

Abstract
Butenolides are well‐known signaling molecules in Gram‐positive bacteria. Here, we describe a novel class of butenolides isolated from a Gram‐negative Pseudomonas strain, the styrolides. Structure elucidation was aided by the total synthesis of styrolide A. Transposon mutagenesis enabled us to identify the styrolide biosynthetic gene cluster, and by using a homology search, we discovered the related and previously unknown acaterin biosynthetic gene cluster in another Pseudomonas species. Mutagenesis, heterologous expression, and identification of key shunt and intermediate products were crucial to propose a biosynthetic pathway for both Pseudomonas‐derived butenolides. Comparative transcriptomics suggests a link between styrolide formation and the regulatory networks of the bacterium.
Keywords: biosynthesis, butenolides, natural products, Pseudomonas
Diversity‐oriented biosynthesis: Styrolides are a novel class of bacterially produced butenolides that represent a structural and biosynthetic link between tetronic acids and γ‐butyrolactones. A comprehensive picture of the biosynthesis of the styrolides and related acaterin is provided.
Bacteria of the genus Pseudomonas are prolific producers of natural products. Together with actinobacteria, myxobacteria, and Bacillus species, their secondary metabolites span a huge space of both structural and functional molecular diversity.1 Gram‐negative pseudomonads can colonize virtually any habitat, and their remarkable ability to adapt to different environments is mirrored and often caused by their biosynthetic capabilities.2 Here, we shed light on a class of molecules that is typically associated with Gram‐positive bacteria, the butenolides. Similarly to γ‐butyrolactone autoregulators,3 butenolides can function as signaling molecules in streptomycetes,4 yet their occurrence in Gram‐negative bacteria is scarce, and both their function and biosynthesis are poorly understood.
We isolated Pseudomonas fluorescens HKI0874 from forest soil, and its cultivation led to the production of two fluorescent compounds with a UV absorption spectrum indicative of extended conjugated π‐systems. Both compounds were sensitive to light as well as elevated temperatures and decomposed readily. Large‐scale fermentation allowed the isolation of two compounds (see the Supporting Information for experimental details) with pseudo‐molecular masses of m/z=217.0860 and 215.0705, consistent with the molecular formulae C13H12O3 and C13H10O3. NMR spectroscopy allowed the structure elucidation of two previously unknown butenolides with a 4‐hydroxystyrene and a butenolide moiety, styrolide A (1) and styrolide B (2; Figure 1 A and Tables S1 and S2 in the Supporting Information).
Figure 1.
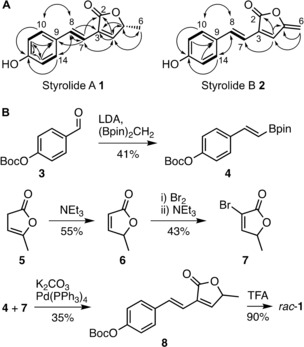
A) Chemical structures of styrolide A (1) and styrolide B (2) with key correlations observed by 2D NMR spectroscopy. Bold lines: 1H‐1H COSY correlations; solid arrows: HMBC correlations. B) Total synthesis of styrolide A (1). Enantiomers of rac‐1 were separated by HPLC on a chiral stationary phase. Boc=tert‐butoxycarbonyl, LDA=lithium diisopropyl amide, (Bpin)2CH2=bis[(pinacolato)boryl]methane, TFA=trifluoroacetic acid.
We synthesized styrolide A (1) to support the structure elucidation and to determine its absolute configuration. Styrene boronic ester 4 was accessed by a boron‐Wittig reaction of Boc‐protected 4‐hydroxybenzaldehyde (3).5 Bromo‐butenolide 7 was obtained by isomerization of α‐angelica lactone 5 and subsequent bromination.6 We coupled boronic ester 4 and bromo‐butenolide 7 under Suzuki conditions.7 Deprotection of coupling product 8 with trifluoroacetic acid yielded racemic styrolide A (rac‐1; Figure 1 B). The spectroscopic data matched that of isolated styrolide A (1; see the Supporting Information). We separated the enantiomers of rac‐1 by HPLC on chiral stationary phase and performed ECD spectroscopy on both enantiomers. Comparison with ECD spectra predicted by DFT allowed us to assign the probable configuration of each enantiomer (Figure S4). Isolated styrolide A (1) had the same retention time as R‐configured synthetic styrolide A (Figure S5). Importantly, the configuration of the C5 atom of styrolide A is identical to that of another Pseudomonas‐derived butenolide, acaterin (9; Figure 3 B).8 Furthermore, styrolide B (2) shares the butenolide exo‐methylene moiety with 4,5‐didehydroacaterin (10). These facts strongly suggest that the biosyntheses of styrolides and acaterins may share the same logic. The latter has remained elusive because of inconsistent biochemical studies and no knowledge of its biosynthetic gene cluster (BGC).9
Figure 3.
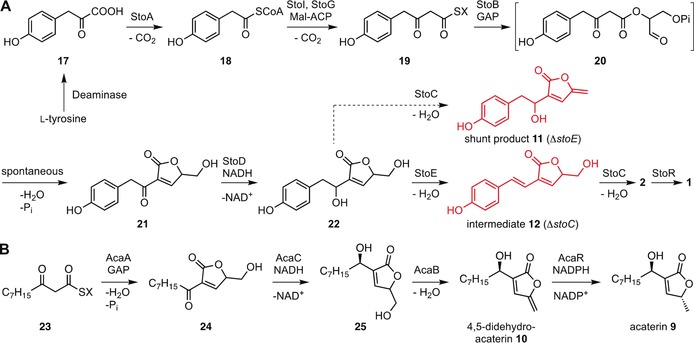
A) Proposed styrolide biosynthesis with isolated shunt product 11 and intermediate 12 highlighted in red. B) Proposed biosynthetic pathway of acaterins 9 and 10. Mal‐CoA=Malonyl‐CoA, GAP=glyceraldehyde‐3‐phosphate, X=ACP, CoA, StoF, or StoH.
In order to find the styrolide BGC, we sequenced the genome of HKI0874 (Illumina HiSeq, 6.96 Mbp, 130 contigs). Yet, genome analysis using antiSMASH 5.010 did not provide any likely BGC. Thus we generated a Tn5‐transposon11 library and screened 2300 Tn5‐mutants using a thin‐layer chromatography (TLC)‐based detection system. Two mutants did not produce fluorescent styrolide B 2 (Figure S6), and LC‐MS analysis confirmed the absence of both styrolides 1 and 2 (Figure S8). The transposon insertion sites determined by HiTAIL PCR12 were found in close proximity to each other on the bacterial chromosome (Figure 2, red arrows). To delineate the styrolide BGC and to identify essential genes, we systematically deleted genes in the vicinity of the insertion sites and heterologously expressed different sets of genes in a P. protegens strain. Impaired or abolished production of styrolide B 2 confirmed that stoB, stoC, stoD, stoE, and stoG are essential for the production in the native host (Figure S8). Styrolide production in the heterologous host, however, required stoA to stoI (Figure S27), which may be due to the absence of an alternative metabolic pathway providing a required biosynthetic precursor.
Figure 2.
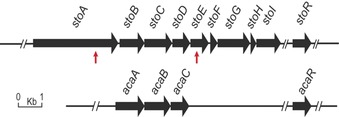
Styrolide BGC (top) with red arrows indicating transposon insertion sites of two Tn5‐mutants and BGC of acaterins (bottom). Acaterin (9) formation additionally requires 4,5‐didehydroacaterin reductase AcaR. StoB, StoC, StoD, and stoR are homologous to AcaA, AcaB, AcaC, and AcaR, respectively.
To propose a biosynthesis of the styrolides, we analyzed the gene deletion mutants for the production of biosynthetic intermediates or shunt products. The strains ΔstoC and ΔstoE produced two different compounds with identical m/z=233 [M+H]+ values, yet with different HPLC retention times and UV spectra (Figure S10). As we speculated these compounds to be different hydration products of styrolide B (2), we computed the UV spectra of all likely candidates 11–16 (Figures S11–S24, Tables S12–S19). Comparison of predicted and observed UV maxima supported our hypothesis and facilitated the isolation of compounds 11 and 12, whose structures were validated by NMR and HRMS analysis (Figures 3 A, S25, and S26, Tables S20 and S21). Both hydration products were crucial in order to understand the ultimate steps of the styrolide biosynthesis. However, the first steps remained elusive. Detailed analysis of structurally related acaterin proved to be key to elucidating the styrolide biosynthesis.
We sequenced the genome of P. jessenii EC‐S101 (Illumina HiSeq, 6.23 Mbp, 35 contigs), a reported producer of acaterins 9 and 10.13 A homology search in this strain using StoB, StoC, and StoD as bait identified similar genes (Table S8). An additional genus‐wide homology search of complete Pseudomonas genomes showed the prevalence of these genes also in other Pseudomonas strains isolated from plants and soil (Figure S7). Importantly, the gene synteny was identical in all genomes, a strong indicator that we had identified BGCs related to the styrolide BGC. Hence, we deleted the candidate genes in P. jessenii EC‐S101. Gene deletion mutants ΔacaA, ΔacaB, and ΔacaC produced neither 9 nor 10 (Figure S9). Thus, we had identified the previously unknown acaterin BGC (Figure 2, bottom) and showed that both acaterins and the styrolides share the same biosynthetic logic. Importantly, heterologous expression of acaA, acaB, and acaC furnished 4,5‐didehydroacaterin (10). Expression of acaA to acaC in conjunction with the reported 4,5‐didehydroacaterin reductase acaR, which is neither located within the acaterin BGC, nor in its proximity,14 additionally yielded acaterin 9 (Figure S28).
Whereas previous studies had clearly shown that a C3 building block is required for the biosynthesis of acaterin, the nature of this precursor was inconclusive.15 Recent reports on the biosynthesis of the Streptomyces‐derived γ‐butyrolactone A‐factor3 and different tetronic acids, for example, RK‐68216 or agglomerin A,17 render the previously proposed acaterin biosynthesis unlikely. The most parsimonious biosynthetic route would utilize glyceraldehyde‐3‐phosphate (GAP) as the common C3 building block, which is condensed with an activated 3‐oxo thioester in both the acaterin as well as the styrolide biosynthesis. Importantly, we excluded tetronic acid intermediates in both the acaterin and styrolide biosynthesis as neither of the two BGCs provides genes coding for enzymes required for tetronic acid reduction.
Hence, we suggest that styrolide biosynthesis (Figure 3 A) starts with the conversion of tyrosine‐derived 4‐hydroxyphenylpyruvic acid (17) into activated 4‐hydroxyphenylacetic acid 18 by the action of StoA, a putative pyruvate‐flavodoxin oxidoreductase (Table S7). Thioester 18 is extended by a C2 group using a malonyl unit (e.g., malonyl‐ACP). This reaction is most likely carried out by StoI, a putative 3‐oxoacyl‐ACP synthase III (ACP=acyl carrier protein), and yields ACP‐bound intermediate 19 (Figure 3, X=ACP). StoG, a putative AMP‐dependent synthetase/ligase, was essential for styrolide biosynthesis. It seems plausible that this enzyme converts an ACP‐bound intermediate into the corresponding CoA‐bound one (Figure 3 A, X=CoA) or into another ACP‐bound intermediate 19 (Figure 3 A, X=StoF or StoH, both belong to the ACP‐like superfamily, Table S7). This may or may not proceed via the intermediacy of a 3‐oxo fatty acid. The key step is then carried out by the A‐factor‐synthase‐like StoB (29.5 % identity with AfsA3), catalyzing the esterification of thioester 19 with GAP. Analogously to the A‐factor biosynthesis,3 a spontaneous Knoevenagel condensation of ester 20 would yield ketone 21, which is then reduced by the predicted short‐chain reductase StoD to allylic alcohol 22. The latter is dehydrated to intermediate 12 by StoE, a putative short‐chain dehydrogenase/reductase. This agrees with our observation that ΔstoE produces shunt product 11, bearing the same exo‐methylene motif as styrolide B (2). Eventually, this exo‐methylene group is installed by StoC, a putative NADH flavin oxidoreductase to yield styrolide B (2). A homologue of the 4,5‐didehydroacaterin reductase AcaR,14b StyR, or possibly also other reductases would then reduce highly unstable and reactive styrolide B (2) to styrolide A (1). Overall, we have strong evidence that glycerol, acetate, and amino acid building blocks are combined during styrolide biosynthesis.
The generation of acaterin (9) in principle represents a shorter variation of the styrolide biogenesis requiring four steps. The shared biosynthetic logic between the two related butenolides further substantiates our proposed biosynthesis. Acaterin biosynthesis commences with the AcaA‐mediated esterification of 3‐oxo thioester 23 derived from fatty acid metabolism, and GAP. The subsequent Knoevenagel condensation furnishes ketone 24 (Figure 3 B). Importantly, AcaA and StoB are homologous and carry out the same type of reaction (Tables S8 and S9). AcaC, a homologue of StoD, reduces 24 to allylic alcohol 25. AcaB, a StoC homologue, installs the exo‐methylene group to furnish 10. The latter is reduced to 9 by the reductase AcaR.
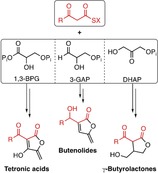
Figure 4 highlights the various biosynthetic strategies available to access five‐membered butenolides, which all rely on condensation of an activated 3‐oxo fatty acid with a C3 building block. Different variants of the latter, all derived from glycolysis, can thus alter the substitution pattern (butenolides vs. γ‐butyrolactones) of the C5 unit or its oxidation level (tetronic acids vs. butenolides/γ‐butyrolactones).
Figure 4.
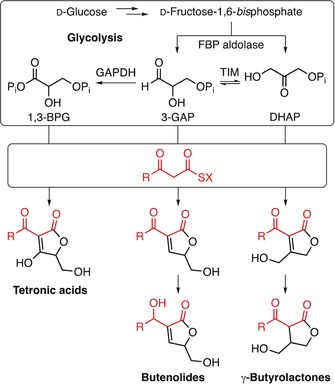
Comparison of the biosynthetic pathways of tetronic acids, butenolides, and γ‐butyrolactones. Different variants of glycolysis‐derived C3 building blocks are condensed with an activated 3‐oxo fatty acid precursor. Additional tailoring enzymes may lead to further diversification.
With an understanding of the styrolide biosynthesis, we investigated their potential function as signaling molecules in this Pseudomonas strain. As basic phenotypic analyses for growth and swarming ability (Figures S30 and S31) did not reveal major differences between the wildtype (WT) and the styrolide‐deficient mutant ΔstoE, we performed a comparative transcriptome analysis (WT vs. ΔstoE). We categorized differentially expressed genes according to their predicted functions (Figure S29). The largest category of genes affected by the absence of styrolides were those involved in amino acid metabolism and transport into the cell. We also observed differential gene expression associated with genes linking primary metabolism with secondary metabolism. Thus, styrolides may play a critical role in orchestrating the supply of building blocks for secondary metabolite biosynthesis, which is currently under investigation.
Overall, we have identified two new butenolides, styrolides A (1) and B (2), from a Gram‐negative P. fluorescens strain. We provide a four‐step synthesis of styrolide A (1) and propose a biosynthesis for styrolides and acaterin based on mutagenesis experiments, isolation of shunt/intermediate products, and heterologous expression of both BGCs. Importantly, we identified three homologous biosynthetic enzymes shared between the sto and aca BGC, with identical gene synteny. A glycerol, an acetate, and an amino acid derived building block are thus combined during styrolide biosynthesis, whereas acaterin is derived from a glycerol and a 3‐oxo fatty acid precursor. This work shows that the class of butenolides, which is well known in Gram‐positive bacteria, is also featured in Gram‐negative pseudomonads.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. Hashidoko for sharing P. jessenii EC‐S101. We thank A. Perner and H. Heinecke (HKI Jena) for MS and NMR measurements. The Biopilot Plant team (HKI Jena) is acknowledged for their support. We are grateful for financial support from the Leibniz Association, the Aventis Foundation (PhD fellowship to M.K.), the Deutsche Forschungsgemeinschaft (DFG) STA1431/2‐1 and CRC1127 ChemBioSys, and the Dr. Illing foundation.
M. Klapper, K. Schlabach, A. Paschold, S. Zhang, S. Chowdhury, K.-D. Menzel, M. A. Rosenbaum, P. Stallforth, Angew. Chem. Int. Ed. 2020, 59, 5607.
References
- 1. Gross H., Loper J. E., Nat. Prod. Rep. 2009, 26, 1408–1446. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Silby M. W., Winstanley C., Godfrey S. A. C., Levy S. B., Jackson R. W., FEMS Microbiol. Rev. 2011, 35, 652–680; [DOI] [PubMed] [Google Scholar]
- 2b. Klapper M., Götze S., Barnett R., Willing K., Stallforth P., Angew. Chem. Int. Ed. 2016, 55, 8944–8947; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9090–9093; [Google Scholar]
- 2c. Klapper M., Braga D., Lackner G., Herbst R., Stallforth P., Cell Chem. Biol. 2018, 25, 659–665; [DOI] [PubMed] [Google Scholar]
- 2d. Arp J., Götze S., Mukherji R., Mattern D. J., Garcia-Altares M., Klapper M., Brock D. A., Brakhage A. A., Strassmann J. E., Queller D. C., Bardl B., Willing K., Peschel G., Stallforth P., Proc. Natl. Acad. Sci. USA 2018, 115, 3758–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kato J. Y., Funa N., Watanabe H., Ohnishi Y., Horinouchi S., Proc. Natl. Acad. Sci. USA 2007, 104, 2378–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Arakawa K., Tsuda N., Taniguchi A., Kinashi H., ChemBioChem 2012, 13, 1447–1457; [DOI] [PubMed] [Google Scholar]
- 4b. Wang W. X., Zhang J. H., Liu X., Li D., Li Y., Tian Y. Q., Tan H. R., J. Biol. Chem. 2018, 293, 20029–20040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coombs J. R., Zhang L., Morken J. P., Org. Lett. 2015, 17, 1708–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Lindström M., Hedenström E., Bouilly S., Velonia K., Smonou I., Tetrahedron: Asymmetry 2005, 16, 1355–1360; [Google Scholar]
- 6b. Ochoa de Echagüen C., Ortuño R. M., Tetrahedron 1994, 50, 12457–12462. [Google Scholar]
- 7.
- 7a. Mathews C. J., Taylor J., Tyte M. J., Worthington P. A., Synlett 2005, 538–540; [Google Scholar]
- 7b. Vasamsetty L., Khan F. A., Mehta G., Tetrahedron 2015, 71, 3209–3215. [Google Scholar]
- 8.
- 8a. Ishigami K., Kitahara T., Tetrahedron 1995, 51, 6431–6442; [Google Scholar]
- 8b. Naganuma S., Sakai K., Hasumi K., Endo A., J. Antibiot. 1992, 45, 1216–1221. [DOI] [PubMed] [Google Scholar]
- 9. Vieweg L., Reichau S., Schobert R., Leadlay P. F., Süssmuth R. D., Nat. Prod. Rep. 2014, 31, 1554–1584. [DOI] [PubMed] [Google Scholar]
- 10. Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S. Y., Medema M. H., Weber T., Nucleic Acids Res. 2019, 310, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Lorenzo V., Herrero M., Jakubzik U., Timmis K. N., J. Bacteriol. 1990, 172, 6568–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y.-G., Chen Y., BioTechniques 2007, 43, 649–656. [DOI] [PubMed] [Google Scholar]
- 13. Hatano E., Hashidoko Y., Deora A., Fukushi Y., Tahara S., Biosci. Biotechnol. Biochem. 2007, 71, 1601–1605. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Oinaka H., Nakano S., Sakane W., Kudo F., Tadashi E., Fujimoto Y., J-STAGE 2006, 48, 241–246; [Google Scholar]
- 14b. Nakano S., Sakane W., Oinaka H., Fujimoto Y., Bioorg. Med. Chem. 2006, 14, 6404–6408. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Sekiyama Y., Araya H., Hasumi K., Endo A., Fujimoto Y., Tetrahedron Lett. 1998, 39, 6233–6236; [Google Scholar]
- 15b. Sekiyama Y., Fujimoto Y., Hasumi K., Endo A., Tetrahedron Lett. 1999, 40, 4223–4226. [Google Scholar]
- 16. Sun Y., Hahn F., Demydchuk Y., Chettle J., Tosin M., Osada H., Leadlay P. F., Nat. Chem. Biol. 2010, 6, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kanchanabanca C., Tao W., Hong H., Liu Y., Hahn F., Samborskyy M., Deng Z., Sun Y., Leadlay P. F., Angew. Chem. Int. Ed. 2013, 52, 5785–5788; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5897–5900. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary