

Abstract
Background
Although Huntington's disease (HD) is caused by a single dominant gene, it is clear that there are genetic modifiers that may influence the age of onset and disease progression.
Objectives
We sought to investigate whether new inflammation‐related genetic variants may contribute to the onset and progression of HD.
Methods
We first used postmortem brain material from patients at different stages of HD to look at the protein expression of toll‐like receptor 4 (TLR4) and triggering receptor expressed on myeloid cells 2 (TREM2). We then genotyped the TREM2 R47H gene variant and 3 TLR4 single nucleotide polymorphisms in a large cohort of HD patients from the European Huntington's Disease Network REGISTRY.
Results
We found an increase in the number of cells expressing TREM2 and TLR4 in postmortem brain samples from patients dying with HD. We also found that the TREM2 R47H gene variant was associated with changes in cognitive decline in the large cohort of HD patients, whereas 2 of 3 TLR4 single nucleotide polymorphisms assessed were associated with changes in motor progression in this same group.
Conclusions
These findings identify TREM2 and TLR4 as potential genetic modifiers for HD and suggest that inflammation influences disease progression in this condition. © 2019 International Parkinson and Movement Disorder Society
Keywords: cognitive decline, Huntington, inflammation, motor symptoms, TLR4, TREM2
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by a CAG trinucleotide expansion in exon 1 of the Huntingtin (HTT) gene,1 which presents with a combination of motor, cognitive, and psychiatric deficits. Despite its clear genetic basis, HD patients show variable ages of onset (AoO) and progression rate, and although CAG repeat length has been shown to correlate with the AoO of motor signs,2, 3, 4, 5, 6, 7, 8, 9, 10 this has been of limited clinical use in predicting AoO for an individual.11 Moreover, patients with similar initial clinical presentations can follow very different clinical courses,12 with variable rates of disease progression that are poorly correlated to CAG repeat length. Hence, the CAG repeat size alone is not sufficient to reliably predict disease onset and progression, and thus there is a need to better define what other factors impact on these 2 aspects of HD.13, 14
Previously, we have shown that tau has such an influence,15 whereas others have reported on a number of other genetic factors that impact on these features of HD. For instance, an abnormal, but relatively short, CAG expansion leading to HD, with a relatively long CAG track in the wild‐type allele has been shown to correlate with more severe clinical features and pathology.16 Genetic polymorphisms adjacent to the CAG repeats have also been shown to influence disease onset3, 4, 10, 17, 18 as have genes related to DNA repair.19, 20, 21
In addition, inflammation has now been shown to be important in many chronic neurodegenerative disorders of the brain such as Alzheimer's disease (AD)22 and Parkinson's disease23 as well as HD.24 The evidence for a key role of inflammation to HD comes from studies looking at microglial activation on imaging and pathologically25 as well as peripheral cytokine profiles,26 which can be found early on in the disease. Inflammation‐related genetic modifiers have also been shown to influence the risk of developing neurodegenerative disorders such as sporadic AD, and this includes the triggering receptor expressed on myeloid cells 2 (TREM2)27, 28, 29, 30, 31, 32, 33 and toll‐like receptor 4 (TLR4).34, 35, 36 We therefore sought to investigate this in HD using both postmortem studies and clinical data from a large cohort of patients. Specifically, we took advantage of tissue microarrays (TMAs) to assess the expression of these proteins in the striatum37 and then looked at TREM2 and TLR4 genetic variants/single nucleotide polymorphisms (SNPs) as genetic modifiers of disease progression in a large cohort of HD patients (N = 830) obtained from the European Huntington's Disease Network (EHDN).
Methods
Ethics Statement
The study was approved by the local research ethics committee and the other sites of the EHDN REGISTRY project.38 The participants and/or the next of kin gave informed written consent for the use of genetic material and brain tissue for research according to International Conference on Harmonisation ‐ Good Clinical Practice (ICH‐GCP) guidelines (http://www.ich.org/LOB/media/MEDIA482.pdf) and the Declaration of Helsinki.
Subjects
Human genetic material, clinical information, and CAG repeat length data were obtained from the EHDN REGISTRY38 (http://www.euro-hd.net/html/registry). In total, data were available from 830 patients who had a clinical and genetically confirmed diagnosis of HD (Table 1). AoO was defined as the age at which their first HD features appeared as judged by a trained neurologist either from the neurological examination or (more frequently) from the patient history as recorded in REGISTRY. Motor, functional, and cognitive features were scored at visits approximately a year apart using the Unified Huntington Disease Rating Scale (UHDRS'99).39 Cognitive assessments included tests of verbal fluency as well as the digit‐symbol modality and Stroop tests (word, color, and interference subtests), all of which are known to be sensitive to the disease process in HD.40
Table 1.
Demographic, Genotypic, and Clinical Characteristics of the European Huntington's Disease Network Huntington's Disease Cohort
| N | 830 |
|---|---|
| Gender, M:F | 413:417 |
| Agea | 50.98 (12.03) |
| CAG repeat length of the expanded allele | 44.27 (4.25) |
| Years since disease onseta | 2.10 (0.93) |
| UHDRS motor scoreb | 32.90 (20.34) |
| UHDRS functional scoreb | 8.42 (3.56) |
| Cognitive scoreb | 157.62 (72.77) |
Group means are shown with standard deviations in parentheses.
At enrollment.
At first visit. Annual change in cognitive performance was assessed based on a composite cognitive score, a sum of individual scores in the verbal fluency, the symbol digit, and all parts of the Stroop test (color, word, and interference). Rate of change (points/year) was calculated by subtracting cognitive score at the first assessment from the score at the last follow‐up assessment (or most complete data set) divided by the time between these assessments in years. The rate of change in motor decline was calculated using the total motor score from the UHDRS'99.
M, male; F, female; UHDRS, Unified Huntington's Disease Rating Scale.
Genotyping
SNP genotyping was undertaken using predesigned assays (Applied Biosystems, Warrington, UK) tagging the R47H variant of TREM2 (SNP: rs75932628) and 3 SNPs of the TLR4 gene (SNP: rs1927911, rs1927914, rs10116253) and run on a Quantstudio 7 Flex Real‐Time PCR System (ThermoFisher, Waltham, MA, USA), according to the manufacturer's instructions. To validate the results, 192 DNA samples randomly selected were regenotyped 3 times in triplicate without any inconsistencies observed among those samples.
Tissue Microarray Preparation
The Cambridge Brain Bank provided anonymous paraffin‐embedded tissue blocks from HD patients (N = 16 [n = 5 grade 3; n = 11 grade 4]) and age‐matched and sex‐matched controls (N = 9) known not to have any neurological or psychiatric disorders (Table 2). Striatal tissue was available for all cases. Demographic data were obtained from the Brain Bank. The pathological severity of HD was scored according to the Vonsattel grading system.41
Table 2.
Demographic Details of the Postmortem Brain Sample Cases
| HD Cases | Grade | Age | Sex |
|---|---|---|---|
| H614 | 3 | 42 | F |
| H659 | 3 | 43 | F |
| H679 | 3 | 51 | M |
| H700 | 3 | 57 | M |
| H709 | 3 | 79 | F |
| H665 | 4 | 70 | F |
| H669 | 4 | 53 | M |
| H671 | 4 | 72 | F |
| H682 | 4 | 40 | M |
| H692 | 4 | 43 | F |
| H693 | 4 | 26 | F |
| H707 | 4 | 39 | M |
| H710 | 4 | 43 | M |
| H718 | 4 | 65 | F |
| H720 | 4 | 68 | M |
| H725 | 4 | 58 | M |
| Mean ± SD | 53.1 ± 14.7 | ||
| Ratio F:M | 8:8 |
| Controls | Age | Sex |
|---|---|---|
| C568 | 69 | M |
| NP16.28 | 68 | M |
| NP16.59 | 60 | F |
| PT88 | 72 | M |
| PT129 | 50 | F |
| PT149 | 56 | M |
| PT151 | 76 | M |
| PT155 | 39 | F |
| PT172 | 45 | F |
| Mean ± SD | 59.4 ± 12.9 | |
| Ratio F:M | 4:5 |
HD, Huntington's disease; SD, standard deviation; F, female; M, male.
After a preliminary hematoxylin and eosin and luxol fast blue staining, all of the blocks were assessed by a neuropathologist to mark the putamen and caudate and sent to the Integrated Systems Engineering (ISENET, Milan, Italy) for TMA assembly.42 A semiautomated tissue array device (Galileo TMA CK4500 platform, ISENET, Milan, Italy) with a needle punch of diameter 0.2 mm was inserted into the marked areas of the donor block within the putamen. Different donor tissue cores were inserted into precored holes in a recipient paraffin wax block according to the array coordinates defined in the predetermined template.
Immunohistochemistry and Quantification
Immunohistochemistry was performed on 10‐μm thick sections from the TMAs (or single‐section slides in the case of cerebral cortex) using TREM2 and TLR4 antibodies and following standard protocols. Deparaffinized and rehydrated tissue sections were incubated overnight at 4°C with the following primary antibodies: mouse monoclonal anti‐TREM2 (1:200; Abcam, Toronto, Canada) and mouse monoclonal anti‐TLR4 (1:100; Abcam, Toronto, Canada). The labeling was revealed with the ABC Elite Vectastain Kit (Vector Laboratories, Peterborough, UK). The sections were then incubated for 2 hours at room temperature with the biotinylated secondary antibody (1:500; Vector Laboratories, Peterborough, UK) and, following washes in phosphate‐buffered saline, horseradish peroxidase Avidin‐D (Vector Laboratories, Peterborough, UK) was added for 1 hour at room temperature and visualized with 3‐3'diaminobenzidine as the chromogen. Controls included staining after omitting the primary antibody and were consistently negative for any staining.
Individual immunolabeled TMA sections were scanned on a Leica Aperio AT2 (Leica Biosystems, Buffalo Grove, IL, USA) at 20× magnification with a resolution of 0.5‐μm per pixel and visualized on ImageScope v12.4.0.7018 (Leica Biosystems, Buffalo Grove, IL, USA). Quantification was performed blinded to case identity. Both a positive cell detection and optical density analyses were performed using QuPath software56 (version 0.1.2). For analysis of the number of cells expressing each marker and the relative optical density in the cortex, 10 images of a 20× field of view per section were taken using a E600 epifluorescence microscope equipped with a DMX1200 digital camera driven by the Automatic Camera Tamer software (Nikon, Melville, NY, USA), and staining was analyzed using the Fiji image analysis software.57 The average value of all images per case was used for statistical analysis.
Statistical Analysis
A χ2 test was used to compare the allele frequency of each variant with that expected for a population in Hardy‐Weinberg equilibrium. Fisher's exact test was used to compare the distribution of genotypes. Only genotyped individuals for whom a complete data set was available for at least 2 visits, a minimum of 1 year apart, were included in the analysis,43, 44 as we have done previously in this cohort.15 Baseline demographic and clinical data were compared between groups using 2‐tailed t tests (2 groups) and analysis of Aariance (more than 2 groups). We first assessed the annual change in cognitive performance based on a composite cognitive score, a sum of individual scores on the verbal fluency and the symbol digit tests as well as all parts of the Stroop test (color, word, and interference). Rate of change (points/year) was calculated by subtracting the cognitive score at the first assessment from the score at the last follow‐up assessment (or most complete data set) divided by the time between these assessments in years, as we have done previously.15, 43, 44 We then measured the rate of change in motor decline, calculated using the total motor score from the UHDRS'99 collected at the same visits as described previously using an equivalent formula. Outliers were identified and the data were winsorized using Tukey's Hinge estimates. The Shapiro‐Wilk test was used to assess the distribution of variables (motor, functional, and cognitive scores). Where data were not normally distributed, a Mann‐Whitney U test was used. A P value <0.05 was defined as statistically significant. Graphs were generated using GraphPad Prism (version 6.04 for Windows; GraphPad, San Diego, CA).
Results
To investigate the potential contribution of TREM2 and TLR4 to HD, we first assessed the protein expression levels in the striatum of HD patients using postmortem tissue. To make this analysis as consistent as possible across different specimens, we built TMAs with striatal tissue from HD patients and controls (Fig. 1a,b). Quantification of the number of cells expressing TREM2 and TLR4 revealed a significant increase in both markers in HD patients when compared with controls (Fig. 1c,d; P < 0.001). Moreover, there was also an increase in relative optical density of TLR4 in the striatum of HD patients when compared with controls (Fig. 1d; P < 0.05). We also sought to look at the expression of these 2 markers in the cortex of a subgroup of patients and controls. Although the number of cases is too low to draw clear conclusions, we did not observe major differences in TREM2 and TLR4 labeling in the cortex between HD cases and controls (Fig. 1c,d).
Figure 1.
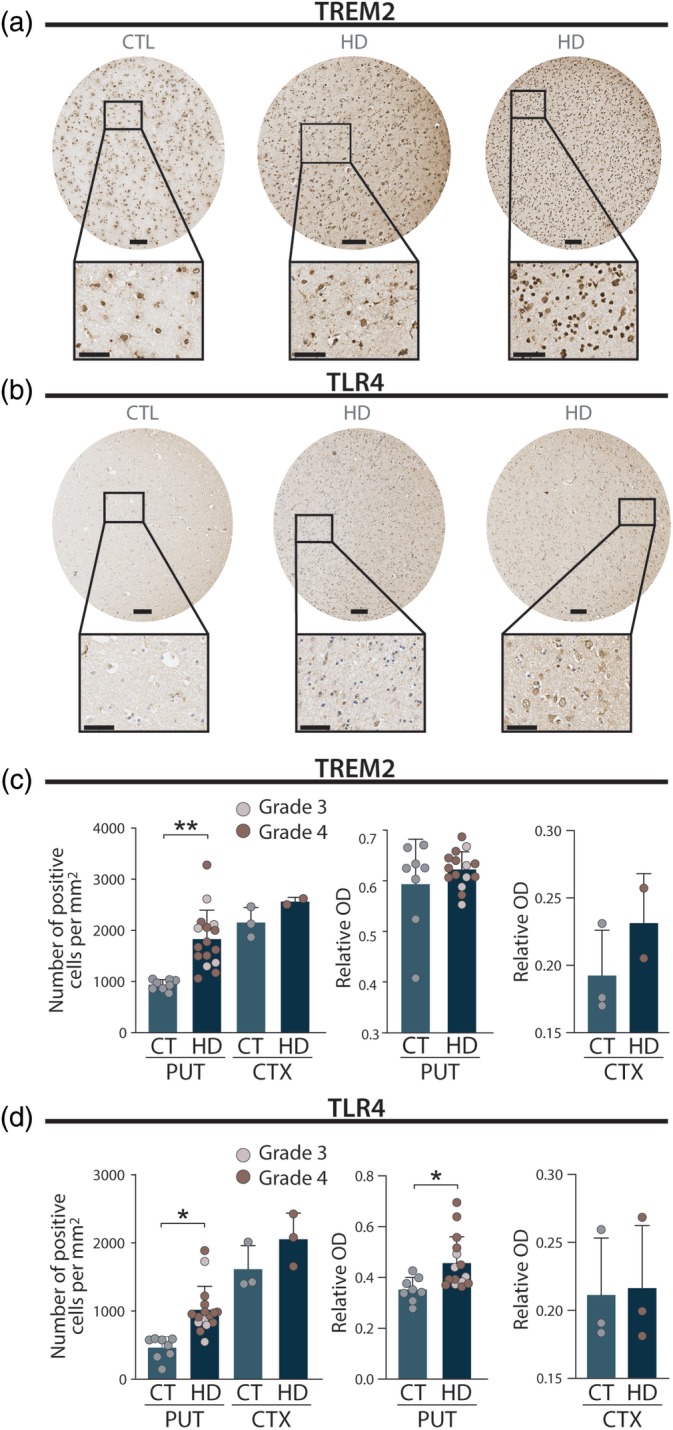
Increased expression of TLR4 and TREM2 in the striatum of HD patients. (a) Representative images of TREM2 immunostaining of putamenal tissue punches from tissue microarrays from HD brains of pathological grades 3 and 4 as well as a control brain. Scale bars = 100 μm in punch, 50 μm in inset. (b) Representative images of TLR4 immunostaining of putamenal tissue punches from tissue microarrays from HD brains of grades 3 and 4 as well as a control brain. Scale bars = 100 μm in punch, 50 μm in inset. (c) Quantification of the number of TREM2‐positive cells per mm2 in control and HD brains. Student's t test: ***P < 0.001, as compared to the control group. (d) Quantification of the number of TLR4‐positive cells per mm2 in control and HD brains. Student's t test: ***P < 0.001, as compared to the control group. CT/CTL, Controls; CTX, Cortex; HD, Huntington's Disease; PUT, Putamen; TLR4, toll‐like receptor 4; TREM2, triggering receptor expressed on myeloid cells 2. [Color figure can be viewed at http://wileyonlinelibrary.com]
We next sought to determine whether TREM2 and TLR4 gene polymorphisms had any impact on disease progression and clinical expression using genotype–phenotype analysis. We thus genotyped 830 HD patients from the EHDN for the TREM2 R47H variant (rs75932628) and 3 TLR4 SNPs (rs1927911, rs1927914, rs10116253) (Table 3). Patients were divided into 2 main groups based on the allelic frequencies as described previously for other genetic variants15, 43, 44; those that were homozygous for the rare allele were combined with heterozygous cases as summarized in Table 3. Complete clinical data for 2 independent assessments at least a year apart were available for all 830 individuals who were then included in the analysis. Although we found no association between AoO and motor and cognitive declines, nor between CAG repeats length and motor and cognitive declines, the already established negative correlation between AoO and CAG repeat length was reproduced in the total population (Kendall's taub ‐0.255, P < 0.0001; Supporting Information Fig. 1).
Table 3.
SNP Analysis
| Gene | SNP | Genotype | N | Motor* | P | N | Functional* | P | N | Cognitive* | P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TLR4 | rs1927914 (G/A) | A/A | 337 | 5.15 (6.56) | 0.039a | 337 | −0.92 (1.53) | 0.828 | 323 | −11.50 (24.88) | 0.686 |
| G carriers | 473 | 3.89 (7.05) | 473 | −0.85 (1.32) | 212 | −12.04 (24.61) | |||||
| TLR4 | rs1927911 (A/G) | G/G | 431 | 4.94 (7.07) | 0.05a | 431 | 0.90 (1.50) | 276 | −12.18 (26.98) | 0.828 | |
| A carriers | 379 | 3.82 (6.91) | 380 | −0.85 (1.31) | 0.511 | 257 | −11.62 (22.05) | ||||
| TLR4 | rs10116253 (T/C) | T/T | 434 | 4.80 (6.79) | 0.087 | 434 | −0.90 (1.49) | 0.604 | 281 | −11.96 (25.29) | 0.547 |
| C carriers | 377 | 3.92 (7.21) | 377 | −0.86 (1.31) | 255 | −11.52 (23.57) | |||||
| TREM2 |
rs75932628 (H47R ‐ C/T) |
C/C | 817 | 4.47 (7.01) | 0.914 | 817 | −0.87 (1.40) | 0.138 | 539 | −11.74 (24.88) | 0.018a |
| T carriers | 13 | 4.69 (11.79) | 13 | −1.25 (2.02) | 9 | −28.36 (26.47) |
Median change (standard error of the ratio change of points/years).
SNP, single nucleotide polymorphism.
Nonparametric comparison showed that there was a significantly higher rate of cognitive decline in TREM2 rs1927911 T carriers when compared with C/C patients, whereas this SNP did not impact on annual changes in functional capacity nor motor change (Fig. 2a; P < 0.05 and Table 3). However, overall motor decline per year was significantly higher in TLR4 rs1927911 G/G when compared with A carriers as well as in TLR4 rs1927914 A/A patients when compared with G carriers (Fig. 2b; P < 0.05 and Table 3). No changes in cognitive nor functional decline were associated with these 2 SNPs. Furthermore, when comparing TLR4 rs10116253 T/T with C carriers, no association with any of the 3 clinical assessments could be detected (Fig. 2b and Table 3). Taken together, these results suggest that TREM2 and TLR4 may be genetic modifiers for disease progression but play different roles in contributing to the clinical phenotypes.
Figure 2.
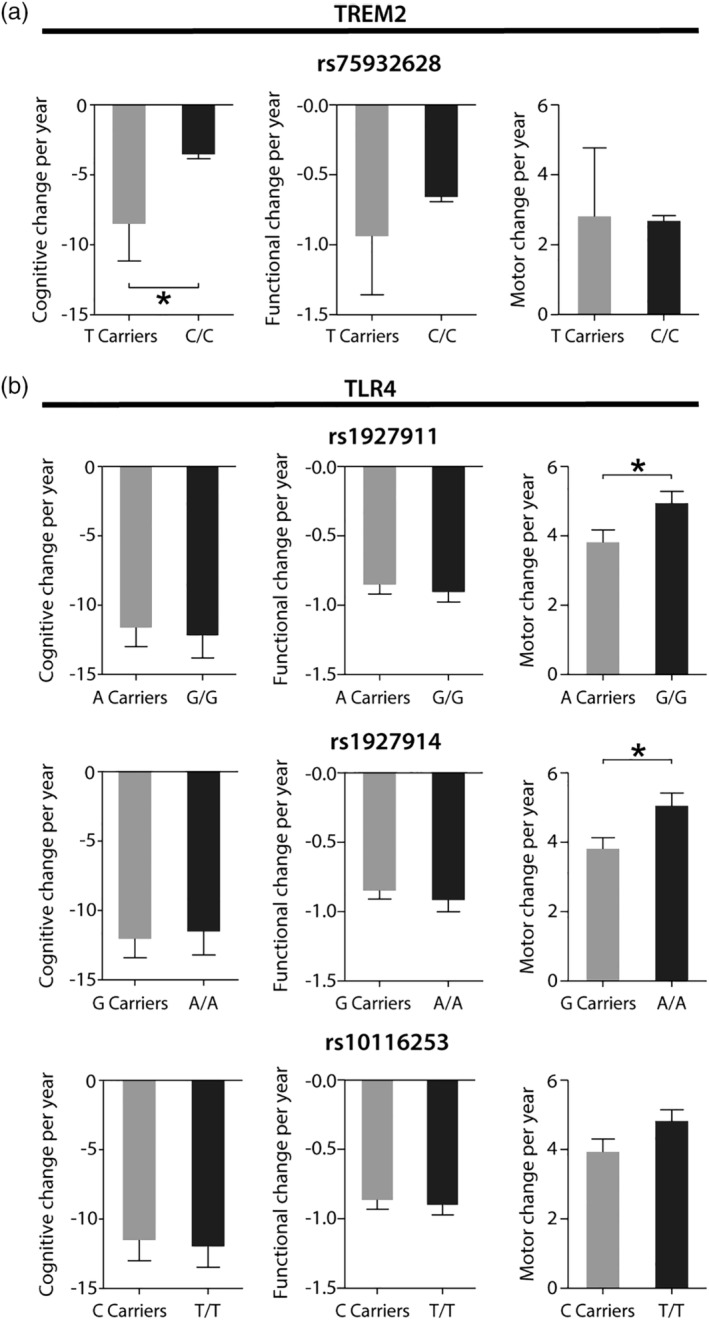
The effect of TREM2 and TLR4 single nucleotide polymorphisms variants on motor and cognitive decline in HD. (a) Graph showing a more severe cognitive decline in T carriers of the rs75932628 single nucleotide polymorphisms variant. Distribution was compared using Mann‐Whitney U test, *P < 0.05. (b) Graph showing a more severe motor decline in G/G carriers of the rs1927911 polymorphism as well as in A/A carriers of the rs1927914 polymorphism. Distribution was compared using Mann‐Whitney U test, *P < 0.05. TLR4, toll‐like receptor 4; TREM2, triggering receptor expressed on myeloid cells 2.
Discussion
This is the first genotype–phenotype study assessing the influence of genes related to inflammation in the progression of HD. We first showed that there is an increased number of cells expressing TREM2 and TLR4 in the striatum of the HD brain, although only TLR4 showed increased protein expression at this site. We then sought to investigate the clinical significance of this by looking into the impact of common variants in these genes on clinical progression in a large cohort of patients with HD. We found that TREM2 rs1927911 was associated with the rate of cognitive decline, whereas TLR4 rs1927911 and TLR4 rs1927914 were both associated with the rate of motor decline. Although we found no association between AoO and motor and cognitive declines, nor between CAG repeat length and motor and cognitive declines, the established negative correlation between AoO and CAG repeat length was reproduced in our population. Furthermore, there was no association between the SNPs TREM2 rs1927911 and TLR4 rs1927911 and TLR4 rs1927914 and AoO and CAG repeat length. This implies that the influence of those genotypes on the rate of cognitive or motor decline is independent of AoO and CAG repeat length.
TLR4, a pattern recognition receptor, has also been associated with misfolded protein clearance. For instance, the uptake of α‐synuclein by microglia has been shown to depend at least in part on TLR4 in models of α‐synucleinopathies.45, 46 This receptor is also responsible for the α‐synuclein‐induced proinflammatory response in astrocytes47 and triggers the amyloid‐β‐induced activation of microglia in AD models.48 As such, it is not unexpected that TLR4 is also involved in the inflammatory response in HD. Consistent with this, the Nuclear Factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) pathway, a key signaling cascade downstream of TLR4, has been shown to interact with mutant huntingtin protein exon 1 in mice.49 Moreover, a recent study reports that N171‐82Q HD mice lacking TLR4 have their lifespan significantly extended.50 The functional impact of the different TLR4 polymorphisms on glial cells and neurons, which seems to impact on motor function rather than cognition, is however not known. Given that we found TLR4 expression to be increased specifically in the putamen of HD patients, this could mean that these SNPs are affecting the expression levels or the affinity of the receptor to the adaptor proteins, thus impacting on motor functions. Nonetheless, TLR4‐deficient mice have been reported to have impaired motor functions, a feature that was attributed to TLR4 neuronal expression in the cerebellum, although the striatum was not assessed pathologically and no cognitive tasks were performed.51
Consistent with the absence of an increase in TREM2 relative optical density in the striatum of HD cases, we found no difference in motor progression in patients carrying the TREM2 R47H variant. The potential role of TREM2 variants as a factor linked to cognitive progression of HD supports the hypothesis that inflammation might also contribute to the cognitive impairments seen in this disorder. TREM2 attenuates macrophage activation52 and microglia expressing the R47H variant have been reported to have a reduced capacity to bind to phospholipids in an AD model, suggesting that TREM2 senses changes in the lipid microenvironment that result from Aβ accumulation and neuronal degeneration, which triggers signals that activate microglial capacity to limit Aβ accumulation.53 As such, similar mechanisms related to the triggered activation of microglia by mutant huntingtin protein could underlie the more severe cognitive decline in patients with the R47H variant. Interestingly, a recent study reports more hyperphosphorylated tau in the cortex of an AD mouse model carrying the human TREM2 R47H variant, which was thought to result from a reduction of microgliosis around amyloid‐β plaques. This in turn could facilitate the local seeding and spreading of tau,54 findings which were also reported in the AD patient brain where a reduced microglial accumulation around plaques associated with higher pathological tau burden was found in TREM2 R47H HD patients.55 However, it still remains unclear as to why TREM2 seems to influence only cognition and not motor decline in our HD study, but it could relate to an effect it may have on the distribution of pathology. Although the postmortem samples available for our study does not allow one to conclude on TREM2 cortical expression levels in HD nor to assess whether such a reduction of microgliosis around mutant huntingtin protein inclusions exists in the cortex of TREM2 R47H HD patient carriers, it will be interesting to look at whether this could underlie the link between the more aggressive cognitive decline suggested by our genotype–phenotype study in TREM2 R47H HD patients.
Although our results implicate TLR4 and TREM2 in the clinical progression of HD, our study has a number of limitations. First, in this study, cognitive data from only N = 9 patients carrying the rs75932628 (H47R ‐ C/T) genotype were available, and thus the interpretation of these results should be done with great caution. Further studies with a larger number of patients carrying that genotype are now needed. Second, for some patients, AoO was defined by retrospective interviews of patients and the patient's history, which can be unreliable. Third, the pathological analysis we performed is very limited in the number of samples and regions assessed, and thus more extensive analyses in larger numbers of brain areas and patients would be useful. In addition, although pathological observations can provide evidence as to whether a factor could be involved in disease pathogenesis, it does not demonstrate causality and as such further in vitro and animal studies will need to be done before conclusions on pathophysiological mechanisms can be made.
In summary, we have shown that TREM2 and TLR4 are linked to the clinical progression and pathology of HD and as such warrant further investigation, including whether therapies modulating these pathways could be useful in slowing down disease progression.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
R.V.: 1A,1B, 1C, 2A, 2B, 2C, 3A, 3B
A.K.: 2A, 2C, 3B
E.M.L.: 1C, 2B, 3B
L.C.: 1C, 3B
K.S.A.: 1C, 2C, 3B
A.L.S.: 1C, 3B
I.B.: 1B, 3B
R.A.B.: 1B, 3B
J.D.‐O.: 1A, 1B, 2A, 2C, 3A, 3B
Financial Disclosures of all authors (for the preceding 12 months)
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme: FP/2007‐2013 NeuroStemcellRepair (no. 602278) to R.A.B. and Fonds du Québec en Recherche, Santé to J.D.O. R.V. has received funding from Alzheimer's Research UK. A.K. has been supported by a PhD scholarship from the Onassis Foundation and a studentship by the Alborada Trust and is currently funded by a grant from the Evelyn Trust. R.A.B. has received grants from National Institute for Health Research( NIHR), Medical Research Council (MRC), Wellcome Trust, Parkinson's UK, Huntington's Disease Association, Rosetrees Trust, Cure Parkinson's Trust, royalties from Wiley and Springer‐Nature, and consultancy monies from Living Cell Technologies (LCT), F‐Prime, Fujifilm Cellular Dynamics International, and Oxford Biomedica. R.A.B. is an NIHR senior investigator. J.D.O. is receiving salary support from Fonds du Québec en Recherche, Santé and Parkinson Québec, and is receiving funding from the Canada Foundation for Innovation (CFI) and Parkinson Canada. The Cambridge Brain Bank is supported in part by NIHR.
Supporting information
Appendix S1: Supporting Information
Supplementary Figure 1 Correlation between CAG repeat length and AoO. Longer CAG repeats lead to early disease onset. Overall, Kendall's taub ‐0.255, p < 0.0001.
Acknowledgments
The authors thank the European Huntington's Disease Network REGISTRY Study Group investigators (listed in Supporting Information) for collecting the data and all participating REGISTRY patients for their time and efforts and the Cambridge Brain Bank for the postmortem tissue, which is supported by a grant to the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. We are grateful to Robert Fincham, Olly Green (Cambridge Brain Bank), Dr. Maria Ban (Neurology Unit at the University of Cambridge), and Pasquale De Blasio (Integrated Systems Engineering, Integrated Systems Engineering S.r.l. (ISENET)) for their technical assistance and input.
Membership of the REGISTRY Investigators of the European Huntington's Disease Network is provided in the Supporting Information.
Relevant conflicts of interests/financial disclosures: Nothing to report.
Contributor Information
Romina Vuono, Email: rv227@kent.ac.uk.
Janelle Drouin‐Ouellet, Email: janelle.drouin-ouellet@umontreal.ca.
References
- 1. Gusella JF, Wexler NS, Conneally PM, et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 1983;306:234–238. [DOI] [PubMed] [Google Scholar]
- 2. Duyao M, Ambrose C, Myers R, et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet 1993;4:387–392. [DOI] [PubMed] [Google Scholar]
- 3. Snell RG, MacMillan JC, Cheadle JP, et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington's disease. Nat Genet 1993;4:393–397. [DOI] [PubMed] [Google Scholar]
- 4. Andrew SE, Goldberg YP, Kremer B, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet 1993;4:398–403. [DOI] [PubMed] [Google Scholar]
- 5. Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, Ross CA. Correlation between the onset age of Huntington's disease and length of the trinucleotide repeat in IT‐15. Hum Mol Genet 1993;2:1547–1549. [DOI] [PubMed] [Google Scholar]
- 6. Trottier Y, Biancalana V, Mandel JL. Instability of CAG repeats in Huntington's disease: relation to parental transmission and age of onset. J Med Genet 1994;31:377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lucotte G, Turpin JC, Riess O, et al. Confidence intervals for predicted age of onset, given the size of (CAG)n repeat, in Huntington's disease. Hum Genet 1995;95:231–232. [DOI] [PubMed] [Google Scholar]
- 8. Ranen NG1, Stine OC, Abbott MH, et al. Anticipation and instability of IT‐15 (CAG)n repeats in parent‐offspring pairs with Huntington disease. Am J Hum Genet 1995;57:593–602. [PMC free article] [PubMed] [Google Scholar]
- 9. Brandt J, Bylsma FW, Gross R, Stine OC, Ranen N, Ross CA. Trinucleotide repeat length and clinical progression in Huntington's disease. Neurology 1996;46:527–531. [DOI] [PubMed] [Google Scholar]
- 10. Vuillaume I, Vermersch P, Destee A, Petit H, Sablonniere B. Genetic polymorphisms adjacent to the CAG repeat influence clinical features at onset in Huntington's disease. J Neurol Neurosurg Psychiatry 1998;64:758–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet 1997;60:1202–1210. [PMC free article] [PubMed] [Google Scholar]
- 12. Kuan WL, Kasis A2, Yuan Y, et al. Modelling the natural history of Huntington's disease progression. J Neurol Neurosurg Psychiatry 2015;86:1143–1149. [DOI] [PubMed] [Google Scholar]
- 13. Holmans PA, Massey TH, Jones L. Genetic modifiers of Mendelian disease: Huntington's disease and the trinucleotide repeat disorders. Hum Mol Genet 2017;26:R83–R90. [DOI] [PubMed] [Google Scholar]
- 14. Moss DJH, Pardiñas AF, Langbehn D, et al. Identification of genetic variants associated with Huntington's disease progression: a genome‐wide association study. Lancet Neurol 2017;16:701–711. [DOI] [PubMed] [Google Scholar]
- 15. Vuono R, Winder‐Rhodes S, de Silva R, et al. The role of tau in the pathological process and clinical expression of Huntington's disease. Brain 2015;138:1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aziz NA, Jurgens CK, Landwehrmeyer GB, et al. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology 2009;73:1280–1285. [DOI] [PubMed] [Google Scholar]
- 17. Farrer LA, Cupples LA, Wiater P, Conneally PM, Gusella JF, Myers RH. The normal Huntington disease (HD) allele, or a closely linked gene, influences age at onset of HD. Am J Hum Genet 1993;53:125–130. [PMC free article] [PubMed] [Google Scholar]
- 18. Rubinsztein DC, Barton DE, Davison BC, Ferguson‐Smith MA. Analysis of the huntingtin gene reveals a trinucleotide‐length polymorphism in the region of the gene that contains two CCG‐rich stretches and a correlation between decreased age of onset of Huntington's disease and CAG repeat number. Hum Mol Genet 1993;2:1713–1715. [DOI] [PubMed] [Google Scholar]
- 19. Bettencourt C, Hensman‐Moss D, Flower M, et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol 2016;79:983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Genetic Modifiers of Huntington's Disease C . Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 2015;162:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goold R, Flower M, Moss DH, et al. FAN1 modifies Huntington's disease progression by stabilising the expanded HTT CAG repeat. Hum Mol Genet 2018;28:650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y) 2018;4:575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Macchi B, Di Paola R, Marino‐Merlo F, Felice MR, Cuzzocrea S, Mastino A. Inflammatory and cell death pathways in brain and peripheral blood in Parkinson's disease. CNS Neurol Disord Drug Targets 2015;14:313–324. [DOI] [PubMed] [Google Scholar]
- 24. Rocha NP, Ribeiro FM, Furr‐Stimming E, Teixeira AL. Neuroimmunology of Huntington's disease: revisiting evidence from human studies. Mediators Inflamm 2016;2016:8653132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pavese N, Gerhard A, Tai YF, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 2006;66:1638–1643 (2006). [DOI] [PubMed] [Google Scholar]
- 26. Björkqvist M, Wild EJ, Thiele J, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med 2008;205:1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol Aging 2013;34:1711 e1715–e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bertram L, Parrado AR, Tanzi RE. TREM2 and neurodegenerative disease. N Engl J Med 2013;369:1565. [DOI] [PubMed] [Google Scholar]
- 29. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013;368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reitz C, Mayeux R, Alzheimer's Disease Genetics, C. TREM2 and neurodegenerative disease. N Engl J Med 2013;369:1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruiz A, Dols‐Icardo O, Bullido MJ, et al. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer's disease and frontotemporal dementia. Neurobiol Aging 2014;35:444 e441–444. [DOI] [PubMed] [Google Scholar]
- 33. Slattery CF, Beck JA, Harper L, et al. R47H TREM2 variant increases risk of typical early‐onset Alzheimer's disease but not of prion or frontotemporal dementia. Alzheimers Dement 2014;10:602–608, e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 2016;17:201–207. [DOI] [PubMed] [Google Scholar]
- 35. Yu JT, Miao D, Cui WZ, et al. Common variants in toll‐like receptor 4 confer susceptibility to Alzheimer's disease in a Han Chinese population. Curr Alzheimer Res 2012;9:458–466. [DOI] [PubMed] [Google Scholar]
- 36. Zhao J, Han X, Xue L, Zhu K, Liu H, Xie A, et al. Association of TLR4 gene polymorphisms with sporadic Parkinson's disease in a Han Chinese population. Neurol Sci 2015;36:1659–1665. [DOI] [PubMed] [Google Scholar]
- 37. Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A 1988;85:5733–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Orth M, Handley OJ, Schwenke C. European Huntington's Disease Network , Handley OJ, et al. Observing Huntington's disease: the European Huntington's Disease Network's REGISTRY. J Neurol Neurosurg Psychiatry 2011;82:1409–1412. [DOI] [PubMed] [Google Scholar]
- 39. Huntington Study Group . Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Disord 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 40. Ho AK, Sahakian BJ, Robbins TW, Barker RA, Rosser AE, Hodges JR. Verbal fluency in Huntington's disease: a longitudinal analysis of phonemic and semantic clustering and switching. Neuropsychologia 2002;40:1277–1284. [DOI] [PubMed] [Google Scholar]
- 41. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 1985;44:559–577. [DOI] [PubMed] [Google Scholar]
- 42. Cardano M, Diaferia GR, Falavigna M, et al. Cell and tissue microarray technologies for protein and nucleic acid expression profiling. J Histochem Cytochem 2013;61:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goris A, Williams‐Gray CH, Clark GR, et al. Tau and alpha‐synuclein in susceptibility to, and dementia in, Parkinson's disease. Ann Neurol 2007;62:145–153. [DOI] [PubMed] [Google Scholar]
- 44. Williams‐Gray CH, Evans JR, Goris A, et al. The distinct cognitive syndromes of Parkinson's disease: 5 year follow‐up of the CamPaIGN cohort. Brain 2009;132:2958–2969. [DOI] [PubMed] [Google Scholar]
- 45. Fellner L, Irschick R, Schanda K, et al. Toll‐like receptor 4 is required for alpha‐synuclein dependent activation of microglia and astroglia. Glia 2013;61:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hughes CD, Choi ML, Ryten M, et al. Picomolar concentrations of oligomeric alpha‐synuclein sensitizes TLR4 to play an initiating role in Parkinson's disease pathogenesis. Acta Neuropathol 2018;137:103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rannikko EH, Weber SS, Kahle PJ. Exogenous alpha‐synuclein induces toll‐like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci 2015;16:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walter S, Letiembre M, Liu Y, et al. Role of the toll‐like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem 2007;20:947–956. [DOI] [PubMed] [Google Scholar]
- 49. Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, Patterson PH, et al. Activation of the IkappaB kinase complex and nuclear factor‐kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci 2004;24:7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griffioen K, Mattson MP, Okun E. Deficiency of toll‐like receptors 2, 3 or 4 extends life expectancy in Huntington's disease mice. Heliyon 2018;4:e00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu JW, Li YF, Wang ZT, Jia WQ, Xu RX. Toll‐like receptor 4 deficiency impairs motor coordination. Front Neurosci 2016;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Turnbull IR, Gilfillan S, Cella M, et al. Cutting edge: TREM‐2 attenuates macrophage activation. J Immunol 2006;177:3520–3524. [DOI] [PubMed] [Google Scholar]
- 53. Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 2015;160:1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Leyns CEG, Gratuze M, Narasimhan S, et al. TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci 2019;22:1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Prokop S, Miller KR, Labra SR, et al. Impact of TREM2 risk variants on brain region‐specific immune activation and plaque microenvironment in Alzheimer's disease patient brain samples. Acta Neuropathol 2019;138:613–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Scientific Reports 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Supplementary Figure 1 Correlation between CAG repeat length and AoO. Longer CAG repeats lead to early disease onset. Overall, Kendall's taub ‐0.255, p < 0.0001.