

Abstract
The oxidation of Fe(CO)5 with the [NO]+ salt of the weakly coordinating perfluoroalkoxyaluminate anion [F‐{Al(ORF)3}2]− (RF=C(CF3)3) leads to stable salts of the 18 valence electron (VE) species [Fe(CO)4(NO)]+ and [Fe(CO)(NO)3]+ with the Enemark–Feltham numbers of {FeNO}8 and {FeNO}10. This finally concludes the triad of heteroleptic iron carbonyl/nitrosyl complexes, since the first discovery of the anionic ([Fe(CO)3(NO)]−) and neutral ([Fe(CO)2(NO)2]) species over 80 years ago. Both complexes were fully characterized (IR, Raman, NMR, UV/Vis, scXRD, pXRD) and are stable at room temperature under inert conditions over months and may serve as useful starting materials for further investigations.
Keywords: carbonyl Ligands, iron(I), nitrogen oxides, vibrational spectroscopy, weakly coordinating anions (WCAs)
Ready to use…! Two novel iron carbonyl/nitrosyl cations in the elusive oxidation state +I are now easily accessible as stable salts of the weakly coordinating anion [F‐{Al(ORF)3}2]−. They may serve as simple model systems for the investigation of the nature of the Fe−NO bond to further understand the (bio‐)chemistry of iron nitrosyl complexes.
Iron and its chemistry might be one of the most fascinating amongst all transition metals—not only because of iron′s highest abundance in the biosphere or its numerous applications in industry and catalysis, but also due to the crucial role of iron‐containing enzymes in biology. All of this while being non‐toxic.1 Especially in regard to the importance of iron nitrosyl intermediates in biochemistry and biomedicine,2 a large variety of model systems has been developed in the recent decades.3 However, from a fundamental perspective, the long‐known flexible bonding motifs and bonding fashions of the NO ligand4 are still under discussion and re‐investigation today.5 And as much as the chemistry of FeII and FeIII is well established, as much is yet to learn about the chemistry of iron in the unusual oxidation state of +I. The synthesis of such compounds is mainly achieved by the reduction of FeII precursors, usually stabilized by bulky strong donor ligands such as [C{Si(CH3)3}3]−,6 cyclic (alkyl)(amino)carbenes7 or N‐heterocyclic carbenes (NHCs).8 But especially in the absence of those strong and sterically demanding ligands, an undisturbed insight to iron(I) systems is scarce. With that in mind, we made use of the versatility of carbon monoxide (CO) as a ligand by starting from Fe(CO)5 as a Fe0 source in combination with an oxidative approach. In order to compensate for the lability of the Fe−CO bond in the resulting iron cations, we utilized the weakly coordinating anion (WCA) [F‐{Al(ORF)3}2]− (RF=C(CF3)3)9, 10 in combination with [NO]+ as oxidant. The inevitable and (in this case) desirable coordination of the resulting NO(g) to the iron center led to the formation of the novel heteroleptic 18 valence electron (VE) iron(I) carbonyl/nitrosyl cations [Fe(CO)4(NO)]+ and [Fe(CO)(NO)3]+ with the respective Enemark–Feltham numbers4 {FeNO}8 and {FeNO}10. Overall, the reports on homoleptic or heteroleptic transition‐metal nitrosyl salts are scarce in literature. Stable salts of homoleptic nitrosyl cations, unlike their carbonyl analogues, were completely unknown until our recent discovery of [Mn(NO)4][WCA].11 In regard to ternary carbonyl/nitrosyl cations, the only currently known examples are [Cr(CO)5(NO)][WCA], reported by us12 and [Co(CO)2(NO)2][WCA].13 And although the neutral Fe(CO)2(NO)2 14 and anionic [Fe(CO)3(NO)]− (the Hieber anion)15 shown in Figure 1 were discovered more than half a decade ago and found their applications as useful starting materials or catalysts,16 as of yet, their cationic counterparts have been mostly untouched.1
Figure 1.
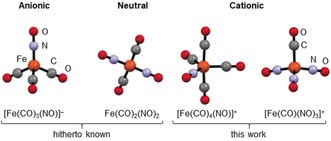
Calculated structures (BP86/def2‐TZVPP‐D3BJ) of the currently known iron carbonyl/nitrosyl complexes.* Only the Hieber anion [Fe(CO)3(NO)]− was hitherto structurally characterized.
Our oxidative syntheses started from NO[F‐{Al(ORF)3}2] and Fe(CO)5 in dichloromethane (CH2Cl2). Conveniently, this reaction can be done with an excess of Fe(CO)5 or even with impure NO[F‐{Al(ORF)3}2], since [Fe(CO)4(NO)][F‐{Al(ORF)3}2] (1) is only poorly soluble in CH2Cl2 (Scheme 1 a). Subsequent washings of the crude product with CH2Cl2 or n‐pentane and a crystallization by vapor diffusion of n‐pentane into an ortho‐difluorobenzene (oDFB) or 1,2,3,4‐tetrafluorobenzene (TFB) solution led to pure brown crystals of 1 in 49 % yield. If starting from pure NO[WCA], usually about 80 % yield of crystalline 1, λ max=450 and 330 nm in oDFB solution (see Supporting Information Figure S23), was obtained. For the synthesis of 2, the stoichiometry is more important. Therefore, the use of solid Fe2(CO)9 or Fe3(CO)12 as the iron source is more practicable. The reaction is best carried out in solution (oDFB, TFB) to promote the CO/NO exchange of the NO atmosphere with the initially formed 1. After several minutes of stirring at room temperature, the brown solution of 1 turned dark green, indicating a complete transformation to 2. Filtration and crystallization by slow vapor diffusion of n‐pentane into the reaction solution led to dark green crystals of pure 2 (80 % yield, λ max=610, 420 and 310 nm in oDFB solution (see Figure S24), Scheme 1 c).
Scheme 1.
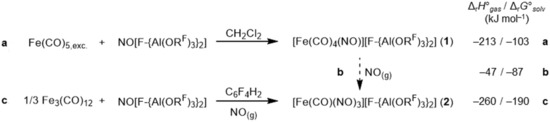
Synthesis of the desired heteroleptic iron carbonyl/nitrosyl cations [Fe(CO)4(NO)]+ and [Fe(CO)(NO)3]+ as [F‐Al(ORF)3}2]− salts 1 and 2. Reactions (a) and (c) give 80 % yield, if starting from pure materials.
Both reactions shown in Scheme 1 work with either combinations of the iron carbonyl source (Fe(CO)5, Fe2(CO)9, Fe3(CO)12) and solvents (CH2Cl2, oDFB, TFB or perfluorohexane C6F14), only reaction times and overall yields may vary. The thermodynamics underlying equations in Scheme 1 were determined as Δr G°solv. in CH2Cl2 starting from Fe(CO)5 at the BP86/def2‐TZVPP‐D3BJ/COSMO level and are in agreement with the experimental findings. Also the colors of the complexes are in good qualitative agreement to TD‐DFT calculations (cf. section S6). However, similar reactions showed that the more accessible alternative [Al(ORF)4]− anion is not suitable for this system: although the [Fe(CO)4(NO)][Al(ORF)4] salt immediately precipitates as a powder in CH2Cl2, upon dissolution in oDFB it readily reacts by anion decomposition to give the thermodynamically favored [Fe(CO)(NO)3][F‐{Al(ORF)3}2] salt and an unidentifiable precipitate. In addition, the crystal structure of [Fe(CO)(NO)3][Al(ORF)4] exhibits the typical overstructure and twinning problems observed for several [Al(ORF)4]− salts with (pseudo‐)tetrahedral cations such as [Mn(NO)4][Al(ORF)4].11 Therefore, we limit our report to the use of the more stable and less‐symmetric [F‐{Al(ORF)3}2]− anion. With this anion, the reactions in Scheme 1 led to phase‐pure crystals of 1 and 2 (cf. section S5 powder XRD; note that 1 crystallizes from CH2Cl2 in space group P21/c and from oDFB/TFB in P−1). In their molecular structures, a differentiation between CO and NO ligands is not possible. The NO ligands were only refined and displayed for visual clarity (Figure 2 c). However, their presence is evident from the averaged Fe−N/C bond lengths 1 (183.2(3) pm; cf. Fe(CO)5: 181.4 pm18/ [Fe(CO)6]2+: 191.1 pm19) and 2 (176.4(3) pm, Table 1) that agree within 2 pm to the DFT calculations (Table 1). All bond angles Fe−N/C−O are close to linear and range from 178 to 180° for 1 and from 176 to 177° for 2 (see section S8). This was also observed for the homoleptic [Mn(NO)4]+ cation, the [F‐{Al(ORF)3}2]− salt of which is isomorphous to 2.
Figure 2.
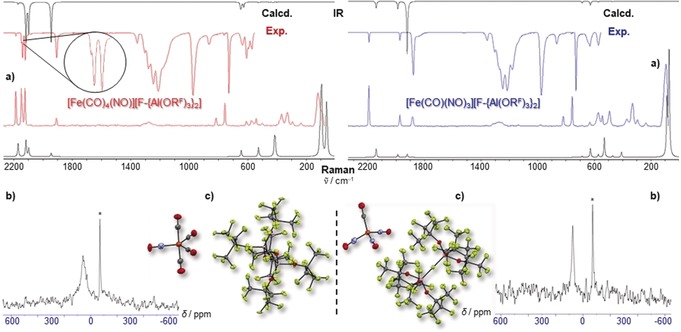
a) Experimental (Exp., red or blue) and calculated (Calcd, black, cations only, C 2v/C 3v symmetry @BP86def2/TZVPP‐D3BJ, no scaling factor was applied) vibrational spectra of 1 and 2, note the 2144 cm−1 shoulder in the expanded CO region of the IR spectrum of 1; b) 14N NMR‐spectra (21.69 MHz, oDFB, 298 K,), * signal from N2 atmosphere; c) molecular structures of 1 (P21/c, R1=4.2 %, wR2=11.9 %) and 2 (P‐1, R1=4.3 %, wR2=10.7 %). Note that the NO ligands are crystallographically indistinguishable from CO and were only colorized for visual purposes; thermal ellipsoids set at 50 % probability.
Table 1.
Experimental and calculated (calcd) data of crystalline [Fe(CO)4(NO)][F‐{Al(ORF)3}2] 1 and [Fe(CO)(NO)3][F‐{Al(ORF)3}2] 2 as well as selected literature‐known compounds; v: very, s: strong, m: medium, w: weak, sh: shoulder.
|
|
[Fe(CO)4(NO)]+ [F‐{Al(ORF)3}2]− |
[Fe(CO)4(NO)]+ calcd.[a] |
[Fe(CO)(NO)3]+ [F‐{Al(ORF)3}2]− |
[Fe(CO)(NO)3]+ calcd.[a] |
[Mn(NO)4]+ [F‐{Al(ORF)3}2]−[11] |
[Co(CO)2(NO)2]+ [B(CF3)4]−[13] |
||
|---|---|---|---|---|---|---|---|---|
|
(CO/NO) IR [cm−1] |
2183 (vvw) 2144 (sh)[b] 2137 (mw)[b] 2119 (m) 1904 (m) |
2168 (vvw) 2114 (vvs) 2114 (vvs) 2096 (vvs) 1905 (w) |
ν(CO) (A 1) ν(CO) (A 1)[b] ν(CO) (B 1)[b] ν(CO) (B 2) ν(NO) (A 1) |
2189 (vw) 1971 (vw) 1876 (s) |
2139 (vvw) 1986 (vvw) 1919 (vvw) |
ν(CO) (A 1) ν(NO) (A 1) ν(NO) (E) |
1855 (vvs) |
2183 (s) 2165 (s) 1960 (m) 1886 (m) |
|
|
|
|
|
|
|
|
|
|
|
(CO/NO) Raman [cm−1] |
2183 (vvs) 2145 (vvs) 2121 (vvs) 1905 (w) |
2169 (vvw) 2114 (vvs) 2096 (vvs) 1905 (w) |
ν(CO) (A 1) ν(CO) (B 1) ν(CO) (B 2) ν(NO) (A 1) |
2190 (s) 1972 (vw) 1881 (vw) |
2139 (vw) 1986 (vw) 1919 (vvw) |
ν(CO) (A 1) ν(NO) (A 1) ν(NO) (E) |
1978 (mw) 1866 (ms) |
2183 (s) 2167 (s) 1962 (m) 1893 (m) |
|
|
|
|
|
|
|
|
|
|
|
d(M‐NO/CO) [pm][c] |
183.2(3) (P21/c) 183.8(4) (P‐1) |
181.9 |
|
176.5(3) |
174.6 |
|
173.4(5) |
–[d] |
|
|
|
|
|
|
|
|
|
|
|
α (M‐N/C‐O) [°][c] |
179(1) (P21/c) 178(1) (P−1) |
179 |
|
177(1) |
178 |
|
178(1) |
–[d] |
|
|
|
|
|
|
|
|
|
|
|
13C NMR Shift [ppm][e] |
191.8 |
196.2 205.7 |
|
184.1 |
200.3 |
|
– |
–[d] |
|
|
|
|
|
|
|
|
|
|
|
14N NMR Shift [ppm][e] |
56 |
43 |
|
73 |
65 |
|
107 |
–[d] |
[a] C 2v/C 3v symmetry @BP86def2/TZVPP‐D3BJ, no scaling factor was applied. For comparison, the experimental d FeC in Fe(CO)5 is 181.4 pm18 and the calculation at the same level of theory gives 180.4 pm; [b] the assignment of the shoulder is ambiguous; [c] distances and angles averaged over all positions (CO and NO); [d] no data are available; [e] oDFB solution, 298 K, calculated shifts referenced to CH3NO2 (14N) and CH3NO2/Si(CH3)4 (13C).
A differentiation between both ligands and an unambiguous characterization of 1 and 2 is possible by IR and Raman spectroscopy. In both cases, the agreement between the simulated (@BP86def2/TZVPP‐D3BJ) and experimental spectra is excellent (Figure 2 a). Generally, the BP86 method is a good compromise between computational demand and the quality of the resulting data (structures, vibrations and NMR parameters, see Table 1) for these systems. The vibrational frequencies of the CO and NO bands of 1 and 2 are surprisingly similar to those of [Co(CO)2(NO)2]+,13 indicating comparable bond strengths and a similar electronic situation. The ν(CO) stretching vibrations of 1 are also very similar to those of the isoelectronic and pseudo‐isostructural [Co(CO)5]+ (2197/2155/2146/2120 cm−1)13 and are in the typical range for monocationic homoleptic carbonyl cations.12, 20 In addition, the four observed CO bands of 1, including the 2144 cm−1 shoulder (Raman: 2145 cm−1) of the A1/B1 vibration at 2137 cm−1 (Figure 2 a), indicate that the NO ligand resides in the equatorial plane. This is in agreement with BP86def2/TZVPP‐D3BJ calculations that give the A 1 and B 1 stretches as isoenergetic and resonating at 2114 cm−1 (Table 1). In addition, DFT supports the equatorial isomer as the global minimum and reveals the axial isomer as being 43 kJ mol−1 higher in energy. Moreover, the C 3v‐symmetric axial isomer would have only three independent CO stretches (calcd at 2158 (A 1), 2113 (A 1), and 2098 (E) cm−1) (see Figure S27). The ν(NO) stretching vibrations of 1 (1905 cm−1) and 2 (1972/1881 cm−1) are in part significantly blue‐shifted compared to those of free NO(g) (cf. 1876 cm−1)12 and similar to [Mn(NO)4]+ (1978/1866/1855 cm−1). Furthermore, the close agreement between the experimental vibrations in the solid state and the ideal, non‐distorted calculated frequencies of [Fe(CO)4(NO)]+ (C 2v) and [Fe(CO)(NO)3]+ (C 3v), indicate that the [F‐{Al(ORF)3}2]− anion truly induces pseudo‐gas‐phase conditions in condensed phases.10
The broad signal in the 14N NMR spectrum (Figure 2 b, left) and the appearance of only one signal in the 13C NMR spectrum (see section S2) for 1 indicate a Berry pseudorotation21 in solution, equilibrating the expected two different sets of CO ligands (Table 1), similar to the related [Co(CO)5]+.22 For 2, the 14N NMR signal is sharper, hinting at a more static structure in solution (Figure 2 b, right).
From the preceding the question arose, where the “electronic truth” in between the two limiting descriptions as M0/NO+ and M+/NO0 (M: metal) might lie. Owing to the linearity of the NO ligands, combined with the ν(NO) vibrational frequencies of complexes 1 and 2 close to that of gaseous NO, the oxidation state of the iron atom appears to be +I, incorporating neutral 3 e− NO donors as ligands. To further evaluate the possible (non‐)innocence of the NO ligand, we conducted AIM charge analyses on the cations of compounds 1 and 2 and compared them to the fully characterized isoelectronic and pseudo‐isostructural analogues [Co(CO)5]+,13 [Mn(NO)4]+[11] as well as [Co(CO)2(NO)2]+[13] (Table 2). Based on their AIM charges, all three metal nitrosyl species appear to be true FeI and MnI complexes with neutral NO ligands and calculated positive charges of about +1 on the metal atoms. Thus, the formal oxidation state and the partial charge on the metal appear to coincide here. The commonly as CoI regarded homoleptic carbonyl cation [Co(CO)5]+ with the stronger CO donor exhibits, as expected, a lower charge of only +0.74 residing on the cobalt atom.
Table 2.
The AIM charges for the isoelectronic and pseudo‐isostructural couples [Fe(CO)4(NO)]+/[Co(CO)5]+ and [Fe(CO)(NO)3]+/[Mn(NO)4]+/[Co(CO)2(NO)2]+.[a]
|
AIM charges at: |
[Fe(CO)4(NO)]+ (C 2v) |
[Fe(CO)4(NO)]+ (C 3v) |
[Co(CO)5]+ |
[Fe(CO)(NO)3]+ |
[Mn(NO)4]+ |
[Co(CO)2(NO)2]+ |
|---|---|---|---|---|---|---|
|
M(=Fe, Co, or Mn) |
+0.93 |
+0.89 |
+0.74 |
+1.05 |
+1.23 |
+0.87 |
|
C |
ax: +1.05 equiv: −1.02 |
ax: +1.06 equiv: −1.03 |
ax: +1.08 equiv: +1.06 |
+1.03 |
– |
+1.06 |
|
O(‐C) |
ax: −1.01 equiv: −1.02 |
ax: −1.01 equiv: −1.02 |
ax: −1.01 equiv: −1.02 |
−1.02 |
– |
−1.02 |
|
N |
+0.24 |
+0.24 |
– |
+0.22 |
+0.18 |
+0.26 |
|
O(‐N) |
−0.26 |
−0.20 |
– |
−0.23 |
−0.24 |
−0.23 |
[a] ax/eq refers to axial and equatorial ligands in a trigonal bipyramid.
Therefore, the AIM analyses and the linearity of the NO ligands in complexes 1 and 2, combined with the ν(NO) vibrational frequencies close to gaseous NO would assign those as true iron(I) complexes.2
In conclusion, we discovered a simple pathway to two novel heteroleptic iron carbonyl/nitrosyl cations, stabilized by a weakly coordinating anion. Both complexes are the first of their kind, exhibit the unusual oxidation state +I, were fully characterized and are accessible as phase pure materials in good to excellent yields. Owning to their simplicity, they may serve as model compounds for further investigations to yield insights into metal NO complexes. We encourage other groups to take over from here on, to pick up on this report not only from a fundamental point of view—but also to apply this knowledge in biochemistry or catalysis in order to allow for a better understanding of the role and nature of the NO ligand.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to thank Dr. Michael Daub for powder‐XRD, Dr. Harald Scherer and Fadime Bitgül for NMR measurements and Daniela Winkler for solution UV/Vis measurements. J.B. gratefully acknowledges financial support by the LGFG Graduate Funding. I.K. thanks the Deutsche Forschungsgemeinschaft DFG for support of this project.
J. Bohnenberger, I. Krossing, Angew. Chem. Int. Ed. 2020, 59, 5581.
Footnotes
The formal FeII cation [Fe(CO)5(NO)]Cl was postulated and assigned in 1968 (IR‐, UV/Vis‐spectroscopy and elemental analysis).17 See the Supporting Information section 7 for a detailed discussion, why we doubt its existence.
Experimental verification of the oxidation state by X‐ray photoelectron spectroscopy (XPS) were deemed unreliable after explicit discussion with experts at the KIT XPS‐site. The problems arise from the difficulty in determining the weak iron peak in XPS at about 706–710 eV in the presence of the intense fluorine peak of the anion with 55 F‐atoms at 688‐89 eV. For Mößbauer spectroscopy—to our knowledge—the nonexistence of true and stable iron(I) reference complexes greatly limits its usefulness.
References
- 1.
- 1a. Bilitewski U., Blodgett J. A. V., Duhme-Klair A.-K., Dallavalle S., Laschat S., Routledge A., Schobert R., Angew. Chem. Int. Ed. 2017, 56, 14360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14552; [Google Scholar]
- 1b. Grubel K., Holland P. L., Angew. Chem. Int. Ed. 2012, 51, 3308; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3364; [Google Scholar]
- 1c. Kovacs J. A., Science 2003, 299, 1024; [DOI] [PubMed] [Google Scholar]
- 1d. Chirik P. J., Angew. Chem. Int. Ed. 2017, 56, 5170; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5252; [Google Scholar]
- 1e. Plietker B., Iron catalysis in organic chemistry. Reactions and applications, Wiley-VCH, Weinheim, 2008; [Google Scholar]
- 1f. Simmons T. R., Artero V., Angew. Chem. Int. Ed. 2013, 52, 6143; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6259; [Google Scholar]
- 1g. Enthaler S., Junge K., Beller M., Angew. Chem. Int. Ed. 2008, 47, 3317; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3363. [Google Scholar]
- 2.
- 2a. Kuypers M. M. M., Marchant H. K., Kartal B., Nat. Rev. Microbiol. 2018, 16, 263; [DOI] [PubMed] [Google Scholar]
- 2b. Keilwerth M., Hohenberger J., Heinemann F. W., Sutter J., Scheurer A., Fang H., Bill E., Neese F., Ye S., Meyer K., J. Am. Chem. Soc. 2019, 141, 17217–17235; [DOI] [PubMed] [Google Scholar]
- 2c. Pacher P., Beckman J. S., Liaudet L., Physiol. Rev. 2007, 87, 315; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Hoffman B. M., Lukoyanov D., Yang Z.-Y., Dean D. R., Seefeldt L. C., Chem. Rev. 2014, 114, 4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Fitzpatrick J., Kim E., Acc. Chem. Res. 2015, 48, 2453; [DOI] [PubMed] [Google Scholar]
- 3b. Berto T. C., Speelman A. L., Zheng S., Lehnert N., Coord. Chem. Rev. 2013, 257, 244; [Google Scholar]
- 3c. In-Iam A., Wolf M., Wilfer C., Schaniel D., Woike T., Klüfers P., Chem. Eur. J. 2019, 25, 1304; [DOI] [PubMed] [Google Scholar]
- 3d. Dong H. T., Speelman A. L., Kozemchak C. E., Sil D., Krebs C., Lehnert N., Angew. Chem. Int. Ed. 2019, 58, 17695–17699; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17859–17863. [Google Scholar]
- 4. Enemark J. H., Feltham R. D., Coord. Chem. Rev. 1974, 13, 339. [Google Scholar]
- 5. Klein J. E. M. N., Miehlich B., Holzwarth M. S., Bauer M., Milek M., Khusniyarov M. M., Knizia G., Werner H.-J., Plietker B., Angew. Chem. Int. Ed. 2014, 53, 1790; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1820. [Google Scholar]
- 6. Zadrozny J. M., Xiao D. J., Atanasov M., Long G. J., Grandjean F., Neese F., Long J. R., Nat. Chem. 2013, 5, 577. [DOI] [PubMed] [Google Scholar]
- 7. Samuel P. P., Mondal K. C., Amin Sk N., Roesky H. W., Carl E., Neufeld R., Stalke D., Demeshko S., Meyer F., Ungur L., et al., J. Am. Chem. Soc. 2014, 136, 11964. [DOI] [PubMed] [Google Scholar]
- 8. Ouyang Z., Du J., Wang L., Kneebone J. L., Neidig M. L., Deng L., Inorg. Chem. 2015, 54, 8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Bihlmeier A., Gonsior M., Raabe I., Trapp N., Krossing I., Chem. Eur. J. 2004, 10, 5041; [DOI] [PubMed] [Google Scholar]
- 9b. Martens A., Weis P., Krummer M. C., Kreuzer M., Meierhöfer A., Meier S. C., Bohnenberger J., Scherer H., Riddlestone I., Krossing I., Chem. Sci. 2018, 9, 7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riddlestone I. M., Kraft A., Schaefer J., Krossing I., Angew. Chem. Int. Ed. 2018, 57, 13982; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14178. [Google Scholar]
- 11. Bohnenberger J., Derstine B., Daub M., Krossing I., Angew. Chem. Int. Ed. 2019, 58, 9586; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9687. [Google Scholar]
- 12. Bohnenberger J., Feuerstein W., Himmel D., Daub M., Breher F., Krossing I., Nat. Commun. 2019, 10, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bernhardt E., Finze M., Willner H., Lehmann C. W., Aubke F., Chem. Eur. J. 2006, 12, 8276. [DOI] [PubMed] [Google Scholar]
- 14. Anderson J. S., Hieber W., Z. Anorg. Allg. Chem. 1932, 208, 238. [Google Scholar]
- 15. Hieber W., Beutner K., Z. Naturforsch. B 1960, 15, 323. [Google Scholar]
- 16.
- 16a. Plietker B., Dieskau A., Eur. J. Org. Chem. 2009, 775; [Google Scholar]
- 16b. Chiou T.-W., Lu T.-T., Wu Y.-H., Yu Y.-J., Chu L.-K., Liaw W.-F., Angew. Chem. Int. Ed. 2015, 54, 14824; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15037; [Google Scholar]
- 16c. Alt I. T., Guttroff C., Plietker B., Angew. Chem. Int. Ed. 2017, 56, 10582; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10718. [Google Scholar]
- 17. Iqbal Z., Waddington T. C., J. Chem. Soc. A 1968, 2958. [Google Scholar]
- 18. Farrugia L. J., Evans C., J. Phys. Chem. A 2005, 109, 8834. [DOI] [PubMed] [Google Scholar]
- 19. Bernhardt E., Bley B., Wartchow R., Willner H., Bill E., Kuhn P., Sham I. H. T., Bodenbinder M., Bröchler R., Aubke F., J. Am. Chem. Soc. 1999, 121, 7188. [Google Scholar]
- 20. Xu Q., Coord. Chem. Rev. 2002, 231, 83. [Google Scholar]
- 21. Berry R. S., J. Chem. Phys. 1960, 32, 933. [Google Scholar]
- 22. Meier S. C., Himmel D., Krossing I., Chem. Eur. J. 2018, 24, 19348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary