

Abstract
The photoinitiated thiol−ene addition reaction is a highly stereo‐ and regioselective, and environmentally friendly reaction proceeding under mild conditions, hence it is ideally suited for the synthesis of carbohydrate mimetics. A comprehensive study on UV‐light‐induced reactions of 2,3‐unsaturated O‐, C‐, S‐ and N‐glycosides with various thiols was performed. The effect of experimental parameters and structural variations of the alkenes and thiols on the efficacy and regio‐ and stereoselectivity of the reactions was systematically studied and optimized. The type of anomeric heteroatom was found to profoundly affect the reactivity of 2,3‐unsaturated sugars in the thiol−ene couplings. Hydrothiolation of 2,3‐dideoxy O‐glycosyl enosides efficiently produced the axially C2‐S‐substituted addition products with high to complete regioselectivity. Moderate efficacy and varying regio‐ and stereoselectivity were observed with 2,3‐unsaturated N‐glycosides and no addition occurred onto the endocyclic double bond of C‐glycosides. Upon hydrothiolation of 2,3‐unsaturated S‐glycosides, the addition of thiyl radicals was followed by elimination of the thiyl aglycone resulting in 3‐S‐substituted glycals.
Keywords: carbohydrate, synthesis, enose, thiyl radical, addition, stereoselective, regioselective
A comprehensive study on radical‐mediated hydrothiolation of 2,3‐unsaturated O‐, C‐, S‐ and N‐glycosides with a broad range of thiols was performed to gain a deeper insight into how experimental parameters and structural variations of the alkenes and thiols influence the efficacy and regio‐ and stereoselectivity of the reactions.

Introduction
Carbohydrates, in the form of oligosaccharides, polysaccharides and glycoconjugates, play a crucial role in cell surface binding events, protein stability and immunology, and represent a highly important class of biomolecules.1 In spite of their central role in biology, carbohydrates remain less exploited compared to proteins and nucleic acids, and are only rarely considered for drug discovery. A major problem with the application of oligosaccharides as biological probes or drug candidates is the sensitivity of the glycosidic bond to enzymatic hydrolysis.2 Stable mimetics have been designed to tackle this problem and the replacement of the interglycosidic oxygen by a sulfur atom to form S‐glycosides is one common tactic to generate stable analogs of native oligosaccharides and glycoconjugates.3, 4, 5
For the synthesis of thio‐linked carbohydrate mimetics, the exocyclic sulfur atom can be efficiently introduced into the sugar backbone via the photoinduced thiol−ene “click” (TEC) reaction using unsaturated pyranoses or furanoses as acceptor substrates.6 The thiol−ene reaction proceeds via a two‐step radical process in mild conditions and often quantitative yields and shows tolerance to a wide range of functional groups.7 This reaction has already been used to couple various thiols to a broad range of unsaturated sugars including glycals,8 1‐ and 2‐substituted glycals,9, 10, 11, 12, 13, 14 exoglycals15, 16, 17, 18 and pyranosyl and furanosyl exomethylene derivatives.10, 19, 20, 21
2,3‐Unsaturated glycosides are useful chiral substrates for further manipulations in organic synthesis and are easily accessible by Ferrier rearrangement of commercially available tri‐O‐acetyl glycals.22, 23, 24 Surprisingly, these types of glycosides were scarcely applied as alkene substrates in the thiol−ene coupling reactions.9, 25 Kushida and coworkers reported that acetone‐sensitized photochemical addition of ethanethiol and 1‐propanethiol to methyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranoside 1 occurred with high stereo‐ and regioselectivity giving the corresponding d‐arabino‐configured 3‐deoxy‐2‐S‐alkyl‐2‐thio‐hexopyranosides 2 a and 2 b in high yields (Scheme 1).25 We have demonstrated that UV‐initiated addition of various acetylated 1‐thiosugars to 2,3‐unsaturated ethyl glycoside 3 using 2,2‐dimethoxy‐2‐phenylacetophenone (DPAP) as the initiator also proceeded with high efficacy and selectivity, providing an easy access to 3‐deoxy‐2‐S‐disaccharides (Scheme 1, 4 a–4 d).9 Hence, 2,3‐unsaturated glycosides can be regarded as very useful yet underexploited acceptor substrates for rapid and stereoselective synthesis of stable analogs of biorelevant sugars such as, among others, 1,2‐α‐mannobiosides.26, 27
Scheme 1.
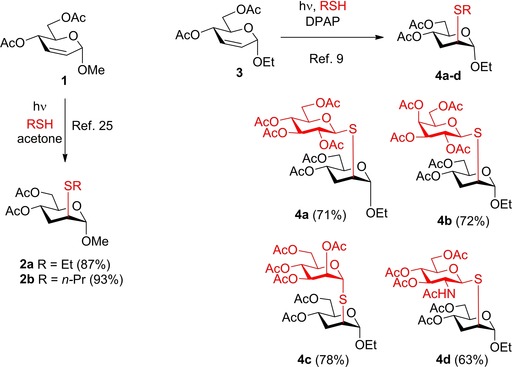
Literature results on hydrothiolation of 2,3‐unsaturated pyranosides. DPAP: 2,2‐dimethoxy‐2‐phenylacetophenone.
To obtain a better view on the scope and limitations of the TEC reaction on unsaturated sugar skeletons, we decided to carry out a detailed study on radical mediated additions of various thiols onto a broad range of 2,3‐unsaturated sugars including O‐glycosides with D‐erythro (3, 5–7) and D‐threo configuration (8) and bearing a C2‐substituent (9, 10), as well as C‐ (11), S‐ (12, 13), and N‐glycosides (14) (Figure 1).
Figure 1.
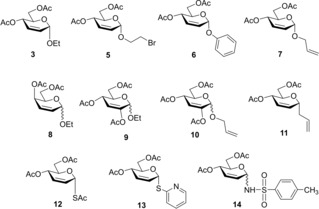
2,3‐Unsaturated glycosides used as alkene reactants.
Results and Discussion
In the first set of experiments, we investigated the effect of the anomeric substituent of enosides on the thiol−ene couplings (Table 1). Therefore, some easily available 2,3‐unsaturated sugars such as ethyl glycoside 3, 2‐bromoethyl glycoside 5, and phenyl glycoside 6 were reacted with 1‐thiosugars (15 and 18) and functionalized alkyl thiols (16 and 17). In the case of addition of 1‐thioglucose 15 to ethyl glycoside 3, the efficacy of various initiation conditions was also studied (Table 1, entries 1–4). In our previous work, 3 and 15 were reacted at room temperature upon photoinitiation to give 4 a as the sole product with 71% yield (Scheme 1).9 Recently, we have found in the case of 2‐acetoxy glycals that the reaction temperature is a crucial factor in terms of efficacy of the thiol−ene coupling, the cooling promotes while the heating inhibits the addition reactions.12, 13 We were curious how cooling and heating affect the conversion of 3. The low‐temperature photoinduced reaction was performed by the previously established standard conditions,12 irradiating at λmax=365 nm for 3×15 min in the presence of the cleavable photoinitiator 2,2‐dimethoxy‐2‐phenylacetophenone (DPAP) (Table 1, Entries 1 and 2). Cooling to 0 °C was beneficial to the conversion, yielding 75% of 4 a, and an even higher, 82% yield was achieved by running the reaction at −80 °C. Changing the method of initiation to thermal activation using azobisisobutyronitrile (AIBN)18 as the radical initiator at 120 °C proved to be detrimental to the reaction, yielding only 11% of 4 a. Recently, Renaud and co‐workers reported that triethlyborane in combination with catechol represents a very efficient initiator system for radical hydrothiolation of allylic double bonds.28 Using Et3B and catechol to initiate a reaction between 3 and 15, the conversion and isolated yield was close to the one observed in the case of photoinitiation at rt; however, the reaction required 3 days instead of 45 min. In view of these results, the most optimal initiation method for completing the thio‐click reaction was the UV irradiaton in the presence of DPAP. Hence, further reactions were performed using photoinitiation.
Table 1.
Hydrothiolation reactions of 2,3‐unsaturated O‐glycosides 3, 5 and 6 under various conditions.
|
Entry |
Solvent |
Alkene |
Thiol [equiv.] |
Conditions[a] |
Product |
Isolated yield [%][b] |
|---|---|---|---|---|---|---|
|
1 2 3 4 |
toluene toluene toluene CH2Cl2 |
3 3 3 3 |
|
0 °C, DPAP, hν −80 °C, DPAP, hν 120 °C, AIBN rt, Et3B, catechol |
|
75 82 11 67 |
|
5 6 |
toluene toluene |
3 |
|
0 °C, DPAP, hν −80 °C, DPAP, hν |
|
69 23 |
|
7 8 |
toluene toluene |
3 |
|
0 °C, DPAP, hν −80 °C, DPAP, hν |
|
46 68 |
|
9 |
toluene |
5 |
|
−40 °C/−80 °C, DPAP, hν |
|
95 |
|
10 |
toluene |
5 |
|
0 °C, DPAP, hν |
|
72 |
|
11 12 |
toluene |
5 |
|
0 °C, DPAP, hν −80 °C, DPAP, hν |
|
32 64 |
|
13 14 15 16 |
toluene toluene toluene toluene |
6 |
|
rt, DPAP, hν 0 °C, DPAP, hν −40 °C, DPAP, hν −80 °C, DPAP, hν |
|
55 56 68 74 |
|
17 18 |
toluene |
6 |
|
rt, DPAP, hν −80 °C, DPAP, hν |
|
69 78 |





[a] The reaction time was 3×15 min for the photoinduced reactions, 6 h for the AIBN mediated reaction and 3 days for the Et3B mediated reaction. [b] By‐product formation was not observed, the low/moderate yields are the results of the low/moderate conversions of the alkenes.
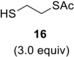
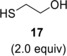
Compound 3 was hydrothiolated with ethane dithiol monoacetate 16 as well at two different temperatures. While the reaction afforded the expected axially 2‐S‐alkylated 19 as the sole product with 69% yield at 0 °C, very low conversion of 3 occurred at −80 °C (entries 5–6). When 3 was reacted with 2‐mercaptoethanol 17 at the above two temperatures, the lower temperature led to the more efficient addition reaction to give 20 in 68% yield (entries 7 and 8).
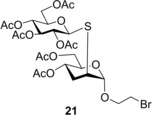
Next, the compatibility of functionalized aglycone on the thiol−ene coupling was examined. It was observed that the bromoethyl glycoside was compatible with the thio‐click reaction. Upon reacting 5 with 1‐thioglucose 15 at either −40 °C or −80 °C the reaction completed with an excellent 95% yield of thiodisaccharide 21. Addition of thiol 16 onto 5 at 0 °C afforded 22 with a good 72% yield, the lower conversion being attributed to the lower reactivity of thiol 16. When 2‐mercaptoethanol 17 was reacted with 5 at the same conditions, the yield of 23 was only 32%, which could be successfully increased to 64% by conducting the reaction at −80 °C.
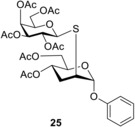
We also studied how changing the alkyl aglycone into an aryl one affects the efficacy of the reaction. Reacting phenyl glycoside 6 to thiol 15 at rt afforded the expected thiodisaccharide 24 with a satisfactory 55% yield. Cooling the reaction to 0 °C showed no significant difference in the conversion, however, cooling to −40 °C increased the yield to 68% and further cooling to −80 °C gave the best results, an excellent 74% yield. Hydrothiolation of compound 6 with 1‐thiogalactose 18 gave 25 with 69% yield at rt and an increased 78% yield at −80 °C. Although changing the alkyl aglycone to an aryl one slightly decreases the conversion of the alkene at rt, efficient additions could be elicited with 1‐thiosugars at lower temperature.
We have found that each reaction showed complete regio‐ and stereoselectivity, however, the reactivity of the thiols proved to be a crucial factor for the conversion and overall yield. Cooling was beneficial to the reactions of carbohydrate thiols and 2‐mercaptoethanol. We assume that in these cases the low temperature stabilizes the intermediate carbon‐centered radical, which is formed in the reversible thiyl addition step, thereby allowing it to react with a thiol in the hydrogen abstraction step.12, 13 In contrast, cooling was detrimental to the reactivity of alkyl thiol 16. These observations are in line with our recent results on hydrothiolation of 2‐acetoxy‐glycals13 and furanose exomethylenes.20, 21 These studies revealed that thiols bearing electron withdrawing substituents, e. g. peracetylated 1‐thiosugars, show high reactivity in the thiol−ene reactions at the −40 to −80 °C temperature interval, while the reactivity of simple alkyl thiols and the thio‐substituted derivative 16 decreased significantly by the excessive cooling, probably because the hydrogen abstraction step is inhibited at such a low temperature due to the less abstractable hydrogen of alkyl thiols.
A chemoselectivity study was carried out on allyl glycoside 7 bearing double bonds both internally and terminally (Scheme 2). The terminal double bond is more reactive than the internal one, offering the possibility to introduce different thiol moieties to the pyranose ring and the aglycone. Reacting 7 with 1.0 equiv. of 1‐thiogalactose 18 at −80 °C we observed exclusive addition onto the allyl moiety affording the thiogalactosylated 26 with 81% yield. Reacting this isolated 26 with 1.2 equiv. of 1‐thioglucose 15 at 0 °C efficient addition occurred onto the internal double bond providing pseudotrisaccharide 27 with an excellent 88% yield. Directly coupling 2.5 equivalents of the β‐1‐thiomannose derivative 28 to carbohydrate 7 resulted in the formation of the expected 29 with a good 75% yield, while less than 5% of 30 was also detected. Performing the reaction at −80 °C the yield of 29 reached 83%. It has been concluded that the terminal unsaturated bond is indeed more reactive, allowing hydrothiolation of the terminal double bond with complete chemoselectivity while the internal double bond remains intact. Hence, this reaction offers a rapid access to various pseudotrisaccharides or multivalent oligosaccharides.
Scheme 2.
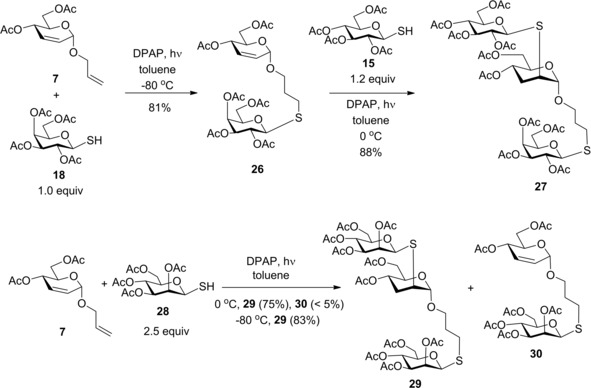
Chemoselectivity study – homo‐ and heterodithioglycosylation of the 2,3‐unsaturated allyl glycoside 7 using different thiol−alkene ratios.
Next, the impact of the enose configuration was studied. The reactions were performed with an inseparable 5 : 1 α : β mixture of ethyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐d‐threo‐hex‐2‐enopyranoside 8 (Scheme 3). Hydrothiolation of the anomeric mixture of 8 with 1‐thioglucose 15 at different temperatures gave two regioisomers, compounds 31 and 32. At rt the overall yield was 78% with isomeric ratio of 31 : 32∼3 : 1. Cooling the reaction to −80 °C increased the overall yield to 80% and increased the regioselectivity in favor of the C‐2 thiolated product to reach a 31 : 32=5 : 1 ratio. The structure of the crystalline major product 31 was confirmed by X‐ray measurements (Figure 2). The constitution and stereochemistry of the minor product was determined on the basis of NMR spectra, and the C3 configuration was confirmed by cross‐peak detected between the H‐3 and H‐1 atoms in the ROESY NMR spectrum of 32 (See Figure S1).
Scheme 3.

The effect of alkene‐configuration on the regioselectivity of thiol−ene reactions.
Figure 2.
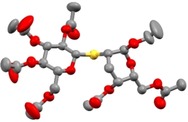
ORTEP view of thiodisaccharide 31; hydrogen atoms are omitted for clarity.
Reacting the unsaturated carbohydrate 8 with the d‐galacto configured thiol 18 in toluene also showed incomplete regioselectivity. At rt the 33 : 34 ratio was 3.8 : 1 with a satisfactory 74% yield. Next the solvent was changed to methanol and the temperature was lowered to −40 °C. We observed that the overall yield decreased slightly to 69%, however, the regioselectivity increased to 6.7 : 1 in favor of the 2‐thiolated product 33. Further cooling the reaction to −80 °C in toluene the ratio of 33 : 34 reached 7.3 : 1 with 73% combined yield.
The regioselectivity of the thiol−ene reactions can be explained by the enhanced stability of the intermediate carbon‐centered radical upon addition of the thiyl radical to the less substituted olefinic carbon atom.29, 30 Although the C2 and C3 carbons are equally substituted in enosides 3 and 5–8 studied above, the addition reactions proceed preferably or exclusively through the C3 centered radical intermediate due to its higher stability. The spatial arrangement of the C4 substituent proved to have a crucial effect on the regio‐ and stereochemical outcome of the reactions. In the D‐erythro cases, as it is shown in Figure 3 on the example of 3, the stable 4 C 1 conformer of the C3‐centered radical intermediate (3‐C3R) can readily form by upper‐face attack of the thiyl radical to C2 of the O H 5 conformational form of the starting enosides leading to exclusive formation of the C2‐thiolated products. In the case of the D‐threo‐configured 8, the axial 4‐OAc group exerts a shielding effect on the β‐side of the double bond, inhibiting the attack of thiyl radicals on the upper‐face and opening up the way for a bottom‐face attack onto C3. Moreover, the 1,3‐syn‐diaxial repulsion between substituents at C‐2 and C‐4 decreases the stability of the resulting radical intermediate 8‐C3R. We assume that these factors result in a lower level of selectivity of the thiol−ene reactions of 8. Although cooling the reactions promotes the formation of the 3‐deoxy‐2‐S‐disaccharides 31 and 33 the regioselectivity was incomplete even at −80 °C.
Figure 3.
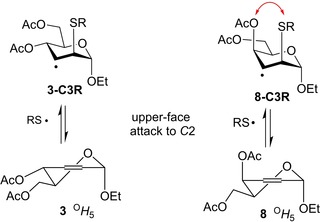
Formation of carbon‐centered radical intermediates upon thiol−ene reactions of enosides 3 and 8.
Importantly, the addition proceeded with complete stereoselectivity at both the C2 and C3 positions providing solely the axially linked 2‐thio‐d‐lyxo (31 and 33) and the axially linked 3‐thio‐d‐xylo (32 and 34) disaccharides, which is in line with the literature results. It was demonstrated in the case of substituted cyclohexenes,30, 31 as well as 1‐ and 2‐substituted glycals9, 10, 11, 12, 13 that the addition occurs preferentially in an anti fashion as the result of a kinetically favored axial attack of the thiyl radical onto the cyclic alkene in its half‐chair conformation together with a stereoselective hydrogen abstraction from the thiol into an axial position.
Next, we studied the hydrothiolation of 2‐substituted 2,3‐unsaturated glycosides to learn the impact of the C2‐substituent on the regio‐ and stereoselectivity and efficacy of the addition (Scheme 4). In this case the starting glycosides 9 and 10 were inseparable anomeric mixtures. Reacting ethyl glycoside 9 with α‐1‐thiomannose 35 at rt moderate conversion occurred to give 36 with 35% yield. Cooling the reaction to −80 °C however increased the isolated yield of 36 to 68%. The D‐gluco configuration of the ethyl glycoside unit was determined by NMR measurements on the basis of the 3 J H2,H3=11.8 Hz and 3 J H3,H4=11.3 Hz coupling constants and the cross‐peak detected between the H‐3 and H‐5 atoms in the ROESY NMR spectrum of 36. Reacting carbohydrate 9 with β‐1‐thioglucose 15 at 0 °C gave 37 with 42% yield. In this case the cooling did not improve the conversion significantly, the reaction repeated at −80 °C gave 37 with 50% yield. Although the isolated 37 contained stereoisomeric impurities, the D‐allo configuration of the major component could be undoubtedly determined by the 1H NMR coupling constants of H‐3 (3 J H2,H3=3 J H3,H4 4.7 Hz) and the ROESY connectivities detected between the H2, H3 and H4 protons of the ethyl glycoside unit (See Figure S1). It can be seen that the regioselectivity of the reaction can be controlled via substitution of position 2 and the hydrothiolation proceeded with full regio‐ and high stereoselectivity. However, surprisingly, the efficacy and the stereochemical result of the reactions strongly depended on the configuration of the thiol. The different stereochemical outcome of the reactions with the different thiosugars can be explained by double stereodifferentiation of the donor and acceptor compounds, which phenomenon is well‐documented in the field of chemical glycosylation.32, 33, 34 Moreover, the results demonstrate that the α anomer of the starting enoside 9 was more reactive than the β one, as we could not detect any thiodisaccharide products bearing β‐ethyl aglycone. (However, it is possible that products were formed from the minor, β‐anomer of 9 in such low amounts that they remained undetected in the reaction mixture.) Upon hydrothiolation of allyl glycoside 10 with 1.2 equiv. of thiol 15 the addition occurred exclusively on the terminal unsaturated bond with an outstanding 90% yield while retaining the anomeric ratio of α : β∼2 : 1 at the O‐glycoside unit.
Scheme 4.
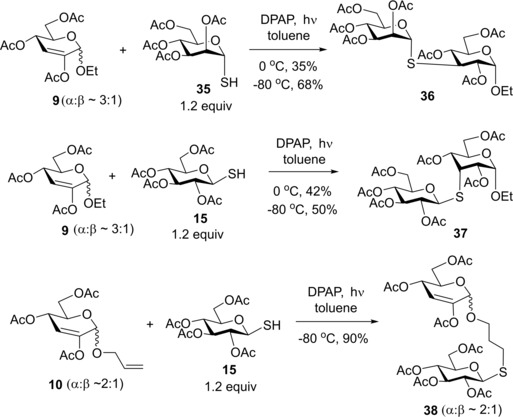
Hydrothiolation of 3‐deoxy‐2,3‐unsaturated glycosides – the impact of the C2‐substituent on the addition.
Following the successful hydrothiolations of O‐allyl glycosides, our focus turned to C‐allyl ones (Scheme 5). The first attempts to selectively couple thiol 15 to the terminal unsaturated bond of 11 were successful, at rt compound 39 was isolated with 40% yield, and increasing the thiol excess to 2.5 equiv. while lowering the temperature to −80 °C raised the yield to 48%. However, attempted hydrothiolations of the internal unsaturated bond of 39 with 8 equivalents of 1‐propanethiol and excess of thiol 15 (2 equiv.) were unsuccessful. Traces of addition product (40) could be detected by MS analysis of the reaction mixtures, however, only unreacted 39 was recovered and no product could be isolated in either case. Afterwards, compound 11 was reacted with 3 equiv. of thiol 15, however, still only compound 39 was isolated. Hydrothiolation of 11 with 3 equiv. of thiol 18 resulted in the exclusive formation of compound 41 along with sulfoxide derivative 42 which was detected in the reaction mixture by MS, and no reaction occurred on the internal unsaturated bond. We have concluded that although the thioladdition can be performed on the more reactive terminal double bond, the conversion is significantly lower than the ones experienced with O‐glycosides, and the internal double bond practically proved intact in the thiol−ene reaction.
Scheme 5.

Hydrothiolation of the 2,3‐unsaturated allyl C‐glycoside 11.
Next, hydrothiolation of 2,3‐unsaturated S‐glycosides was taken into focus (Scheme 6). Surprisingly, when 1‐acetylthio derivative 12 was reacted with thiol 35 at rt, the expected addition product could not be detected. Instead, moderate conversion of the starting compounds and formation of a complex mixture were observed from which the 3‐thio‐substituted glycals 43 and 44 along with disulfide 45 were isolated. We tested whether the allyl rearrangement of 12 producing 43 can be elicited by UV‐light irradiation without any thiol, however, no reaction occurred. Afterwards, thioacetic acid was reacted with 1‐thioenose 12, and compound 43 was isolated again as the sole product. Subjecting the arylthio enoside 13 to hydrothiolation with thiol 35, only allyl rearrangement and formation of disulfide were observed again to give 3‐thioglycosylated glycal 44 (40%) and disulfide 45.
Scheme 6.

Thiol−ene coupling reactions of 2,3‐unsaturated S‐glycosides.
The proposed mechanism of this rearrangement is shown on Scheme 7. After addition of the thiyl radical, the formed 1,3‐dithio C2‐centered radical II rapidly decomposes either by homolytic cleavage of the C3−SR2 bond to give the starting enoside and thiyl radical or by homolytic cleavage of the C1−SR1 bond resulting in glycal III and the aglycone‐derived thiyl radical (R1S radical). Addition of the latter R1S radical onto enoside I leads to glycal V via intramolecular decomposition of the 1,3‐dithio C2‐centered radical intermediate IV. Similar failure of radical mediated hydrothiolation of S‐allyl cysteine derivative has been reported previously by Dondoni, Marra and co‐workers.35
Scheme 7.
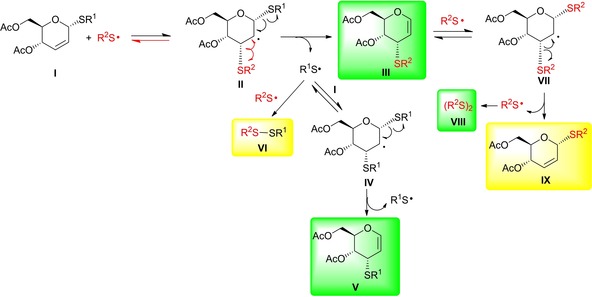
Proposed mechanism for hydrothiolation of 2,3‐unsaturated thioglycosides (Structure of products observed are highlighted in green, further possible products are highlighted in yellow).
Finally, thiol−ene coupling reactions of the 2,3‐unsaturated N‐glycoside 14 was studied (Scheme 8). Compound 14 consisted of an inseparable anomeric mixture of α : β∼3 : 1 which posed difficulties in the purification of the addition products. Reacting compound 14 with thiol 15 at rt and 0 °C no reaction was detected. Running the reaction at −40 °C resulted in the formation of a multicomponent mixture, of which only compound 46 could be isolated and the starting compound was also recovered mainly in α‐anomeric form. Reacting 14 with 1‐thioxylose 47 also resulted in a multicomponent mixture consisting of compounds 48 and 49 as the major components. Interestingly enough, only hydrothiolation products arisen from the β‐anomer of 14 have been isolated from the reactions above. It is known that the addition of thiyl radicals requires electron‐rich double bonds. The results suggest that the electron density of the double bond might be significantly different in the α and β anomers of the starting enoside. The electron withdrawing effect of the sulfonyl group might be more pronounced in the α anomer leading to its decreased reactivity in the thiol−ene coupling reaction. Similarly to the thiol−ene couplings of 2‐acetoxy‐2,3‐unsaturated‐O‐glycosides, the stereochemical outcome of the reactions was controlled by the configuration of the thiols.
Scheme 8.

Hydrothiolation of 2,3‐unsaturated N‐glycoside 14.
Conclusions
The effect of the anomeric heteroatom of enosides on the radical‐mediated thiol−ene coupling reactions was studied for the first time. It has been demonstrated that photoinitiated thiol−ene additions of 2,3‐unsaturated‐O‐glycosides proceed with high to complete regioselectivity and stereoselectivity in favor of the C‐2‐axially‐linked products. Additions onto D‐erythro‐configured 2,3‐dideoxy‐2,3‐unsaturated‐O‐glycosides occurred with exclusive regio‐ and stereoselectivity to afford the D‐arabino‐configured C‐2 thioalkylated or thioglycosylated products. The photoinduced thiol−ene coupling proved to be compatible with the 2‐bromoethyl aglycone of 5, and proceeded efficiently both with alkyl and aryl glycosides. Cooling the reaction mixture was generally beneficial to the conversion, however, lower conversions were observed at low temperature with thiol 16. Hydrothiolation reactions of the D‐threo‐configured glycoside 8 led to a mixture of the C‐2‐thio and C‐3‐thio products revealing that the regioselectivity of additions can be modified by changing the configuration of the unsaturated glycosides, and the level of regioselectivity was also found to be controlled by the configuration of the thiols. We have found that the addition occurred with complete axial stereoselectivity at both the C2 and C3 positions, moreover, both the yields and regioselectivity could be effectively increased by cooling. Substitution at C2 position of 9 led to reversed regioselectivity and slightly decreased reactivity, probably due to steric congestion. In this case the stereochemistry of the reaction seems to be unpredictable: addition of α‐1‐thiomannose occurred with d‐gluco selectivity while addition of β‐1‐thioglucose provided the d‐allo configured product.
Changing the glycosidic oxygen to carbon, sulfur and nitrogen profoundly affects the reactivity of the C2−C3 double bond. In the case of the C‐allyl glycoside, the lack of anomeric oxygen slightly decreases the reactivity of the terminal double bond and completely inhibits the radical hydrothiolation of the internal one. The S‐allyl derivatives are not useful alkenes in thiol−ene reactions, as they suffer allyl rearrangement upon hydrothiolation due to the high lability of the forming 1,3‐dithio carbon‐centered radical intermediate.
The reactivity of the unsaturated N‐glycoside 14 and the 2‐substituted O‐glycoside 9 proved to be highly dependent on the anomeric configuration of the enosides and also on the stereochemistry of thiols.
If only moderate/low yields were observed in the thiol−ene reactions, it was the result of moderate/low conversion of the alkene. In these cases, a disulfide by‐product was always formed from the thiol but not in significant amounts, and the unreacted thiol and alkene could be recovered from the reaction mixture.
Our results demonstrate that the photoinduced addition of thiols to 2,3‐unsaturated O‐glycosides is a mild, atom economic and efficient method for the synthesis of carbohydrate mimetics with diverse regio‐ and stereoselectivity. Hydrothiolation reactions of N‐glycosides are worth further studying because they provide an opportunity to synthesize valuable glycomimetics.
Experimental Section
General method for photoinitiated free radical thioladdition
Ethyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranoside 3,36 2‐bromoethyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranoside 5,37 phenyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranoside 6,36 allyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranoside 7,38 ethyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α,β‐d‐threo‐hex‐2‐enopyranoside 8,39 ethyl 2,4,6‐tri‐O‐acetyl‐3‐deoxy‐α,β‐d‐erythro‐hex‐2‐enopyranoside 9,40, 41 allyl 2,4,6‐tri‐O‐acetyl‐3‐deoxy‐α,β‐d‐erythro‐hex‐2‐enopyranoside 10,41 3‐(4,6‐Di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranosyl)‐1‐propene 11 42 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐1‐thio‐1‐S‐acetyl‐α‐d‐erythro‐hex‐2‐enopyranose 12,43 pyperidin‐2‐yl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐1‐thio‐α‐d‐erythro‐hex‐2‐enopyranoside 13,44 N‐(4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α,β‐d‐erythro‐hex‐2‐enopyranosyl) p‐toluenesulfonamide 14,45 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranose 15,46 mono‐S‐acetyl‐ethanedithiol 16,47 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐galactopyranose 18,48 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐mannopyranose 28,49 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐mannopyranose 35 50 and 2,3,4‐tri‐O‐acetyl‐1‐thio‐β‐d‐xylopyranose 47 51 were prepared according to the literature procedures. 2‐Mercaptoethanol (17) and initiator 2,2‐dimethoxy‐2‐phenylacetophenone (DPAP) were purchased from Sigma Aldrich Chemical Co. Optical rotations were measured at room temperature with a Perkin‐Elmer 241 automatic polarimeter. TLC was performed on Kieselgel 60 F254 (Merck) with detection by UV‐light (254 nm) and immersing into sulfuric acidic ammonium−molibdenate solution or 5% ethanolic sulfuric acid followed by heating. Flash column chromatography was performed on Silica gel 60 (Merck 0.040–0.063 mm), column chromatography was performed on Silica gel 60 (Merck 0.063–0.200 mm). Organic solutions were dried over Na2SO4 or MgSO4, and concentrated in vacuum. One‐ and two‐dimensional 1H, 13C, COSY and HSQC spectra were recorded with Bruker DRX‐360 (1H: 360 MHz; 13C: 90 MHz), Bruker DRX‐400 (1H: 400 MHz; 13C: 100 MHz) and Avance II 500 (1H: 500.13 MHz; 13C: 125.76 MHz) spectrometers at 25 °C. Chemical shifts are referenced to Me4Si (0.00 ppm for 1H) and to the residual solvent signals (CDCl3: 77.1, CD3OD: 49.3 for 13C). MALDI‐TOF MS analyses of the compounds were carried out in the positive reflectron mode using a BIFLEX III mass spectrometer (Bruker, Germany) equipped with delayed‐ion extraction. 2,5‐Dihydroxybenzoic acid (DHB) was used as matrix and F3CCOONa as cationising agent in DMF. ESI‐TOF HRMS spectra were recorded by a microTOF−Q type QqTOFMS mass spectrometer (Bruker) in the positive ion mode using MeOH as the solvent. CCDC deposition number for compound 31: 1941430.
The photocatalytic reactions were carried out in a borosilicate vessel by irradiation with a 75 W Hg‐lamp giving maximum emission at 365 nm. The mercury lamp was placed in a water‐cooled immersion mantle. The samples to be irradiated were placed in a 25–50 mL borosilicate flask located ∼2 cm away from the lamp. The samples were not purged to remove the oxygen and were not stirred during the reaction. In case of low‐temperature reactions, the reaction flask was submerged in a cooling bath (acetone‐liquid nitrogen mixture in a Dewar flask). During irradiation, the entire set‐up was covered in aluminum foil. The corresponding alkene, thiol and DPAP (DPAP, 0.1 equiv./alkene) were dissolved in the given solvent. The reaction mixture was cooled to the given temperature and was irradiated with UV‐light for 15 min. After irradiation another 0.1 equiv. of DPAP dissolved in the given solvent, was added to the reaction mixture and the irradiation continued for another 15 min. The addition of 0.1 equiv. of DPAP and the irradiation was repeated one more time. The solvent was evaporated in vacuo and the crude product was purified by column chromatography or flash column chromatography.
Ethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐ glucopyranosyl)‐2‐thio‐α‐d‐arabino‐hexopyranoside (4 a)9
A: Compound 3 (0.3 mmol, 86 mg) in toluene (2.0 mL) was reacted with thiol 15 (0.36 mmol, 131 mg, 1.2 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75 : 25) to give compound 4 a (141 mg, 75%) as colourless foam. The reaction was performed in the same scale at −80 °C to 4 a with 82% yield.
B: Compound 3 (0.3 mmol, 86 mg), thiol 15 (0.36 mmol, 131 mg, 1.2 equiv.) and AIBN (0.03 mmol, 5 mg, 0.1 equiv) were dissolved in dry toluene (3.0 mL). The reaction mixture was put in a pressure cylinder and was heated to 120 °C for 6 hours. Afterwards the solvent was evaporated in vacuo. The crude product was purified by flash chromatography to give compound 4 a (21 mg, 11%).
C: Compound 3 (1.0 mmol, 258 mg), thiol 15 (1.2 mmol, 438 mg, 1.2 equiv.), triethylborane solution (15% in n‐hexane) (1.2 mmol, 0.784 mL, 1.2 equiv) and catechol (1.2 mmol, 132 mg, 1.2 equiv) were dissolved in dry dichloromethane (2.0 mL). The reaction mixture was stirred at room temperature for three days. Afterwards the solvent was evaporated in vacuo. The crude product was purified by flash chromatography to give compound 4 a (420 mg, 67%). [α]D 20+10.6 (c 1.18, CHCl3), lit. [9] [α]D 20+10.5.
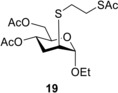
Ethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2‐acetylthioethyl)‐2‐thio‐ α‐d‐arabino‐hexopyranoside (19)
A: Compound 3 (0.5 mmol, 129 mg) in toluene (3 mL) was reacted with thiol 16 (1.5 mmol, 0.204 mg, 3.0 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8 : 2) to give compound 19 (123 mg, 69%) as yellow syrup.
B: The reaction was repeated at −80 °C with 3.0 equiv. of thiol 16 to give compound 19 with 23% yield. [α]D 20+76.4 (c 0.25, CHCl3); R f=0.48 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.05 (dd, J=8.7 Hz, J=15.2 Hz, 1H, H‐4), 4.82 (s, 1H, H‐1), 4.21 (dd, J=5.6 Hz, J=11.9 Hz, 1H, H‐6a), 4.15 (d, J=10.6 Hz, 1H, H‐6b), 3.98–3.94 (m, 1H, H‐5), 3.80–3.72 (m, 1H, OCH 2a−CH3), 3.59–3.51 (m, 1H, OCH 2b−CH3), 3.07 (dd, J=6.7 Hz, J=8.9 Hz, 3H, H‐2, CH 2−SAc), 2.77–2.73 (m, 2H, SCH 2), 2.20–2.18 (m, 2H, H‐3a,b), 2.35, 2.09, 2.05 (3×s, 9H, 3×Ac−CH 3), 1.25 (t, J=6.9 Hz, 3H, OCH2−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 195.3 (1C, SCOCH3), 170.9, 169.8 (2C, 2×OCOCH3), 99.2 (1C, C‐1), 69.0 (1C, C‐5), 65.4 (1C, C‐4), 63.3 (1C, OCH2−CH3), 63.2 (1C, C‐6), 44.2 (1C, C‐2), 31.8 (1C, SCH2), 30.7, (2C, C‐3, SAc−CH3), 29.3 (CH2−SAc), 21.1, 20.8 (2C, 2×Ac−CH3), 15.1 (1C, OCH2−CH3) ppm; MALDI‐TOF HRMS: m/z calcd for C16H26NaO7S2 [M+Na]+ 417.1018, found 417.1014.
Ethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2‐hydroxyethyl)‐2‐thio‐α‐ d‐arabino‐hexopyranoside (20)
A: Compound 3 (0.5 mmol, 129 mg) in toluene (3 mL) was reacted with thiol 17 (1.0 mmol, 0.078 mL, 2.0 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8 : 2) to give compound 20 (78 mg, 46%) as yellow syrup.
B: The reaction was repeated at −80 °C under the same conditions to give compound 20 (116 mg, 68%) as yellow syrup. [α]D 20+60.0 (c 0.14, CHCl3); R f=0.19 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.07 (dd, J=7.4 Hz, J=16.6 Hz, 1H, H‐4), 4.82 (d, J=1.5 Hz, 1H, H‐1), 4.21 (dd, J=5.5 Hz, J=12.0 Hz, 1H, H‐6a), 4.16 (dd, J=2.6 Hz, J=11.9 Hz, 1H, H‐6b), 3.98–3.95 (m, 1H, H‐5), 3.80–3.72 (m, 3H, OCH 2a,b, OCH 2a−CH3), 3.58–3.52 (m, 1H, OCH 2b−CH3), 3.03 (d, J=1.7 Hz, 1H, H‐2), 2.83–2.76 (m, 2H, SCH 2), 2.43 (s, 1H, OH), 2.18 (dd, J=4.4 Hz, J=7.2 Hz, 2H, H‐3a,b), 2.09, 2.05 (2×s, 6H, 2×Ac−CH 3), 1.25 (t, J=7.1 Hz, 3H, OCH2−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 171.0, 170.0 (2C, 2×COCH3), 99.4 (1C, C‐1), 69.1 (1C, C‐5), 65.6 (1C, C‐4), 63.4 (1C, OCH2−CH3), 63.2 (1C, C‐6), 61.1 (1C, CH2OH), 44.3 (1C, C‐2), 35.5 (1C, SCH2), 30.2, (1C, C‐3), 21.1, 20.9 (2C, 2×Ac−CH3), 15.1 (1C, OCH2−CH3) ppm; MALDI‐TOF MS: m/z calcd for C14H24NaO7S [M+Na]+ 359.114, found 359.167.
2‐Bromoethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐ acetyl‐β‐d‐glucopyranosyl)‐2‐thio‐α‐d‐arabino‐hexopyranoside (21)
Compound 5 (0.57 mmol, 192 mg) in toluene (2.0 mL) was reacted with thiol 15 (0.68 mmol, 249 mg, 1.2 equiv.) at −80 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75 : 25) to give compound 21 (378 mg, 95%) as white powder. [α]D 20+14.1 (c 0.29, CHCl3); R f=0.18 (n‐hexane/acetone 7 : 3); M.p.: 124–126 °C; 1H NMR (400 MHz, CDCl3) δ 5.23 (t, J=9.3 Hz, 1H, H‐3′), 5.07 (dd, J=10.0 Hz, J=20.1 Hz, 2H, H‐2′, H‐4′), 4.92 (s, 1H, H‐1), 4.88 (dd, J=5.1 Hz, J=9.3 Hz, 1H, H‐4), 4.65 (d, J=10.0 Hz, 1H, H‐1′), 4.27 (dd, J=4.8 Hz, J=12.3 Hz, 1H, H‐6′a), 4.21–4.11 (m, 3H, H‐6a,b, H‐6′b), 4.03–3.97 (m, 2H, H‐5, OCH 2a), 3.86 (dt, J=5.8 Hz, J=11.3 Hz, 1H, OCH 2b), 3.74–3.72 (m, 1H, H‐5′), 3.55 (t, J=5.8 Hz, 2H, CH 2−Br), 3.30 (s, 1H, H‐2), 2.28–2.21 (m, 1H, H‐3a), 2.14–2.10 (m, 1H, H‐3b), 2.10, 2.09, 2.07, 2.05, 2.03, 2.01 (6×s, 18H, 6×Ac−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.9, 170.6, 170.2, 169.7, 169.5, 169.4 (6C, 6×COCH3), 100.5 (1C, C‐1), 82.8 (1C, C‐1′), 76.2 (1C, C‐5′), 73.7 (1C, C‐3′), 69.5 (1C, C‐2′), 69.4 (1C, C‐5), 68.2 (1C, C‐4′), 68.0, (1C, OCH2),65.1 (1C, C‐4), 63.0 (1C, C‐6), 62.0 (1C, C‐6′), 41.8 (1C, C‐2), 30.5 (1C, C‐3), 30.4 (1C, CH2−Br), 21.1, 20.9, 20.8, 20.7, 20.6 (6C, 6×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C26H37BrNaO15S [M+Na]+ 723.0934, found 723.0910.
2‐Bromoethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2‐ acetylthioethyl)‐2‐thio‐α‐d‐arabino‐hexopyranoside (22)
Compound 5 (0.6 mmol, 202 mg) in toluene (2.0 mL) was reacted with thiol 16 (1.8 mmol, 0.245 mg, 3.0 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 85 : 15) to give compound 22 (204 mg, 72%) as yellow syrup. R f=0.16 (n‐hexane/acetone 85 : 15); 1H NMR (400 MHz, CDCl3) δ 5.04 (td, J=8.9 Hz, J=6.0 Hz, 1H, H‐4), 4.88 (d, J=2.1 Hz, 1H, H‐1), 4.18 (d, J=4.2 Hz, 2H), 4.07–3.97 (m, 2H), 3.94–3.83 (m, 1H), 3.54 (t, J=6.0 Hz, 2H CH 2−Br), 3.15 (td, J=4.3 Hz, J=2.0 Hz, 1H, H‐2), 3.11–3.04 (m, 2H), 2.79–2.73 (m, 2H), 2.35 (s, 3H, CH 3), 2.23–2.17 (m, 2H, H‐3a,b), 2.09 (s, 3H, CH 3), 2.06 (s, 3H, CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 195.3 (1C, SCOCH3), 170.8, 169.8 (2C, 2×OCOCH3), 99.8 (1C, C‐1), 69.5(1C, C‐5), 67.9(1C, OCH2), 65.3(1C, C‐4), 63.1 (1C, C‐6), 43.8(1C, C‐2), 31.8 (1C, SCH2), 30.7 (1C, SAc−CH3), 30.4, 29.9, 29.3 (3C, 2×SCH2 and CH2−Br), 21.1, 20.9 (2C, 2×OAc−CH3) ppm; MALDI‐TOF MS: m/z calcd for C16H25BrNaO7S2 [M+Na]+ 495.012, found 495.08.
2‐Bromoethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2‐hydroxyethyl)‐2‐ thio‐α‐d‐arabino‐hexopyranoside (23)
A: Compound 5 (0.92 mmol, 309 mg) in toluene (2.0 mL) was reacted with thiol 17 (1.84 mmol, 0.128 mL, 2.0 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8 : 2) to give compound 23 (115 mg, 32%) as yellow syrup.
B: The reaction was repeated at the same scale at −80 °C to give 23 with 64% yield. [α]D 20+59.3 (c 0.27, CHCl3); R f=0.25 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.14–4.99 (m, 1H, H‐4), 4.88 (d, J=2.1 Hz, 1H, H‐1), 4.18 (d, J=4.2 Hz, 2H), 4.07–3.98 (m, 1H), 3.90–3.83 (m, 1H), 3.76 (t, J=6.0 Hz, 2H, OCH 2), 3.54 (t, J=5.9 Hz, 2H, CH 2−Br), 3.15–3.06 (m, 1H, H‐2), 2.81 (t, J=6.0 Hz, 2H), 2.21–2.16 (m, 2H, H‐3a,b), 2.12 (d, J=4.9 Hz, 1H), 2.09 (d, J=3.1 Hz, 3H, CH 3), 2.06 (s, 3H, CH 3), 1.26 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.8, 170.0 (2C, 2×COCH3), 99.9 (1C, C‐1), 69.5 (1C, C‐5), 67.8 (1C, OCH2), 65.3(1C, C‐4), 63.1(1C, C‐6) 61.1(1C, OCH2), 43.9(1C, C‐2), 35.3 (1C, OCH2), 30.4, 30.0 (2C, SCH2 and CH2−Br), 21.1, 20.8 (2C, 2×Ac−CH3) ppm; MALDI‐TOF MS: m/z calcd for C14H23BrNaO7S [M+Na]+ 437.025, found 437.121.
Phenyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐ glucopyranosyl)‐2‐thio‐α‐d‐arabino‐hexopyranoside (24)
A: Compound 6 (0.435 mmol, 133 mg) in toluene (2.0 mL) was reacted with thiol 15 (0.522 mmol, 190 mg, 1.2 equiv.) at rt according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75 : 25) to give compound 24 (150 mg, 55%) as yellow syrup.
B: The reaction was repeated at 0 °C, to give 24 with 56% yield C: The reaction was repeated at −40 °C, the yield of 24 was 68%.
D: The reaction was repeated at −80 °C, the yield of 24 was 74%. [α]D 20 −12.7 (c 0.33, CHCl3); R f=0.25 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 7.39–7.21 (m, 2H, arom), 7.09–7.00 (m, 3H, arom), 5.63 (s, 1H, H‐1), 5.23 (t, J=9.4 Hz, 1H), 5.17–5.03 (m, 2H), 4.95 (td, J=10.3 Hz, J=4.8 Hz, 1H), 4.62 (d, J=9.8 Hz, 1H, H‐1′), 4.17 (dt, J=12.4 Hz, J=4.8 Hz, 2H), 4.10 (dd, J=7.8 Hz, J=2.4 Hz, 1H), 3.99 (ddd, J=9.8 Hz, J=5.6 Hz, J=2.4 Hz, 1H), 3.73 (ddd, J=10.0 Hz, J=4.1 Hz, J=2.3 Hz, 1H), 3.59–3.44 (m, 1H), 2.46 (ddd, J=13.0 Hz, J=10.7 Hz, J=4.6 Hz, 1H, H‐3a), 2.24 (dt, J=13.1 Hz, J=4.3 Hz, 1H, H‐3b), 2.08 (s, 3H, Ac−CH 3), 2.07–2.05 (m, 1H, H‐2), 2.04 (s, 3H, Ac−CH 3), 2.00 (s, 6H, 2×Ac−CH 3), 1.98 (s, 3H, Ac−CH 3), 1.78 (s, 3H, Ac−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.6, 170.5, 170.0, 169.5, 169.4, 169.3 (6C, 6×COCH3), 156.1 (1C, CAr−O), 129.5, 129.4, 122.4, 116.4 (5C, arom), 97.9 (1C, C‐1), 82.5 (1C, C‐1′), 76.1, 73.6, 69.5, 69.1, 67.9, 64.7 (6C, skeleton carbons), 62.6, 61.6 (2C, C‐6, C‐6′), 41.9 (1C, C‐2), 30.4 (1C, C‐3), 20.9, 20.7, 20.5, 20.5, 20.2 (6C, 6×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C30H38NaO15S [M+Na]+ 693.1829, found 693.1819.
Phenyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐ galactopyranosyl)‐2‐thio‐α‐d‐arabino‐hexopyranoside (25)
A: Compound 6 (0.36 mmol, 110 mg) in toluene (2.0 mL) was reacted with thiol 18 (0.432 mmol, 157 mg, 1.2 equiv.) at rt according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75 : 25) to give compound 25 (166 mg, 69%) as colourless syrup. B: The reaction was repeated at −80 °C to give 25 with 78% yield. [α]D 20+30.8 (c 0.13, CHCl3); R f=0.29 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 7.36–7.23 (m, 1H), 7.13–7.00 (m, 3H, arom), 5.64 (s, 1H, H‐1), 5.41 (d, J=3.3 Hz, 1H), 5.30 (t, J=9.9 Hz, 1H), 5.05 (dd, J=10.0 Hz, J=3.3 Hz, 1H), 4.98 (td, J=10.0 Hz, J=4.7 Hz, 1H), 4.63 (d, J=10.0 Hz, 1H, H‐1′), 4.18–4.00 (m, 4H), 3.94 (t, J=6.6 Hz, 1H), 2.51–2.40 (m, 1H), 2.33–2.22 (m, 1H), 2.19–2.12 (m, 1H), 2.08, 2.05, 2.04, 2.00, 1.99, 1.98 (6 s, 6×3H, 6×Ac−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.3, 170.1, 169.9, 169.7, 169.6 (6C, COCH3), 156.3 (1C, CAr−O), 129.6, 129.5, 122.6, 117.1, 116.5 (5C, arom), 98.3 (1C, C‐1), 83.5 (1C, C‐1′), 74.8, 71.8, 69.6, 67.2, 66.6, 64.8 (6C, skeleton carbons), 62.7, 61.4 (2C, C‐6), 42.4 (1C, C2), 30.4 (1C, C‐3), 21.0, 20.8, 20.7, 20.6, 20.6, 20.5 (6C, 6×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C30H38NaO15S [M+Na]+ 693.1829, found 693.1814.
3‐(2,3,4,6‐Tetra‐O‐acetyl‐1‐thio‐β‐d‐galactopyranosyl)‐n‐propyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranoside (26)
Compound 7 (0.75 mmol, 200 mg) in toluene (2.0 mL) was reacted with thiol 18 (0.75 mmol, 273 mg, 1.0 equiv.) at −80 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75 : 25) to give compound 26 (385 mg, 81%) as yellow syrup. [α]D 20+28.9 (c 0.26, CHCl3); R f=0.1 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.96–5.79 (m, 2H, CH=CH), 5.43 (d, J=3.3 Hz, 1H, H‐1), 5.31 (dd, J=9.7 Hz, J=1.5 Hz, 1H), 5.23 (t, J=10.0 Hz, 1H), 5.07–5.03 (m, 2H), 4.50 (d, J=9.9 Hz, 1H, H‐1′), 4.26 (dd, J=12.1 Hz, J=5.3 Hz, 1H), 4.20 (d, J=2.5 Hz, 1H, H1‐S), 4.18–4.11 (m, 2H), 4.12–4.04 (m, 1H), 3.95 (t, J=6.7 Hz, 1H), 3.87 (dt, J=9.8 Hz, J=6.1 Hz, 1H, CH 2O), 3.59 (dt, J=9.8 Hz, J=6.1 Hz, 1H, CH 2O), 2.80 (qt, J=12.8 Hz, J=7.2 Hz, 2H, CH 2S), 2.16 (s, 3H, CH 3), 2.11 (s, 3H, CH 3), 2.09 (s, 3H, CH 3), 2.07 (s, 3H, CH 3), 2.04 (s, 3H, CH 3), 2.00–1.90 (m, 2H, CH 2) ppm; 13C NMR (100 MHz, CDCl3) δ 170.8, 170.4, 170.3, 170.2, 170.1, 169.5 (6C, 6×COCH3), 129.2, 127.8, (2C CH=CH) 94.5 (1C, C‐1), 84.3 (1C, C‐1′), 74.5, 71.9, 67.3, 67.2 (4C, skeleton carbons), 67.0 (1C, CH2O) 67.0, 65.3 (2C, skeleton carbons), 63.0, 61.5 (2C, 2C‐6, C‐6′), 30.0 (1C, CH2S), 27.1 (1C, CH2), 21.0, 20.8, 20.7, 20.6 (6C, 6×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C27H38NaO15S [M+Na]+ 657.1829, found 657.1829.
3‐(2,3,4,6‐Tetra‐O‐acetyl‐1‐thio‐β‐d‐galactopyranosyl)‐n‐propyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐2‐thio‐α‐d‐arabino‐hexopyranoside (27)
Compound 26 (0.55 mmol, 353 mg) in toluene (2.0 mL) was reacted with thiol 15 (0.66 mmol, 240 mg, 1.2 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 7 : 3) to give compound 27 (482 mg, 88%) as white crystals. [α]D 20+5.5 (c 0.29, CHCl3); R f=0.19 (n‐hexane/acetone 7 : 3); M.p.: 54–57 °C; 1H NMR (400 MHz, CDCl3) δ 5.41 (dd, J=3.4 Hz, J=1.2 Hz, 1H, H‐1), 5.20 (td, J=9.7 Hz, J=6.2 Hz, 2H), 5.06 (d, J=7.2 Hz, 1H), 5.05–5.00 (m, 2H), 4.88 (td, J=9.8 Hz, J=5.2 Hz, 1H), 4.80 (d, J=1.8 Hz, 1H), 4.60 (d, J=10.0 Hz, 1H), 4.49 (d, J=9.9 Hz, 1H), 4.40–4.02 (m, 6H), 3.93 (td, J=6.7 Hz, J=1.2 Hz, 1H), 3.85 (ddd, J=9.5 Hz, J=5.3 Hz, J=2.7 Hz, 1H), 3.77–3.73 (m, 1H), 3.70 (ddd, J=10.0 Hz, J=5.0 Hz, J=2.4 Hz, 1H), 3.51 (dt, J=9.7 Hz, J=6.1 Hz, 1H), 3.21 (td, J=4.1 Hz, J=1.8 Hz, 1H), 2.88–2.69 (m, 2H), 2.20–2.10 (m. 2 H), 2.14 (s, 3H, Ac−CH 3), 2.07 (s, 3H, Ac−CH 3), 2.05 (s, 6H, 2×Ac−CH 3), 2.04 (s, 3H, Ac−CH 3), 2.02 (s, 3H, Ac−CH 3), 2.01 (s, 3H, Ac−CH 3), 2.00 (s, 3H, Ac−CH 3), 1.98 (s, 3H, Ac−CH 3), 1.96 (s, 3H, Ac−CH 3), 1.95–1.88 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 170.9, 170.5, 170.4, 170.2, 170.1, 170.1, 169.6, 169.6, 169.5, 169.4 (10C, 10×COCH3), 99.9 (1C, C‐1), 84.2, 83.1 (2C, C‐1′, C‐1”), 76.1, 74.4, 73.7, 71.9, 69.6, 69.1, 68.3, 67.3, 67.2„ 65.0 (10C, skeleton carbons), 66.1 (1C, OCH2), 63.0, 62.0, 61.3 (3C, C‐6, C‐6, C‐6”), 42.5 (1C, C‐2), 30.7, 29.7, 27.0 (3C, C‐3 and 2×CH2), 21.0, 20.8, 20.8, 20.7, 20.6 (10C, 10×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C41H58NaO24S2 [M+Na]+ 1021.2657, found 1021.2663.
3‐(2,3,4,6‐Tetra‐O‐acetyl‐1‐thio‐β‐d‐mannopyranosyl)‐n‐propyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐mannopyranosyl)‐2‐thio‐α‐d‐arabino‐hexopyranoside (29) and 3‐(2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐mannopyranosyl)‐n‐propyl 4,6‐di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranoside (30)
A: Compound 7 (0.6 mmol, 162 mg) in toluene (3.0 mL) was reacted with thiol 28 (1.5 mmol, 546 mg, 2.5 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 65 : 35) to give compound 29 (448 mg, 75%) as colourless syrup. Compound 30 was detected in MS but could not be isolated in pure form.
B: The reaction was repeated at −80 °C to give 29 with 83% yield.
Data for compound 29:
[α]D 20+107.5 (c 0.20, CHCl3); R f=0.18 (n‐hexane/acetone 65 : 35); 1H NMR (400 MHz, CDCl3) δ 5.38–5.21 (m, 7H), 5.01 (td, J=9.8, 5.4 Hz, 1H), 4.82 (s, 1H), 4.39–4.06 (m, 9H), 3.89 (ddd, J=9.9 Hz, J=4.7 Hz, J=3.3 Hz, 1H), 3.79 (dt, J=9.7 Hz, J=6.2 Hz, 1H), 3.52 (dt, J=9.7 Hz, J=5.8 Hz, 1H), 3.20 (td, J=3.8 Hz, J=1.5 Hz, 1H), 2.79–2.66 (m, 2H), 2.24–2.20 (m 2H, H‐3a,b), 2.17 (s, 6H, 2×Ac−CH 3), 2.12 (s, 3H, Ac−CH 3), 2.10 (s, 3H, Ac−CH 3), 2.09 (s, 3H, Ac−CH 3), 2.06 (s, 9H, 3×Ac−CH 3), 2.01 (s, 6H, 2×Ac−CH 3), 1.95 (t, J=6.7 Hz, 2H CH 2) ppm; 13C NMR (100 MHz, CDCl3) δ 170.8, 170.5, 170.0, 169.8, 169.8, 169.7, 169.7, 169.6 (10C, 10×COCH3), 98.8 (1C, C‐1), 82.6, 82.6 (2C, C‐1′, C‐1“), 71.1, 70.9, 69.4, 69.4, 69.3, 69.2, 69.1, 66.2, 66.0, 64.8, (10C, skeleton carbons), 66.0 (1C, OCH2), 63.0, 62.4, 62.3 (3C, C‐6, C‐6′, C‐6”), 44.9 (1C, C‐2), 29.3, 28.2 (2C, 2×CH2), 21.0, 20.9, 20.9, 20.7, 20.7, 20.6 (10C, 10×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C41H58NaO24S2 [M+Na]+ 1021.2657, found 1021.2664.
Ethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐ glucopyranosyl)‐2‐thio‐α‐d‐lyxo‐hexopyranoside (31) and ethyl 4,6‐di‐O‐acetyl‐2‐deoxy‐3‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐ glucopyranosyl)‐2‐thio‐α‐d‐xylo‐hexopyranoside (32)
A: Compound 8 (1.0 mmol, 258 mg) in toluene (2.0 mL) was reacted with thiol 15 (437 mg, 1.2 mmol, 1.2 equiv) at rt according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8 : 2) to give compounds 31 (354 mg, 58%) as white crystals and 32 (124 mg, 20%) as colourless syrup.
B: The reaction was repeated at −80 °C to give 31 and 32 in a 5 : 1 ratio with 80% combined yield.
Data for compound 31:
[α]D 20+103.5 (c 0.17, CHCl3); R f=0.17 (n‐hexane/acetone 7 : 3); M.p: 147–149 °C; 1H NMR (400 MHz, CDCl3) δ 5.22 (t, J=9.4 Hz, 1H, H‐3′), 5.07 (dt, J=1.9 Hz, J=9.8 Hz, 2H, H‐2′, H‐4′), 4.95 (s, 1H, H‐1), 4.86 (s, 1H, H‐4), 4.44 (d, J=10.0 Hz, 1H, H‐1′), 4.27 (dd, J=4.8 Hz, J=12.4 Hz, 1H, H‐6′a), 4.16–4.08 (m, 4H, H‐5, H‐6a,b, H‐6′b), 3.78–3.69 (m, 2H, H‐5′, OCH 2a−CH3), 3.58–3.50 (m, 1H, OCH 2b−CH3), 3.08 (dd, J=1.2 Hz, J=2.9 Hz, 1H, H‐2), 2.47 (ddd, J=3.2 Hz, J=5.3 Hz, J=15.2 Hz, 1H, H‐3a), 2.14–2.08 (m, 1H, H‐3b), 2.09, 2.08, 2.05, 2.03, 2.01 (5×s, 18H, 6×Ac−CH 3), 1.25 (t, J=7.1 Hz, 3H, −OCH2−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.6, 170.5, 170.3, 170.1, 169.5, 169.3 (6C, 6×COCH3), 100.5 (1C, C‐1, 1 J C1,H1=172.7 Hz), 83.2 (1C, C‐1′, 1 J C1′,H1′=152.6 Hz), 76.3 (1C, C‐5′), 73.7 (1C, C‐3′), 69.0 (1C, C‐2′), 68.3 (1C, C‐4′), 66.5 (1C, C‐5), 65.8 (1C, C‐4), 63.3, (1C, OCH2−CH3), 63.2 (1C, C‐6), 62.0 (1C, C‐6′), 38.5 (1C, C‐2), 28.4 (1C, C‐3), 21.3, 20.8, 20.7, 20.6 (6C, 6×Ac−CH3), 15.1 (1C, OCH2−CH3) ppm; ESI‐HRMS: m/z calcd for C26H38NaO15S [M+Na]+ 645.1829, found 645.1832.
Data for compound 32:
[α]D 20 −12.0 (c 0.40, CHCl3); R f=0.18 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.27 (t, J=9.3 Hz, 1H, H‐3′), 5.15 (t, J=9.7 Hz, 1H, H‐4′), 5.08 (t, J=9.7 Hz, 1H, H‐2′), 4.88 (d, J=3.0 Hz, 1H, H‐1), 4.83 (s, 1H, H‐4), 4.65 (d, J=10.1 Hz, 1H, H‐1′), 4.52 (t, J=6.4 Hz, 1H, H‐5), 4.25 (dd, J=4.5 Hz, J=12.4 Hz, 1H, H‐6′a), 4.19–4.10 (m, 3H, H‐6a,b, H‐6′b), 3.74 (ddd, J=2.2 Hz, J=4.3 Hz, J=9.8 Hz, 1H, H‐5′), 3.71–3.65 (m, 1H, OCH 2a−CH3), 3.50–3.42 (m, 2H, H‐3, OCH 2b−CH3), 2.35 (ddd, J=3.7 Hz, J=6.1 Hz, J=14.7 Hz, 1H, H‐2a), 2.12, 2.08, 2.06, 2.03, 2.02 (6×s, 18H, 6×Ac−CH 3), 1.78 (d, J=14.3 Hz, 1H, H‐2b), 1.21 (t, J=7.1 Hz, 3H, −OCH2−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.8, 170.7, 170.5, 170.4, 169.5, 169.4 (6C, 6×COCH3), 95.6 (1C, C‐1, 1 J C1,H1=165.5 Hz), 82.9 (1C, C‐1′, 1 J C1′,H1′=154.2 Hz), 76.3 (1C, C‐5′), 74.1 (1C, C‐3′), 70.2 (1C, C‐4), 69.4 (1C, C‐2′), 68.4 (1C, C‐4′), 63.7 (1C, C‐6), 62.9, (1C, OCH2−CH3), 62.7 (1C, C‐5), 62.1 (1C, C‐6′), 36.4 (1C, C‐3), 29.9 (1C, C‐2), 21.1, 20.9, 20.8, 20.7 (6C, 6×Ac−CH3), 15.1 (1C, OCH2−CH3) ppm; ESI‐HRMS: m/z calcd for C26H38NaO15S [M+Na]+ 645.1829, found 645.1826.
Ethyl 4,6‐di‐O‐acetyl‐3‐deoxy‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐ galactopyranosyl)‐2‐thio‐α‐d‐lyxo‐hexopyranoside (33) and ethyl 4,6‐di‐O‐acetyl‐2‐deoxy‐3‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐ galactopyranosyl)‐2‐thio‐α‐d‐xylo‐hexopyranoside (34)
A: Compound 8 (0.79 mmol, 203 mg) in toluene (2.0 mL) was reacted with thiol 18 (350 mg, 0.96 mmol, 1.2 equiv) at rt according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8 : 2) to give compounds 33 (291 mg, 59%) as colourless syrup and 34 (77 mg, 15%) as colourless syrup.
B: The reaction was repeated at −40 °C in methanol to give compounds 33 and 34 with 69% overall yield, 33 to 34 ratio was 6.7 : 1.
C: The reaction was repeated at −80 °C to give compounds 33 and 34 with 73% overall yield, 33 to 34 ratio was 7.3 : 1.
Data for compound 33:
[α]D 20+25.2 (c 0.31, CHCl3); R f=0.26 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.43 (d, J=2.2 Hz, 1H, H‐4′), 5.27 (t, J=10.0 Hz, 1H, H‐2′), 5.05 (dd, J=3.3 Hz, J=10.0 Hz, 1H, H‐3′), 4.96 (s, 1H, H‐1), 4.87 (s, 1H, H‐4), 4.44 (d, J=9.9 Hz, 1H, H‐1′), 4.17–4.05 (m, 5H, H‐5, H‐6a,b, H‐6′a,b), 3.96 (t, J=6.4 Hz, 1H, H‐5′), 3.78–3.72 (m, 1H, OCH 2a−CH3), 3.60–3.54 (m, 1H, OCH 2b−CH3), 3.13 (dd, J=1.0 Hz, J=3.2 Hz, 1H, H‐2), 2.48 (ddd, J=3.1 Hz, J=5.3 Hz, J=15.2 Hz, 1H, H‐3a), 2.21–2.15 (m, 1H, H‐3b), 2.18, 2.16, 2.10, 2.06, 2.05, 1.99 (6×s, 18H, 6×Ac−CH 3), 1.25 (t, J=7.0 Hz, 3H, OCH2−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.4, 170.3, 170.2, 170.1, 169.6 (6C, 6×Ac−CO), 100.4 (1C, C‐1, 1 J C1,H1=171.9 Hz), 83.7 (1C, C‐1′, 1 J C1′,H1′=153.5 Hz), 74.7 (1C, C‐5′), 71.8 (1C, C‐3′), 67.3 (1C, C‐4′), 66.5 (1C, C‐5), 66.4 (1C, C‐2′), 65.8 (1C, C‐4), 63.3, (1C, OCH2−CH3), 63.2 (1C, C‐6), 61.4 (1C, C‐6′), 38.5 (1C, C‐2), 28.4 (1C, C‐3), 21.3, 20.8, 20.7, 20.6 (6C, 6×Ac−CH3), 15.1 (1C, −OCH2−CH3) ppm; ESI‐HRMS: m/z calcd for C26H38NaO15S [M+Na]+ 645.1829, found 645.1832.
Data for compound 34:
[α]D 20 −41.7 (c 0.06, CHCl3); R f=0.27 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.44 (t, J=2.9 Hz, 1H, H‐4′), 5.27 (t, J=10.0 Hz, 1H, H‐2′), 5.09 (dd, J=3.2 Hz, J=9.9 Hz, 1H, H‐3′), 4.88 (s, 2H, H‐1, H‐4), 4.60 (d, J=10.0 Hz, 1H, H‐1′), 4.52 (t, J=6.2 Hz, 1H, H‐5), 4.20–4.09 (m, 4H, H‐6a,b, H‐6′a,b), 3.95 (t, J=6.6 Hz, 1H, H‐5′), 3.72–3.65 (m, 1H, OCH 2a−CH3), 3.48–3.44 (m, 1H, OCH 2b−CH3), 3.41–3.39 (m, 1H, H‐3), 2.38 (ddd, J=3.5 Hz, J=5.6 Hz, J=14.6 Hz, 1H, H‐2a), 2.15, 2.12, 2.08, 2.04, 1.99 (5×s, 18H, 6×Ac−CH 3), 1.82 (d, J=14.5 Hz, 1H, H‐2b), 1.22 (t, J=7.0 Hz, 3H, OCH2−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.4, 170.1, 169.6 (6C, 6×COCH3), 95.7 (1C, C‐1, 1 J C1,H1=167.1 Hz), 83.5 (1C, C‐1′, 1 J C1′,H1′=154.0 Hz), 74.8 (1C, C‐5′), 72.0 (1C, C‐3′), 70.1 (1C, C‐4), 67.3 (1C, C‐4′), 66.8 (1C, C‐2′), 63.6 (1C, C‐6), 62.8, (1C, OCH2−CH3), 62.7 (1C, C‐5), 61.3 (1C, C‐6′), 36.7 (1C, C‐3), 30.0 (1C, C‐2), 21.1, 20.9, 20.8, 20.7 (6C, 6×Ac−CH3), 15.1 (1C, −OCH2−CH3) ppm; ESI‐HRMS: m/z calcd for C26H38NaO15S [M+Na]+ 645.1829, found 645.1837.
Ethyl 2,3,6‐tri‐O‐acetyl‐3‐S‐(2,3,4,6‐tetra‐O‐acetyl‐α‐d‐ mannopyranosyl)‐3‐thio‐α‐d‐glucopyranoside (36)
A: Compound 9 (0.5 mmol, 158 mg) in toluene (2.0 mL) was reacted with thiol 35 (0.6 mmol, 218 mg, 1.2 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8 : 2) to give compound 36 (119 mg, 35%) as colourless syrup.
B: The reaction was repeated at −80 °C to give 36 with 68% yield. [α]D 20+93.9 (c 0.23, CHCl3); R f=0.31 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.40 (s, 1H, H‐1′), 5.33 (t, J=9.5 Hz, 1H, H‐4′), 5.21–5.18 (m, 2H, H‐2′, H‐3′), 5.04 (t, J=10.4 Hz, 1H, H‐4), 5.01 (d, J=3.3 Hz, 1H, H‐1), 4.79 (dd, J=3.4 Hz, J=11.8 Hz, 1H, H‐2), 4.40–4.30 (m, 2H, H‐5′, H‐6′a), 4.19–4.10 (m, 2H, H‐6a, H‐6′b), 4.04 (dd, J=2.5 Hz, J=12.3 Hz, 1H, H‐6b), 3.90 (ddd, J=2.6 Hz, J=4.7 Hz, J=9.7 Hz, 1H, H‐5), 3.76–3.68 (m, 1H, OCH 2a−CH3), 3.57–3.49 (m, 1H, OCH 2b−CH3), 3.46 (t, J=11.3 Hz, 1H, H‐3), 2.16, 2.14, 2.12, 2.08, 2.05, 2.00 (6×s, 21H, 7×Ac−CH 3), 1.21 (t, J=7.1 Hz, 3H, OCH2−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.8, 170.0, 169.9, 169.7, 169.6 (7C, 7×COCH3), 95.1 (1C, C‐1, 1 J C1,H1=171.9 Hz), 82.6 (1C, C‐1′, 1 J C1′,H1′=172.5 Hz), 71.5 (1C, C‐2′), 70.3 (2C, C‐2, C‐4), 69.6 (1C, C‐5′), 69.1 (1C, C‐3′), 68.7 (1C, C‐5), 66.2 (1C, C‐4′), 63.9, (1C, OCH2−CH3), 62.5 (1C, C‐6), 62.1 (1C, C‐6′), 46.2 (1C, C‐3), 21.0, 20.8, 20.7, 20.6 (7C, 7×Ac−CH3), 15.0 (1C, −OCH2−CH3) ppm; MALDI‐TOF HRMS: m/z calcd for C28H40NaO17S [M+Na]+ 703.1884, found 703.1793.
Ethyl 2,3,6‐tri‐O‐acetyl‐3‐S‐(2,3,4,6‐tetra‐O‐acetyl‐α‐d‐ glucopyranosyl)‐3‐thio‐α‐d‐allopyranoside (37)
A: Compound 9 (0.5 mmol, 158 mg) in toluene (2.0 mL) was reacted with thiol 15 (0.6 mmol, 218 mg, 1.2 equiv.) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8 : 2) to give compound 37 (140 mg, 42%) as colourless syrup.
B: The reaction was repeated at −80 °C to give 37 with 50% yield. [α]D 20+30.8 (c 0.49, CHCl3); R f=0.27 (n‐hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3) δ 5.13 (t, J=9.3 Hz, 1H, H‐3′), 5.04–4.97 (m, 3H, H‐2, H‐4, H‐4′), 4.39 (t, J=9.6 Hz, 1H, H‐2′), 4.84 (d, J=3.4 Hz, 1H, H‐1), 4.32 (dd, J=4.6 Hz, J=12.0 Hz, 1H, H‐6′a), 4.25 (dd, J=4.9 Hz, J=12.3 Hz, 1H, H‐6a), 4.15 (dd, J=2.2 Hz, J=11.4 Hz, 1H, H‐6′b), 4.13 (d, J=7.5 Hz, 1H, H‐1′), 4.13–4.10 (m, 1H, H‐5′), 4.03 (dd, J=1.5 Hz, J=12.4 Hz, 1H, H‐6b), 3.95 (t, J=4.7 Hz, 1H, H‐3), 3.75–3.71 (m, 1H, OCH 2a), 3.61–3.55 (m, 2H, OCH 2b, H‐5), 2.16, 2.12, 2.09, 2.08, 2.01, 2.00 (6×s, 21H, 7×Ac−CH 3), 1.26 (t, J=7.0 Hz, 3H, CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.2, 170.0, 169.8, 169.4, 169.0 (7C, 7×COCH3), 95.5 (1C, C‐1, 1 J C1,H1=169.0 Hz), 85.9 (1C, C‐1′, 1 J C1′,H1′=156.4 Hz), 75.8 (1C, C‐5), 73.9 (1C, C‐3′), 70.6 (1C, C‐2′), 68.0, 67.9 (2C, C‐2, C‐4), 67.3 (1C, C‐4′), 64.6 (1C, C‐5′), 63.8 (1C, −OCH2), 62.3 (1C, C‐6′), 61.9 (1C, C‐6), 46.5 (1C, C‐3), 20.8, 20.7, 20.6, 20.5 (7C, 7×Ac−CH3), 14.8 (1C, CH3), ppm; MALDI‐TOF MS: m/z calcd for C28H40NaO17S [M+Na]+ 703.188, found 703.215.
3‐(2,3,4,6‐Tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranosyl)‐n‐propyl 2,4,6‐tri‐O‐acetyl‐3‐deoxy‐α,β‐d‐erythro‐hex‐2‐enopyranoside (38)
Compound 10 (0.5 mmol, 164 mg) in toluene (2.0 mL) was reacted with thiol 15 (0.6 mmol, 218 mg, 1.2 equiv.) at −80 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/ethyl acetate 55 : 45) to give compound 38 (314 mg, 90%) as colourless syrup, with anomeric ratio α : β=2 : 1. R f=0.39 (n‐hexane/acetone 7 : 3).
NMR data for the anomers from the spectrum of the anomeric mixture.
Data for compound 38α given from the spectrum of the anomeric mixture:
1H NMR (400 MHz, CDCl3) δ 5.72 (d, J=1.9 Hz, 1H, H‐3), 5.46 (dd, J=1.2 Hz, J=9.4 Hz, 1H, H‐4), 5.25–5.20 (m, 1H, H‐3′), 5.11–5.00 (m, 3H, H‐1, H‐2′, H‐4′), 4.51 (d, J=10.0 Hz, 1H, H‐1′), 4.27–4.20 (m, 3H, H‐6a,b, H‐6′a), 4.15–4.10 (m, 2H, H‐5, H‐6′b), 3.86 (dt, J=6.0 Hz, J=12.0 Hz, 1H, OCH 2a), 3.74–3.71 (m, 1H, H‐5′), 3.64–3.59 (m, 1H, OCH 2b), 2.83–2.69 (m, 2H, SCH 2), 2.18, 2.11, 2.08, 2.06, 2.03, 2.01 (6×s, 21H, 7×CH 3), 1.92 (ddd, J=6.8 Hz, J=13.6 Hz, J=16.5 Hz, 2H, CH 2) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.6, 170.3, 170.2, 170.1, 169.5, 168.2 (7C, 7×COCH3), 146.3 (1C, C‐2), 115.4 (1C, C‐3), 94.1 (1C, C‐1), 83.6 (1C, C‐1′), 75.9 (1C, C‐5′), 73.8 (1C, C‐3′), 69.8 (1C, C‐2′), 68.3 (1C, C‐4′), 67.3 (1C, OCH2), 67.2 (1C, C‐5), 65.3 (1C, C‐4), 62.6 (1C, C‐6), 62.1 (1C, C‐6′), 29.8, (1C, SCH2), 26.7 (1C, CH2), 21.0, 20.9, 20.8, 20.7, 20.6 (7C, 7×CH3) ppm.
Data for compound 38β given from the spectrum of the anomeric mixture:
1H NMR (400 MHz, CDCl3) δ 5.78 (d, J=4.5 Hz, 1H, H‐3), 5.33 (t, J=4.3 Hz, 1H, H‐4), 5.25–5.20 (m, 2H, H‐1, H‐3′), 5.11–5.00 (m, 2H, H‐2′, H‐4′), 4.51 (d, J=10.0 Hz, 1H, H‐1′), 4.27–4.20 (m, 3H, H‐6a,b, H‐6′a), 4.15–4.10 (m, 2H, H‐5, H‐6′b), 3.86 (dt, J=6.0 Hz, J=12.0 Hz, 1H, OCH 2a), 3.74–3.71 (m, 1H, H‐5′), 3.64–3.59 (m, 1H, OCH 2b), 2.83–2.69 (m, 2H, SCH 2), 2.18, 2.10, 2.08, 2.06, 2.03, 2.01 (6×s, 21H, 7×CH 3), 1.92 (ddd, J=6.8 Hz, J=13.6 Hz, J=16.5 Hz, 2H, CH 2) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.6, 170.3, 170.2, 170.1, 169.5, 168.2 (7C, 7×COCH3), 148.1 (1C, C‐2), 112.5 (1C, C‐3), 93.9 (1C, C‐1), 84.0 (1C, C‐1′), 75.8 (1C, C‐5′), 73.8 (1C, C‐3′), 72.7 (1C, C‐5), 69.9 (1C, C‐2′), 68.3 (1C, C‐4′), 66.9 (1C, OCH2), 65.6 (1C, C‐4), 63.1 (1C, C‐6), 62.2 (1C, C‐6′), 29.8, (1C, SCH2), 27.1 (1C, CH2), 21.0, 20.9, 20.8, 20.7, 20.6 (7C, 7×CH3) ppm.
ESI‐HRMS: m/z calcd for C29H40NaO17S [M+Na]+ 715.1884, found 715.1870.
3‐(4,6‐Di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranosyl)‐propyl 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranoside (39)
A: Compound 11 (0.86 mmol, 220 mg) in toluene (7.0 mL) was reacted with thiol 15 (0.86 mmol, 313 mg, 1.0 equiv.) at rt according to the general method. The crude product was purified by flash chromatography (CH2Cl2/acetone 95 : 5) to give compound 39 (213 mg, 40%) as colourless syrup.
B: The reaction was repeated with 2.5 equiv. of thiol 15 (2.15 mmol, 782 mg) at −80 °C to give compound 39 (308 mg, 58%) as colourless syrup. R f=0.48 (n‐hexane/acetone 6 : 4); 1H NMR (400 MHz, CDCl3) δ 5.90 (ddd, J gem=10.4 Hz, J=2.5 Hz, J gem=1.5 Hz, 1H, CH=CH), 5.78 (ddd, J=10.4 Hz, J=3.0 Hz, J=2.0 Hz, 1H, CH=CH), 5.22 (t, J=9.4 Hz, 1H), 5.14–5.11 (m, 1H), 5.11–5.00 (m, 2H), 4.50 (d, J=10.0 Hz, 2H, H‐1′), 4.29–4.24 (m, 1H), 4.23 (t, J=2.1 Hz, 1H), 4.15 (d, J=3.0 Hz, 1H), 4.13–4.10 (m, 1H), 3.92 (td, J=6.4 Hz, J=3.6 Hz, 1H), 3.73 (ddd, J=10.0 Hz, J=4.8 Hz, J=2.4 Hz, 1H), 2.84–2.65 (m, 2H, CH 2−S), 2.10, 2.08, 2.08, 2.06, 2.03, 2.01 (6 s, 6×3H, 6 Ac−CH 3), 1.84–1.60 (m, 4H CH 2−C) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.5, 170.3, 170.1, 169.3, 169.3 (6C, 6×COCH3), 133.3, 123.5 (2C, C‐2, C‐3), 83.4 (1C, C‐1′), 75.8, 73.8, 71.1, 69.8, 69.7, 68.3, 65.0 (7C, skeleton carbons), 62.7, 62.1 (2C, C‐6, C‐6′), 31.8, 29.4, 25.8 (3C, 3×CH2 linker), 21.0, 20.8, 20.7, 20.7, 20.5, 20.5 (6C, 6×Ac−CH3) ppm; ESI‐HRMS MS: m/z calcd for C27H38NaO14S [M+Na]+ 641.1880, found 641.1876.
3‐(4,6‐Di‐O‐acetyl‐2,3‐dideoxy‐α‐d‐erythro‐hex‐2‐enopyranosyl)‐propyl 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐galactopyranoside (41)
Compound 11 (1.0 mmol, 254 mg) in toluene (3.0 mL) was reacted with thiol 18 (3.0 mmol, 1.09 g, 3.0 equiv.) at −40 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 7 : 3) to give compound 41 (301 mg, 49%) as yellow syrup. Compound 42, sulfoxide derivative of 41, was detected from the reaction mixture by MS. [α]D 20+6.43 (c 0.14, CHCl3); R f=0.24 (n‐hexane/acetone 7 : 3); 1H NMR (360 MHz, CDCl3) δ 5.90 (dt, J gem=10.3 Hz, J=1.7 Hz, 1H, CH=CH), 5.79 (dt, J gem=10.4 Hz, J=2.4 Hz, 1H, CH=CH), 5.43 (d, J=3.2 Hz, 1H), 5.23 (t, J=9.9 Hz, 1H), 5.12 (ddt, J=6.5 Hz, J=3.2 Hz, J=1.7 Hz, 1H), 5.05 (dd, J=10.0 Hz, J=3.4 Hz, 1H), 4.50 (d, J=9.9 Hz, 1H, H‐1), 4.23 (tt, J=12.3 Hz, J=6.4 Hz, 2H), 4.17–4.07 (m, 3H), 4.01–3.80 (m, 2H), 2.88–2.68 (m, 2H, CH 2S), 2.16 (s, 4H), 2.13–2.02 (m, 12H, 4×Ac−CH 3), 2.01 (s, 3H), 1.75 (dddd, J=29.5 Hz, J=22.1 Hz, J=10.0 Hz, J=4.4 Hz, 4H, CH 2−CH 2) ppm; 13C NMR (90 MHz, CDCl3) δ 170.7, 170.3, 170.3, 170.1, 169.9, 169.5 (6C, 6×COCH3), 133.2, 123.5 (2C, C=C), 83.9 (1C, C‐1′), 74.4, 71.8, 71.1, 69.7, 67.2, 67.2, 64.9 (7C, skeleton carbons), 62.7, 61.4 (2C, C‐6, C‐6′), 31.9, 29.7, 25.8, (3C, 3×CH2 linker), 21.0, 20.8, 20.6, 20.5 (6C, 6×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for 41C 27H38NaO14S [M+Na]+ 641.1880, found 641.1876. MALDI‐TOF MS m/z calcd for 42C 27H38NaO15S [M+Na]+ 657.182, found 657.010.
4,6‐Di‐O‐acetyl‐3‐S‐acetyl‐3‐thio‐1,5‐anhydro‐d‐ribo‐hex‐1‐enitol (43), 4,6‐di‐O‐acetyl‐3‐S‐(2,3,4,6‐tetra‐O‐acetyl‐α‐d‐ mannopyranosyl)‐3‐thio‐1,5‐anhydro‐d‐ribo‐hex‐1‐enitol (44) and bis‐(2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐D‐mannopyranosyl) ‐1,1′‐disulfide (45)
Compound 12 (0.6 mmol, 170 mg) in toluene (5.0 mL) was reacted with thiol 35 (0.9 mmol, 322 mg, 1.5 equiv.) at room temperature according to the general method. The crude product was purified by flash chromatography (n‐hexane/ethyl acetate 7 : 3) to give compound 43 (27 mg, 16%) as colourless syrup, compound 44 (60 mg, 18%) as colourless syrup, and 45 11, 52 (14 mg, 3%) as white foam.
Compound 44 was also obtained from 13 with 40% yield by reacting 13 and 35 according to the general method.
Data for compound 43:
[α]D 20+202.9 (c 0.13, CHCl3); R f=0.31 (n‐hexane/ethyl acetate 6 : 4); 1H NMR (400 MHz, CDCl3) δ 6.42 (d, J=5.9 Hz, 1H, H‐1), 5.36 (dd, J=9.8 Hz, J=4.7 Hz, 1H), 4.79 (t, J=5.8 Hz, 1H), 4.50–4.44 (m, 1H), 4.42–4.33 (m, 1H), 4.32–4.28 (m, 1H), 4.19–4.07 (m, 1H), 2.32 (s, 3H, SAc−CH 3), 2.09, 1.99 (2 s, 2×3H, 2 OAc−CH 3); 13C NMR (100 MHz, CDCl3) δ 170.8, 169.4 (2C, 2×COCH3), 145.6 (1C, C‐1), 97.6 (1C, C‐2), 72.2, 66.0 (2C, C‐4, C‐5), 62.1 (1C, C‐6), 39.4 (1C, C‐3), 30.2 (1C, SAc−CH3), 20.8, 20.7 (2C, 2×OAc−CH3) ppm; ESI‐HRMS: m/z calcd for C12H16NaO6S [M+Na]+ 311.0565, found 311.0559.
Data for compound 44:
[α]D 20+311.9 (c 0.28, CHCl3); R f=0.78 (n‐hexane/ethyl acetate 6 : 4); 1H NMR (400 MHz, CDCl3) δ 6.45 (d, J=6.0 Hz, 1H, H‐1), 5.39–5.31 (m, 2H) 5.30–5.26 (m, 1H), 5.16 (dd, J=8.8 Hz, J=5.0 Hz, 1H), 4.94 (t, J=5.7 Hz, 1H), 4.41–4.24 (m, 6H), 4.11 (dd, J=9.5 Hz, J=4.5 Hz, 1H), 4.06 (d, J=4.8 Hz, 1H), 2.18, 2.16, 2.11, 2.10, 2.06, 2.00 (6 s, 6×3H, 6 Ac−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 170.1, 170.0, 170.0, 169.8 (6C, 6×COCH3), 145.3 (1C, C‐1), 97.9 (1C, C‐2), 81.0 (1C, C‐1′), 71.4, 71.2, 69.7, 69.6, 67.7, 66.2, (1C, skeletal,carbons), 62.3, 62.1 (2C, C‐6, C‐6′), 39.8 (1C, C‐3), 21.1, 20.9, 20.8, 20.7 (6C, 6×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for, C24H32NaO14S [M+Na]+ 599.1410, found 599.1403.
N‐(4,6‐Di‐O‐acetyl‐2‐deoxy‐3‐thio‐3‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐β‐d‐ribo‐hexopyranosyl) p‐toluenesulfonamide (46)
Compound 14 (0.5 mmol, 192 mg) in toluene (2.0 mL) and MeOH (1.0 mL) was reacted with thiol 15 (0.75 mmol, 273 mg, 1.5 equiv.) at −80 °C according to the general method. The crude product was purified by flash chromatography (toluene/acetone 9 : 1) to give compound 46 (62 mg, 16%) as colourless syrup. [α]D 20+57.4 (c 0.19, CHCl3); R f=0.28 (n‐hexane/acetone 6 : 4); 1H NMR (400 MHz, CDCl3) δ 7.78–7.25 (m, 4H, arom), 5.49 (d, J=9.9 Hz, 1H, NH), 5.18 (t, J=9.4 Hz, 1H, H‐3′), 5.10 (dt, J=2.0 Hz, J=10.3 Hz, 1H, H‐4′), 5.07 (t, J=9.7 Hz, 1H, H‐1), 4.97 (t, J=9.6 Hz, 1H, H‐2′), 4.71 (dd, J=4.3 Hz, J=9.7 Hz, 1H, H‐4), 4.53 (d, J=10.0 Hz, 1H, H‐1′), 4.21–4.11 (m, 2H, H‐6′a,b), 4.00 (dd, J=4.6 Hz, J=12.1 Hz, 1H, H‐6a), 3.87–3.81 (m, 3H, H‐3, H‐5, H‐6b), 3.67–3.62 (m, 1H, H‐5′), 2.41 (s, 3H, Ph−CH 3), 2.32–2.28 (m, 1H, H‐2a), 2.12–2.08 (m, 1H, H‐2b), 2.10, 2.06, 2.05, 2.03, 2.01, 2.00 (6×s, 18H, 6×Ac−CH 3) ppm; 13C NMR (100 MHz, CDCl3) δ 170.9, 170.6, 170.3, 169.8, 169.5, 169.4 (6C, 6×COCH3), 143.7, 138.7 (2C, 2×Cq arom), 129.4, 127.2 (4C, arom), 84.1 (1C, C‐1′, 1 J C1′,H1′=153.7 Hz), 79.1 (1C, C‐1, 1 J C1,H1=155.3 Hz), 76.2 (1C, C‐5′), 73.8 (1C, C‐3′), 72.1 (1C, C‐5), 70.5 (1C, C‐2′), 68.5 (1C, C‐4), 68.1 (1C, C‐4′), 62.7 (1C, C‐6), 62.0 (1C, C‐6′), 42.9 (1C, C‐3), 37.8 (1C, C‐2), 21.6 (1C, Ph−CH3), 20.9, 20.8, 20.7, 20.6 (6C, 6×Ac−CH3) ppm; MALDI‐TOF HRMS: m/z calcd for C31H41NNaO16S2 [M+Na]+ 747.1867, found 747.1859.
N‐(4,6‐Di‐O‐acetyl‐2‐deoxy‐3‐thio‐3‐S‐(2,3,4‐tri‐O‐acetyl‐β‐d‐xylopyranosyl)‐β‐d‐arabino‐hexopyranosyl) p‐toluenesulfonamide (48) and N‐(4,6‐di‐O‐acetyl‐3‐deoxy‐ 2‐thio‐2‐S‐(2,3,4‐tri‐O‐acetyl‐β‐d‐xylopyranosyl)‐β‐d‐ arabino‐hexopyranosyl) p‐toluenesulfonamide (49)
Compound 14 (0.5 mmol, 192 mg) in toluene (2.0 mL) and MeOH (1.0 mL) was reacted with thiol 47 (0.75 mmol, 219 mg, 1.5 equiv.) at −40 °C according to the general method. The crude product was purified by flash chromatography (toluene/acetone 9 : 1) to give compound 48 (54 mg, 16%) as colourless syrup and compound 49 (27 mg, 8%) as colourless syrup.
Data for compound 48:
[α]D 20 −17.1 (c 0.24 CHCl3); R f=0.24 (toluene/acetone 9 : 1); 1H NMR (400 MHz, CDCl3) δ 7.83–7.27 (m, 4H, arom), 5.48 (d, J=10.1 Hz, 1H, NH), 5.14 (t, J=7.7 Hz, 1H, H‐3′), 4.92–4.84 (m, 3H, H‐1, H‐2′, H‐4′), 4.75–4.69 (m, 2H, H‐1′, H‐4), 4.19 (dd, J=4.7 Hz, J=11.9 Hz, 1H, H‐5′a), 4.01 (dd, J=4.6 Hz, J=12.3 Hz, 1H, H‐6a), 3.75 (dd, J=2.3 Hz, J=12.2 Hz, 1H, H‐6b), 3.52 (ddd, J=2.5 Hz, J=4.6 Hz, J=9.7 Hz, 1H, H‐5), 3.40 (dd, J=8.1 Hz, J=11.9 Hz, 1H, H‐5′b), 3.07 (td, J=4.3 Hz, J=12.6 Hz, 1H, H‐3), 2.45–2.41 (m, 1H, H‐2a), 2.42 (s, 3H, Ph−CH 3), 2.18, 2.07, 2.05, 2.04, 2.01 (5×s, 15H, 5×Ac−CH 3), 1.81 (td, J=11.0 Hz, J=13.1 Hz, 1H, H‐2b) ppm; 13C NMR (100 MHz, CDCl3) δ 170.7, 169.9, 169.8, 169.4 (5C, 5×COCH3), 143.9, 138.4 (2C, 2×Cq arom), 129.5, 127.4 (4C, arom), 82.4 (1C, C‐1′, 1 J C1′,H1′=161.5 Hz), 81.7 (1C, C‐1, 1 J C1,H1=151.2 Hz), 76.0 (1C, C‐5), 71.3 (1C, C‐3′), 69.9 (1C, C‐2′), 68.3 (1C, C‐4′), 67.6 (1C, C‐4), 64.6 (1C, C‐5′), 62.6 (1C, C‐6), 45.3 (1C, C‐3), 39.4 (1C, C‐2), 21.7 (1C, Ph−CH3), 20.9, 20.8 (5C, 5×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C28H37NNaO14S2 [M+Na]+ 698.1553, found 698.1572.
Data for compound 49:
[α]D 20 −44.3 (c 0.23, CHCl3); R f=0.26 (toluene/acetone 9 : 1); 1H NMR (500 MHz, CDCl3) δ 7.80–7.27 (m, 4H, arom), 6.19 (d, J=9.7 Hz, 1H, NH), 5.16 (t, J=8.6 Hz, 1H, H‐3′), 5.02 (dd, J=1.7 Hz, J=9.7 Hz, 1H, H‐1), 4.99–4.94 (m, 1H, H‐4′), 4.92 (t, J=8.6 Hz, 1H, H‐2′), 4.74 (td, J=4.8 Hz, J=11.0 Hz, 1H, H‐4), 4.58 (d, J=8.9 Hz, 1H, H‐1′), 4.22 (dd, J=5.2 Hz, J=11.6 Hz, 1H, H‐5′a), 3.99 (dd, J=5.1 Hz, J=12.1 Hz, 1H, H‐6a), 3.90 (dd, J=2.3 Hz, J=12.1 Hz, 1H, H‐6b), 3.61 (ddd, J=2.4 Hz, J=5.1 Hz, J=9.8 Hz, 1H, H‐5), 3.40 (dd, J=9.4 Hz, J=11.6 Hz, 1H, H‐5′b), 3.28 (s, 1H, H‐2), 2.47–2.43 (m, 1H, H‐3a), 2.42 (s, 3H, Ph−CH 3), 2.17, 2.05, 2.04, 2.03, 2.02 (5×s, 15H, 5×Ac−CH 3), 2.00–1.98 (m, 1H, H‐3b) ppm; 13C NMR (125 MHz, CDCl3) δ 170.7, 169.9, 169.7, 169.6 (5C, 5×COCH3), 143.6, 138.8 (2C, 2×Cq arom), 129.5, 127.2 (4C, arom), 82.8 (1C, C‐1′), 82.5 (1C, C‐1), 76.9 (1C, C‐5), 72.3 (1C, C‐3′), 69.4 (1C, C‐2), 68.3 (1C, C‐4′), 66.0 (1C, C‐5′), 64.2 (1C, C‐4), 62.7 (1C, C‐6), 45.2 (1C, C‐2), 36.9 (1C, C‐3), 21.6 (1C, Ph−CH3), 21.0, 20.8, 20.7 (5C, 5×Ac−CH3) ppm; ESI‐HRMS: m/z calcd for C28H37NNaO14S2 [M+Na]+ 698.1553, found 698.1526.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was funded by: the National Research, Development and Innovation Office of Hungary (TÉT_15_IN‐1‐2016‐0071, K109208 and K119509), the Premium Postdoctoral Program of HAS (PPD 461038), Gedeon Richter's Talentum Foundation (1103 Budapest, Gyömrői út 19–21.), the “Debrecen Venture Catapult Program” (EFOP‐3.6.1‐16‐2016‐00022 and EFOP‐3.6.3‐VEKOP‐16‐2017‐00009, János Bolyai Fellowship of the Hungarian Academy of Sciences (M. Csávás) and the ÚNKP‐19‐3 New National Excellence Program of the Ministry for Innovation and Technology. The research was also supported by the EU and co‐financed by the European Regional Development Fund under the projects GINOP‐2.3.2‐15‐2016‐00008, GINOP‐2.3.3‐15‐2016‐00021 and GINOP‐2.3.3‐15‐2016‐00004. The authors gratefully acknowledge Tobias Stürzer and Attila Bényei for performing the X‐ray spectroscopy experiments.
V. Kelemen, M. Csávás, J. Hotzi, M. Herczeg Poonam, B. Rathi, P. Herczegh, N. Jain, A. Borbás, Chem. Asian J. 2020, 15, 876.
References
- 1. Wang L.-X., Davis B. G., Chem. Sci. 2013, 4, 3381–3394, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ernst B., Magnani J. L., Nat. Rev. Drug Discov. 2009, 8, 661–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Driguez H., ChemBioChem 2001, 2, 311–318. [DOI] [PubMed] [Google Scholar]
- 4. Szilágyi L., Varela O., Curr. Org. Chem. 2006, 10, 1745–1770. [Google Scholar]
- 5. Koester D. C., Holkenbrink A., Werz D. B., Synthesis 2010, 19, 3217–3242. [Google Scholar]
- 6.
- 6a. Witczak Z. J., Bielski R., Click Chemistry in Glycoscience: New Developments and Strategies, Wiley, Weinheim, 2013; [Google Scholar]
- 6b. McSweeney L., Dénès F., Scanlan E. M., Eur. J. Org. Chem. 2016, 12, 2080–2095. [Google Scholar]
- 7. Dondoni A., Marra A., Chem. Soc. Rev. 2012, 41, 573–586. [DOI] [PubMed] [Google Scholar]
- 8. Staderini S., Chambery A., Marra A., Dondoni A., Tetrahedron Lett. 2012, 53, 702–704. [Google Scholar]
- 9. Lázár L., Csávás M., Herczeg M., Herczegh P., Borbás A., Org. Lett. 2012, 14, 4650–4653. [DOI] [PubMed] [Google Scholar]
- 10. Lázár L., Csávás M., Hadházi Á., Herczeg M., Tóth M., Somsák L., Barna T., Herczegh P., Borbás A., Org. Biomol. Chem. 2013, 11, 5339–5350. [DOI] [PubMed] [Google Scholar]
- 11. Lázár L., Juhász L., Batta G., Borbás A., Somsák L., New J. Chem. 2017, 41, 1284–1292. [Google Scholar]
- 12.
- 12a. Eszenyi D., Kelemen V., Balogh F., Bege M., Csávás M., Herczegh P., Borbás A., Chem. Eur. J. 2018, 24, 4532–4536; [DOI] [PubMed] [Google Scholar]
- 12b. Eszenyi D., Lázár L., Borbás A., McCourt R. in Carbohydrate Chemistry: Proven Synthetic Methods, Vol. 4 (Eds.: P. Kováč, C. Vogel, P. Murphy), CRC Press, Weinheim, 2017, pp. 33–44. [Google Scholar]
- 13. Kelemen V., Bege M., Eszenyi D., Debreczeni N., Bényei A., Stürzer T., Herczegh P., Borbás A., Chem. Eur. J. 2019, 25, 14555–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lázár L., Borbás A., Somsák L., Carbohydr. Res. 2018, 470, 8–12. [DOI] [PubMed] [Google Scholar]
- 15. Lázár L., Csávás M., Tóth M., Somsák L., Borbás A., Chem. Pap. 2015, 69, 889–895. [Google Scholar]
- 16. Richard M., Didierjean C., Chapleur Y., Pellegrini-Moïse N., Eur. J. Org. Chem. 2015, 12, 2632–2645. [Google Scholar]
- 17. József J., Juhász L., Illyés T. Z., Csávás M., Borbás A., Somsák L., Carbohydr. Res. 2015, 413, 63–69. [DOI] [PubMed] [Google Scholar]
- 18. Gervay J., Flaherty T. M., Holmes D., Tetrahedron 1997, 53, 16355–16364. [Google Scholar]
- 19. Fiore M., Marra A., Dondoni A., J. Org. Chem. 2009, 74, 4422–4425. [DOI] [PubMed] [Google Scholar]
- 20. Bege M., Bereczki I., Herczeg M., Kicsák M., Eszenyi D., Herczegh P., Borbás A., Org. Biomol. Chem. 2017, 15, 9226–9233. [DOI] [PubMed] [Google Scholar]
- 21. Bege M., Kiss A., Kicsák M., Bereczki I., Baksa V., Király G., Szemán-Nagy G., Szigeti M. Z., Herczegh P., Borbás A., Molecules 2019, 24, 2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Ferrier R. J., Hoberg J. O., Adv. Carbohydr. Chem. Biochem. 2003, 58, 55–119; [DOI] [PubMed] [Google Scholar]
- 22b. Ferrier R. J., Zubkov O. A., Org. React. 2003, 62, 569–736; [Google Scholar]
- 22c. Ferrier R. J., Top. Curr. Chem. 2001, 215, 153–175. [Google Scholar]
- 23.
- 23a. Gómez A. M., Lobo F., Uriel C., López J. C., Eur. J. Org. Chem. 2013, 32, 7221–7262; [Google Scholar]
- 23b. Gómez A. M., Miranda S., López J. C., Carbohydr. Chem. 2017, 42, 210–247. [Google Scholar]
- 24. Ansari A. A., Lahiri R., Vankar Y. D., Arkivoc 2013, 2, 316–362. [Google Scholar]
- 25. Araki Y., Matsuura K., Ishido Y., Kushida K., Chem. Lett. 1973, 383–386. [Google Scholar]
- 26. Csávás M., Demeter T., Herczeg M., Timári I., Kövér K. E., Herczegh P., Borbás A., Tetrahedron Lett. 2014, 55, 6983–6986. [Google Scholar]
- 27. Csávás M., Malinovská L., Illyés T. Z., Wimmerová M., Borbás A., Carbohydr. Res. 2017, 437, 1–8. [DOI] [PubMed] [Google Scholar]
- 28. Povie G., Tran A.-T., Bonnaffé D., Habegger J., Hu Z., Le Narvor C., Renaud P., Angew. Chem. Int. Ed. 2014, 53, 3894–3898. [DOI] [PubMed] [Google Scholar]
- 29. Mayo F. R., Walling C., Chem. Rev. 1940, 27, 351–412. [Google Scholar]
- 30. Dénès F., Pichowicz M., Povie G., Renaud P., Chem. Rev. 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- 31. LeBel N. A., Czaja R. F., DeBoer A., J. Org. Chem. 1969, 34, 3112–3126. [Google Scholar]
- 32. Masamune S., Choy W., Petersen J. S., Sita L. R., Angew. Chem. Int. Ed. Engl. 1985, 24, 1–30. [Google Scholar]
- 33. Spijker N. M., van Boeckel A. A., Angew. Chem. Int. Ed. Engl. 1991, 30, 180–183. [Google Scholar]
- 34.
- 34a. Fraser-Reid B., López J. C., Gomez A. M., Uriel C., Eur. J. Org. Chem. 2004, 7, 1387–1395; [Google Scholar]
- 34b. Guillemineau M., Auzanneau F.-I., Carbohydr. Res. 2012, 357, 132–138. [DOI] [PubMed] [Google Scholar]
- 35. Fiore M., Lo Conte M., Pacifico S., Marra A., Dondoni A., Tetrahedron Lett. 2011, 52, 444–447. [Google Scholar]
- 36. Yadav J. S., Subba Reddy B. V., Reddy J. S. S., J. Chem. Soc., Perkin Trans. 1 2002, 21, 2390–2394. [Google Scholar]
- 37. Ferrier R. J., Petersen P. M., Tetrahedron 1990, 46, 1–11. [Google Scholar]
- 38.
- 38a. Krohn K., Florke U., Gehle D., J. Carbohydr. Chem. 2002, 21, 431–443; [Google Scholar]
- 38b. Naik P. U., Nara S. J., Harjani J. R., Salunkhe M. M., J. Mol. Catalysis A: Chemical 2005, 234, 35–43. [Google Scholar]
- 39.
- 39a. Grynkiewicz G., Priebe W., Zamojski A., Carbohydr. Res. 1979, 68, 33–41; [Google Scholar]
- 39b. Grugel H., Albrecht F., Minuth T., Boysen M. M. K., Org. Lett. 2012, 14, 3780–3783; [DOI] [PubMed] [Google Scholar]
- 39c. Rokade S. M., Bhate P. M., Carbohydr. Res. 2015, 415, 28–30. [DOI] [PubMed] [Google Scholar]
- 40. Varela O., de Fina G. M., de Lederkremer R. M., Carbohydr. Res. 1987, 167, 187–196. [Google Scholar]
- 41. Gupta P., Kumari N., Agarwal A., Vankar Y. D., Org. Biomol. Chem. 2008, 6, 3948–3956. [DOI] [PubMed] [Google Scholar]
- 42. Ichikawa Y., Isobe M., Konobe M., Goto T., Carbohdyr. Res. 1987, 171, 193–199. [Google Scholar]
- 43. Igarashi K., Honma T., J. Org. Chem. 1970, 35, 606–610. [Google Scholar]
- 44. Takeda K., Nakamura H., Ayabe A., Akiyama A., Harigaya Y., Mizuno Y., Tetrahedron Lett. 1994, 35, 125–128. [Google Scholar]
- 45. Colinas A., Bravo R. D., Carbohydr. Res. 2007, 342, 2297–2302. [DOI] [PubMed] [Google Scholar]
- 46. Horton D., Methods Carbohydr. Chem. 1963, 2, 433–437. [Google Scholar]
- 47. Wiesler W. T., Caruthers M. H., J. Org. Chem. 1996, 61, 4272–4281. [DOI] [PubMed] [Google Scholar]
- 48. Cerny M., Stanek J., Pacak J., Monatsh. Chem. 1963, 94, 290–294. [Google Scholar]
- 49. Ghosh T., Santra A., Misra A. K., Beilstein J. Org. Chem. 2013, 9, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matta K. L., Girotra R. N., Barlow J. J., Carbohydr. Res. 1975, 43, 101–109. [DOI] [PubMed] [Google Scholar]
- 51. Stanek J., Sindlerova M., Cerny M., Collect. Czech. Chem. Commun. 1965, 30, 297–303. [Google Scholar]
- 52.
- 52a. Sivapriya K., Chandrasekaran S., Carbohydr. Res. 2006, 341, 2204–2210; [DOI] [PubMed] [Google Scholar]
- 52b. Ge J.-T., Zhou L., Zhao F.-L., Dong H., J. Org. Chem. 2017, 82, 12613–12623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary