

Abstract
Synonymous mutations do not change the amino acid but do change the synonymous codon usage. In genomes of different organisms, the gene conversion process is biased toward GC, which is irrespective of mutation bias. In the coding region, this trend is especially obvious and it is possibly caused by the preference on G/C-ending codons over the A/T-ending ones. If the G/C-ending codons are advantageous, then the synonymous mutations that change A/T to G/C would be “optimal” compared to the opposite ones. In theory, one should observe signals of positive selection on these optimal synonymous mutations. The recently released single-nucleotide polymorphism (SNP) data from the 1001 genome project of Arabidopsis thaliana provided researchers with an unprecedented opportunity to verify this assumption. I fully take advantage of the SNP data from 1,135 A thaliana lines and came to the conclusion that synonymous mutations in natural populations are not strictly neutral: the synonymous mutations that increase GC content (from A/T to G/C) tend to have higher derived allele frequencies (DAFs) and, therefore, are likely to be positively selected. My current study broadens our knowledge of the selection patterns of synonymous mutations and should be appealing to evolutionary biologists.
One sentence summary: In 1135 genomes of Arabidopsis thaliana, the synonymous mutations that increase the GC content tend to have higher derived allele frequencies (DAFs) and are likely to be positively selected.
Keywords: Synonymous mutations, derived allele frequency (DAF), GC content, Arabidopsis thaliana, natural selection
Introduction
Synonymous mutations are those mutations in the coding sequence (CDS) that do not change the amino acids (AAs). However, this does not mean that synonymous mutations are free from natural selection.1-3 It was reported that some synonymous mutations could affect messenger RNA (mRNA) splicing4 and the splicing patterns might consequently affect the biological processes.5 These splicing-related synonymous mutations were likely to be selected against.4 Another well-known impact of synonymous mutation is the influence on synonymous codon usage. The G/C-ending synonymous codons seem to appear more frequently in the genome compared to the neutral expectation.6 Theories were established to explain the preference for G/C-ending codons. One potential advantage of the G/C-ending synonymous codons is that they are decoded at higher rates during mRNA translation elongation 7-10 due to the putatively higher transfer RNA (tRNA) availability. Accordingly, G/C-ending synonymous codons usually have higher codon adaptation index (CAI)11 or tRNA adaptation index (tAI)12 values than their A/T-ending counterpart. Codon adaptation index describes the relative usage of synonymous codons in the genome and tAI incorporates the tRNA copy number of the corresponding codon. Faster translation rates might provide higher probability for the G/C-ending codons to be selectively maintained by natural selection.
In addition, there are also other potential impacts of synonymous mutations, such as the changes in thermodynamic stability of the secondary structures of mRNA (termed minimum free energy [MFE]) or methylation contexts. RNA structures could affect many aspects of RNA biology, such as the RNA-binding proteins (RBP) binding efficiency and the movement of ribosomes on RNAs. If a synonymous mutation altered the structure of RNAs, this change could be either deleterious, beneficial, or neutral and consequently subjected to natural selection.
Despite the established hypotheses explaining the putative advantage of G/C-ending codons, it would be interesting to directly verify the selection force acting on those synonymous mutations that change the codon usage patterns. If the G/C-ending codons are advantageous, then the synonymous mutations that change A/T to G/C would be “optimal” compared to the opposite ones. In theory, signals of positive selection should be observed on these “optimal” synonymous mutations.
I fully take advantage of the single-nucleotide polymorphism (SNP) data from 1135 Arabidopsis thaliana lines (the 1001 genome project). If all synonymous mutations are strictly neutral, then the different types of mutations (eg, A-to-C, G-to-T, T-to-C) would exhibit similar degrees of derived allele frequencies (DAFs). If particular types of synonymous changes (eg, T-to-C) are more advantageous than the opposite ones (C-to-T), then the optimal synonymous changes should be positively selected and exhibit relatively higher DAF.
Moreover, it is necessary to test whether the observed pattern on synonymous DAF is affected by population structures. Population structure reflects the ancestry of different groups within the population. If an expected pattern is unbiased, it should not be affected by population structure and should be observed across all the representative subpopulations.
In this study, by analyzing the frequency spectrum of the synonymous mutations, I draw to the conclusion that synonymous mutations in natural populations are not strictly neutral: the synonymous mutations that increase GC content (from A/T to G/C) tend to have higher DAF and therefore are likely to be positively selected. This pattern is not affected by the population structure as it is observed in all representative subpopulations. My current study suggests that synonymous mutations are not strictly neutral and should not be automatically ignored in evolutionary analyses.
Materials and Methods
Data collection
I download the SNP data as well as the genome-wide Fst values (window size 10 000 bp) of the 1,135 natural inbred lines of A thaliana from the 1001 genome project (http://1001genomes.org/data/GMI-MPI/releases/v3.1).13 The annotation of each SNP is included in the downloaded variant calling format (vcf) files. Note that I only used the SNPs in the main chromosomes and discarded the very few SNPs in the chloroplast or mitochondrial genomes. Furthermore, the admixture groups defined by the original work13 were retrieved. Some subpopulations had only a few alternative alleles so that the allele frequency might be very low and fluctuating due to sampling bias. In the subpopulation analyses, I only chose 3 representative subpopulations with at least 10 alternative alleles to ensure that the allele frequencies were not affected by sampling bias.
Inferring the DAF using an outgroup
I aligned the CDSs sequences between A thaliana (TAIR 10 version, link: https://www.arabidopsis.org/download_files/Sequences/TAIR10_blastsets/TAIR10_cds_20101214_updated) and Arabidopsis lyrata (from Ensembl Plant, link: ftp://ftp.ensemblgenomes.org/pub/release-46/plants/fasta/arabidopsis_lyrata/cds/) using the BLAST+ package.14 The best-matched pairs of CDSs in 2 species are regarded as orthologous genes. For each polymorphic site in A thaliana, the orthologous site was extracted from the orthologous gene of A lyrata.
If the nucleotide in A lyrata is the same as the reference allele of A thaliana (most cases), then DAF is the alternative allele frequency.
If the nucleotide in A lyrata is the same as the alternative allele of A thaliana, then DAF is the reference allele frequency.
If the nucleotide in A lyrata does not match either the reference or alternative allele in A thaliana (very few cases), then these SNP sites are discarded.
According to these classification criteria, the direction of mutation is from ancestral to derived. However, A lyrata may experience the same type of selection on synonymous changes. It should be mentioned that using only one species as an outgroup does not provide a certain ancestral inference. More closely related species could be used in future analyses to give a more accurate ancestral inference.
The allele counts used to calculate allele frequencies are provided in the SNP file. The vcf format contains the position and mutation-type columns as well as the “info” column. The “info” column includes information of annotation (genic or intergenic, coding or noncoding, missense or synonymous) and allele counts (the reference and alternative allele counts from the reads mapped to this position).
Classification of synonymous mutations
Synonymous mutations were classified into 3 categories according to whether they increase (from A/T to G/C), decrease (from G/C to A/T), or maintain (the remaining) the GC content. Among the 12 types of mutations, A-to-C, T-to-C, A-to-G, and T-to-G increase the GC content. C-to-A, C-to-T, G-to-A, and G-to-T decrease the GC content. C-to-G, G-to-C, A-to-T, and T-to-A do not affect the global GC content.
Regression analysis
The regression analysis was performed using “lm(Y ~ X1 + X2 + . . . + Xn)” in the R platform (http://www.R-project.org/). Y is the DAF of each synonymous mutation; X1 is the change of CAI value caused by the synonymous mutation (from −1 to 1); X2 is the change of tAI value (from −1 to 1); X3 is whether the mutation increase CG content (1 = increase, −1 = decrease, and 0 = unchanged); X4 is the GC content of host gene (normalized to −1 to 1 by a “B = 2*A−1” transfer); X5 is whether the SNP site is immediately upstream/downstream or exactly at a methylation site (1 = yes and −1 = no; if yes, that means this SNP may affect the methylation status); X6 is the MFE (per Kb) of host genes (normalized to −1 to 1), which was calculated by software RNAfold15 with default parameters. Lower MFE values indicated a more stable secondary structure. As MFE was directly affected by the length of sequences, we used MFE per Kb to cancel the length effect. The regression analysis will provide us an estimate of the relative contribution of X1, X2, . . ., Xn to Y, termed regression coefficient. Note that X1, X2, . . ., Xn all range from −1 to 1, so that their regression coefficients are comparable.
Codon adaptation index11 and tAI12 were defined by early studies, which were the parameters for codon bias and described the synonymous codon preference and tRNA availability of each codon. My group and other group(s) have previously done works investigating the selection patterns on CAI and tAI16-18 and the calculations were performed with the same pipeline. I folded the CDSs of A thaliana using RNAfold.15 The MFE value of each CDS sequence was extracted from the output file. The MFE per kilobase was calculated for each CDS.
Statistical analysis
All statistical analyses and graphic work were conducted in the R environment (http://www.R-project.org/). When comparing 2 sets of mutations (eg, missense vs synonymous mutations), if synonymous mutations have a globally higher DAF spectrum than missense mutations, then this would indicate a stronger purifying selection on missense mutations. In other words, synonymous mutations are “less harmful” (more advantageous) than missense mutations. Likewise, when comparing different sets of synonymous mutations, the group with higher DAF is likely to be advantageous and positively selected. The statistical tests (comparing DAF values) could be the non-parameter tests like Wilcoxon rank sum tests.
Results and Discussion
Variations in the 1135 natural inbred lines of Arabidopsis thaliana
A total number of 11 609 631 SNP sites and 1 271 972 indels are included in the vcf file that I have downloaded (see Materials and Methods section). According to the annotation provided in the vcf file, 1 135 084 SNPs are missense (nonsynonymous), 795 623 SNPs are synonymous, and 27 813 SNPs are nonsense mutations (that introduce in-frame stop codons in the main CDS). Apart from the mutations in the coding region, 319 647 SNPs take place in 5′ UTR and 465 647 SNPs are located in 3′ UTR. The remaining variations are non-exonic, including intronic and intergenic variations.
Synonymous mutations that increase GC content have higher DAF
Synonymous mutations were classified into 3 categories (see Materials and Methods section). I first looked at the fractions of the 12 types of mutations (Figure 1A). The most prevalent mutation type is C-to-T and G-to-A. This is reasonable as transitions take place more frequently than transversions. Next, I defined the DAF using the information provided in the SNP files and an outgroup (see Materials and Methods section). I found that those synonymous mutations that increase the GC content (from A/T to G/C) have significantly higher DAF than other synonymous mutations (Figure 1B, Wilcoxon rank-sum tests). This indicates that these “optimal” mutations are likely to be positively selected. To make the relationship between mutation and selection clearer, I tested the Spearman correlation between the fractions shown in Figure 1A and the median DAF plotted in Figure 1B (each of the 12 boxes in Figure 1B has a median value plotted horizontally in the boxes). No correlation was observed between these 2 aspects (Figure 1C), suggesting that my observed pattern on DAF might not be caused by the mutation bias.
Figure 1.

Landscape of synonymous mutations among the 1135 lines. The mutations that increase the GC content (from A/T to G/C) are labeled in orange. The mutations that decrease the GC content (from G/C to A/T) are labeled in purple. (A) Fractions (Y-axis, frequencies) of the 12 mutation types among all derived synonymous mutations. Here, for example, for A-to-C mutations, fraction(A-C) = number of A-C mutations/number of total mutations (B) derived allele frequencies (DAFs) of the 12 mutation types. P values are calculated from Wilcoxon rank-sum tests. (C) Spearman correlation between the fraction (X-axis, the fraction mentioned in Figure 1A) and median DAF (as shown in Figure 1B) of the 12 mutation types.
Relative contribution of different features to the frequency spectrum
To better decipher the relative contribution of different features to the observed allele frequency spectrum of synonymous mutations, I perform multiple regression analysis: Y ~ X1 + . . . + Xn (see Materials and Methods section). First, I listed multiple variables that might potentially affect the DAF spectrum, for example, whether the mutation increases CG content (1 = increase, −1 = decrease, and 0 = unchanged), the GC content of host genes, CAI, tAI, methylation status, and folding energy. It is clearly shown that “whether the mutation increase CG content” is correlated with the DAF of synonymous mutations (Figure 2A) and the changes of CAI/tAI caused by the mutation also contributed positively to the DAF spectrum (Figure 2B).
Figure 2.

Regression analysis of relative contribution of different features to the DAF spectrum. (A) Scatterplot of CG content of host genes (X-axis) and changes in CG status (Y-axis) against DAF (Z-axis). (B) Scatterplot of changes in CAI (X-axis) and tAI (Y-axis) against DAF (Z-axis). (C) Regression coefficients in the analysis. CAI indicates codon adaptation index; DAFs, derived allele frequencies; tAI, tRNA adaptation index.
In the multiple regression analysis, the regression coefficients showed us that CAI, tAI, and the change in CG content have a remarkably larger contribution to the frequency spectrum compared to other features (Figure 2C). This indicates a role of GC content in determining the frequency spectrum. It could also be inferred from this result that the selection patterns on CG content itself might be related to the CAI and tAI parameters. Note that the contribution of MFE to DAF is negative, suggesting that the synonymous mutations on genes with lower MFE (stronger structure) tend to have higher DAF. One speculation is that the “more structured” genes have already experienced many structural changes during evolution so that the newly emerged structural changes on them tend to be less harmful. At this stage, this observation is correlative, and the detailed reason remains unexplored. One certain thing is that the increase in CG content positively contributes to the DAF spectrum.
No correlation is observed between Fst and the prevalence of optimal synonymous mutations
It is necessary to discuss and test whether the observed pattern on synonymous DAF is affected (or caused) by population structures. Fst values describe the difference in allele frequencies among populations from different locations. Higher Fst values might indicate higher divergence across different natural populations (Figure 3A). My concern is (1) if the prevalence of optimal mutations is correlated (no matter positive or negative correlation) with Fst values, then when I look at the DAF I should also consider the Fst values. The putative solution is to divide the genome into several groups with high/medium/low Fst values and then test the pattern (which has been done in Figure 1) within each Fst group. (2) If the prevalence of optimal mutations is not correlated with Fst, then that means the pattern observed in Figure 1 is not affected by the population structure.
Figure 3.
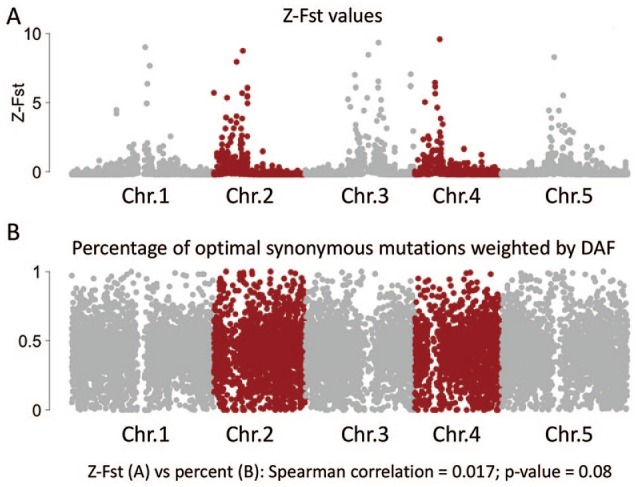
No correlation is observed between Fst and the prevalence of optimal mutations. (A) Z-Fst values with 10 000 window size along the chromosomes. The Fst values are scaled by Z-score. (B) Fraction of optimal synonymous mutations (among all synonymous mutations) weighted by DAF. The fractions are plotted with 10 000 window size along the chromosomes. The Spearman correlation between Fst and this fraction is insignificant. DAF indicates derived allele frequency.
With the same window size (10 000 bp) of Fst values provided by the 1001 genome project (see Materials and Methods section), I calculated the fraction of “optimal” synonymous mutations (among all synonymous mutations) weighted by DAF in each 10 000 bp window (Figure 3B; here “optimal” refers to A/T to G/C synonymous mutations). I intended to use this parameter to measure the prevalence of optimal synonymous mutations within each window. Higher fractions might indicate a stronger positive selection on optimal synonymous mutations (Figure 3B). However, the genome-wide Fst shown in Figure 3A and the prevalence of optimal mutations in Figure 3B are not correlated even under such a huge sample size (Figure 3). This result might exclude the possibility that the higher DAF observed on optimal synonymous mutations (Figure 1B) is affected (or caused) by population structures.
The biased DAF spectrum is observed in all representative subpopulations
To further prove the robustness of the observed DAF spectrum of different synonymous mutations, I set out to test this pattern in different subpopulations. The admixture groups defined by the original work13 were retrieved (Figure 4A). I chose 3 representative subpopulations with adequate accessions with alternative alleles (Figure 4A and B). To correct the direction of mutations by using the outgroup A lyrata, I further discarded the few sites with derived reference alleles so that the alternative alleles of those remaining sites are derived alleles. In the 3 representative subpopulations, the DAF distributions of different synonymous mutation types are illustrated (Figure 4B). Those synonymous mutations that increase the GC content (orange) have remarkably higher DAF than others. Notably, this pattern appears to be stronger in the “Italy/Balkan/Caucasus” subpopulation (Figure 4B), which is possibly due to the mixed origins of these accessions. Although the reason for different selection strength across subpopulations remains speculative and uncertain, what is clear is the global pattern that the synonymous mutations that increase the GC content have higher DAF.
Figure 4.

The DAF spectrum is consistent across different subpopulations. (A) The map labeled with sample collection information (from Figure 5A of Alonso-Blanco).13 (B) DAF spectrum of the 12 mutation types in 3 representative subpopulations. The name of the subpopulation is colored with the same color shown in panel A. DAF indicates derived allele frequency.
As mentioned in the Introduction section, the advantage of these optimal synonymous mutations (from A/T to G/C) is likely conferred by the faster translation rate during tRNA decoding. The proposed advantage might act as the selection force shaping the DAF spectrum of synonymous mutations. Nevertheless, the pure evolutionary analyses in this study did not include any functional tests. The observed differences in DAF of those mutations serve as evidence to speculate the advantage of the optimal synonymous mutations. At this stage, the link between the biased DAF and function remains to be explored.
Conclusions
By analyzing the frequency spectrum of the synonymous mutations, I draw to the conclusion that synonymous mutations in natural populations are not strictly neutral: the synonymous mutations that increase GC content (from A/T to G/C) tend to have higher DAF and therefore are likely to be positively selected.
Acknowledgments
I thank all members of my Lab for their constructive suggestions to this project.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was financially supported by the National Natural Science Foundation of China (Grant no. 31770213). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: LW designed and supervised this research. LW analyzed the data and wrote this article.
ORCID iD: Lai Wei  https://orcid.org/0000-0003-1000-7559
https://orcid.org/0000-0003-1000-7559
Data Accessibility: All data used in this study have been described in the Materials and Methods section, which are free to access.
References
- 1. Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12:32-42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chu D, Wei L. Nonsynonymous, synonymous and nonsense mutations in human cancer-related genes undergo stronger purifying selections than expectation. BMC Cancer. 2019;19:359. doi: 10.1186/s12885-019-5572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chu D, Wei L. Parsing the synonymous mutations in the maize genome: isoaccepting mutations are more advantageous in regions with codon co-occurrence bias. BMC Plant Biol. 2019;19:422. doi: 10.1186/s12870-019-2050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Supek F, Minana B, Valcarcel J, Gabaldon T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324-1335. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 5. Zhang YC, Chen ZY, Zhang LJ, Jiang P, Li W. N-6-methyladenosine could indirectly modulate translation in human cancer cells via cis-elements. Transl Cancer Res. 2019;8:1931-1938. doi: 10.21037/tcr.2019.09.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harrison RJ, Charlesworth B. Biased gene conversion affects patterns of codon usage and amino acid usage in the saccharomyces sensu stricto group of yeasts. Mol Biol Evol. 2011;28:117-129. doi: 10.1093/molbev/msq191. [DOI] [PubMed] [Google Scholar]
- 7. Comeron JM. Selective and mutational patterns associated with gene expression in humans: influences on synonymous composition and intron presence. Genetics. 2004;167:1293-1304. doi: 10.1534/genetics.104.026351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dana A, Tuller T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014;42:9171-9181. doi: 10.1093/nar/gku646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu CH, Dang Y, Zhou Z, et al. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell. 2015;59:744-754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chu D, Wei L. Human cancer cells compensate the genes unfavorable for translation by N-6-methyladenosine modification and enhance their translation efficiency. Transl Cancer Res. 2019;8:499-508. doi: 10.21037/tcr.2019.03.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharp PM, Li WH. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281-1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036-5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alonso-Blanco C. 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell. 2016;166:481-491. doi: 10.1016/j.cell.2016.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429-3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chu D, Wei L. Characterizing the heat response of Arabidopsis thaliana from the perspective of codon usage bias and translational regulation. J Plant Physiol. 2019;240:153012. doi: 10.1016/j.jplph.2019.153012. [DOI] [PubMed] [Google Scholar]
- 17. Chu D, Wei L. Reduced C-to-U RNA editing rates might play a regulatory role in stress response of Arabidopsis. J Plant Physiol. 2020;244:153081. doi: 10.1016/j.jplph.2019.153081. [DOI] [PubMed] [Google Scholar]
- 18. Sabi R, Tuller T. Modelling the efficiency of Codon-tRNA interactions based on codon usage bias. DNA Res. 2014;21:511-526. doi: 10.1093/dnares/dsu017. [DOI] [PMC free article] [PubMed] [Google Scholar]