN | Titin | Autosomal Recessive | Infantile or childhood |
‐Limb‐girdle pattern ‐Severe and progressive, leading to permanent loss of ambulation |
Develop early in course | Variable |
Clinical features associated with a primary mutation of these genes causing an EDMD phenotype are based on a single reported case.
Not well‐established.
In 1986, the molecular era began for EDMD when the first locus for this disease was mapped to the Xq27‐28 region by Thomas and colleagues.25 The gene was subsequently cloned and assigned the symbol EMD.17, 26, 27 The encoded protein, emerin, is a 254 amino acid nuclear envelope protein found in both muscle as well as several other tissues.28 A mutation in EMD leads to a complete cessation of emerin production and results in what is now called EDMD1.17, 27, 28, 29
In 1999, Bonne et al. mapped the locus for EDMD2 to chromosome 1q11‐q23, and the LMNA gene which lies within that interval was found to be the associated gene.18, 30 Mutations in LMNA result in disruption of the lamin A/C (LMNA) proteins, most typically with an autosomal dominant inheritance pattern, leading to EDMD2.29 Missense mutations are frequently seen in EDMD2 as opposed to other laminopathies.31 De novo mutations are common (76% in one study).10 Autosomal recessive mutations in LMNA have also been associated with EDMD, and these have been assigned to the subtype EDMD3.10, 18, 32, 33
Together, mutations in LMNA and EMD are the most common genetic causes of EDMD, accounting for around 40% of cases.34, 35
The discovery of a pair of additional genes occurred in 2007, when the synaptic nuclear envelope genes SYNE1 and SYNE2, encoding Nesprin‐1 and Nesprin‐2, respectively, were found to be associated with EDMD and subsequently assigned to the respective subtypes EDMD4 and EDMD5.23 Nesprins contribute to nuclear envelope localization and structural integrity.36, 37
In 2009, a fifth gene, FHL1, located on Xq26.3, was identified as causing an EDMD phenotype,20 now known as EDMD6. The encoded protein, four and a half LIM domains 1 (FHL1), is unusual among EDMD‐associated proteins in its cellular localization, which is at the sarcomere and sarcolemma rather than the nuclear envelope.38, 39
Two years later, another EDMD gene, TMEM43, was discovered.21, 40 This gene encodes LUMA, another nuclear membrane protein that binds with both emerin and LMNA, and is involved with the structural organization of the nuclear membrane and maintenance of nuclear shape. Mutant LUMA can result in abnormally‐shaped nuclei. LUMA may also play a role in the proper localization of emerin, and has an important interaction with SUN2 (see below), as there is evidence that mutant LUMAs may bind SUN2 to impede its nuclear localization and facilitate its destruction.21 TMEM43‐associated EDMD has been assigned to the subtype EDMD7.
SUN1, encoded by a gene of the same name, was originally studied as a protein that accumulates in the setting of LMNA mutations,41 but more recently variants in SUN1 were reported to worsen cellular defects in the setting of primary EDMD mutations.42 In 2014, a report showed both primary mutations and modifying variants for SUN1 and SUN2, with potential primary mutations documented in one family for each gene.43
Mutations in TTN have been associated with various phenotypes, including distal tibial myopathy, limb‐girdle muscular dystrophy (LGMD R10 titin‐related, previously known as LGMD2J),44, 45 and dilated cardiomyopathy.46, 47 Recent reports indicate that the EDMD phenotype is also associated with TTN mutations, including recessive truncating mutations.19, 48 Some of these patients have been reported to have cardiomyopathy,48 while others have not.19
5. PATHOPHYSIOLOGY
EDMD typically results from a structural or functional defect of one or more proteins comprising the nuclear envelope (Figure 1), thus giving rise to the term “nuclear envelopathy”.49 A potential unifying disease mechanism may be loss of protein importation into the nucleus.50, 51 The nuclear envelope is composed of an inner and outer nuclear membrane as well as a nuclear lamina, which, collectively, form a structural framework for the nucleus. A deficiency or mutation affecting any of the proteins providing this framework can result in a loss of the structural integrity of the nucleus, which can be particularly problematic for tissues that are frequently under stress, including cardiac and skeletal muscle. Such proteins include emerin, LMNA, nesprin‐1, nesprin‐2, LUMA, SUN1, and SUN2, which are encoded by the EMD, LMNA, SYNE1, SYNE2, TMEM43, SUN1, and SUN2 genes, respectively.17, 18, 20, 21, 22, 23, 43, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66 Specifically, the linker of nucleoskeleton‐and‐cytoskeleton (LINC) bridging complex located at the nuclear envelope is believed to tether the nucleo and cyto‐skeletons, and is composed of emerin, LMNA, nesprin‐1 and nesprin‐2, SUN1 and SUN2.35, 67 An exception is FHL1, a protein encoded by the gene of the same name, which localizes to the sarcomere and the sarcolemma; at the former, it contributes to sarcomere assembly.38, 39
Figure 1.
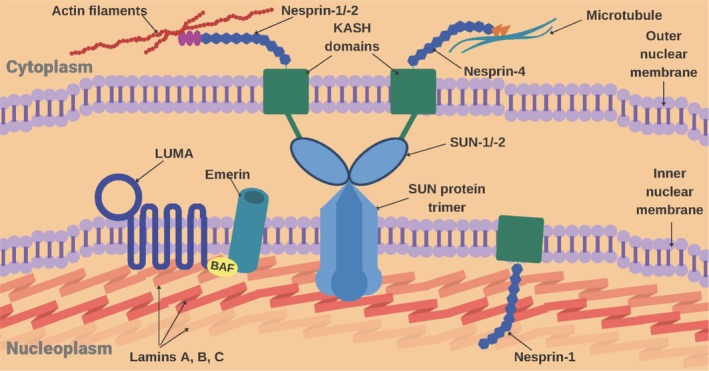
Schematic diagram of nuclear membrane indicating locations of proteins known to be associated with EDMD. Known protein interactions are shown (Courtesy Raghav Kalra)
Mutant forms of emerin show diminished transport to the inner nuclear membrane,68 and have been associated with decreased nuclear invagination and abnormalities in nuclear Ca++ transients.69
In the case of LMNA mutations, and the associated effects on LMNA, there also appears to be an effect on myocytes and muscle regeneration, as these proteins are expressed in mature myocytes, skeletal muscle stem cells, and satellite cells.70 Thus, mutations in LMNA can lead to impaired muscle regeneration, and ultimately a progressive muscular dystrophy.71 There is evidence for apoptosis in the atrioventricular (AV) nodal cells and cardiac conduction defects in mice heterozygous for mutations in LMNA.72
A defect in some of these nuclear proteins can lead to mislocalization of one of its binding partners. For example, overexpression of mutant LUMA can disrupt the nuclear localization of emerin and SUN2, leading to downstream effects that are similar to those seen in emerin deficiency. Similarly, nesprin deficiency has been shown to result in altered localization of emerin and lamins,73, 74 while a deficiency in A‐type lamins or overexpression of their mutant form can lead to mislocalization of lamina‐associated polypeptide 2 (LAP2), Nup (Nucleoporin) 153, and lamin B.21 Mutant forms of SUN1 show impaired interactions with both LMNA and emerin.42
6. GENOTYPE–PHENOTYPE CORRELATIONS
The cellular localization of proteins associated with EDMD at the nuclear envelope is a distinct biological feature of this class of diseases, distinguishing EDMD from other categories of muscular dystrophies. However, our knowledge of how various defects at the nuclear envelope lead to widespread destruction of myofibers, merging into the classic clinical features as well as clinical features distinct for specific EDMD subtypes, is unclear. Some genotype–phenotype correlations have been observed that may help with diagnosis and patient counseling. Key phenotypic patterns distinct for each subtype are noted in Table 1. A list of known mutations is provided in Supplemental Table 1.
6.1. EDMD1 and EDMD2
There is significant overlap in the phenotypes resulting from mutations in the two most common genes associated with EDMD, EMD (EDMD1) and LMNA (EDMD2), suggesting a functional relationship between their protein products (emerin and LMNA, respectively).29 However, there are also clear clinical distinctions between the two subtypes. On the whole, patients with EDMD2 have a more severe disease course than those with EDMD1, including more severe and progressive wasting of the biceps brachii.10, 75. Hypertrophy of the quadriceps and extensor digitorum brevis musculature has also been reported in EDMD2, but not in EDMD1; this appears to be true hypertrophy rather than pseudohypertrophy as connective tissue infiltration is not prominent on muscle biopsy.3, 10 In EDMD1, skeletal muscle symptoms usually occur before cardiac involvement, while cardiac symptoms are more likely to be an initial manifestation in EDMD2. Additionally, as in most X‐linked conditions, the full clinical spectrum of EDMD1, with skeletal muscle symptoms and cardiac involvement, is only seen in men. Female carriers do not typically present with muscle signs or symptoms, although approximately 20% of them may develop cardiac abnormalities, sometimes requiring treatments such as pacemaker implantation.76 The cardiac complications of EDMD1 consist most prominently of AV conduction defects, including complete heart block, atrial paralysis, and atrial flutter.77 Autopsy studies of EDMD1 showed gradual replacement of myocardium by fibrous and adipose tissue, most prominently in the atria and AV node, and affecting the ventricles at later stages.78, 79
A notable distinguishing feature of EDMD2 is the much broader, more diverse spectrum of disease compared with that seen in EDMD1, to the point where several patients with LMNA mutations are assigned to non‐EDMD disease categories. These alternate phenotypes include a congenital muscular dystrophy with a distinct “dropped head” syndrome caused by severe neck muscle weakness,80 autosomal dominant dilated cardiomyopathy with conduction defect,81 familial partial lipodystrophy (FPLD),82, 83 an autosomal recessive axonal Charcot–Marie‐Tooth (CMT) neuropathy phenotype,84 and Hutchinson‐Gilford progeria.85, 86 Of note, a subtype of autosomal dominant LGMD, LGMD1B, was previously associated with LMNA mutations.87 However, a recent reclassification of the LGMDs recognizes laminopathy as a separate category of muscle disease.88
The two subtypes also differ in that the severity of skeletal muscle symptoms in EDMD2 is less predictable than in EDMD1. In general, most patients with EDMD2 have a slowly progressive course during the first 3 decades of life with more rapid progression thereafter. However, in some cases, symptoms present very early (before the age of 3) and muscle weakness and contractures can progress more rapidly. Most patients experience difficulties walking and require ambulatory support, occasionally losing ambulation by the fourth decade.10 In one study, three of five patients with the early and rapidly progressive phenotype became nonambulatory between the ages of 8 and 13 years.10, 89, 90
The phenotypic variability seen with LMNA mutations does not appear to be directly associated with the type of mutation, evidenced by the significant inconsistency in severity between families, as well as among members of the same family, with identical mutations.10 For example, a single nucleotide deletion (c.959Tdel in exon 6) shared by members of the same family has been shown to lead to diverse clinical phenotypes, including pure dilated cardiomyopathy (DCM) with conduction defects, DCM with EDMD‐like skeletal muscle abnormalities, and DCM with LGMD‐like skeletal muscular dystrophy.91
7. EDMD3
EDMD3, associated with an LMNA mutation but a recessive trait of inheritance, is much less common than its autosomal dominant counterpart. The first described case manifested a very severe muscle disease with onset at 14 months of age, characterized by marked contractures, prominent and progressive muscle weakness and atrophy, loss of ambulation, normal intellect, and no cardiac involvement.33 However, four members of a family with EDMD3 did in fact develop cardiac sequelae, including supraventricular and/or ventricular arrhythmias.92
7.1. EDMD4 and EDMD5
The clinical phenotypes related to SYNE1 or SYNE2 mutations can be quite variable, ranging from nearly asymptomatic hyperCKemia to a muscular dystrophy with severe DCM requiring heart transplantation.37 Within this spectrum, most patients with EDMD4, associated with SYNE1 mutations, manifest with gradually progressive muscle weakness and atrophy, joint contractures, and no significant cardiac involvement.93, 94 EDMD5, on the other hand, classically presents with muscle weakness and cardiac features (eg, arrhythmia, DCM, heart failure) but no notable contractures.12
8. EDMD6
EDMD6 is caused by mutations in FHL1. Skeletal muscle hypertrophy has been observed for EDMD6; this appears to be a true hypertrophy rather than a pseudohypertrophy, although interstitial tissue was present on some muscle biopsies.20 Other potential symptoms include vocal cord paresis with dysphonia, facial weakness, ptosis, dysphagia, and respiratory compromise. Female carriers may have cardiac defects, either in isolation or along with mild skeletal muscle involvement.12, 20, 76 Mutations in FHL1 have also been associated with other myopathies, including reducing body myopathy, scapuloperoneal myopathy, X‐linked myopathy with postural muscle atrophy, and hypertrophic cardiomyopathy.95, 96
9. EDMD7
EDMD7, caused by a heterozygous mutation in TMEM43, is an autosomal dominant subtype that was initially described in 2011.21 Two Japanese individuals with this mutation were identified, both of whom manifested with adult‐onset disease. Only one of these patients had a clearly defined clinical phenotype, characterized by proximal muscle weakness and atrophy, as well as cardiac involvement (atrial fibrillation and bradycardia, requiring pacemaker implantation).21
9.1. EDMD due to SUN1 and SUN2 mutations
Only a single case each of primary mutation(s) causing EDMD has been described for SUN1 and SUN2.43 The putative SUN1 patient had onset at age 10 of mild muscle weakness, spine rigidity, moderate serum CK elevations, and sparing of cardiac involvement early in the course. Detailed phenotype information is not available for the SUN2 patient.
9.2. EDMD due to TTN mutations
The EDMD phenotype associated with mutations in TTN has generally consisted of progressive limb‐girdle weakness, early‐onset contractures, and sparing of the facial, bulbar, and oculomotor musculature. As noted above, the presence of cardiomyopathy appears to be variable.19, 48 The disease process tends to begin in infancy or childhood, and ultimately results in permanent loss of ambulation between 13 and 36 years of age.19
10. DIAGNOSTIC TESTING
Due to its rarity and the phenotypic overlap with other forms of muscular dystrophy such as congenital muscular dystrophy and LGMD, diagnosing EDMD may be challenging.97 Clinical clues include neck extensor weakness, the classic pattern of contractures, a cardiomyopathy, bradyarrhythmias, and tachyarrhythmias.97
10.1. Creatine kinase levels
In patients with skeletal muscle involvement, creatine kinase (CK) levels can range from normal to 15 times the upper limit of normal. In those with exclusive cardiac involvement, CK levels are generally normal.10 Thus, elevated CK levels may be helpful in the diagnostic evaluation but normal CK levels do not exclude the diagnosis of EDMD.
10.2. Electrodiagnosis
Overall, electromyography (EMG) findings in EDMD are similar to those seen in other myopathies, including low amplitude, short duration motor unit action potentials and early recruitment patterns.98 However, the needle examination may also reveal “irregular” motor unit action potentials consisting of high amplitudes, increased polyphasia, and normal or long durations; this can cause confusion as these are generally regarded to be neurogenic features.98 This range of EMG findings likely reflects the muscle fiber changes (such as hypertrophy and splitting) and overall fiber size variability (with both hypertrophy and atrophy) that can occur in a slowly progressive myopathy.98, 99, 100 Abnormal spontaneous activity tends to be abundant101, 102 with myotonic discharges reported in one case,102 although abnormal spontaneous activity is not universally present103 or may be present only in selected muscles.104
No systematic studies have been published regarding the use of electrical impedance myography in the diagnostic evaluation of EDMD.
10.3. Muscle imaging studies
Skeletal muscle imaging can be a helpful adjunctive tool to be used alongside other diagnostic modalities. A handful of articles indicate that distinct patterns of muscle involvement may be seen on muscle imaging studies in the setting of EDMD, sometimes suggesting specific disease subtypes.
A muscle MRI study of 22 patients with laminopathies, including 5 with EDMD2, showed fatty infiltration of the semimembranosus, long and short heads of the biceps femoris, adductor magnus, and vasti muscles, with relative sparing of the rectus femoris. Only one of these patients showed a similar pattern of fatty infiltration in the calf muscles.105
With regard to distinguishing EDMD from other muscle diseases, one study showed that muscle CT imaging can differentiate EDMD from collagen VI‐related myopathies. This can be especially useful for clinicians, as both of these myopathies can present with significant contractures. In Bethlem or Ullrich myopathy, fatty infiltration was more likely to be seen in the rectus femoris, while posterior thigh muscles were more prominently infiltrated in EDMD. The patients with EDMD also displayed more severe involvement of the posterior calf muscles.106
Two studies examined the question of whether skeletal muscle imaging can distinguish among different subtypes of EDMD. One study examined patterns of muscle abnormalities in 42 patients with EDMD, 10 with EDMD1 and 32 with EDMD2. In both subtypes, paraspinal muscles, adductors, glutei, quadriceps, biceps femoris, semitendinosus, semimembranosus, soleus, and gastrocnemius were affected. The peroneal muscles were more frequently involved in patients with EDMD1 compared with EDMD2, suggesting that these muscles may help distinguish the two.107 The second study suggests that patterns of gastrocnemius involvement may help distinguish patients with EDMD2 from other subtypes. Sixteen patients (9 with EDMD2, 4 with EDMD1, and 3 who appeared to have other subtypes) underwent MRI imaging of the leg muscles. All patients with EDMD2 had involvement of the medial gastrocnemius, with relative sparing of the lateral head. In contrast, none of the patients with EDMD1 or other subtypes showed this pattern.108
The imaging literature is thus intriguing but sparse to date. No systematic studies have been published regarding the use of ultrasound in the diagnostic evaluation of EDMD. Further studies are needed to define fully the role of skeletal muscle imaging in the diagnostic evaluation of EDMD.
10.4. Muscle pathology
Traditionally, muscle biopsy was used in the diagnostic evaluation of patients suspected of having EDMD. In those patients with skeletal muscle involvement, muscle biopsies typically demonstrate dystrophic or other myopathic features, including a variation in muscle fiber size, a marked increase in internal nuclei, and, occasionally, a mild increase in endomysial connective tissue and necrotic fibers.10 Disruption of myofibrillar architecture has been reported, particularly in EDMD 1,49 EDMD 2,49 and EDMD 6.109 Abnormally shaped nuclei can be seen as well.21 However, these structural findings are not specific to EDMD, and thus muscle biopsy has limitations with regard to diagnosing EDMD.
Immunohistochemistry can yield useful diagnostic findings in some EDMD subtypes. In EDMD1, for example, the anti‐emerin antibody shows absence of staining of the inner nuclear membrane.27, 28 This finding can be seen not only in muscle, but also in peripheral leukocytes, skin fibroblasts, and buccal cells.110 In the skeletal muscle of female carriers, emerin protein levels are variable, ranging from <5% of normal to normal levels.111
Immunohistochemistry is not as useful in EDMD2, as staining for LMNA is normal in these patients. Of note, however, reduced LUMA staining has been demonstrated in these patients, in addition to those with EDMD7.21
In EDMD7 due to TMEM43 mutations, there may be reduced nuclear staining not only of LUMA, but also of emerin and SUN2, presumably due to the interaction that LUMA has with these other nuclear membrane proteins.21
Distinct, clinically useful immunohistochemistry findings have not been established in the other EDMD subtypes.112
10.5. Genetic Testing
The gold standard for establishing a subtype‐specific diagnosis for EDMD is genetic testing. Genetic testing is commercially available for the genes associated with this condition: EMD, LMNA, FHL1, SYNE1, SYNE2, TMEM43, SUN1, SUN2, and TTN. Specific genes and associated phenotypes are described above. Most disease‐focused genetic tests are currently designed as targeted sequence capture panels, based on next generation sequencing technology.113, 114 To capture slightly larger mutations such as exon‐level deletions, the sequencing panel is often supplemented by specific deletion‐duplication testing as well.115 The older Sanger sequencing technology is currently used only for selected applications such as verification of a specific single nucleotide mutation. Exome sequencing will capture many pathogenic mutations also, but does not consistently detect larger deletions and duplications.
11. MANAGEMENT
As there are currently no disease modifying therapies available for EDMD, management consists of appropriate clinical monitoring and symptomatic treatment. There are currently no systematic articles on the use of exercise, creatine, or coenzyme Q10 in EDMD. Given the complex, multi‐organ system complications seen in EDMD, patients should ideally be monitored in either a multidisciplinary clinic or in a setting in which coordination and communication among different specialists is seamless.
11.1. Genetic counseling
Genetic counseling should be offered to all patients and their families to help them better understand the recurrence risks for future children as well as potential risks for other individuals in the extended family. Genetic counselors can also guide families regarding family planning options to curtail the risk of recurrence, such as preimplantation genetic diagnosis. Genetic counseling also allows for carriers to pursue appropriate monitoring for potentially life‐threatening cardiac complications.89
11.2. Contractures
Physical therapy with an emphasis on stretching is the initial management strategy for this symptom; however, severe contractures may require surgical interventions, such as elongation of the Achilles tendon for ankle contractures. Procedures often need to be repeated to achieve a sustained benefit. However, the therapeutic effects of surgical procedures appear to last longer if the surgery is done after the adolescent growth spurt.10, 76, 89 Overall, outcomes for ankle contractures seem to be the most favorable, while surgical treatment for elbow contractures is more complex and results are frequently not long‐lasting. Surgical intervention for neck contractures, typically requiring internal fixation with rods, can be considered, although the potential risks (including loss of ambulation) should be weighed against the benefits before pursuing this option.
11.3. Cardiac screening
All EDMD patients should have thorough cardiac evaluations at diagnosis, including a physical examination, electrocardiogram (ECG), echocardiogram, and Holter monitor.116, 117 For LMNA mutations in particular, the high penetrance, especially with regard to cardiac symptoms (nearly complete by the age of 60 years), and potential severity (initial manifestation can be sudden cardiac death [SCD]) must be appreciated; therefore, everyone who harbors these mutations requires a comprehensive cardiac assessment and ongoing monitoring.118, 119
ECG abnormalities include low amplitude P waves with prolonged PR intervals. Tachyarrhythmias, such as atrial fibrillation, atrial flutter, as well as supraventricular and ventricular arrhythmias, are commonly seen, most likely due to progressive atrial, ventricular, and AV node fibrosis.120 There is a high risk of progression of these electrocardiographic abnormalities to complete heart block and/or atrial standstill/paralysis121, 122, 123; the latter may be associated with junctional escape rhythms.120
Echocardiograms are particularly useful to screen for dilated or hypertrophic cardiomyopathy. The severity of cardiac complications does not correlate with the degree of skeletal muscle weakness. Signs of cardiac fibrosis are not typically seen early in the course of LMNA‐associated EDMD, but subtle signs of ventricular dysfunction may be seen on echocardiogram and cardiac MRI.124 Cardiac MRI is particularly useful for the detection of cardiac fibrosis; however, many children will require sedation for such studies.124
Female carriers of X‐linked EDMD, either confirmed or those at risk, should be informed about the risk of cardiomyopathy and the symptoms of cardiac failure. In those without cardiac symptoms, cardiac evaluations should begin at diagnosis, with periodic monitoring thereafter. If any cardiac symptoms arise, a complete baseline cardiac evaluation, including a clinical examination, ECG, echocardiogram, and Holter monitor should be performed, followed by annual follow‐up evaluations.76
11.4. Cardiac pharmacotherapy
Angiotensin‐converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) are recommended for all patients with decreased ejection fractions (EFs).117 Other medications commonly used to treat heart failure should also be considered in the appropriate clinical setting of progressive cardiac dysfunction; however, beta blockers should be used with caution due to the high prevalence of AV block.
11.5. Pacemaker/implantable cardioverter defibrillator placement
National guidelines recommend pacemaker placement on EDMD patients with any degree of AV block, including first‐degree block, due to known progression to complete AV block over time.125, 126 A newer approach, cardiac resynchronization therapy (CRT), also known as a biventricular pacemaker, is worth considering in certain situations due to the potential for ventricular dysfunction116; however, this device has not been studied extensively in EDMD. Cardiac complications of LMNA mutations have been examined in greater depth than those of EMD mutations; there is a 30% risk of sudden death in patients with the former. Several reports recommend placement of an implantable cardioverter defibrillator (ICD) concurrently with a pacemaker in the setting of LMNA mutations due to the high risk of fatal tachyarrhythmias.15, 127, 128 Another source recommends ICD placement as soon as ventricular tachycardia is detected.129
11.6. Antithromboembolic prophylaxis
The efficacy of antiplatelet or anticoagulation prophylaxis for atrial fibrillation and atrial flutter has not been studied in the context of EDMD; however, due to the risk of cerebral thromboembolism and myocardial infarction, prophylaxis is recommended barring contraindication.11, 129
11.7. Heart transplantation
Heart transplantations have been performed on patients with EDMD who experience progressive heart failure.11, 130, 131, 132, 133 Thus, this therapeutic option should be considered under the appropriate circumstances in patients with EDMD.
11.8. Respiratory management
Respiratory dysfunction is generally not a prominent component of EDMD, but can become a factor during the disease course; therefore, respiratory monitoring and periodic pulmonary function testing should be performed. In those patients with respiratory failure, respiratory support can be used, particularly at night, preferably with noninvasive devices.
11.9. Investigational therapies
Investigational therapies for EDMD have been largely directed at the laminopathy subtype (EDMD2), due in part to the larger proportion of patients represented by this subtype, but also due to the availability of a robust mouse model for laminopathy134; in contrast, a mouse model that was developed for emerin deficiency has a more subtle phenotype.135
For laminopathies, several preclinical studies have shown promising results, some focusing primarily on the cardiac phenotype. One therapeutic strategy targets the activation of the mitogen‐activated protein kinase (MAPK) pathway, particularly the extracellular‐signal regulated kinase (ERK) branch.136 Candidate therapies that inhibit MAPK/ERK activity have included PD98059,137 selumetinib,138 “molecule 8”,139 and the ACE inhibitor benazepril.140 ARRY‐371797 targets a different branch of the MAPK cascade, p38α, and prevented cardiac complications in a mouse model of laminopathy.141 Another strategy used temsirolimus to inhibit the mammalian target of rapamycin (mTOR) pathway, which also appears to be involved in the pathogenesis of laminopathies.142 Alleviation of oxidative stress in laminopathies has been achieved with N‐acetyl cysteine.143 Lastly, nicotinamide riboside, a natural precursor of nicotinamide adenine dinucleotide (NAD+), improved cardiac function in a laminopathy mouse model.144 Among these candidates, ARRY‐371797 is currently under investigation in a Phase 3 study focusing on patients with cardiomyopathy caused by LMNA mutations.145
As in many other hereditary neuromuscular diseases, genetically sophisticated treatments are under investigation for EDMD. A recent study showed that, at least in cell culture, antisense oligonucleotide‐mediated skipping of exon 5 of LMNA can be successfully used, and, therefore, shows promise as a potential therapeutic approach for patients with dominant mutations in this exon.146, 147 Other molecular strategies that have a possible role in EDMD, such as adeno‐associated virus (AAV)‐based gene replacement and gene‐editing techniques, have not been formally studied in this disease thus far.146
12. CONFLICT OF INTEREST
Scott A. Heller has no conflicts of interest to disclose. Peter B. Kang has served as a consultant for AveXis and ChromaDex. He has served on an advisory board for Sarepta Therapeutics. He has received honoraria from Wiley for serving as an associate editor for Muscle & Nerve and from Wolters Kluwer for contributing material to UpToDate.
13. ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Supplemental Table 1 Mutations associated with Emery‐Dreifuss muscular dystrophy (EDMD)
Heller SA, Shih R, Kalra R, Kang PB. Emery‐Dreifuss muscular dystrophy. Muscle Nerve. 2020;61:436–448. 10.1002/mus.26782
Funding information The authors are grateful for the support of EDMD International, Inc
REFERENCES
- 1. Cestan R, Lajonne NJ. Dystrophie musculaire. Iconogr Salpetriere. 1902;155:35. [Google Scholar]
- 2. Becker PE, Kiener F. A new x‐chromosomal muscular dystrophy. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1955;193(4):427‐448. [DOI] [PubMed] [Google Scholar]
- 3. Emery AE, Dreifuss FE. Unusual type of benign x‐linked muscular dystrophy. J Neurol Neurosurg Psychiatry. 1966;29(4):338‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rowland LP, Fetell M, Olarte M, Hays A, Singh N, Wanat FE. Emery‐Dreifuss muscular dystrophy. Ann Neurol. 1979;5(2):111‐117. [DOI] [PubMed] [Google Scholar]
- 5. Mah JK, Korngut L, Fiest KM, et al. A systematic review and meta‐analysis on the epidemiology of the muscular dystrophies. Can J Neurol Sci. 2016;43(1):163‐177. [DOI] [PubMed] [Google Scholar]
- 6. Hughes MI, Hicks EM, Nevin NC, Patterson VH. The prevalence of inherited neuromuscular disease in Northern Ireland. Neuromuscul Disord. 1996;6(1):69‐73. [DOI] [PubMed] [Google Scholar]
- 7. Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in‐depth analysis of a muscle clinic population. Brain. 2009;132(Pt 11):3175–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. El‐Tallawy HN, Khedr EM, Qayed MH, Helliwell TR, Kamel NF. Epidemiological study of muscular disorders in Assiut, Egypt. Neuroepidemiology. 2005;25(4):205‐211. [DOI] [PubMed] [Google Scholar]
- 9. Chung B, Wong V, Ip P. Prevalence of neuromuscular diseases in Chinese children: a study in southern China. J Child Neurol. 2003;18(3):217‐219. [DOI] [PubMed] [Google Scholar]
- 10. Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery‐Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48(2):170‐180. [PubMed] [Google Scholar]
- 11. Boriani G, Gallina M, Merlini L, et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery‐Dreifuss muscular dystrophy: a long‐term longitudinal study. Stroke. 2003;34(4):901‐908. [DOI] [PubMed] [Google Scholar]
- 12. Madej‐Pilarczyk A. Clinical aspects of Emery‐Dreifuss muscular dystrophy. Nucleus. 2018;9(1):268‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eymard B, Ferreiro A, Ben Yaou R, Stojkovic T. Muscle diseases with prominent joint contractures: main entities and diagnostic strategy. Rev Neurol (Paris). 2013;169(8–9):546‐563. [DOI] [PubMed] [Google Scholar]
- 14. Kang PB, Lidov HG, David WS, et al. Diagnostic value of electromyography and muscle biopsy in arthrogryposis multiplex congenita. Ann Neurol. 2003;54(6):790‐795. [DOI] [PubMed] [Google Scholar]
- 15. van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta‐analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83(1):79‐83. [DOI] [PubMed] [Google Scholar]
- 16. OMIM . https://www.ncbi.nlm.nih.gov/omim. Accessed September 27, 2019.
- 17. Bione S, Maestrini E, Rivella S, et al. Identification of a novel X‐linked gene responsible for Emery‐Dreifuss muscular dystrophy. Nat Genet. 1994;8(4):323‐327. [DOI] [PubMed] [Google Scholar]
- 18. Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery‐Dreifuss muscular dystrophy. Nat Genet. 1999;21(3):285‐288. [DOI] [PubMed] [Google Scholar]
- 19. De Cid R, Ben Yaou R, Roudaut C, et al. A new titinopathy: childhood‐juvenile onset Emery‐Dreifuss‐like phenotype without cardiomyopathy. Neurology. 2015;85(24):2126‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gueneau L, Bertrand AT, Jais JP, et al. Mutations of the FHL1 gene cause Emery‐Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85(3):338‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang WC, Mitsuhashi H, Keduka E, et al. TMEM43 mutations in Emery‐Dreifuss muscular dystrophy‐related myopathy. Ann Neurol. 2011;69(6):1005‐1013. [DOI] [PubMed] [Google Scholar]
- 22. Starr DA, Fridolfsson HN. Interactions between nuclei and the cytoskeleton are mediated by SUN‐KASH nuclear‐envelope bridges. Annu Rev Cell Dev Biol. 2010;26:421‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Q, Bethmann C, Worth NF, et al. Nesprin‐1 and ‐2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16(23):2816‐2833. [DOI] [PubMed] [Google Scholar]
- 24. Bonne G, Quijano‐Roy S. Emery‐Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol. 2013;113:1367‐1376. [DOI] [PubMed] [Google Scholar]
- 25. Thomas NS, Williams H, Elsas LJ, Hopkins LC, Sarfarazi M, Harper PS. Localisation of the gene for Emery‐Dreifuss muscular dystrophy to the distal long arm of the X chromosome. J Med Genet. 1986;23(6):596‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bione S, Small K, Aksmanovic VM, et al. Identification of new mutations in the Emery‐Dreifuss muscular dystrophy gene and evidence for genetic heterogeneity of the disease. Hum Mol Genet. 1995;4(10):1859‐1863. [DOI] [PubMed] [Google Scholar]
- 27. Nagano A, Koga R, Ogawa M, et al. Emerin deficiency at the nuclear membrane in patients with Emery‐Dreifuss muscular dystrophy. Nat Genet. 1996;12(3):254‐259. [DOI] [PubMed] [Google Scholar]
- 28. Manilal S, Nguyen TM, Sewry CA, Morris GE. The Emery‐Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet. 1996;5(6):801‐808. [DOI] [PubMed] [Google Scholar]
- 29. Brown CA, Lanning RW, McKinney KQ, et al. Novel and recurrent mutations in lamin A/C in patients with Emery‐Dreifuss muscular dystrophy. Am J Med Genet. 2001;102(4):359‐367. [DOI] [PubMed] [Google Scholar]
- 30. Wydner KL, McNeil JA, Lin F, Worman HJ, Lawrence JB. Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescence in situ hybridization. Genomics. 1996;32(3):474‐478. [DOI] [PubMed] [Google Scholar]
- 31. Maggi L, D'Amico A, Pini A, et al. LMNA‐associated myopathies: the Italian experience in a large cohort of patients. Neurology. 2014;83(18):1634‐1644. [DOI] [PubMed] [Google Scholar]
- 32. Felice KJ, Schwartz RC, Brown CA, Leicher CR, Grunnet ML. Autosomal dominant Emery‐Dreifuss dystrophy due to mutations in rod domain of the lamin A/C gene. Neurology. 2000;55(2):275‐280. [DOI] [PubMed] [Google Scholar]
- 33. Raffaele Di Barletta M, Ricci E, et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery‐Dreifuss muscular dystrophy. Am J Hum Genet. 2000;66(4):1407‐1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bonne G, Yaou RB, Beroud C, et al. 108th ENMC International Workshop, 3rd Workshop of the MYO‐CLUSTER project: EUROMEN, 7th International Emery‐Dreifuss Muscular Dystrophy (EDMD) Workshop, 13‐15 September 2002, Naarden, The Netherlands. Neuromuscul Disord. 2003;13(6):508‐515. [DOI] [PubMed] [Google Scholar]
- 35. Meinke P, Nguyen TD, Wehnert MS. The LINC complex and human disease. Biochem Soc Trans. 2011;39(6):1693‐1697. [DOI] [PubMed] [Google Scholar]
- 36. Kandert S, Luke Y, Kleinhenz T, et al. Nesprin‐2 giant safeguards nuclear envelope architecture in LMNA S143F progeria cells. Hum Mol Genet. 2007;16(23):2944‐2959. [DOI] [PubMed] [Google Scholar]
- 37. Zhang X, Xu R, Zhu B, et al. Syne‐1 and Syne‐2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development. 2007;134(5):901‐908. [DOI] [PubMed] [Google Scholar]
- 38. Brown S, McGrath MJ, Ooms LM, Gurung R, Maimone MM, Mitchell CA. Characterization of two isoforms of the skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at focal adhesions and the isoform slimmer in the nucleus of myoblasts and cytoplasm of myotubes suggests distinct roles in the cytoskeleton and in nuclear‐cytoplasmic communication. J Biol Chem. 1999;274(38):27083‐27091. [DOI] [PubMed] [Google Scholar]
- 39. McGrath MJ, Cottle DL, Nguyen MA, et al. Four and a half LIM protein 1 binds myosin‐binding protein C and regulates myosin filament formation and sarcomere assembly. J Biol Chem. 2006;281(11):7666‐7683. [DOI] [PubMed] [Google Scholar]
- 40. Mukai T, Mori‐Yoshimura M, Nishikawa A, et al. Emery‐Dreifuss muscular dystrophy‐related myopathy with TMEM43 mutations. Muscle Nerve. 2019;59(2):E5‐E7. [DOI] [PubMed] [Google Scholar]
- 41. Chen CY, Chi YH, Mutalif RA, et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell. 2012;149(3):565‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li P, Meinke P, Huong le TT, Wehnert M, Noegel AA. Contribution of SUN1 mutations to the pathomechanism in muscular dystrophies. Hum Mutat. 2014;35(4):452‐461. [DOI] [PubMed] [Google Scholar]
- 43. Meinke P, Mattioli E, Haque F, et al. Muscular dystrophy‐associated SUN1 and SUN2 variants disrupt nuclear‐cytoskeletal connections and myonuclear organization. PLoS Genet. 2014;10(9):e1004605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haravuori H, Vihola A, Straub V, et al. Secondary calpain3 deficiency in 2q‐linked muscular dystrophy: titin is the candidate gene. Neurology. 2001;56(7):869‐877. [DOI] [PubMed] [Google Scholar]
- 45. Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal‐muscle protein titin. Am J Hum Genet. 2002;71(3):492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30(2):201‐204. [DOI] [PubMed] [Google Scholar]
- 47. Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chauveau C, Bonnemann CG, Julien C, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23(4):980‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Astejada MN, Goto K, Nagano A, et al. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol. 2007;26(3):159‐164. [PMC free article] [PubMed] [Google Scholar]
- 50. Busch A, Kiel T, Heupel WM, Wehnert M, Hubner S. Nuclear protein import is reduced in cells expressing nuclear envelopathy‐causing lamin A mutants. Exp Cell Res. 2009;315(14):2373‐2385. [DOI] [PubMed] [Google Scholar]
- 51. Kelkar P, Walter A, Papadopoulos S, et al. Nesprin‐2 mediated nuclear trafficking and its clinical implications. Nucleus. 2015;6(6):479‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ben Yaou R, Toutain A, et al. Multitissular involvement in a family with LMNA and EMD mutations: role of digenic mechanism? Neurology. 2007;68(22):1883‐1894. [DOI] [PubMed] [Google Scholar]
- 53. Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7(12):940‐952. [DOI] [PubMed] [Google Scholar]
- 54. Clements L, Manilal S, Love DR, Morris GE. Direct interaction between emerin and lamin A. Biochem Biophys Res Commun. 2000;267(3):709‐714. [DOI] [PubMed] [Google Scholar]
- 55. Dechat T, Pfleghaar K, Sengupta K, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22(7):832‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Haque F, Mazzeo D, Patel JT, et al. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem. 2010;285(5):3487‐3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holaska JM, Wilson KL. Multiple roles for emerin: implications for Emery‐Dreifuss muscular dystrophy. Anat Rec A Discov Mol Cell Evol Biol. 2006;288(7):676‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holt I, Ostlund C, Stewart CL, Man N, Worman HJ, Morris GE. Effect of pathogenic mis‐sense mutations in lamin A on its interaction with emerin in vivo. J Cell Sci. 2003;116(Pt 14):3027‐3035. [DOI] [PubMed] [Google Scholar]
- 59. Isermann P, Lammerding J. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol. 2013;23(24):R1113‐R1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin‐deficient cells. J Cell Biol. 2005;170(5):781‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Markiewicz E, Tilgner K, Barker N, et al. The inner nuclear membrane protein emerin regulates beta‐catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25(14):3275‐3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mislow JM, Holaska JM, Kim MS, et al. Nesprin‐1alpha self‐associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 2002;525(1–3):135‐140. [DOI] [PubMed] [Google Scholar]
- 63. Rowat AC, Lammerding J, Ipsen JH. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophys J. 2006;91(12):4649‐4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wheeler MA, Davies JD, Zhang Q, et al. Distinct functional domains in nesprin‐1alpha and nesprin‐2beta bind directly to emerin and both interactions are disrupted in X‐linked Emery‐Dreifuss muscular dystrophy. Exp Cell Res. 2007;313(13):2845‐2857. [DOI] [PubMed] [Google Scholar]
- 65. Wilson KL, Foisner R. Lamin‐binding Proteins. Cold Spring Harb Perspect Biol. 2010;2(4):a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang Q, Ragnauth CD, Skepper JN, et al. Nesprin‐2 is a multi‐isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J Cell Sci. 2005;118(Pt 4):673–687. [DOI] [PubMed] [Google Scholar]
- 67. Zhou C, Li C, Zhou B, et al. Novel nesprin‐1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum Mol Genet. 2017;26(12):2258‐2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pfaff J, Rivera Monroy J, Jamieson C, et al. Emery‐Dreifuss muscular dystrophy mutations impair TRC40‐mediated targeting of emerin to the inner nuclear membrane. J Cell Sci. 2016;129(3):502‐516. [DOI] [PubMed] [Google Scholar]
- 69. Shimojima M, Yuasa S, Motoda C, et al. Emerin plays a crucial role in nuclear invagination and in the nuclear calcium transient. Sci Rep. 2017;7:44312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lattanzi G, Cenni V, Marmiroli S, et al. Association of emerin with nuclear and cytoplasmic actin is regulated in differentiating myoblasts. Biochem Biophys Res Commun. 2003;303(3):764‐770. [DOI] [PubMed] [Google Scholar]
- 71. Frock RL, Kudlow BA, Evans AM, et al. C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20(4):486‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Holaska JM. Emerin and the nuclear lamina in muscle and cardiac disease. Circ Res. 2008;103(1):16‐23. [DOI] [PubMed] [Google Scholar]
- 73. Raharjo WH, Enarson P, Sullivan T, Stewart CL, Burke B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery‐Dreifuss muscular dystrophy. J Cell Sci. 2001;114(Pt 24):4447‐4457. [DOI] [PubMed] [Google Scholar]
- 74. Sullivan T, Escalante‐Alcalde D, Bhatt H, et al. Loss of A‐type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147(5):913‐920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Emery AE. X‐linked muscular dystrophy with early contractures and cardiomyopathy (Emery‐Dreifuss type). Clin Genet. 1987;32(5):360‐367. [DOI] [PubMed] [Google Scholar]
- 76. Madej‐Pilarczyk A, Kochanski A. Emery‐Dreifuss muscular dystrophy: the most recognizable laminopathy. Folia Neuropathol. 2016;54(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 77. Hsu DT. Cardiac manifestations of neuromuscular disorders in children. Paediatr Respir Rev. 2010;11(1):35‐38. [DOI] [PubMed] [Google Scholar]
- 78. Fishbein MC, Siegel RJ, Thompson CE, Hopkins LC. Sudden death of a carrier of X‐linked Emery‐Dreifuss muscular dystrophy. Ann Intern Med. 1993;119(9):900‐905. [DOI] [PubMed] [Google Scholar]
- 79. Verhaert D, Richards K, Rafael‐Fortney JA, Raman SV. Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging. 2011;4(1):67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Quijano‐Roy S, Mbieleu B, Bonnemann CG, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. 2008;64(2):177‐186. [DOI] [PubMed] [Google Scholar]
- 81. Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction‐system disease. N Engl J Med. 1999;341(23):1715‐1724. [DOI] [PubMed] [Google Scholar]
- 82. Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan‐type familial partial lipodystrophy. Hum Mol Genet. 2000;9(1):109‐112. [DOI] [PubMed] [Google Scholar]
- 83. Shackleton S, Lloyd DJ, Jackson SN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24(2):153‐156. [DOI] [PubMed] [Google Scholar]
- 84. De Sandre‐Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear‐envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot‐Marie‐Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70(3):726‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson‐Gilford progeria syndrome. Nature. 2003;423(6937):293‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A‐type lamins. Nature. 2003;423(6937):298‐301. [DOI] [PubMed] [Google Scholar]
- 87. Muchir A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9(9):1453‐1459. [DOI] [PubMed] [Google Scholar]
- 88. Straub V, Murphy A, Udd B, group Lws . 229th ENMC international workshop: limb girdle muscular dystrophies ‐ Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul Disord. 2018;28(8):702‐710. [DOI] [PubMed] [Google Scholar]
- 89. Madej‐Pilarczyk A, Marchel M, Ochman K, Cegielska J, Steckiewicz R. Low‐symptomatic skeletal muscle disease in patients with a cardiac disease ‐ Diagnostic approach in skeletal muscle laminopathies. Neurol Neurochir Pol. 2018;52(2):174‐180. [DOI] [PubMed] [Google Scholar]
- 90. Vytopil M, Benedetti S, Ricci E, et al. Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet. 2003;40(12):e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C, Mestroni L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation. 2000;101(5):473‐476. [DOI] [PubMed] [Google Scholar]
- 92. Jimenez‐Escrig A, Gobernado I, Garcia‐Villanueva M, Sanchez‐Herranz A. Autosomal recessive Emery‐Dreifuss muscular dystrophy caused by a novel mutation (R225Q) in the lamin A/C gene identified by exome sequencing. Muscle Nerve. 2012;45(4):605‐610. [DOI] [PubMed] [Google Scholar]
- 93. Chen Z, Ren Z, Mei W, et al. A novel SYNE1 gene mutation in a Chinese family of Emery‐Dreifuss muscular dystrophy‐like. BMC Med Genet. 2017;18(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fanin M, Savarese M, Nascimbeni AC, et al. Dominant muscular dystrophy with a novel SYNE1 gene mutation. Muscle Nerve. 2015;51(1):145‐147. [DOI] [PubMed] [Google Scholar]
- 95. Cowling BS, Cottle DL, Wilding BR, D'Arcy CE, Mitchell CA, McGrath MJ. Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: a comprehensive review of the clinical, histological and pathological features. Neuromuscul Disord. 2011;21(4):237‐251. [DOI] [PubMed] [Google Scholar]
- 96. Wilding BR, McGrath MJ, Bonne G, Mitchell CA. FHL1 mutants that cause clinically distinct human myopathies form protein aggregates and impair myoblast differentiation. J Cell Sci. 2014;127(Pt 10):2269–2281. [DOI] [PubMed] [Google Scholar]
- 97. Menezes MP, Waddell LB, Evesson FJ, et al. Importance and challenge of making an early diagnosis in LMNA‐related muscular dystrophy. Neurology. 2012;78(16):1258‐1263. [DOI] [PubMed] [Google Scholar]
- 98. Rowinska‐Marcinska K, Szmidt‐Salkowska E, Fidzianska A, et al. Atypical motor unit potentials in Emery‐Dreifuss muscular dystrophy (EDMD). Clin Neurophysiol. 2005;116(11):2520‐2527. [DOI] [PubMed] [Google Scholar]
- 99. Hausmanowa‐Petrusewicz I. The Emery‐Dreifuss disease. Neuropatol Pol. 1988;26(3):265‐281. [PubMed] [Google Scholar]
- 100. Zivari Adab H, Firoozabadi SM, Chalavi S, Maghooli K. Simulation and analysis of needle electromyogram in Emery‐Dreifuss muscular dystrophy by using line source model. Conf Proc IEEE Eng Med Biol Soc. 2008;2008:338‐342. [DOI] [PubMed] [Google Scholar]
- 101. Heuss D, Claus D, Neundorfer B. Fibrillations in regenerating muscle in dystrophic myopathies. Clin Neuropathol. 1996;15(4):200‐208. [PubMed] [Google Scholar]
- 102. Kissel JT, Dimberg EL, Emslie‐Smith AM, Selcen D, Keegan BM. A 49‐year‐old man with contractures, weakness, and cardiac arrhythmia. Neurology. 2009;72(23):2036‐2043. [DOI] [PubMed] [Google Scholar]
- 103. Miller RG, Layzer RB, Mellenthin MA, Golabi M, Francoz RA, Mall JC. Emery‐Dreifuss muscular dystrophy with autosomal dominant transmission. Neurology. 1985;35(8):1230‐1233. [DOI] [PubMed] [Google Scholar]
- 104. Petty RK, Thomas PK, Landon DN. Emery‐Dreifuss syndrome. J Neurol. 1986;233(2):108‐114. [DOI] [PubMed] [Google Scholar]
- 105. Lin HT, Liu X, Zhang W, et al. Muscle magnetic resonance imaging in patients with various clinical subtypes of LMNA‐related muscular dystrophy. Chin Med J. 2018;131(12):1472‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Deconinck N, Dion E, Ben Yaou R, et al. Differentiating Emery‐Dreifuss muscular dystrophy and collagen VI‐related myopathies using a specific CT scanner pattern. Neuromuscul Disord. 2010;20(8):517‐523. [DOI] [PubMed] [Google Scholar]
- 107. Diaz‐Manera J, Alejaldre A, Gonzalez L, et al. Muscle imaging in muscle dystrophies produced by mutations in the EMD and LMNA genes. Neuromuscul Disord. 2016;26(1):33‐40. [DOI] [PubMed] [Google Scholar]
- 108. Mercuri E, Counsell S, Allsop J, et al. Selective muscle involvement on magnetic resonance imaging in autosomal dominant Emery‐Dreifuss muscular dystrophy. Neuropediatrics. 2002;33(1):10‐14. [DOI] [PubMed] [Google Scholar]
- 109. Selcen D, Bromberg MB, Chin SS, Engel AG. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology. 2011;77(22):1951‐1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Manilal S, Sewry CA, Man N, Muntoni F, Morris GE. Diagnosis of X‐linked Emery‐Dreifuss muscular dystrophy by protein analysis of leucocytes and skin with monoclonal antibodies. Neuromuscul Disord. 1997;7(1):63‐66. [DOI] [PubMed] [Google Scholar]
- 111. Manilal S, Recan D, Sewry CA, et al. Mutations in Emery‐Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum Mol Genet. 1998;7(5):855‐864. [DOI] [PubMed] [Google Scholar]
- 112. Le Thanh P, Meinke P, Korfali N, et al. Immunohistochemistry on a panel of Emery‐Dreifuss muscular dystrophy samples reveals nuclear envelope proteins as inconsistent markers for pathology. Neuromuscul Disord. 2017;27(4):338‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5(12):1574‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Park J, Oh HM, Park HJ, et al. Usefulness of comprehensive targeted multigene panel sequencing for neuromuscular disorders in Korean patients. Mol Genet Genomic Med. 2019;e947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ankala A, da Silva C, Gualandi F, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77(2):206‐214. [DOI] [PubMed] [Google Scholar]
- 116. Sommerville RB, Vincenti MG, Winborn K, et al. Diagnosis and management of adult hereditary cardio‐neuromuscular disorders: a model for the multidisciplinary care of complex genetic disorders. Trends Cardiovasc Med. 2017;27(1):51‐58. [DOI] [PubMed] [Google Scholar]
- 117. Feingold B, Mahle WT, Auerbach S, et al. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation. 2017;136(13):e200‐e231. [DOI] [PubMed] [Google Scholar]
- 118. Hausmanowa‐Petrusewicz I, Madej‐Pilarczyk A, Marchel M, Opolski G. Emery‐Dreifuss dystrophy: a 4‐year follow‐up on a laminopathy of special interest. Neurol Neurochir Pol. 2009;43(5):415‐420. [PubMed] [Google Scholar]
- 119. Pasotti M, Klersy C, Pilotto A, et al. Long‐term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52(15):1250‐1260. [DOI] [PubMed] [Google Scholar]
- 120. Hong JS, Ki CS, Kim JW, et al. Cardiac dysrhythmias,cardiomyopathy and muscular dystrophy in patients with Emery‐Dreifuss muscular dystrophy and limb‐girdle muscular dystrophy type 1B. J Korean Med Sci. 2005;20(2):283‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Achmad C, Zada A, Affani M, et al. A novel de novo mutation in Lamin A/C gene in Emery Dreifuss Muscular Dystrophy patient with atrial paralysis. J Atr Fibrillation. 2017;9(6):1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wozakowska‐Kaplon B, Bakowski D. Atrial paralysis due to progression of cardiac disease in a patient with Emery‐Dreifuss muscular dystrophy. Cardiol J. 2011;18(2):189‐193. [PubMed] [Google Scholar]
- 123. Buckley AE, Dean J, Mahy IR. Cardiac involvement in Emery Dreifuss muscular dystrophy: a case series. Heart. 1999;82(1):105‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Smith GC, Kinali M, Prasad SK, et al. Primary myocardial dysfunction in autosomal dominant EDMD. A tissue doppler and cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 2006;8(5):723‐730. [DOI] [PubMed] [Google Scholar]
- 125. Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 Guidelines for Device‐Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices) developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;51(21):e1‐e62. [DOI] [PubMed] [Google Scholar]
- 126. Epstein AE, DiMarco JP, Ellenbogen KA, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device‐based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2013;61(3):e6‐e75. [DOI] [PubMed] [Google Scholar]
- 127. Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354(2):209‐210. [DOI] [PubMed] [Google Scholar]
- 128. Anselme F, Moubarak G, Savoure A, et al. Implantable cardioverter‐defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm. 2013;10(10):1492‐1498. [DOI] [PubMed] [Google Scholar]
- 129. Finsterer J, Stollberger C, Keller H. Arrhythmia‐related workup in hereditary myopathies. J Electrocardiol. 2012;45(4):376‐384. [DOI] [PubMed] [Google Scholar]
- 130. Merchut MP, Zdonczyk D, Gujrati M. Cardiac transplantation in female Emery‐Dreifuss muscular dystrophy. J Neurol. 1990;237(5):316‐319. [DOI] [PubMed] [Google Scholar]
- 131. Rees W, Schuler S, Hummel M, Hetzer R. Heart transplantation in patients with muscular dystrophy associated with end‐stage cardiomyopathy. J Heart Lung Transplant. 1993;12(5):804‐807. [PubMed] [Google Scholar]
- 132. Kichuk Chrisant MR, Drummond‐Webb J, Hallowell S, Friedman NR. Cardiac transplantation in twins with autosomal dominant Emery‐Dreifuss muscular dystrophy. J Heart Lung Transplant. 2004;23(4):496‐498. [DOI] [PubMed] [Google Scholar]
- 133. Dell'Amore A, Botta L, Martin Suarez S, et al. Heart transplantation in patients with Emery‐Dreifuss muscular dystrophy: case reports. Transplant Proc. 2007;39(10):3538‐3540. [DOI] [PubMed] [Google Scholar]
- 134. Arimura T, Helbling‐Leclerc A, Massart C, et al. Mouse model carrying H222P‐Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14(1):155‐169. [DOI] [PubMed] [Google Scholar]
- 135. Melcon G, Kozlov S, Cutler DA, et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15(4):637‐651. [DOI] [PubMed] [Google Scholar]
- 136. Muchir A, Pavlidis P, Decostre V, et al. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery‐Dreifuss muscular dystrophy. J Clin Invest. 2007;117(5):1282‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Muchir A, Shan J, Bonne G, et al. Inhibition of extracellular signal‐regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A‐type lamins. Hum Mol Genet. 2009;18(2):241‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Muchir A, Kim YJ, Reilly SA, Wu W, Choi JC, Worman HJ. Inhibition of extracellular signal‐regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery‐Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet Muscle. 2013;3(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wu W, Chordia MD, Hart BP, et al. Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by lamin A/C gene mutation. Bioorg Med Chem. 2017;25(3):1004‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Muchir A, Wu W, Sera F, Homma S, Worman HJ. Mitogen‐activated protein kinase kinase 1/2 inhibition and angiotensin II converting inhibition in mice with cardiomyopathy caused by lamin A/C gene mutation. Biochem Biophys Res Commun. 2014;452(4):958‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Muchir A, Wu W, Choi JC, et al. Abnormal p38alpha mitogen‐activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet. 2012;21(19):4325‐4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Choi JC, Muchir A, Wu W, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012;4(144):144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rodriguez BM, Khouzami L, Decostre V, et al. N‐acetyl cysteine alleviates oxidative stress and protects mice from dilated cardiomyopathy caused by mutations in nuclear A‐type lamins gene. Hum Mol Genet. 2018;27(19):3353‐3360. [DOI] [PubMed] [Google Scholar]
- 144. Vignier N, Chatzifrangkeskou M, Morales Rodriguez B, et al. Rescue of biosynthesis of nicotinamide adenine dinucleotide protects the heart in cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet. 2018;27(22):3870‐3880. [DOI] [PubMed] [Google Scholar]
- 145. https://clinicaltrials.gov. Volume September 27, 2019September 27, 2019.
- 146. Dowling JJ, DG H, Cohn RD, Campbell C. Treating pediatric neuromuscular disorders: the future is now. Am J Med Genet A. 2018;176(4):804‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Scharner J, Figeac N, Ellis JA, Zammit PS. Ameliorating pathogenesis by removing an exon containing a missense mutation: a potential exon‐skipping therapy for laminopathies. Gene Ther. 2015;22(6):503‐515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 Mutations associated with Emery‐Dreifuss muscular dystrophy (EDMD)