

Abstract
The toxic bicyclic octapeptide α‐amanitin is mostly found in different species of the mushroom genus Amanita, with the death cap (Amanita phalloides) as one of the most prominent members. Due to its high selective inhibition of RNA polymerase II, which is directly linked to its high toxicity, particularly to hepatocytes, α‐amanitin received an increased attention as a toxin‐component of antibody‐drug conjugates (ADC) in cancer research. Furthermore, the isolation of α‐amanitin from mushrooms as the sole source severely restricts compound supply as well as further investigations, as structure–activity relationship (SAR) studies. Based on a straightforward access to the non‐proteinogenic amino acid dihydroxyisoleucine, we herein present a robust total synthesis of α‐amanitin providing options for production at larger scale as well as future structural diversifications.
Keywords: amatoxins, asymmetric synthesis, total synthesis, tryptathionine, α-amanitin
A new route to the toxic bicyclic octapeptide α‐amanitin is presented. The key steps of the convergent [5+1+2]‐synthesis are the preformation of the thioether building block and access to the enantiomerically pure non‐proteinogenic amino acids 6‐hydroxytryptophan and (3R,4R)‐l‐4,5‐dihydroxyisoleucine on a multigram scale. The peptide fragment based methodology is the first convergent α‐amanitin synthesis fully performed in solution phase.
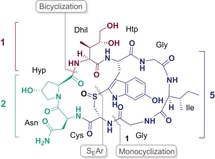
Amatoxins are ribosomally synthesized toxic bicyclic octapeptides mainly found in different species of the fungus genus Amanita, most notably Amanita phalloides.1, 2, 3, 4 These compounds are selective inhibitors of eukaryotic RNA polymerase II [α‐amanitin, K d=10−9 m], which ultimately leads to apoptosis.4, 5 The fungal toxins were first isolated by Heinrich Wieland and Rudolf Hallermayer in 1941 and their structure was elucidated in the 1950s and 1960s (Wieland et al.).6, 7 All amatoxins contain eight l‐configured amino acids and a tryptathionine linkage between the side chains of tryptophan and cysteine, with an (R)‐configured sulfoxide moiety.1, 3 However, the amanitins differ in their hydroxylation pattern of the amino acid side chains, which modulates their toxicity. The effect of the differently hydroxylated amino acids on the amatoxin toxicity was investigated in various studies.8, 9, 10, 11
In the past 30 years several approaches were made to synthesize amatoxin derivatives.8, 12, 13, 14, 15 Zanotti et al. (1987) described the first synthesis of an amaninamide derivative starting from the linear octapeptide followed by thioether formation using the Savige‐Fontana reaction and subsequent macrocyclization.8 The Savige‐Fontana methodology is based on an N‐terminal Hpi (3a‐hydroxypyrollo[2,3‐b]indole) moiety and a trityl side‐chain protected cysteine residue. Simultaneous deprotection of the cysteine residue and activation of the Hpi moiety with trifluoroacetic acid (Tfa) leads to the formation of the thioether.14, 16 Other routes to tryptathionine involved the reaction of tryptophan with a cysteine‐sulfenyl iodide or chloride. Until now, these methodologies were only used for the synthesis of phalloidin and its derivatives.17, 18, 19
The most intensively studied amatoxin is α‐amanitin (1) (Figure 1). It contains the hydroxylated amino acids 6‐ hydroxytryptophan (Htp), trans‐4‐hydroxyproline (Hyp) and (3R,4R)‐4,5‐dihydroxyisoleucine (Dhil).1, 2 Due to its small molecular size, good solubility in aqueous buffers and its inhibition of RNA polymerase II, it received an increased attention as an antibody‐drug conjugate (ADC) in cancer research in the past years.14, 20, 21 In a preclinical study in 2011 conducted by the German Cancer Research Center in Heidelberg (Germany) naturally occurring α‐amanitin was conjugated with the chimerized anti‐EpCAM monoclonal antibody chiHEA125 to form the antibody‐drug conjugate chiHEA125‐Ama.22 This therapeutic ADC targets the human epithelial adhesion molecule (EpCAM), which is overexpressed in the majority of cancer cells. The study showed that chiHEA125‐Ama has a potent antitumor activity against pancreatic cancer cells. Compared with chiHEA125 two injections of chiHEA125‐Ama with a dosage of 50 or 100 μg kg−1 (in relation to α‐amanitin), showed significant tumor regression in mice.22 Based on its promising properties for drug development there is an increasing demand for α‐amanitin, which cannot be satisfied by biotechnological methods or from nature. Only last year the first total synthesis of α‐amanitin has been accomplished by Perrin and co‐workers, applying a sophisticated Savige‐Fontana methodology as a key step for the formation of the tryptathionine bridge.23
Figure 1.
Structure of α‐Amanitin (1). The numbers in blue, turquoise and red refer to the number of amino acids of the building blocks (depicted blue, turquoise and red) employed in this total synthesis strategy [5+1+2].
Herein, we report an alternative and robust total synthesis of α‐amanitin with the aim to be convergent and scalable in solution phase synthesis. Therefore, we applied a peptide fragment based methodology and developed straightforward syntheses to the central Dhil and Htp building blocks.
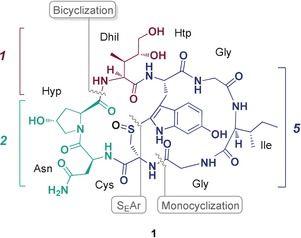
Scheme 1 shows the general retrosynthetic strategy for the total synthesis of α‐amanitin (1) referred to as [5+1+2]‐strategy. Sulfide oxidation of the tryptathionine linkage was envisioned to be introduced close to the final stages of the synthesis route, when the bicyclic structure was established by macrocyclization between Dhil and Hyp of monocyclic octapeptide 2. This amanitin precursor should be accessible by stepwise couplings of the monocyclic building block 5 with a Dhil derivative 6 and an Asn‐Hyp dipeptide 4. It was important for reasons of scalability that both, the Dhil derivative 6 and the monocyclic tryptathionine‐containing building block 5 could be synthesized on a multi‐gram scale in solution phase. In our synthesis strategy, 5 is synthesized by preformation of the pseudo‐orthogonally protected tryptathione linkage 7 between a 6‐hydroxytryptophan (Htp) derivative (8) and cysteine.
Scheme 1.
Retrosynthetic analysis of α‐amanitin (1). Final assembly of α‐amanitin from three building blocks and enantioselective synthesis of the hydroxylated amino acid building blocks.
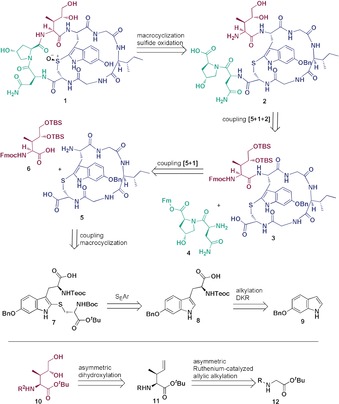
The synthesis of 6‐hydroxytryptophan was not trivial and finally we decided for a dynamic kinetic resolution (DKR) route employing a chiral tridentate ligand.24, 25 Starting with 6‐benzyloxyindol (9), alkylation using l‐serine, Ac2O and AcOH (Scheme 2) was performed, which rendered racemic tryptophan 13.26 After removal of the acetyl group the racemate was treated with the chiral ligand 16 and Ni(NO3)2⋅6 H2O under basic conditions, which led to nickel(II) complex 14 with a diastereomeric ratio (dr) of 99 % (see Supporting Information). Disassembly of the nickel(II) complex 14 under acidic conditions resulted in enantiomerically pure 6‐benzyloxy‐l‐tryptophan (15). Finally, the free amino acid was trapped with TeocOSu, which provided N‐Teoc‐6‐benzyloxy‐l‐tryptophan (8).
Scheme 2.
Synthesis of N‐Teoc‐6‐benzyloxy‐l‐tryptophan (8) by dynamic kinetic resolution: a) l‐serine, Ac2O, AcOH, 75 °C, 2 h, 82 %; b) 40 % NaOH, MeOH/H2O, c) chiral ligand 16, Ni(NO3)2 6H2O, MeOH, reflux, 16 h, 84 % over two steps, d) 6 m HCl, MeOH, 70 °C, 2 h, e) TeocOSu, Et3N, DMF, 60 °C, 4 d, quant.
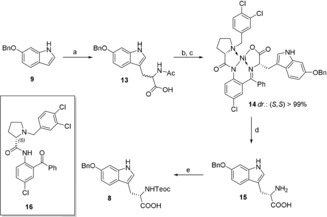
With 6‐hydroxytryptophan derivative 8 in hand, the tryptathionine linkage was readily established by treatment of a fully protected l‐cystine derivative (18) with sulfuryl chloride (SO2Cl2) (Scheme 3).17 In the course of the reaction, cleavage of the disulfide afforded the highly reactive sulfenyl chloride monomer as an intermediate, which underwent a SEAr reaction with Htp (8). We realized that protection of the C‐terminus of 8 was not necessary for quantitative tryptathionine formation and that slow titration with the sulfenyl chloride solution prevents cysteine double substitution at Htp. Coupling of OSu‐ester preactivated tryptathione 7 with a H‐Gly‐Ile‐Gly‐OH tripeptide (see Supporting Information) afforded linear pentapeptide 19.
Scheme 3.
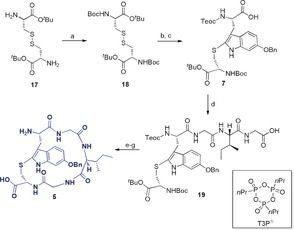
Synthesis of the C‐ and N‐terminally deprotected monocyclic tryptathionine peptide 5: a) Boc2O, NaHCO3, dioxane/H2O, r.t., 16 h, 99 %; b) SO2Cl2, CHCl3, r.t., 1 h; c) 8, NaHCO3, CHCl3, 0 °C to r.t., quant., d) H‐Gly‐Ile‐Gly‐OH, CO(OSu)2, collidine, acetonitrile/H2O, r.t., 2 h; e) PTSA, THF, r.t., 2 h, 70 % over 2 steps; f) T3P, DIPEA, DMF, r.t., 16 h, 70 %, g) TFA/H2O (7:3), r.t., 2 h, quant.
After Boc‐deprotection with para‐toluenesulfonic acid (PTSA), macrocyclization with propanephosphonic acid anhydride (T3P) was performed. Both Teoc and tBu protecting groups could be simultaneously cleaved under strong acidic conditions using TFA, rendering monocyclic building block 5 with no decomposition of the thioether observed.
A particular synthetic challenge was the (3R,4R)‐4,5‐l‐dihydroxyisoleucine derivative 6, which was finally accomplished in seven steps starting from glycine tert‐butyl ester (23). Quantitative trifluoroacetylation yielded Tfa‐Gly‐OtBu (12) followed by ruthenium‐catalyzed asymmetric allylic alkylation with 22, in analogy to a method of Kazmaier et al.27, 28 While the original protocol of asymmetric alkylation employed (S)‐3‐(benzyloxy)‐1‐butene as alkylating agent, we used a chiral allylic carbonate (22), which was synthesized by Sharpless epoxidation of (E)‐crotyl alcohol 20 followed by reductive elimination promoted by a zinc‐copper‐couple.29, 30 The chirality transfer promoted by allylic carbonate 22 mainly led to the anti‐directed formation of a fully protected 4,5‐didehydroisoleucine derivative 11 with a diastereomeric ratio (dr=86:14) and an enantiomeric excess (ee=98 %), superior to initial original reaction conditions (see Supporting Information). The amino group was deprotected by reduction of the trifluoroacetyl group with sodium borohydride31 and refurnished with the Fmoc‐protecting group to give 25. The C‐terminal tBu protecting group proved to be essential preventing lactone formation, the main threat during dihydroxylation of the fully protected didehydroisoleucine derivative 25. The AD reaction was performed by a simple Upjohn dihydroxylation with potassium osmate and NMO. The stereoselectivity of the reaction was controllable solely by the applied solvent mixture. While dihydroxylation in a biphasic system of H2O/CHCl3 led to the formation of mainly the desired (2S,3R,4R)‐diastereomer 10 in a 2.5:1 ratio, the undesired (2S,3R,4S)‐diastereomer was mainly formed in a mixture of H2O/tBuOH. Surprisingly, adding (DHQD)2PHAL ligand to the reaction mixture led to an impairment of the stereoselectivity towards the wrong enantiomer. Finally, the hydroxy groups of the side chain of 10 were protected with the tert‐butyldimethylsilyl (TBS) protecting group, allowing for the mild orthogonal cleavage of tBu in nearly quantitative yield in presence of an excess of TMSOTf, which resulted in the formation of Dhil building block 6 (Scheme 4).
Scheme 4.
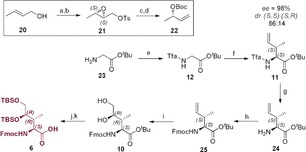
Synthesis of side chain‐protected Fmoc‐(3R,4R)‐4,5‐l‐dihydroxyisoleucine (6) a) (+)‐DIPT, Ti(OiPr)4, TBHP, DCM, −20 °C, 4 h; b) TsCl, Et3N, DMAP, DCM, −10 °C, 30 h, 67 %; c) NaI, Zn(Cu), THF, 70 °C, 2 h; d) Boc2O, NaH, THF, 0 °C to r.t., 16 h, 75 %; e) ethyl trifluoroacetate, NEt3, MeOH, r.t., 16 h, quant.; f) LHMDS, ZnCl2, PPh3, [(p‐cymene)RuCl2]2, 22, THF, −72 °C to r.t., 16 h, 88 %; g) NaBH4, MeOH, r.t., 1 h; h) FmocOSu, Et3N, dioxane, r.t., 4 h, 82 %; i) K2OsO4⋅H2O, NMO, CHCl3/H2O, r.t., 6 h, 40 %; j) TBSCl, pyridine/DMF(1:9), r.t., 24 h, 95 %; k) TMSOTf, 2,6‐lutidine, 0 °C to r.t., 2 h, 90 %.
Parallel to the synthesis of Dhil and the monocyclic tryptathionine building block, C‐terminally 9‐fluorenylmethyl ester (Fm)‐protected dipeptide 4 was synthesized in solution phase (see Supporting Information) serving as the third building block for the [5+1+2]‐assembly. First, Dhil derivative 6 was coupled to the N‐ and C‐terminally unprotected monocyclic pentapeptide 5 applying a protocol activating the carboxylic function of Dhil as an active ester and concomitantly increasing the nucleophilicity of the amino group of 5 by silylation with N‐methyl‐N‐trimethylsilylacetamide (MSA) (Scheme 5).32, 33 This way no oligomerized side‐products were formed. The resulting monocyclic hexapeptide 3 could then be coupled to the previously synthesized C‐terminally Fm‐protected dipeptide 4 in a one pot reaction. The final cyclization was performed after simultaneous Fmoc‐ and Fm‐cleavage, followed by TBS deprotection with TBAF yielding monocyclic octapeptide 2. After bicyclization using HATU to 26, the benzyl protecting group was cleaved from the Htp side‐chain of 26 applying a hard acid and soft nucleophile system.34 Finally, the oxidation to the sulfoxide was performed using mCPBA according to Perrin et al.23 affording the target molecule α‐amanitin (1). The oxidation step resulted to be necessary after benzyl‐deprotection and not vice versa, because of sulfoxide instability under strongly acidic conditions.
Scheme 5.
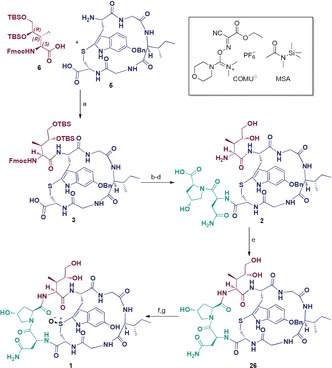
Fragment couplings and cyclizations to the target molecule α‐Amanitin (1): a) MSA, COMU, DIPEA, DMA, 0 °C to r.t. 3 h; b) (a and b: onepot), H‐Asn‐Hyp‐OFm⋅HCl (4), HATU, DIPEA, DMF, 0 °C to r.t., 2 h; c) Et2NH, DMF, 1 h, r.t.; d) 1 m TBAF, THF, r.t., 2 h, 77 % over four steps; e) HATU, DIPEA, DMF, r.t., 16 h, 68 %, f) BF3⋅OEt2, EtSH, r.t., 2 h; g) mCPBA, iPrOH/EtOH (2:1), r.t., 30 min, 35 % over two steps.
The final characterization of synthetic α‐amanitin was performed by CD spectroscopy. The recorded spectrum coincided well with that of the natural sample (Figure 2). The 1H‐ and 2D‐NMR‐data and the co‐injection on RP‐C18 chromatography (see Supporting Information) of the synthetic α‐amanitin were in accordance with the data of the natural one, proving undoubtedly that the α‐amanitin synthesis was successful.
Figure 2.
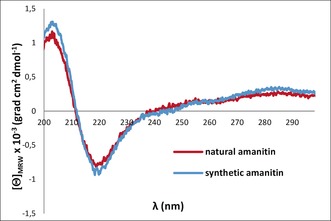
CD spectra of natural and synthetic α‐amanitin (1).
In conclusion, our herein presented synthetic strategy towards α‐amanitin is clearly different from the ground breaking work of Perrin et al.23 in several aspects: with regard to the amino acid building blocks, we developed an independent access to 6‐OH‐Htp and Dhil, both non‐trivial enterprises. For the latter amino acid, we could rely on methodologies developed by Kazmaier et al. which we could engineer into a seven‐step synthesis with high enantiomeric excess, which so far is the shortest access to this type of amino acid. Unlike the work of Perrin et al., we employed a [5+1+2]‐strategy under preformation of the thioether bond thus rendering a (pseudo‐orthogonally) protected tryptathionine. Finally, the synthesis constitutes the first α‐amanitin synthesis fully performed in solution phase. We believe that the convergent and robust synthesis, including the industrially exploitable syntheses of the amino acids Htp and Dhil, will be valuable for future therapeutic purposes in cancer therapy, when larger amounts of α‐amanitin are required.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Christian Ewers and Dr. Gerhard Jas for support of the project. Furthermore, the work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation RTG 2473 “Bioactive Peptides”, project number 392923329).
M.-A. J. Siegert, C. H. Knittel, R. D. Süssmuth, Angew. Chem. Int. Ed. 2020, 59, 5500.
In memory of Gerhard Jas
References
- 1. Wieland T., Faulstich H., Crit. Rev. Biochem. 1978, 5, 185–260. [DOI] [PubMed] [Google Scholar]
- 2. Wieland T., Int. J. Pept. Protein Res. 1983, 22, 257–276. [DOI] [PubMed] [Google Scholar]
- 3. Wienland T., Faulstich H., Experientia 1991, 47, 1186–1193. [DOI] [PubMed] [Google Scholar]
- 4. Walton J. D., Hallen-Adams H. E., Luo H., Biopolymers 2010, 94, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cochet-Meilhac M., Chambon P., Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1974, 353, 160–184. [DOI] [PubMed] [Google Scholar]
- 6. Wieland T., Schmidt G., Justus Liebigs Ann. Chem. 1952, 577, 215–233. [Google Scholar]
- 7. Wieland T., Gebert U., Justus Liebigs Ann. Chem. 1966, 700, 157–173. [DOI] [PubMed] [Google Scholar]
- 8. Zanotti G., Möhringer C., Wieland T., Int. J. Peptide Protein Res. 1987, 30, 450–459. [DOI] [PubMed] [Google Scholar]
- 9. Zanotti G., Petersen G., Int. J. Pept. Res. 1992, 551–558. [PubMed] [Google Scholar]
- 10. Shoham G., Lipscomb W. N., Wieland T., J. Am. Chem. Soc. 1989, 111, 4791–4809. [Google Scholar]
- 11. Buku A., Wieland T., Bodenmüller H., Faulstich H., Experientia 1980, 36, 33–34. [DOI] [PubMed] [Google Scholar]
- 12. Zanotti G., D′auria G., Paolillo L., Trivellone E., Int. J. Pept. Protein Res. 1988, 32, 9–20. [PubMed] [Google Scholar]
- 13. Zanotti G., Wieland T., Dauria G., Paolillo L., Trivellone E., Int. J. Pept. Protein Res. 1990, 35, 263–270. [DOI] [PubMed] [Google Scholar]
- 14. May J. P., Perrin D. M., Chem. Eur. J. 2008, 14, 3404–3409. [DOI] [PubMed] [Google Scholar]
- 15. Zhao L., May J. P., Blanc A., Dietrich D. J., Loonchanta A., Matinkhoo K., Pryyma A., Perrin D. M., ChemBioChem 2015, 16, 1420–1425. [DOI] [PubMed] [Google Scholar]
- 16. May J. P., Fournier P., Pellicelli J., Patrick B. O., Perrin D. M., J. Org. Chem. 2005, 70, 8424–8430. [DOI] [PubMed] [Google Scholar]
- 17. Anderson M. O., Shelat A. A., Guy R. K., J. Org. Chem. 2005, 70, 4578–4584. [DOI] [PubMed] [Google Scholar]
- 18. Schuresko L. A., Lokey R. S., Angew. Chem. Int. Ed. 2007, 46, 3547–3549; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3617–3619. [Google Scholar]
- 19. Yao G., Joswig J.-O., Keller B. G., Süssmuth R. D., Chem. Eur. J. 2019, 25, 8030–8034. [DOI] [PubMed] [Google Scholar]
- 20. Pahl A., Lutz C., Hechler T., Drug Discovery Today Technol. 2018, 30, 85–89. [DOI] [PubMed] [Google Scholar]
- 21. Anderl J., Faulstich H., Hechler T., Kulke M., in Antib.-Drug Conjug. (Ed.: L. Ducry), Humana Press, Totowa, NJ, 2013, pp. 51–70. [Google Scholar]
- 22. Moldenhauer G., Salnikov A. V., Lüttgau S., Herr I., Anderl J., Faulstich H., J. Natl. Cancer Inst. 2012, 104, 622–634. [DOI] [PubMed] [Google Scholar]
- 23. Matinkhoo K., Pryyma A., Todorovic M., Patrick B. O., Perrin D. M., J. Am. Chem. Soc. 2018, 140, 6513–6517. [DOI] [PubMed] [Google Scholar]
- 24. Zhou S., Wang J., Chen X., Aceña J. L., Soloshonok V. A., Liu H., Angew. Chem. Int. Ed. 2014, 53, 7883–7886; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8017–8020. [Google Scholar]
- 25. Nian Y., Wang J., Zhou S., Wang S., Moriwaki H., Kawashima A., Soloshonok V. A., Liu H., Angew. Chem. Int. Ed. 2015, 54, 12918–12922; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13110–13114. [Google Scholar]
- 26. Blaser G., Sanderson J. M., Batsanov A. S., Howard J. A. K., Tetrahedron Lett. 2008, 49, 2795–2798. [Google Scholar]
- 27. Bayer A., Kazmaier U., Chem. Eur. J. 2014, 20, 10484–10491. [DOI] [PubMed] [Google Scholar]
- 28. Bayer A., Kazmaier U., Org. Lett. 2010, 12, 4960–4963. [DOI] [PubMed] [Google Scholar]
- 29. Balmer E., Germain A., Jackson W. P., Lygo B., J. Chem. Soc. Perkin Trans. 1 1993, 399–400. [Google Scholar]
- 30. Gao Y., Klunder J. M., Hanson R. M., Masamune H., Ko S. Y., Sharpless K. B., J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar]
- 31. Weygand F., Frauendorfer E., Chem. Ber. 1970, 103, 2437–2449. [DOI] [PubMed] [Google Scholar]
- 32. Suresh Babu V. V., Vasanthakumar G.-R., Tantry S. J., Tetrahedron Lett. 2005, 46, 4099–4102. [Google Scholar]
- 33. Process for the Manufacture of Persilylated Peptides, Callens R., Delplanche T., EP2060580A1, 2009.
- 34. Fuji K., Ichikawa K., Node M., Fujita E., J. Org. Chem. 1979, 44, 1661–1664. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary