

Abstract
Posttranslational modifications (PTMs) are important in the regulation of protein function, trafficking, localization, and marking for degradation. This work describes the development of peptide activity/affinity‐based probes for the discovery of proteins that recognize novel acyl‐based PTMs on lysine residues in the proteome. The probes contain surrogates of ϵ‐N‐acyllysine by introduction of either hydrazide or thioamide functionalities to circumvent hydrolysis of the modification during the experiments. In addition to the modified PTMs, the developed chemotypes were analyzed with respect to the effect of peptide sequence. The photo cross‐linking conditions and subsequent functionalization of the covalent adducts were systematically optimized by applying fluorophore labeling and gel electrophoresis (in‐gel fluorescence measurements). Finally, selected probes, containing the ϵ‐N‐glutaryllysine and ϵ‐N‐myristoyllysine analogues, were successfully applied for the enrichment of native, endogenous proteins from cell lysate, recapitulating the expected interactions of SIRT5 and SIRT2, respectively. Interestingly, the latter mentioned was able to pull down two different splice variants of SIRT2, which has not been achieved with a covalent probe before. Based on this elaborate proof‐of‐concept study, we expect that the technology will have broad future applications for pairing of novel PTMs with the proteins that target them in the cell.
Keywords: activity-based probes, affinity-based probes, histone deacetylases, photo-affinity labeling, posttranslational modifications, sirtuins
A new strategy for activity‐based probes is reported to address the members of the proteome that target ϵ‐N‐acyllysine posttranslational modifications, which are highly prevalent and constitute a continuously expanding collection of chemical groups. In addition, this work provides the first investigation of the importance of peptide sequence on performance of ϵ‐N‐acyllysine‐containing photo cross‐linking probes, screening the efficiency of a combined series of 36 probe constructs against all 7 human sirtuins.
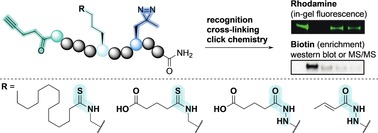
Introduction
Posttranslational modifications (PTMs) are important in the regulation of protein function, trafficking, localization, and marking for degradation.1 The effects and mechanisms of several types of chemical modification to protein side chains have been studied extensively, with particular attention dedicated to phosphorylation,2 ubiquitinylation,3 and acetylation4 thus far. However, it has recently become evident that lysine side chains may be decorated with a variety of different acyl groups in addition to ϵ‐N‐acetyllysine (Kac).5 Examples of these novel acyl PTMs include ϵ‐N‐malonyllysine (Kmal),6 ϵ‐N‐succinyllysine (Ksuc),6, 7 ϵ‐N‐glutaryllysine (Kglut),8 ϵ‐N‐crotonyllysine (Kcr),9 ϵ‐N‐phosphoglyceryllysine (Kpg),10 ϵ‐N‐myristoyllysine (Kmyr),11 ϵ‐N‐methylglutaconyllysine (Kmgc),12 and ϵ‐N‐(3‐hydroxy‐3‐methylglutaryl)‐lysine (Khmg).12 Except for Kpg, the above‐mentioned PTMs have all been shown to be regulated by members of the histone deacylase (HDAC) enzyme family. The human genome encodes 18 HDACs, the Zn2+‐dependent histone deacetylases (HDAC1‐11)13 and the NAD+‐dependent sirtuins (SIRT1‐7).14 Though originally classified as deacetylases, recent studies have revealed that SIRT5 targets Kmal, Ksuc, and Kglut,6, 7c–7e, 8a SIRT2 and SIRT6 regulate the levels of Kmyr,11b SIRT4 regulates Kmgc and Khmg,12, 15 Zn2+‐dependent HDAC1‐3 appear to be the major regulators of Kcr,16 and HDAC11 preferentially cleaves Kmyr.17
To complement the rapid rate of discovery of PTMs enabled by advanced mass spectrometry‐based proteomic investigations, we decided to develop a catalogue of chemical tools for matching novel PTMs with their respective targeting proteins. We envisioned that this could be achieved by applying the concept of photo cross‐linkable activity/affinity‐based probes (ABPs),18 containing different ϵ‐N‐acyllysine residues for recognition of the PTM of interest. Examples of such ABPs have been reported19 but the inherent susceptibility of these substrate‐based probes, to PTM removal by HDAC/SIRT activity, calls for alternative probe designs. Here, we describe an updated version of the previously reported probe concept by introducing amide bond analogues to avoid premature hydrolysis of the interrogated PTMs (Figure 1).
Figure 1.
Overview of the developed methodology. Upon recognition of the modified lysine, UV irradiation activates the photo cross‐linker forming a covalent bond between probe and protein. Conjugation through click chemistry then allows visualization by in‐gel fluorescence or streptavidin enrichment followed by western blotting.
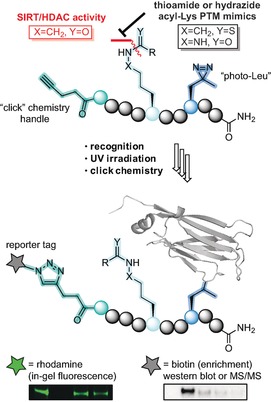
The chosen amide bond analogues were either the thioamide functional group (X=CH2, Y=S), which has been employed in mechanism‐based SIRT inhibitors20 or the δ‐azalysine‐based hydrazides described by Cole and co‐workers (X=NH, Y=O).21 Both were selected due to their ability to form so‐called stalled intermediates with the NAD+ co‐substrate in the sirtuin active site, resulting in decreased rates of hydrolysis.20g Encouragingly, Cen and co‐workers recently reported on an ABP, containing a ϵ‐N‐thioacetyllysine that could successfully label overexpressed SIRT2 in HEK293 cells, providing further impetus for this concept.22
Results and Discussion
Design and synthesis of photo cross‐linking probe collection
For a proof‐of‐concept probe collection, decameric peptides were chosen to allow several amino acids to flank the modified lysine residue. To assess the effect of the peptide sequence together with the PTM of interest, the four selected sequences were based around lysine residues that are known to be decorated with the different PTMs investigated in this study. Thus, in order to investigate Kac and Kcr the histone sequences around histone 4‐Lys12 (H4K12) and histone 3‐Lys9 (H3K9) were chosen as they both have been shown to carry Kac4a, 4b, 23 and Kcr.9 To date only two proteins have been shown to carry the Kmyr modification on ϵ‐amino groups of lysine residues—namely tumor necrosis factor‐α (TNF‐α)11a and interleukin 1α (IL‐1α)24—and we decided to base the probe sequence around TNF‐α K20. Glutarylation of numerous lysine residues has been identified on multiple proteins. Carbamoyl phosphate synthetase‐1 (CPS‐1) is among the proteins that has been most extensively investigated in this context8a and we therefore chose to design our probe sequence around CPS‐1 K1356.
Previous investigations by Sieber and co‐workers showed that the commonly used benzophenone photo cross‐linker gave rise to high levels of non‐specific binding.25 Furthermore, Li and co‐workers found that using the diazirine photo cross‐linker „photo‐Leu“ in close proximity to their modified lysine was preferred.19d In light of this insight regarding the choice and position of photo cross‐linker, as well as preliminary results from our own laboratory (unpublished), we designed the collection of probe sequences outlined in Scheme 1 A. In addition to the mentioned ϵ‐amide bond analogues, we also prepared the native oxoamide versions of the probes for comparison (motifs A–C in Scheme 1 B). Finally, four well‐described acyl‐based PTMs to lysine were chosen (Kac, Kcr, Kmyr, and Kglut) (Scheme 1 C) and a collection of 36 probes out of the full matrix of 48 combinations was synthesized (Table S1, Supporting Information (SI)). For control experiments and validation of specificity, we also synthesized a non‐acylated probe based on sequence 1 and a number of competitor probes that contain PTMs but are devoid of photo cross‐linker and click handle (sequences 1C, 3C, and 4C; Scheme 1 D and Table S2, SI).
Scheme 1.
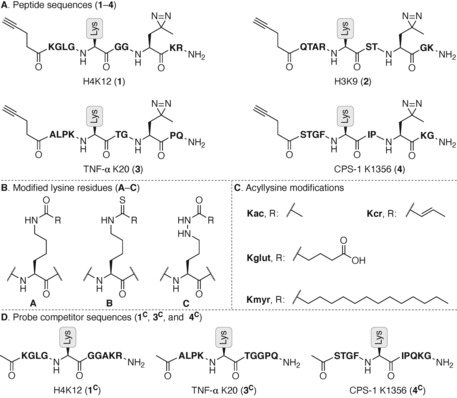
Nomenclature for the series of probes and competitors prepared in this study: (A) Sequences of the photo cross‐linkable and clickable decamer peptide probes based on H4, H3, TNF‐α, and CPS‐1 sequences (1 – 4, respectively). (B) Structures of the posttranslationally modified lysine residues A–C. (C) Structures and abbreviations for the investigated PTMs. (D) Competitor sequences, lacking photo cross‐linker and click handle, are named similar to the probes but marked with a “C” in superscript 1C, 3C, 4C.
Briefly, a versatile α‐N‐Fmoc‐δ‐N‐Boc‐ϵ‐N‐Teoc‐(δ‐aza)lysine building block was designed for introduction of hydrazide lysine‐mimicking side chains on solid support (Scheme S1, SI) and thioamide‐containing building blocks were prepared using Lawesson's reagent26 (Scheme S2, SI). After several failed attempts to prepare the thiocrotonylated building block, potentially due to instability of the α,β‐unsaturated thioamide functionality,27 it was discarded from our investigation. A small selection of additional probe constructs were furthermore omitted from the series based on the results of initial screens (see explanations vide infra). Thus, 36 different probes, systematically covering peptide sequences, lysine mimics, and PTMs (see Table S1), were synthesized by standard automated Fmoc solid‐phase peptide synthesis (SPPS). Though, coupling of the photo‐leucine building block and introduction of the acyl modifications in the hydrazide‐based probes were performed manually (Schemes S3 and S4, SI). The competitor sequences were similarly synthesized (Schemes S5 and S6) and all peptides were purified to >95 % homogeneity by preparative reverse‐phase HPLC separation and lyophilization of the fractions that contained pure product, according to MALDI‐TOF MS.
Optimization of ABP methodology using in‐gel fluorescence
With the collection of probes in hand, we first optimized the conditions for cross‐linked adduct formation and subsequent click chemistry using the native amide‐containing control probes based on H4K12 (1A sequences). We optimized the enzyme–probe conjugate formation by applying sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and in‐gel fluorescence measurements (SI, Figures S1–S5). For the photo cross‐linking we applied UV irradiation (365 nm) for 10 min at 0 °C.19d First, we assessed the choice of ligand for the CuI‐catalyzed azide‐alkyne 3+2 cycloaddition (CuAAC). In our hands, the more polar ligands exhibited superior performance than TBTA. Although different ligand effects may be observed depending on the used buffers and reducing agents as well, this finding was in agreement with a recent study of hydrophilic ligands,28 and we therefore chose BTTAA for further experiments (see SI, Figure S1 for western blot analysis, IUPAC names, and chemical structures). We then investigated the efficiency of different copper‐ligand ratios on the CuAAC. Based on labeling of sirtuins 2 and 3 (Figure S2), the optimal ratio of CuSO4‐BTTAA was selected to be 1:2. Next, we addressed the amount of probe and the probe‐fluorophore ratio, using recombinant enzymes (1 μm) in the presence of HeLa whole cell lysate (2 μg μL−1) to better mimic a cellular environment for the labeling reaction. The experiments revealed dose‐dependent and selective labeling of SIRT1, SIRT2, and SIRT5 (Figure 2 A and Figure S3). Based on those results, it was decided to continue with 25 μm of probe and 20 equivalents of fluorophore relative to this concentration, except when using the Kmyr probes where 12.5 μm of probe was considered sufficient. As evident from Figure 2 A, the labeling experiments using the acetylated probe (1A‐Kac) led to significant labeling of multiple proteins in the lysate in addition to the recombinant SIRT3.
Figure 2.
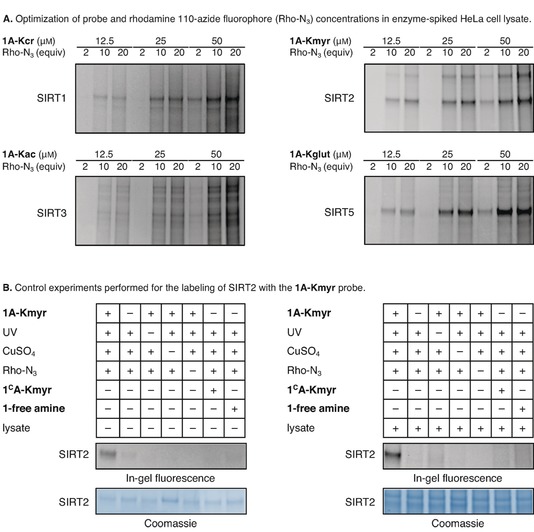
Optimization of covalent adduct formation and control experiments applying in‐gel fluorescence: (A) Optimization of probe concentration and probe‐fluorophore ratio required for labeling of SIRT1‐3 and SIRT5 (1 μM enzyme concentrations). (B) Control experiments shown for 1A‐Kmyr (12.5 μM) showing that labeling is indeed dependent on all components added. See the Supporting Information for full gel images (Figures S3–S5).
After this optimization, we performed a series of control experiments to confirm that the fluorescent labeling of the enzymes relied on addition of probe, UV irradiation, CuSO4, and fluorophore (Figure 2 B and Figures S4 and S5). Furthermore, it was confirmed that the non‐cross‐linking peptides Ac‐H48‐17K12(myr)‐NH2 (1CA‐Kmyr) and Ac‐H48‐17K12(glut)‐NH2 (1CA‐Kglut) do not lead to labeling, which allows for the use of these peptides as competitors. Finally, the importance of the lysine modification was further confirmed by applying a probe without a PTM but still containing the photo cross‐linker and alkyne moieties (1‐free amine, Table S2), which did not lead to fluorescent bands.
Addressing the importance of PTM and peptide sequence for sirtuin labeling
Encouraged by the above control experiments, we then systematically screened for the effect of peptide sequence (1–4) and modified lysine motif (A–C) on the ability of the probes to interact with the different sirtuins. It was gratifying to observe that thioamide (1–4B)‐ and hydrazide (1–3C)‐containing probes were generally as well recognized by the enzymes as the native oxoamides (1–4A) (see Figures S6–S21, Table S3, and Figure 3 for extracted examples using Kmyr). The screening of probe sequence 1 against a mixture of SIRT1‐3 (Figure S6) revealed that the 1C‐Kac hydrazide‐based probe performed similarly to the native 1A‐Kac probe, while the 1B‐Kac thioacetylated probe significantly enhanced the labeling efficiency. Therefore, it was decided to focus on thioacetylated probes rather than the hydrazide versions of the acetylated probes. Somewhat surprisingly in light of the recent work by Cen and co‐workers,22 however, the thioacetylated probes appeared to substantially label most sirtuins, including SIRT4 and SIRT5, which are not believed to target Kac6a, 8a, 12 (Figure S10–S17). Although, the observed differences in selectivity between our more elaborate screening of probes and the previous report may be explained by differences in experimental conditions, we conclude that probes containing this modification may be of limited utility and speculate that perhaps the hydrazide versions could rather be revisited after all. Nevertheless, we decided to focus the present investigation on non‐Kac modifications.
Figure 3.
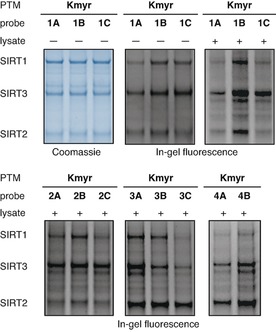
In‐gel fluorescence showing efficiency of SIRT1–3 labeling using myristoylated probes. Coomassie‐stained gel is shown in blue‐scale and in‐gel fluorescence is shown in gray‐scale. Screening of the labeling efficiency of different myristoylated probes (12.5 μM) against recombinant SIRT1‐3 (1 μM) in pure buffer or buffer containing HeLa cell lysate (2 μg μL−1). The concentration of SIRT2 was slightly lower than expected based on the protein concentration provided by the vendor, which we corrected for in later experiments. See the Supporting Information (Figures S6–S21) for full gel images and further discussion.
Interestingly, the ϵ‐N‐glutaryl‐(δ‐aza)lysine‐containing probe (1C‐Kglut) exhibited decreased labeling efficiency of SIRT5 compared to 1A‐Kglut (Figure S14). This observation was presumably due to zwitter ion formation or intramolecular hydrogen bonding between the basic NH group of the hydrazide and the terminal carboxylate. Based on this finding, we decided not to pursue the remaining hydrazide versions of the glutarylated probes. The thioglutarylated probes, on the other hand, are highly promising for the study of SIRT5, showing increased labeling compared to the glutarylated probes, and therefore potentially also for other proteins that may recognize Kglut (Figure S14–S17).
The crotonylated hydrazide‐based probes (1–3C‐Kcr) robustly labeled SIRT4, which is an interesting observation because SIRT4 has never been connected with this PTM before and therefore requires further investigation. In addition, future work employing these chemotypes will include investigation of recognition domains such as YEATS29 as well as class I zinc‐dependent HDACs, which are believed to be the main regulators of Kcr.16
The ϵ‐N‐myristoyllysine‐containing probe series efficiently labeled SIRT1‐3 as expected30 and also gave rise to substantial fluorescent bands for SIRT7 (Figures S20 and S21), which is in agreement with a recent study.31 In general, we found that the PTMs more significantly affect probe efficacy than the peptide sequence; albeit, with some exceptions. Therefore, we speculated that adding the hydrazide‐based series of probes with sequence 4 would provide limited information and decided not to include 4C probes in the collection. Interestingly, the Kmyr‐containing peptide sequences based on TNF‐α, which has been shown to be targeted by sirtuins,11b appeared to label SIRT2 with some degree of selectivity over other sequences. This was particularly pronounced when using the probes containing the thioamide or hydrazide Kmyr mimics (Figure 3).
Stability of probes containing amide bond analogues and competition experiments
Based on the above screening results, and because the PTMs in question have been shown to regulate the respective enzymes,8a, 11b we selected the TNF‐α sequence (3) for probes containing Kmyr residues and the CPS1 sequence (4) for glutarylated probes for further experiments. First, we challenged the performance of probes containing different lysine residues (A–C) by addition of NAD+, the co‐substrate of sirtuins. This clearly resulted in a significant decrease in fluorescence intensity for the oxoamide‐containing probes and in the case of Kmyr, a complete abolishment of sirtuin labeling (Figure 4). However, the experiment gratifyingly showed retention of fluorescent bands for the thioamide bond‐containing probes 3B‐Kmyr and 4B‐Kglut as well as the δ‐azalysine mimic 3C‐Kmyr (Figure 4 A, B and Figure S22).
Figure 4.
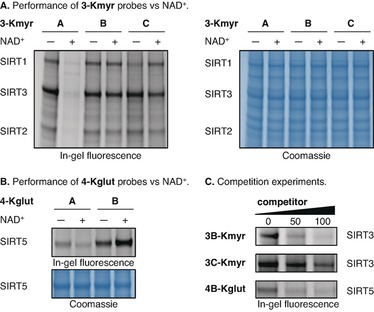
(A) Testing the performance of the myristoyl probes without or with NAD+ (500 μM) in the presence of recombinant SIRT1‐3 (1 μM of each). (B) Testing the performance of the glutaryl probes without or with NAD+ (500 μM) in the presence of recombinant SIRT5 (1 μM). (C) Competition of 3B‐Kmyr (12.5 μM), 3C‐Kmyr (12.5 μM) and 4B‐Kglut (25 μM) against recombinant enzymes (1 μM) with 3CB‐Kmyr, 3C‐Kmyr, and 4CB‐Kglut, respectively. See the Supporting Information for full gel images (Figures S22 and S23) and further discussion.
Interestingly, the glutarylated probe 4B‐Kglut even exhibited more intense labeling of SIRT5 in the presence of NAD+, which could be explained by extended residence time in the enzyme active site due to formation of a stalled intermediate between probe and co‐substrate. Taken together, this strongly indicates that compromised structural integrity of probes that are based on native PTMs (type A) could give rise to ambiguous results if applied in a biological environment and shows that this challenge can be solved by applying non‐cleavable amide bond analogues (i.e., thioamides or hydrazides). Importantly, we further showed by competition experiments, that the novel PTM mimics indeed bind to the sirtuin enzymes through specific recognition that can be outcompeted by non‐cross‐linking, PTM‐modified peptides (Figure 4 C, Figure S23 and supporting discussion). The thioamide probe (3B‐Kmyr) was more efficiently outcompeted than the hydrazide probe (3C‐Kmyr) and was therefore selected for the further experiments.
Employing photo cross‐linking probes for the enrichment of native enzymes from cell lysates
With these encouraging proof‐of‐concept results from in‐gel fluorescence experiments, we next ventured into trapping of native enzymes from whole cell lysates. Here, we employed coupling of biotin to the protein‐ABP conjugate instead of the fluorophore, followed by enrichment on streptavidin‐coated beads, and protein identification using western blot analysis. Most ABPs developed for investigation of sirtuins have only been evaluated using recombinant enzymes or with overexpressing cell lines thus far.19b, 22 A notable exception to this is work from Li and co‐workers, who have demonstrated enrichment of endogenous SIRT3 and SIRT5 from HeLa whole cell lysate using probes based on Kac and Kmal, respectively.19d We show here the first examples of successful pull‐down of endogenous interaction partners for the Kmyr and Kglut posttranslational modifications. Thus, probes 3B‐Kmyr and 4B‐Kglut were incubated with native HEK293 whole cell lysate and different amounts of the corresponding competitor probes. After elution from the streptavidin‐coated beads the samples were analyzed by western blotting (Figure 5).
Figure 5.
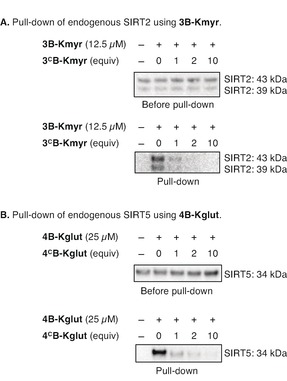
Pull‐down of endogenous proteins from HEK293 whole cell lysate: (A) Pull‐down of endogenous SIRT2 using 3B‐Kmyr. Top membrane shows samples before pull‐down. Bottom membrane shows samples after pull‐down, including dose‐dependent competition of the adduct formation. (B) Pull‐down of endogenous SIRT5 using 4B‐Kglut. Top membrane shows samples before pull‐down. Bottom membrane shows samples after pull‐down, including dose‐dependent competition of the adduct formation. *All blots are performed in duplicates and can be seen in full in Figures S25 and S26.
Importantly, an extensive amount of experiments proved necessary to optimize this protocol, which eventually showed that the choice of beads was of tremendous importance. Thus, at first sight, we achieved robust enrichment of several enzymes using both agarose beads and Dynabeads. However, the control experiments revealed this to be accompanied by substantial non‐specific binding to the beads, which we unfortunately failed to eliminate, even after extensive washing steps (see Figure S24 for details). Finally, a type of streptavidin magnetic beads that could be washed properly was identified and successfully employed for enrichment (Figure S24C).
Satisfyingly, both probes efficiently trapped and pulled down the reported interaction partners SIRT2 and SIRT5, respectively, when employing the optimized conditions (Figure 5). With the myristoylated probe (3B‐Kmyr) we enriched two isoforms of SIRT2 and the labeling was successfully abolished by competition using 3CB‐Kmyr for both bands. This constitutes the first demonstration of a versatile photo cross‐linking probe that is able to enrich two different endogenous and physiologically relevant splice variants of SIRT2 [i.e., isoform 1 (43 kDa) and isoform 2 (39 kDa],32 which underscores the utility of our new probe design (Figure 5 A). Affinity purification of both splice variants of SIRT2 from HL60 cell lysate has previously been demonstrated. However, this was achieved by using beads coated with immobilized SirReal2, which is a selective inhibitor of SIRT2 with long residence time in the active site.33
We also attempted to enrich HDAC11 from MCF‐7 cell lysate, where this enzyme is abundantly expressed. Unfortunately, these efforts failed, which we suspect may be due to the low binding affinities (high K M values) recorded for Kmyr substrates with HDAC11.17
Finally, the glutarylated probe (4B‐Kglut) was able to form a cross‐linked adduct with and enrich endogenous SIRT5 and this adduct formation was also outcompeted by the corresponding 4CB‐Kglut (Figure 5 B).
Conclusions
In summary, we have developed a new strategy for photo cross‐linking probes to address the members of the proteome that target ϵ‐N‐acyllysine posttranslational modifications, which are highly prevalent and constitute a continuously expanding collection of chemical groups. We have harnessed design principles from mechanism‐based inhibitors of the sirtuin enzymes, because this may allow for extended residence time in the targeting enzyme's pocket and, importantly, will preserve the integrity of the probe during the course of the experiment. Our results are highly encouraging, as they corroborate the hypothesis that we can install enzymatically stable amide bond analogues, while retaining the ability of native interaction partners in cell lysates to recognize the modification with high selectivity and specificity. In addition, we provide the first investigation of the importance of peptide sequence on performance of ϵ‐N‐acyllysine‐containing photo cross‐linking probes, screening the efficiency of a combined series of 36 probe constructs against all 7 human sirtuins.
The developed methodology provides basis for detailed chemoproteomics studies by combining our protocol with stable isotope labeling with amino acids in cell culture (SILAC) tandem mass spectrometry‐based methods.34 This will broaden the potential of the technology beyond identification of the endogenous “eraser” enzymes (i.e., HDACs or sirtuins) as demonstrated in this proof‐of‐concept study. With the enhanced structural integrity of the probes, we expect that potential reader domains can be identified as well.
Finally, the relatively simple design allows for easy preparation of novel probes containing different PTMs. Thus, with the continuously expanding landscape of acyl modifications to lysine residues in the human proteome,35 discovered by tandem mass spectrometry proteomics methods, we expect this technology to find broad applications in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Andreas S. Madsen, Dr. Nima Rajabi, Dr. Carlos Moreno‐Yruela, Ms. Kathrin S. Troelsen, Ms. Julie E. Bolding, Ms. Katrine Krydsfeldt, and Ms. Janne M. Colding for helpful discussions and technical assistance. This work was supported by the Danish Independent Research Council‐Natural Sciences (Grant No. 6108‐00166B), the Villum Foundation (Blok stipend 341/300‐123012), the Carlsberg Foundation (2013‐01‐0333 and CF15‐011), the Novo Nordisk Foundation (NNF15OC0017334), and the European Research Council (ERC‐CoG‐725172‐SIRFUNCT).
M. Bæk, P. Martín-Gago, J. S. Laursen, J. L. H. Madsen, S. Chakladar, C. A. Olsen, Chem. Eur. J. 2020, 26, 3862.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.10315502.v1).
References
- 1. Aebersold R., Agar J. N., Amster I. J., Baker M. S., Bertozzi C. R., Boja E. S., Costello C. E., Cravatt B. F., Fenselau C., Garcia B. A., Ge Y., Gunawardena J., Hendrickson R. C., Hergenrother P. J., Huber C. G., Ivanov A. R., Jensen O. N., Jewett M. C., Kelleher N. L., Kiessling L. L., Krogan N. J., Larsen M. R., Loo J. A., Ogorzalek Loo R. R., Lundberg E., MacCoss M. J., Mallick P., Mootha V. K., Mrksich M., Muir T. W., Patrie S. M., Pesavento J. J., Pitteri S. J., Rodriguez H., Saghatelian A., Sandoval W., Schlüter H., Sechi S., Slavoff S. A., Smith L. M., Snyder M. P., Thomas P. M., Uhlén M., Van Eyk J. E., Vidal M., Walt D. R., White F. M., Williams E. R., Wohlschlager T., Wysocki V. H., Yates N. A., Young N. L., Zhang B., Nat. Chem. Biol. 2018, 14, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Cohen P., Nat. Cell Biol. 2002, 4, E127–130; [DOI] [PubMed] [Google Scholar]
- 2b. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M., Cell 2006, 127, 635–648; [DOI] [PubMed] [Google Scholar]
- 2c. Linding R., Jensen L. J., Ostheimer G. J., van Vugt M. A., Jorgensen C., Miron I. M., Diella F., Colwill K., Taylor L., Elder K., Metalnikov P., Nguyen V., Pasculescu A., Jin J., Park J. G., Samson L. D., Woodgett J. R., Russell R. B., Bork P., Yaffe M. B., Pawson T., Cell 2007, 129, 1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Teixeira L. K., Reed S. I., Annu. Rev. Biochem. 2013, 82, 387–414; [DOI] [PubMed] [Google Scholar]
- 3b. Chernorudskiy A. L., Gainullin M. R., Sci. Signaling 2013, 6, pe22; [DOI] [PubMed] [Google Scholar]
- 3c. Swatek K. N., Komander D., Cell Res. 2016, 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y., Mol. Cell 2006, 23, 607; [DOI] [PubMed] [Google Scholar]
- 4b. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M., Science 2009, 325, 834; [DOI] [PubMed] [Google Scholar]
- 4c. Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., Grueter C. A., Harris C., Biddinger S., Ilkayeva O. R., Stevens R. D., Li Y., Saha A. K., Ruderman N. B., Bain J. R., Newgard C. B., R. V. Farese Jr , Alt F. W., Kahn C. R., Verdin E., Nature 2010, 464, 121–125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P., Science 2010, 327, 1004–1007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Choudhary C., Weinert B. T., Nishida Y., Verdin E., Mann M., Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550; [DOI] [PubMed] [Google Scholar]
- 4f. Verdin E., Ott M., Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Hirschey M. D., Cell Metab. 2011, 14, 718–719; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Olsen C. A., Angew. Chem. Int. Ed. 2012, 51, 3755–3756; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3817–3819; [Google Scholar]
- 5c. Lin H., Su X., He B., ACS Chem. Biol. 2012, 7, 947–960; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Olsen C. A., ChemMedChem 2014, 9, 434–437. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., Lin H., Science 2011, 334, 806–809; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Zhang Z., Tan M., Xie Z., Dai L., Chen Y., Zhao Y., Nat. Chem. Biol. 2011, 7, 58–63; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Xie Z., Dai J., Dai L., Tan M., Cheng Z., Wu Y., Boeke J. D., Zhao Y., Mol. Cell. Proteomics 2012, 11, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., Zwaans B. M., Tishkoff D., Ho L., Lombard D., He T. C., Dai J., Verdin E., Ye Y., Zhao Y., Mol. Cell. Proteomics 2011, 10, M111.012658; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Zhou Y., Zhang H., He B., Du J., Lin H., Cerione R. A., Hao Q., J. Biol. Chem. 2012, 287, 28307–28314; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M., Skinner M. E., Lombard D. B., Zhao Y., Mol. Cell 2013, 50, 919–930; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Weinert B. T., Scholz C., Wagner S. A., Iesmantavicius V., Su D., Daniel J. A., Choudhary C., Cell Rep. 2013, 4, 842–851; [DOI] [PubMed] [Google Scholar]
- 7e. Wagner G. R., Payne R. M., J. Biol. Chem. 2013, 288, 29036–29045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., Peterson B. S., Xu G., Ilkayeva O. R., Muehlbauer M. J., Braulke T., Muhlhausen C., Backos D. S., Olsen C. A., McGuire P. J., Pletcher S. D., Lombard D. B., Hirschey M. D., Zhao Y., Cell Metab. 2014, 19, 605–617; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Carrico C., Meyer J. G., He W., Gibson B. W., Verdin E., Cell Metab. 2018, 27, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., Lu Z., Ye Z., Zhu Q., Wysocka J., Ye Y., Khochbin S., Ren B., Zhao Y., Cell 2011, 146, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moellering R. E., Cravatt B. F., Science 2013, 341, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Stevenson F. T., J. Exp. Med. 1992, 176, 1053–1062; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R., Hang H. C., Hao Q., Lin H., Nature 2013, 496, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anderson K. A., Huynh F. K., Fisher-Wellman K., Stuart J. D., Peterson B. S., Douros J. D., Wagner G. R., Thompson J. W., Madsen A. S., Green M. F., Sivley R. M., Ilkayeva O. R., Stevens R. D., Backos D. S., Capra J. A., Olsen C. A., Campbell J. E., Muoio D. M., Grimsrud P. A., Hirschey M. D., Cell Metab. 2017, 25, 838–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gregoretti I., Lee Y.-M., Goodson H. V., J. Mol. Biol. 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Haigis M. C., Sinclair D. A., Annu. Rev Pathol 2010, 5, 253–295; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Anderson K. A., Madsen A. S., Olsen C. A., Hirschey M. D., Biochim. Biophys. Acta Bioenergetics 2017, 1858, 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pannek M., Simic Z., Fuszard M., Meleshin M., Rotili D., Mai A., Schutkowski M., Steegborn C., Nat. Commun. 2017, 8, 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Madsen A. S., Olsen C. A., Angew. Chem. Int. Ed. 2012, 51, 9083–9087; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9217–9221; [Google Scholar]
- 16b. Wei W., Liu X., Chen J., Gao S., Lu L., Zhang H., Ding G., Wang Z., Chen Z., Shi T., Li J., Yu J., Wong J., Cell Res. 2017, 27, 898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Kutil Z., Novakova Z., Meleshin M., Mikesova J., Schutkowski M., Barinka C., ACS Chem. Biol. 2018, 13, 685–693; [DOI] [PubMed] [Google Scholar]
- 17b. Moreno-Yruela C., Galleano I., Madsen A. S., Olsen C. A., Cell Chem. Biol. 2018, 25, 849–856; [DOI] [PubMed] [Google Scholar]
- 17c. Cao J., Sun L., Aramsangtienchai P., Spiegelman N. A., Zhang X., Huang W., Seto E., Lin H., Proc. Natl. Acad. Sci. USA 2019, 116, 5487–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Cravatt B., Sorensen E. J., Curr. Opin. Chem. Biol. 2000, 4, 663–668; [DOI] [PubMed] [Google Scholar]
- 18b. Adam G. C., Sorensen E. J., Cravatt B. F., Mol. Cell. Proteomics 2002, 1, 781–790; [DOI] [PubMed] [Google Scholar]
- 18c. Speers A. E., Cravatt B. F., Chem. Biol. 2004, 11, 535–546; [DOI] [PubMed] [Google Scholar]
- 18d. Jessani N., Cravatt B. F., Curr. Opin. Chem. Biol. 2004, 8, 54–59; [DOI] [PubMed] [Google Scholar]
- 18e. Heal W. P., Dang T. H., Tate E. W., Chem. Soc. Rev. 2011, 40, 246–257; [DOI] [PubMed] [Google Scholar]
- 18f. Hu X., Zheng W., Prog. Mol. Biol. Transl. Sci. 2018, 154, 1–24. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Bao X., Wang Y., Li X., Li X. M., Liu Z., Yang T., Wong C. F., Zhang J., Hao Q., Li X. D., eLife 2014, 3, e02999; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Kalesh K. A., Tate E. W., Org. Biomol. Chem. 2014, 12, 4310–4313; [DOI] [PubMed] [Google Scholar]
- 19c. Liu Z., Yang T., Li X., Peng T., Hang H. C., Li X. D., Angew. Chem. Int. Ed. 2015, 54, 1149–1152; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1165–1168; [Google Scholar]
- 19d. Yang T., Liu Z., Li X. D., Chem. Sci. 2015, 6, 1011–1017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19e. Yang T., Li X. M., Bao X., Fung Y. M., Li X. D., Nat. Chem. Biol. 2016, 12, 70–72. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Fatkins D. G., Monnot A. D., Zheng W., Bioorg. Med. Chem. Lett. 2006, 16, 3651–3656; [DOI] [PubMed] [Google Scholar]
- 20b. Smith B. C., Denu J. M., J. Biol. Chem. 2007, 282, 37256–37265; [DOI] [PubMed] [Google Scholar]
- 20c. Smith B. C., Hallows W. C., Denu J. M., Chem. Biol. 2008, 15, 1002–1013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. He B., Du J., Lin H., J. Am. Chem. Soc. 2012, 134, 1922–1925; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20e. Jing H., Hu J., He B., Negron Abril Y. L., Stupinski J., Weiser K., Carbonaro M., Chiang Y. L., Southard T., Giannakakou P., Weiss R. S., Lin H., Cancer Cell 2016, 29, 297–310; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20f. Rajabi N., Auth M., Troelsen K. R., Pannek M., Bhatt D. P., Fontenas M., Hirschey M. D., Steegborn C., Madsen A. S., Olsen C. A., Angew. Chem. Int. Ed. 2017, 56, 14836–14841; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15032–15037; [Google Scholar]
- 20g. Jiang Y., Liu J., Chen D., Yan L., Zheng W., Trends Pharmacol. Sci. 2017, 38, 459–472; [DOI] [PubMed] [Google Scholar]
- 20h. Rajabi N., Galleano I., Madsen A. S., Olsen C. A., Prog. Mol. Biol. Transl. Sci. 2018, 154, 25–69. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Dancy B. C., Ming S. A., Papazyan R., Jelinek C. A., Majumdar A., Sun Y., Dancy B. M., W. J. Drury 3rd , Cotter R. J., Taverna S. D., Cole P. A., J. Am. Chem. Soc. 2012, 134, 5138–5148; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Bhat S., Hwang Y., Gibson M. D., Morgan M. T., Taverna S. D., Zhao Y., Wolberger C., Poirier M. G., Cole P. A., J. Am. Chem. Soc. 2018, 140, 9478–9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graham E., Rymarchyk S., Wood M., Cen Y., ACS Chem. Biol. 2018, 13, 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Beck H. C., Nielsen E. C., Matthiesen R., Jensen L. H., Sehested M., Finn P., Grauslund M., Hansen A. M., Jensen O. N., Mol. Cell. Proteomics 2006, 5, 1314–1325; [DOI] [PubMed] [Google Scholar]
- 23b. Garcia B. A., Hake S. B., Diaz R. L., Kauer M., Morris S. A., Recht J., Shabanowitz J., Mishra N., Strahl B. D., Allis C. D., Hunt D. F., J. Biol. Chem. 2007, 282, 7641–7655. [DOI] [PubMed] [Google Scholar]
- 24. Stevenson F. T., Bursten S. L., Fanton C., Locksley R. M., Lovett D. H., Proc. Natl. Acad. Sci. USA 1993, 90, 7245–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kleiner P., Heydenreuter W., Stahl M., Korotkov V. S., Sieber S. A., Angew. Chem. Int. Ed. 2017, 56, 1396–1401; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1417–1422. [Google Scholar]
- 26.
- 26a. Lecher H. Z., Greenwood R. A., Whitehouse K. C., Chao T. H., J. Am. Chem. Soc. 1956, 78, 5018–5022; [Google Scholar]
- 26b. Thomsen I., Clausen K., Scheibye S., Lawesson S.-O., Org. Synth. 1984, 62, 158; [Google Scholar]
- 26c. Ozturk T., Ertas E., Mert O., Chem. Rev. 2007, 107, 5210–5278. [DOI] [PubMed] [Google Scholar]
- 27. Alper H., Brandes D. A., Organometallics 1991, 10, 2457–2467. [Google Scholar]
- 28. Besanceney-Webler C., Jiang H., Zheng T., Feng L., Soriano del Amo D., Wang W., Klivansky L. M., Marlow F. L., Liu Y., Wu P., Angew. Chem. Int. Ed. 2011, 50, 8051–8056; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8201–8206. [Google Scholar]
- 29.
- 29a. Andrews F. H., Shinsky S. A., Shanle E. K., Bridgers J. B., Gest A., Tsun I. K., Krajewski K., Shi X., Strahl B. D., Kutateladze T. G., Nat. Chem. Biol. 2016, 12, 396–398; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Li Y., Sabari B. R., Panchenko T., Wen H., Zhao D., Guan H., Wan L., Huang H., Tang Z., Zhao Y., Roeder R. G., Shi X., Allis C. D., Li H., Mol. Cell 2016, 62, 181–193; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29c. Zhao D., Guan H., Zhao S., Mi W., Wen H., Li Y., Zhao Y., Allis C. D., Shi X., Li H., Cell Res. 2016, 26, 629–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Feldman J. L., Baeza J., Denu J. M., J. Biol. Chem. 2013, 288, 31350–31356; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Madsen A. S., Andersen C., Daoud M., Anderson K. A., Laursen J. S., Chakladar S., Huynh F. K., Colaco A. R., Backos D. S., Fristrup P., Hirschey M. D., Olsen C. A., J. Biol. Chem. 2016, 291, 7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.
- 31a. Tong Z., Wang M., Wang Y., Kim D. D., Grenier J. K., Cao J., Sadhukhan S., Hao Q., Lin H., ACS Chem. Biol. 2017, 12, 300–310; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31b. Tong Z., Wang Y., Zhang X., Kim D. D., Sadhukhan S., Hao Q., Lin H., ACS Chem. Biol. 2016, 11, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rack J. G., VanLinden M. R., Lutter T., Aasland R., Ziegler M., J. Mol. Biol. 2014, 426, 1677–1691. [DOI] [PubMed] [Google Scholar]
- 33. Schiedel M., Rumpf T., Karaman B., Lehotzky A., Gerhardt S., Ovadi J., Sippl W., Einsle O., Jung M., Angew. Chem. Int. Ed. 2016, 55, 2252–2256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2293–2297. [Google Scholar]
- 34. Li X., Foley E. A., Molloy K. R., Li Y., Chait B. T., Kapoor T. M., J. Am. Chem. Soc. 2012, 134, 1982–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.
- 35a. Huang H., Zhang D., Wang Y., Perez-Neut M., Han Z., Zheng Y. G., Hao Q., Zhao Y., Nat. Commun. 2018, 9, 3374; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35b. Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y., Liu W., Kim S., Lee S., Perez-Neut M., Ding J., Czyz D., Hu R., Ye Z., He M., Zheng Y. G., Shuman H. A., Dai L., Ren B., Roeder R. G., Becker L., Zhao Y., Nature 2019, 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary