

Abstract
The synthesis of complex molecules from simple, renewable carbon units is the goal of a sustainable economy. Here we explored the biocatalytic potential of the thiamine‐diphosphate‐dependent (ThDP) oxalyl‐CoA decarboxylase (OXC)/2‐hydroxyacyl‐CoA lyase (HACL) superfamily that naturally catalyzes the shortening of acyl‐CoA thioester substrates through the release of the C1‐unit formyl‐CoA. We show that the OXC/HACL superfamily contains promiscuous members that can be reversed to perform nucleophilic C1‐extensions of various aldehydes to yield the corresponding 2‐hydroxyacyl‐CoA thioesters. We improved the catalytic properties of Methylorubrum extorquens OXC by rational enzyme engineering and combined it with two newly described enzymes—a specific oxalyl‐CoA synthetase and a 2‐hydroxyacyl‐CoA thioesterase. This enzymatic cascade enabled continuous conversion of oxalate and aromatic aldehydes into valuable (S)‐α‐hydroxy acids with enantiomeric excess up to 99 %.
Keywords: biocatalysis, C1 building blocks, C−C coupling, oxalyl-CoA decarboxylase, thiamine diphosphate
The thiamine‐diphosphate‐dependent (ThDP) oxalyl‐CoA decarboxylase (OXC)/2‐hydroxyacyl‐CoA lyase (HACL) superfamily contains promiscuous members that can perform nucleophilic C1‐extensions of aldehydes to yield 2‐hydroxyacyl‐CoA thioesters. An enzymatic cascade consisting of OXC, oxalyl‐CoA synthetase, and a 2‐hydroxyacyl‐CoA thioesterase converted oxalate and aromatic aldehydes into (S)‐α‐hydroxy acids with enantiomeric excess up to 99 %.
One of biotechnology's central goals is the synthesis of multicarbon compounds under mild and sustainable conditions from renewable resources. This requires biocatalysts that enable selective C−C bond formation (“carboligation”) between two carbon units. Thiamine diphosphate (ThDP)‐dependent enzymes display high catalytic and substrate promiscuity with respect to C−C bond breaking and forming reactions and they catalyze carboligation reactions at high rates and with excellent stereo‐ and enantioselectivity.1 Several biocatalytic applications have been developed that rely on ThDP‐dependent carboligases; notable examples are pyruvate decarboxylase (PDC),2 benzoylformate decarboxylase (BFD),3 benzaldehyde lyase,4 transketolase (TK),5a–5c and branched‐chain alpha‐ketoacid decarboxylase (KdcA).5d Their broad catalytic repertoire makes ThDP‐dependent enzymes promising starting points for the development of biocatalysts for C−C bond forming reactions.
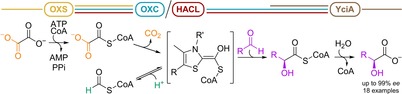
In the context of a methanol‐ and/or formate‐based economy, carboligations with one‐carbon units are of particular interest.6 The potential of ThDP‐dependent enzymes for synthetic one‐carbon fixation has been showcased by the computationally designed enzyme formolase, which condenses three formaldehyde molecules into dihydroxyacetone.7 Here, we focused on the superfamily of ThDP‐dependent oxalyl‐CoA decarboxylase (OXC)/2‐hydroxyacyl‐CoA lyase (HACL). The OXC/HACL superfamily comprises of decarboxylating members (OXCs), as well as non‐decarboxylating members (HACLs). Both catalyze the ThDP‐dependent cleavage of formyl‐CoA from their respective acyl‐CoA thioester substrates (Scheme 1). OXCs catalyze the decarboxylation of the C2‐compound oxalyl‐CoA to formyl‐CoA,8 whereas HACLs cleave a 2‐hydroxyacyl‐CoA into a fatty aldehyde that is shortened by a C1‐unit.9 In OXC and HACL, catalysis has been proposed to proceed through the same covalent intermediate 1 on the ThDP cofactor (Scheme 1).9, 10 After release of CO2 or aldehyde, the remaining formyl‐CoA moiety forms 1. Analogous to other ThDP‐dependent enzymes that form similar ThDP carbanion/enamine intermediates, we speculated that 1 can act as nucleophile in a carboligation reaction with an electrophilic carbon center, essentially reversing the native OXC/HACL reactions. This would enable nucleophilic C1‐extension reactions employing formyl‐CoA or oxalyl‐CoA as the donor, which can in turn be produced from the cheap carbon sources formate6b and oxalate, respectively. Recently, Chou et al. demonstrated that HACL catalyzes the acyloin condensation of formyl‐CoA with aldehyde acceptor substrates.11 However, the study focused on HACL and formyl‐CoA, thus the carboligation potential of OXC with oxalyl‐CoA as the donor remains unknown.
Scheme 1.
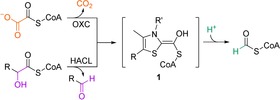
OXC and HACL form the α‐hydroxyl‐CoA‐ThDP carbanion/enamine intermediate (1) by decarboxylation of oxalyl‐CoA and cleavage of a 2‐hydroxyacyl‐CoA, respectively. In the second half reaction, 1 is protonated and released as formyl‐CoA.
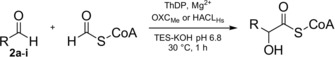
To further explore the carboligase potential within the OXC/HACL superfamily, we recombinantly produced human HACL (HACLHs), as well as OXC from Methylorubrum extorquens (OXCMe). HACLHs expressed very poorly at 25 °C. Lowering the temperature to 15 °C and using the Escherichia coli strain ArcticExpress, protein production was improved, but was still rather low (approximately 2 mg protein per L culture), especially compared to OXC (approximately 30 mg L−1). The low expression and/or stability limit the use of human HACL for biocatalytic applications. To test their carboligation activity, the enzymes were incubated with formyl‐CoA and various aldehyde acceptors. 2‐Hydroxyacyl‐CoA thioesters were analyzed by LC‐MS and the products verified by MS/MS fragmentation (Supporting Information, Figure S1). Product formation was detected in the presence of formaldehyde, acetaldehyde, propionaldehyde, glycolaldehyde, succinic semialdehyde, benzaldehyde, and phenylacetaldehyde with both enzymes (Table 1). Glyceraldehyde, glyoxylate, and acetone (not shown) were not accepted by OXCMe and HACLHs. As expected from its physiological role as fatty acyl‐CoA lyase, HACLHs showed high activity with the aliphatic aldehydes acetaldehyde and propionaldehyde. On the other hand, OXCMe showed very high activity with benzaldehyde, indicating a distinct catalytic spectrum between the two enzymes. In summary, HACLHs and OXCMe are able to perform nucleophilic C1‐extension reactions of different aldehydes with formyl‐CoA as C1‐donor.
Table 1.
Comparison of the aldehyde substrate scope of OXCMe and HACLHs.[a]
|
Aldehyde |
R |
Product name |
OXCMe [c] |
HACLHs [c] |
|---|---|---|---|---|
|
2 a |
H |
glycolyl‐CoA |
100 |
11 |
|
2 b |
Me |
lactyl‐CoA |
74 |
100 |
|
2 c |
CH2Me |
2‐hydroxybutyryl‐CoA |
5 |
100 |
|
2 d |
CH2OH |
glyceryl‐CoA |
1 |
100 |
|
2 e |
CHOHCH2OH |
erythronyl‐CoA |
n.d.[b] |
n.d.[b] |
|
2 f |
COOH |
tartronyl‐CoA |
n.d.[b] |
n.d.[b] |
|
2 g |
(CH2)2COOH |
2‐hydroxyglutaryl‐CoA |
1 |
100 |
|
2 h |
Ph |
mandelyl‐CoA |
100 |
3 |
|
2 i |
CH2Ph |
3‐phenyllactyl‐CoA |
22 |
100 |
[a] The reaction contained 2 a–2 i (10 mm), formyl‐CoA (1 mm), OXCMe or HACLHs (5 μm). Products were analyzed by LC‐MS after 1 h reaction time. [b] Product not detected. [c] Relative activity in %. Relative activity refers to the comparison of OXCMe and HACLHs for each aldehyde substrate.
Next, we investigated whether both enzymes would also be able to perform decarboxylating carboligations with oxalyl‐CoA as C1‐donor. We argued that the release of CO2 should provide a strong thermodynamic driving force towards carboligation, analogous to the reactions reported for glyoxylate carboligase,12 PDC,13 BFD,14 TK,5a, 5b and KdcA.15 HACLHs possessed only very low oxalyl‐CoA decarboxylation activity (k cat<1 min−1, see Figure S2 in the Supporting Information), in contrast to OXCMe (k cat=98±3 s−1, see Table 2 and Figure S3 in the Supporting Information), which confirms OXCMe’s physiological function as oxalyl‐CoA decarboxylase.
Table 2.
Steady‐state kinetic parameters of oxalyl‐CoA decarboxylation catalyzed by OXCMe.[a]
|
Mutation |
k cat [s−1] |
K M [μm] |
k cat/K M [s−1 m −1] |
|---|---|---|---|
|
wild‐type |
98±3 |
105±11 |
9.3×105 |
|
Y497A |
1.32±0.04 |
180±17 |
7.3×103 |
|
S568A |
5.53±0.11 |
23±2 |
1.1×105 |
|
Y497A S568A |
0.32±0.01 |
103±15 |
3.1×103 |
[a] Michaelis–Menten graphs are shown in Figure S3 in the Supporting Information.
Given the high activity towards benzaldehyde, we chose it as model substrate to study the decarboxylating carboligation of OXCMe in more detail. When started with oxalyl‐CoA and benzaldehyde, OXCMe as expected produced mandelyl‐CoA at a rate of 4 min−1 (Supporting Information, Figure S4). However, mandelyl‐CoA formation was preceded by the formation of formyl‐CoA, suggesting that OXCMe first rapidly decarboxylated oxalyl‐CoA into formyl‐CoA, followed by slow carboligation of formyl‐CoA with benzaldehyde (Supporting Information, Figure S4).
Decarboxylation of oxalyl‐CoA proceeds via the formation of 1, which can be either protonated and released as formyl‐CoA or undergo a nucleophilic attack on benzaldehyde to form mandelyl‐CoA. We speculated that mandelyl‐CoA formation may be increased by suppressing the unwanted protonation reaction (and subsequent release of formyl‐CoA) that competes with carboligation.
Structural, biochemical, and theoretical studies on OXC from Oxalobacter formigenes (OXCOf; 63 % sequence identity to OXCMe) demonstrated that the protonation of 1 is mediated by a water molecule that is coordinated by several polar side chains, notably a tyrosine and a serine, which are conserved in OXCMe (Tyr497 and Ser568).10 In OXCMe variants Y497A and S568A, formyl‐CoA formation was decreased 20‐ to 50‐fold, while the K M of both variants was largely unaffected (Table 2 and Supporting Information, Figure S3). When starting with oxalyl‐CoA and benzaldehyde, OXCMe‐Y497A showed a 5‐fold increased mandelyl‐CoA production rate (20 min−1; Supporting Information, Figure S4). The mutation Y497A thus increased the ratio of carboligation to decarboxylation by a factor of approximately 400 compared to the wildtype, suggesting that we successfully redirected activity of the enzyme towards carboligation by suppressing the protonation of 1.
Hydrolysis of mandelyl‐CoA leads to mandelic acid, which serves as a building block for various drugs as well as a resolving agent in chiral resolution processes.16 We set out to identify a thioesterase capable of hydrolyzing the non‐natural metabolite mandelyl‐CoA. We tested PaaI, TesB, and YciA from E. coli, which have been shown to hydrolyze a broad range of acyl‐CoA thioesters.17 YciA showed relatively high mandelyl‐CoA thioesterase activity (k obs≈1.5 s−1), low activity with formyl‐CoA (k obs≈0.06 s−1), and no activity with oxalyl‐CoA (Figure 1 B and Supporting Information, Figure S5). When used in combination with OXCMe, the two enzymes formed an enzymatic cascade for the conversion of oxalyl‐CoA and benzaldehyde into mandelic acid and free CoA (data not shown).
Figure 1.
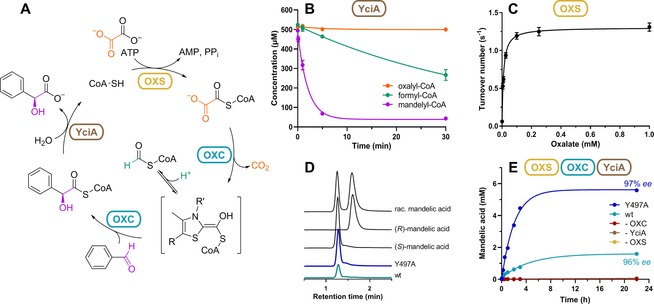
A) The OXS–OXCMe–YciA cascade converts benzaldehyde and oxalate into mandelic acid and CO2 under consumption of ATP. B) Thioesterase activity of YciA (2 μm) towards oxalyl‐CoA, formyl‐CoA, and racemic mandelyl‐CoA (0.5 mm each). The negative control without enzyme is shown in Figure S6 in the Supporting Information. Error bars show standard deviation of two replicates. C) Michaelis–Menten graph of OXS with oxalate as substrate. D) Chromatograms of chiral HPLC analyzing the reaction products of the complete cascade with either wildtype OXCMe (wt) or Y497A. The chiral HPLC traces of commercial mandelic acid standards are shown at the top. E) Mandelic acid formation of the cascade over time. The reaction contained 2 h (25 mm), disodium oxalate (10 mm), ATP (10 mm), CoA (0.5 mm), OXS (5 μM), OXC (5 μM), YciA (2 μM). ee of (S)‐mandelic acid is indicated for the last time point (22 h).
We noticed that subsequent regeneration of free CoA into oxalyl‐CoA would allow us to close a catalytic cycle for the continuous production of mandelic acid (Figure 1 A). To establish such a catalytic cycle, we obtained an oxalyl‐CoA synthetase AMP‐forming (OXS) homologue from M. extorquens,18 and determined the enzyme's steady‐state kinetic parameters (k cat=1.30±0.02 s−1; K M(oxalate)=9±1 μm; Figure 1 C). We then used OXS in combination with YciA and OXCMe to continuously produce mandelic acid from oxalate and benzaldehyde. When we replaced OXCMe by the Y497A variant, the mandelic acid production rate increased 5‐fold and the conversion increased 4‐fold (Figure 1 E). This was likely caused by the decreased formation of the unwanted side product formyl‐CoA (and its further hydrolysis by YciA). Chiral LC‐MS revealed that (S)‐mandelic acid was produced with an enantiomeric excess of 97 % (Figure 1 D). Since YciA showed no stereospecificity in the hydrolysis of mandelyl‐CoA (Figure 1 B), the stereochemistry is exclusively determined by OXCMe.
Next, we tested the substrate scope of the catalytic cycle by replacing benzaldehyde with substituted variants 2 i–2 k (Figure 2 A). Under limiting ATP concentrations (10 mm), the expected products 3 a–3 d were formed at varying yields (57–93 %) and ee (44–99 %, Figure 2 B). Notably, also sterically demanding 2 k was converted to 3 d with high yield, albeit with moderate ee. The broad substrate scope of the OXS–OXC–YciA cascade was further confirmed by an extended screen, in which product formation was detected for ten other aromatic and three heteroaromatic aldehydes (3 e–3 r, Figure 2 C). To test if the cascade can be scaled up, we performed the reaction on a semi‐preparative scale (0.625 mmol 2 h). We added an ATP regeneration system comprising of creatine phosphate, creatine kinase, and adenylate kinase. With catalytic amounts of ADP (0.5 mm) this five enzyme one‐pot cascade produced 3 a with a yield of 53 %. Taken together, these results indicate that the established enzymatic cascade can be employed to produce various aromatic α‐hydroxy acids with moderate to high (S)‐selectivity.
Figure 2.
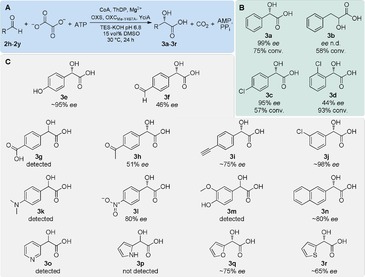
Scope of the OXS–OXCMe‐Y497A–YciA cascade for the synthesis of aromatic (S)‐α‐hydroxy acids. Chromatograms and time courses of the reactions are shown in Figure S7 in the Supporting Information. A) The reactions contained 2 h–2 y (25 mm), disodium oxalate (10 mm), ATP (10 mm), CoA (0.5 mm), OXS (5 μm), OXCMe‐Y497A (5 μm), YciA (2 μm). Products were analyzed by chiral LC‐MS after 24 h reaction time. ee’s were estimated based on extracted ion counts. B) For 3 a–3 d conversion and ee were quantified by comparison to a commercial standard. For 3 b separation of the enantiomers could not be achieved; n.d, not determined. C) 3 e–3 r were analyzed qualitatively. Where chromatographic separation allowed, the ee was estimated, assuming identical ionization of the two enantiomers.
We demonstrated that members of the OXC/HACL superfamily are able to catalyze C1‐carboligation reactions between 1—formed either through decarboxylation of oxalyl‐CoA or deprotonation of formyl‐CoA—and several aldehydes to yield chiral 2‐hydroxyacyl‐CoA thioesters. These nucleophilic C1‐extension reactions expand the spectrum of ThDP‐dependent enzymes as versatile biocatalysts for C−C bond forming reactions.19
What determines substrate specificity in the OXC/HACL superfamily? The observed variances in the aldehyde scope of OXCMe and HACLHs are likely caused by differences in the C‐terminal domain, which forms a “lid” on top of the active site.10a While the bottom part of the active site is virtually identical between OXC and HACL (with exception of Tyr133 and Glu134 in OXCMe that are replaced by Phe and Gln in HACLHs), the C‐terminal lid‐domain shows a high variability between individual superfamily members. Further characterization of the OXC/HACL superfamily could reveal more carboligases with aldehyde preference for a desired application.
We engineered OXCMe towards improved carboligation rate at the expense of the formyl‐CoA formation rate by replacing Tyr497 with Ala. Interestingly, the mutation Y497A did not affect the enantioselectivity for (S)‐mandelyl‐CoA. This is reminiscent of engineering efforts on PDC from Zymomonas mobilis, where Glu473 positions a water molecule that acts as proton donor for the ThDP carbanion/enamine intermediate. Mutating this amino acid to glutamine led to a 100‐fold preference of carboligation over aldehyde release, under full retention of enantioselectivity.13 Considering the high demand of mandelic acid and its derivatives in the (R) configuration,16 it would be interesting to engineer OXCMe towards inverted enantioselectivity. This has been achieved for other ThDP‐dependent carboligases.20
The rational engineering of OXCMe‐Y497A enabled a three‐enzymatic cascade comprising of OXS, a newly identified oxalyl‐CoA synthetase, and YciA, an efficient mandelyl‐CoA thioesterase with only minor formyl‐ and oxalyl‐CoA hydrolysis activities. The OXCMe‐mediated production of aromatic (S)‐α‐hydroxy acids in high ee from aldehydes and oxalate offers an alternative to hydrogen‐cyanide‐based syntheses catalyzed by nitrilases.21 However, the requirements of CoA in catalytic amounts and an ATP regeneration system may limit the potential for a synthetic application on the larger scale. To this end, employing whole‐cell catalysts may prove to be advantageous, providing not only the cascade enzymes but also ATP regeneration and a CoA pool without the addition of purified enzymes and cofactors.22
Altogether, our study expands the spectrum of ThDP‐dependent transformations by nucleophilic C1‐extensions, which gives access to α‐hydroxy acids that are valuable chiral building blocks and showcases ways to establish in vitro and in vivo platforms for the continuous production of these compounds in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank L. Schulz for cloning of oxc and B. Heinrich for NMR measurements. This work was supported by the German Ministry of Education and Research Grant FormatPlant (part of BioEconomy 2030, Plant Breeding Research for the Bioeconomy). A provisional patent application has been filed through the Max‐Planck‐Innovation based on the results presented here.
S. Burgener, N. S. Cortina, T. J. Erb, Angew. Chem. Int. Ed. 2020, 59, 5526.
Contributor Information
Simon Burgener, Email: simon.burgener@mpi-marburg.mpg.de.
Prof. Dr. Tobias J. Erb, Email: toerb@mpi-marburg.mpg.de.
References
- 1. Kluger R., Tittmann K., Chem. Rev. 2008, 108, 1797–1833. [DOI] [PubMed] [Google Scholar]
- 2. Schörken U., Sprenger G. A., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1998, 1385, 229–243. [DOI] [PubMed] [Google Scholar]
- 3. Demir A. S., Dünnwald T., Iding H., Pohl M., Müller M., Tetrahedron: Asymmetry 1999, 10, 4769–4774. [Google Scholar]
- 4. Demir A. S., Sesenoglu O., Eren E., Hosrik B., Pohl M., Janzen E., Kolter D., Feldmann R., Dünkelmann P., Müller M., Adv. Synth. Catal. 2002, 344, 96–103. [Google Scholar]
- 5.
- 5a. Payongsri P., Steadman D., Strafford J., MacMurray A., Hailes H. C., Dalby P. A., Org. Biomol. Chem. 2012, 10, 9021–9029; [DOI] [PubMed] [Google Scholar]
- 5b. L'enfant M., Bruna F., Lorilliere M., Ocal N., Fessner W. D., Pollegioni L., Charmantray F., Hecquet L., Adv. Synth. Catal. 2019, 361, 2550–2558; [Google Scholar]
- 5c. Ranoux A., Karmee S. K., Jin J. F., Bhaduri A., Caiazzo A., Arends I. W. C. E., Hanefeld U., ChemBioChem 2012, 13, 1921–1931; [DOI] [PubMed] [Google Scholar]
- 5d. Germer P., Gauchenova E., Walter L., Müller M., ChemCatChem 2019, 11, 4276–4280. [Google Scholar]
- 6.
- 6a. Olah G. A., Angew. Chem. Int. Ed. 2013, 52, 104–107; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 112–116; [Google Scholar]
- 6b. Yishai O., Lindner S. N., de la Cruz J. G., Tenenboim H., Bar-Even A., Curr. Opin. Chem. Biol. 2016, 35, 1–9. [DOI] [PubMed] [Google Scholar]
- 7. Siegel J. B., Smith A. L., Poust S., Wargacki A. J., Bar-Even A., Louw C., Shen B. W., Eiben C. B., Tran H. M., Noor E., Gallaher J. L., Bale J., Yoshikuni Y., Gelb M. H., Keasling J. D., Stoddard B. L., Lidstrom M. E., Baker D., Proc. Natl. Acad. Sci. USA 2015, 112, 3704–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Quayle J. R., Biochem. J. 1963, 89, 492–503; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Berthold C. L., Moussatche P., Richards N. G., Lindqvist Y., J. Biol. Chem. 2005, 280, 41645–41654. [DOI] [PubMed] [Google Scholar]
- 9. Foulon V., Sniekers M., Huysmans E., Asselberghs S., Mahieu V., Mannaerts G. P., Van Veldhoven P. P., Casteels M., J. Biol. Chem. 2005, 280, 9802–9812. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Berthold C. L., Toyota C. G., Moussatche P., Wood M. D., Leeper F., Richards N. G., Lindqvist Y., Structure 2007, 15, 853–861; [DOI] [PubMed] [Google Scholar]
- 10b. Sheng X., Liu Y. J., Zhang R., RSC Adv. 2014, 4, 35777–35788. [Google Scholar]
- 11. Chou A., Clomburg J. M., Qian S., Gonzalez R., Nat. Chem. Biol. 2019, 15, 900–906. [DOI] [PubMed] [Google Scholar]
- 12. Kaplun A., Binshtein E., Vyazmensky M., Steinmetz A., Barak Z., Chipman D. M., Tittmann K., Shaanan B., Nat. Chem. Biol. 2008, 4, 113–118. [DOI] [PubMed] [Google Scholar]
- 13. Meyer D., Walter L., Kolter G., Pohl M., Müller M., Tittmann K., J. Am. Chem. Soc. 2011, 133, 3609–3616. [DOI] [PubMed] [Google Scholar]
- 14. Dünkelmann P., Kolter-Jung D., Nitsche A., Demir A. S., Siegert P., Lingen B., Baumann M., Pohl M., Müller M., J. Am. Chem. Soc. 2002, 124, 12084–12085. [DOI] [PubMed] [Google Scholar]
- 15. Berthold C. L., Gocke D., Wood M. D., Leeper F. J., Pohl M., Schneider G., Acta Crystallogr. Sect. D 2007, 63, 1217–1224. [DOI] [PubMed] [Google Scholar]
- 16. Gröger H., Adv. Synth. Catal. 2001, 343, 547–558. [Google Scholar]
- 17.
- 17a. Sonntag F., Buchhaupt M., Schrader J., Appl. Microbiol. Biotechnol. 2014, 98, 4533–4544; [DOI] [PubMed] [Google Scholar]
- 17b. Kuznetsova E., Proudfoot M., Sanders S. A., Reinking J., Savchenko A., Arrowsmith C. H., Edwards A. M., Yakunin A. F., FEMS Microbiol. Rev. 2005, 29, 263–279; [DOI] [PubMed] [Google Scholar]
- 17c. Zhuang Z., Song F., Zhao H., Li L., Cao J., Eisenstein E., Herzberg O., Dunaway-Mariano D., Biochemistry 2008, 47, 2789–2796. [DOI] [PubMed] [Google Scholar]
- 18. Schneider K., Skovran E., Vorholt J. A., J. Bacteriol. 2012, 194, 3144–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Müller M., Gocke D., Pohl M., FEBS J. 2009, 276, 2894–2904; [DOI] [PubMed] [Google Scholar]
- 19b. Giovannini P. P., Bortolini O., Massi A., Eur. J. Org. Chem. 2016, 4441–4459. [Google Scholar]
- 20.
- 20a. Westphal R., Hahn D., Mackfeld U., Waltzer S., Beigi M., Widmann M., Vogel C., Pleiss J., Muller M., Rother D., Pohl M., ChemCatChem 2013, 5, 3587–3594; [Google Scholar]
- 20b. Rother neé Gocke D., Kolter G., Gerhards T., Berthold C. L., Gauchenova E., Knoll M., Pleiss J., Muller M., Schneider G., Pohl M., ChemCatChem 2011, 3, 1587–1596. [Google Scholar]
- 21. Gong J. S., Lu Z. M., Li H., Shi J. S., Zhou Z. M., Xu Z. H., Microb. Cell Fact. 2012, 11, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Endo T., Koizumi S., Adv. Synth. Catal. 2001, 343, 521–526. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary