

Abstract
1‐Aminopyrenes with three ω‐hydroxylated N‐alkylsulfonamido or alkylsulfonyl residues in positions 3, 6, and 8 were prepared, O‐phosphorylated, and applied for reductive amination of oligosaccharides. The dyes (ϵ≈20 000 m −1 cm−1) with six negative charges (pH≥8) and low m/z ratios enable labeling and fluorescence detection of reducing sugars (glycans) related to the most structurally and functionally diverse class of natural products. Under excitation with a 488 nm laser, the new glycoconjugates emit yellow light of about 560 nm, outperforming (with respect to brightness and faster electrophoretic mobilities) the corresponding APTS derivatives (benchmark dye with green emission in conjugates).
Keywords: chromophores, donor–acceptor systems, electrophoresis, fluorescent probes, glycoconjugates
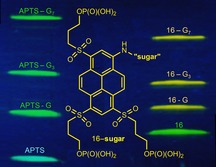
Bright 1‐aminopyrenes (ϵ≈20.000 m −1 cm−1) with six negative charges (at pH≥8) and high mobility in the electric field are designed for labeling and fluorescence detection of glycans. The right and left lanes in the figure show the gel electrophoresis bands of new and reference dyes (lowest bands) and their conjugates.
Glycosylation is an enzymatically driven and highly diverse transformation of proteins, lipids, and other noncarbohydrates. The products of glycosylation (glycoconjugates) have a new chemical bond formed between a carbohydrate (glycan; donor) and another molecule (acceptor). Glycoconjugates represent one of the most structurally and functionally diverse class of natural products involved in fundamental biochemical processes in living matter.1 Only few specific functions of these complex and carbohydrate‐rich molecules have been well understood so far.2 Further progress in glycomics and glycobiology depends on the advances in analytic techniques applicable to complex carbohydrates. Carbohydrates do not absorb visible light, and for the sensitive detection by emission they need to be labeled with a fluorescent tag.3 Capillary gel electrophoresis (CGE) is an important method for analyzing glycoconjugates including glycoproteins, glycopeptides, and “released” (enzymatically cleaved from the acceptors) N‐ or O‐glycans.4 The net electrical charge is required for separation of the analytes by CGE. The native carbohydrates, except sialic or glucuronic acids, sulfated or phosphorylated derivatives, are uncharged and cannot be separated by their mass to charge ratio (electrophoresis). Importantly, the features of an “ideal” fluorescent label required for CGE—the electrical charge, emissive properties, and the reactive group—can be incorporated in one fluorescence dye with (multiple) charges and the amino group reacting with aldehyde residues in reducing sugars (Scheme 1). Combined with laser‐induced fluorescence detection (LIF), this method allows fast and very fine resolution of analytes according to their mass to charge ratio (m/z) and hydrodynamic radius.5 A high throughput analysis of fluorescent glycan derivatives is performed on commercial DNA sequencers equipped with a CGE‐LIF module.5c The fluorescent label (R3‐NH2 in Scheme 1) applicable in CGE‐LIF must have an amino group suitable for reductive amination, high electrophoretic mobility, and “brightness” (which is the product of the fluorescence quantum yield and absorption coefficient at the excitation wavelength).
Scheme 1.
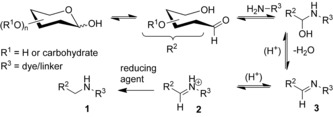
Reductive amination of mono‐ and oligosaccharides.
APTS (Scheme 2) emerged as a unique dye for the reductive amination and detection of glycans.5, 6, 7 The fluorescence of APTS derivatives is captured in the “green” color channel of the standard DNA sequencers.5c Conjugates of APTS are excitable with an argon laser (emission lines 488 nm and 514 nm).5c, 5e However, the performance of APTS as a fluorescent tag providing only one emission color, moderate brightness, and three negative charges, is limited. To facilitate further progress in glycomics, glycobiology, and analytical chemistry of complex carbohydrates, we developed bright fluorescent dyes with an aromatic amino group, multiple negative charges and yellow emission.
Scheme 2.
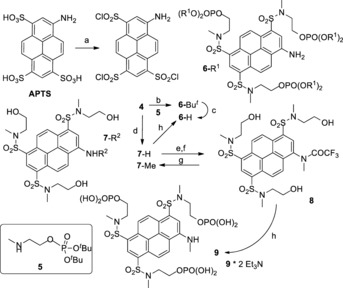
Triple O‐phosphorylated 1‐aminopyrene trisulfonamides: a) ClSO3H, 65 °C, 3 h; b) 5, Et3N, MeCN, r.t., overnight; c) CF3CO2H, r.t., 1 h, then 1 m aq. Et3N/H2CO3 (pH 8–9), overnight; d) CH3NHCH2CH2OH, aq. MeCN, r.t., overnight; e) (CF3CO)2O, CH2Cl2, Et3N, then CH3I, Cs2CO3, DMF, 70 °C, 40 min; f) MeOH, NaHCO3; g) aq. NaOH, MeOH, r.t.; h) POCl3, (MeO)3PO, r.t., 3 h, then aq. Et3N/H2CO3 buffer (pH 8–9), overnight.
The high‐performing fluorescent tags applicable in the reductive amination and CGE‐LIF of glycans must have an amino group for which the pK a of the conjugated acid is in the range of 3–4 for the efficient reaction (Scheme 1) at pH≈3. The glycan conjugates should have a net charge of −3…−6 at pH 8 (pH of the buffer solution in CGE) to provide high electrophoretic mobility, should be soluble in aqueous buffers, and should be stable against reduction with boranes or borohydrides over a wide pH range (3–8). The absorption at 488 nm (ϵ 488) or 505 nm (ϵ 505) determined the excitation efficiency with an argon ion laser or solid‐state laser; high brightness and the minimal cross‐talk with the “APTS channel” in the detector (low emission of conjugates at 520 nm) are also required. These features are set by the reaction conditions in Scheme 1, and the properties of the standard DNA‐sequencing equipment used for the separation and detection of the fluorescent glycan derivatives. APTS is a reference dye with green emission in conjugates with oligosaccharides (see Figure 2 and the Supporting Information). We designed new dyes with yellow emission in conjugates, trying to minimize the interference with an APTS detection window.
Figure 2.

Gel electrophoresis (migration from “north” to “south”, pH 8.3); detection by emission (excitation at 365 nm). From bottomn to top. Lane 1: APTS (lowest; blue), APTS+G, APTS+G3, APTS+G7 (green). Lane 2: dye 16 (green), 16+G, 16+G3, 16+G7 (yellow). Lane 3: APTS, APTS+M, APTS+M2‐2O/APTS+M2‐3O (unresolved), APTS+M2‐4O, APTS+M3 and APTS+M4. Lane 4: dye 16, 16+M, 16+M2‐3O, 16+M2‐2O/16+M2‐4O (unresolved), 16+M3 and 16+M4. Lane 5: APTS, APTS‐labeled 3′‐ and 6′‐sialyllactoses. Lane 6: dye 16 and its conjugates with 3′‐ and 6′‐sialyllactoses.
To achieve this goal, we converted the sulfonic acid residues in APTS (Scheme 2) into more powerful electron acceptors—sulfonamides. Sulfonamides (represented by dyes 6–9 in Scheme 2) have higher values of the Hammett σ‐constants (σ m=0.53, σ p=0.60 for SO2NH2)8 than ionized sulfonic acid residues in APTS (σ m=0.05, σ p=0.09).9 The presence of an electron‐donating (N‐alkyl)amino group and the acceptor groups in “active” positions (3, 6, and 8) of the pyrene system leads to the “push–pull” dyes10 emitting blue‐green (APTS), green (6‐R1, 7‐H), or yellow (7‐Me, 9) light. The spectral properties of the dyes are given in Table 1 (see also the Supporting Information).
Table 1.
Spectral properties of the dyes and their m/z ratios.
|
Dye (m/z) |
Absorption λ max [nm] (ϵ, m −1 cm−1) |
Emission λ max [nm] (Φ fl)[a] |
Solvent |
Fluor. lifetime τ [ns] |
|---|---|---|---|---|
|
APTS [b] (151) |
424 (20 600) |
500 (0.95) |
aq. PBS |
– |
|
APTS‐G6 [c] |
455 (17 000) |
511 |
H2O |
– |
|
6‐H (144) |
471 |
544 (0.88) |
H2O |
5.9 |
|
7‐H |
477 (22 400) |
535 (0.96) |
MeOH |
5.6 |
|
7‐Me |
493 (23 000) |
549 (0.97) |
MeOH |
5.9 |
|
9 |
502 |
563 (0.85) |
H2O |
3.6 |
|
13 |
502 (23 400) 509 (19 500) |
550 (0.88) 563 (0.67) |
MeOH H2O |
6.3 6.4 |
|
15 |
486 (21 000) |
534 (0.80)[d,e] |
MeOH |
4.9 |
|
16 (137) |
477 (19 600) |
542 (0.92) |
TEAB[f,g] |
5.8 |
[a] Absolute values of the fluorescence quantum yields (if not stated otherwise). [b] Data from Ref. 6b, abs. measured in H2O, emission measured in aq. phosphate buffer at pH 7.4. [c] Conjugate with maltohexaose (G6), data from Ref. 5b. [e] Excitation at 375 nm. [e] Rhodamine 6G as a reference dye with Φ fl=0.9. [f] Aq. Et3N*H2CO3, pH 8–8.5. [g] Fluorescein as a reference dye with Φ fl=0.9 in 0.1 m NaOH.
To provide multiple negative charges and high electrophoretic mobility at pH 8, primary phosphates (R‐OPO3H2) are preferred over phosphonates,11a because their first and second pK a values are in the range of 1.5–1.9 and 6.3–6.8, respectively.11b In the electrophoresis buffer solution, one primary phosphate group introduces two negative charges (phosphonates are less acidic and not fully ionized at pH 8). APTS (Scheme 2) was converted into the relatively stable 1,3,6‐tris(chlorosulfonyl)pyrene‐8‐amine (4) and then to sulfonamides (6‐tBu, 7‐H) by reaction with the corresponding amine (5 or CH3NHCH2CH2OH).
Direct phosphorylation of three hydroxyl groups in 7‐H or 8 (reagent h in Scheme 2) followed by hydrolysis12 afforded dyes 6‐H (16 %) or 9 bearing six negative charges at pH 8. To improve the yield, we prepared O‐phosphorylated N‐(methylamino)ethanol 5, let it react with compound 4, isolated the intermediate 6‐tBu, and then converted it into dye 6‐H (83 %). Trying to increase the polarity of the aminopyrene chromophore even further, we introduced alkyl sulfonyl groups into positions 3, 6, and 8 of 1‐aminopyrene and prepared dyes 13, 15, and 16 (Scheme 3). Alkyl sulfonyl substituents have higher values of the Hammett σ‐constants (σ m=0.56–0.66, σ p=0.68–0.77 for SO2Alkyl)8 than sulfonamides (σ m=0.53, σ p=0.60 for SO2NH2).8 This indicates that they are more powerful acceptors than sulfonamides (compounds 6‐R1, 7‐R2, and 9 in Scheme 2).
Scheme 3.
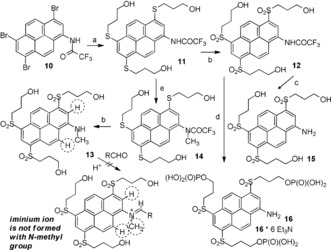
Triple O‐phosphorylated 1,3,6‐tris[(3‐hydroxypropyl)sulfonyl]‐pyrene‐8‐amine 16: a) HS(CH2)3OH, DIPEA, Pd2(dba)3, XantPhos, DMF, 100 °C, overnight; b) aq. H2O2, Na2WO4, AcOH, r.t., overnight; c) aq. NaOH, MeOH; d) POCl3, (MeO)3PO, r.t., then aq. Et3N/H2CO3 buffer (pH 8–9); e) CH3I, Cs2CO3, DMF.
We found that the trifluoroacetyl residue is a better protecting group for 1‐aminopyrene than acetyl,13 because it is acid‐stable but can be easily cleaved under mild basic conditions. Bromination of 1‐(trifluoroacetylamino)pyrene led to tribromide 10 (Scheme 3), an important precursor to the functionally substituted aminopyrenes. Palladium‐catalyzed cross‐coupling14 of tribromide 10 with 3‐mercapto‐1‐propanol afforded triol 11 with three alkyl aryl sulfide residues. Oxidation of 11 with hydrogen peroxide in acetic acid in the presence of sodium tungstate15 led to trisulfonyl derivative 12. Deprotection of the amino group in compound 12 by heating with aq. NaOH in methanol gave amine 15 (model compound). Another model dye with an N‐methyl group (13) was prepared from intermediate 11 by N‐methylation of the trifluoroacetylamino group and mild hydrolysis of the amide group. Phosphorylation of 12 followed by hydrolysis led to aminopyrene 16 with three primary phosphate groups attached to alkyl sulfonyl residues.The phosphate groups in dyes 6‐H, 9, and 16 are hydrolytically stable over a broad pH range, from pH 3 (reductive amination conditions) to pH 8.3 (electrophoresis) and beyond.
The model pyrenes with N‐methylamino groups (7‐Me, 9 in Scheme 2, and 13 in Scheme 3) are structurally similar to the final products formed in Scheme 1 upon reductive amination of carbohydrates; they make it possible to measure the red‐shifts in the absorption and emission spectra and extinction coefficients (7‐Me, 13 in Table 1). Importantly, dyes 7‐Me, 9, and 13 did not participate in the reductive amination of glucose (even under harsh conditions). This result may be explained by the additional steric hindrance associated with the planar iminium ion (Scheme 3), in which two aromatic hydrogen atoms adjacent to the reaction center are expected to “repulse“ the N‐methyl group.
The photophysical properties of APTS, its conjugate with maltohexaose (APTS‐G6), and the new pyrenes, are given in Table 1. The dyes in Table 1 form two groups: compounds with a primary amino group (APTS, 6‐H, 7‐H, 15, and 16) and dyes with a secondary amino group (APTS‐G6, 7‐Me, 9, and 13). Pyrenes in the first group absorb at 424 nm (APTS) to 486 nm (15). Compounds in the second group are related to the products formed in the course of reductive amination (Scheme 1); their absorption maxima are red‐shifted and found in the range from 455 nm (APTS‐G6) to 509 nm (13) (in aqueous solutions). For example, N‐methylation (6‐H→9) shifted the absorption maximum to the red by 31 nm, while the emission underwent bathofluoric shift of “only” 19 nm. Thus, the Stokes shift was reduced from 73 nm (6‐H) to 61 nm (9). Importantly, within each group the spectrum of APTS or APTS‐G6 conjugate is separated from the other spectra of the same group by 42–69 nm (absorption maxima) and by 38–54 nm (emission maxima): new dyes absorb and emit at longer wavelengths. This feature is important, as the glycan conjugates of dyes 6‐H and 16 are intended to have minimal emission in the APTS detection window. Comparing sulfonamides (6‐H, 7‐Me) with structurally related alkyl sulfones (13, 16) we observed the red shift of only 6–9 nm in absorption, while the positions and shapes of emission bands remained the same (Table 1 and Figure 1). The electron‐donating capacity of the amino group as a single donor is limited, and this can explain only the small red‐shift observed by transition from sulfonamides to alkyl sulfones.
Figure 1.
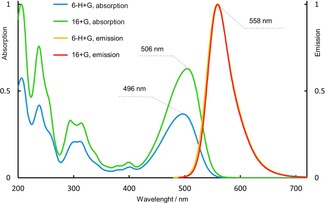
Absorption and emission spectra (aq. Et3N/H2CO3 buffer) of conjugates with glucose prepared from dyes 6‐H and 16.
The brightness of a glycan label is termed as a product of the extinction coefficient (at 488 nm, as an excitation wavelength) and the fluorescence quantum yield. The fluorescence quantum yields of dyes 6‐H and 16 (Table 1) are 0.88 and 0.92, respectively. For APTS conjugates, the extinction coefficient at the maximum (455 nm) is 17 000 m −1 cm−1, and the absorption at 488 nm is ca. 35 % of the maximal value.5b For all new N‐alkylated pyrenes (7‐Me, 9, 13, 6‐H+G, 16+G) we can assume the extinction coefficient at 488 nm to be about 18 000 m −1 cm−1. Therefore, the conjugates of the new dyes are ca. three times brighter than APTS derivatives (under excitation with the 488 nm laser).
We used APTS as a reference dye and compounds 6‐H and 16 as new reagents, and obtained their conjugates with glucose (G) and oligomers (maltotriose G3 and maltoheptaose G7), mannose (M), and oligomers [2‐O‐, 3‐O‐ and 4‐O‐(α‐d‐mannopyranosyl)‐d‐mannoses (M2‐2O, M2‐3O and M2‐4O), mannotriose (M3), mannotetraose (M4)], as well as 3′‐ and 6′‐sialyllactoses. Due to the presence of the strong acceptors—sulfonamido and alkylsulfonyl groups—dyes 6‐H and 16 undergo reductive alkylation more reluctantly than APTS. The detailed procedures for reductive amination16 and yields of the individual conjugates17 are given in Supporting Information. Figure 1 shows absorption and emission spectra of glucose conjugates prepared from dyes 6‐H and 16. The absorption spectra are very similar, and the emission spectra are practically identical. Therefore, we preferentially used dye 16 in reductive amination. Figure 2 shows the gel electrophoresis results obtained with APTS and its conjugates (lanes 1, 3, 5), as well as compound 16 and its conjugates with reducing sugars (lanes 2, 4, 6). The conjugates of dye 16 move faster than the corresponding conjugates of APTS. Due to higher net charge of dyes 6‐H, 16, and their conjugates, the distances between yellow bands related to them are shorter than the distances between green bands of APTS conjugates (Figure 2, Figure S5 and S6). In the case of mannobiose isomers, the selectivity profile of dye 16 is different from that of APTS (Figure 2). Conjugates of M2‐2O and M2‐3O with APTS move as one spot (lane 3, fourth spot from the top), and APTS+M2‐4O moves slower (lane 3, third spot from top). Conjugates 16+M2‐2O and 16+M2‐4O move as one spot, and conjugate 16+M2‐3O moves faster (lane 4 in Figure 2). Each conjugate was also analyzed separately (Figure S6). Both dyes (APTS and 16) easily resolve 3′‐ and 6′‐isomers of sialyllactoses (lanes 5 and 6).
The conjugates of the new dyes are ca. three times brighter than APTS derivatives (excitation with the 488 nm laser). The results obtained with dimers of mannose indicate that the selectivity profile of dye 16 is different from that of APTS, and this feature may be useful for the analysis of complex glycan mixtures. Figures 2, S5, and S6 show that all conjugates of dyes 6‐H and 16 move faster than the corresponding APTS analogues.18 Therefore, dyes 6‐H and 16 with six negative charges may reveal “heavy” glycans undetectable with APTS due to very long retention times caused by the relatively low charge (−3) and the limited brightness. Access to the DNA sequencer with a CGE‐LIF unit will make it possible to evaluate the crosstalk between the emission signals of APTS, on one hand, and the dyes 6‐H and 16, on the other hand (also in conjugates), and their applicability for calibration of the retention times in CGE.4b
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
L.T. is grateful to Max Planck Institute for Dynamics of Complex Technical Systems (Magdeburg) for financial support. We thank Dr. M. Fomin (MPIBPC) for discussions and help with software. We thank J. Bienert (MPIBPC), Dr. H. Frauendorf, Dr. M. John, and co‐workers (Institut für organische und biomolekulare Chemie, Georg‐August‐Universität, Göttingen, Germany) for recording NMR and mass spectra.
E. A. Savicheva, G. Y. Mitronova, L. Thomas, M. J. Böhm, J. Seikowski, V. N. Belov, S. W. Hell, Angew. Chem. Int. Ed. 2020, 59, 5505.
Contributor Information
Elizaveta A. Savicheva, http://www.mpibpc.gwdg.de/abteilungen/200
Dr. Vladimir N. Belov, Email: vladimir.belov@mpibpc.mpg.de.
References
- 1.
- 1a. Turnbull J. E., Field R. A., Nat. Chem. Biol. 2007, 3, 74–77; [DOI] [PubMed] [Google Scholar]
- 1b. Cummings R. D., Pierce J. M., Chem. Biol. 2014, 21, 1–15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Varki A., Glycobiology 2017, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, 2009, pp. 1–36, 101–114, 617–632, 661–678; [PubMed] [Google Scholar]
- 2b. Roseman S., J. Biol. Chem. 2001, 276, 41527–41542. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Ruhaak L. R., Zauner G., Huhn C., Bruggink C., Deelder A. M., Wuhrer M., Anal. Bioanal. Chem. 2010, 397, 3457–3481; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Villadsen K., Martos-Maldonado M. C., Jensen K. J., Thygesen M. B., ChemBioChem 2017, 18, 574–612; [DOI] [PubMed] [Google Scholar]
- 3c. Ruhaak L. R., Xu G., Li Q., Goonatilleke E., Lebrilla C. B., Chem. Rev. 2018, 118, 7886–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Novotny M. V., Alley W. R. Jr., Curr. Opin. Chem. Biol. 2013, 17, 832–840; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Lu G., Crihfield C. L., Gattu S., Veltri L. M., Holland L. A., Chem. Rev. 2018, 118, 7867–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Guttman A., Pritchett T., Electrophoresis 1995, 16, 1906–1911; [DOI] [PubMed] [Google Scholar]
- 5b. Evangelista R. A., Liu M.-S., Chen F.-T. A., Anal. Chem. 1995, 67, 2239–2245; [Google Scholar]
- 5c. Laroy W., Contreras R., Callewaert N., Nat. Protoc. 2006, 1, 397–405; [DOI] [PubMed] [Google Scholar]
- 5d. Volpi N., Capillary electrophoresis of carbohydrates. From monosaccharides to complex polysaccharides, Humana Press, New York, 2011, pp. 1–51; [Google Scholar]
- 5e. Callewaert N., Geysens S., Molemans F., Contreras R., Glycobiology 2001, 11, 275–281. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Suzuki H., Müller O., Guttman A., Karger B. L., Anal. Chem. 1997, 69, 4554–4559; [DOI] [PubMed] [Google Scholar]
- 6b. Sharrett Z., Gamsey S., Hirayama L., Vilozny B., Suri J. T., Wessling R. A., Singaram B., Org. Biomol. Chem. 2009, 7, 1461–1470; [DOI] [PubMed] [Google Scholar]
- 6c. Ruhaak L. R., Hennig R., Huhn C., Borowiak M., Dolhain R. J. E. M., Deelder A. M., Rapp E., Wuhrer M., J. Proteome Res. 2010, 9, 6655–6664; [DOI] [PubMed] [Google Scholar]
- 6d. Bunz S.-C., Cutillo F., Neusüß C., Anal. Bioanal. Chem. 2013, 405, 8277–8284. [DOI] [PubMed] [Google Scholar]
- 7. Pabst M., Kolarich D., Pöltl G., Dalik T., Lubec G., Hofinger A., Altmann F., Anal. Biochem. 2009, 384, 263–273. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Hansch C., Leo A., Taft R. W., Chem. Rev. 1991, 91, 165–195; [Google Scholar]
- 8b. McDaniel D. H., Brown H. C., J. Org. Chem. 1958, 23, 420–427. [Google Scholar]
- 9. Zollinger H., Büchler W., Wittwer C., Helv. Chim. Acta 1953, 36, 1711–1722; larger values for SO3 − (σ m=0.30 and σ p=0.35) are mentioned in ref. [8a] as a private communication of Viktor Palm. [Google Scholar]
- 10. Bureš F., RSC Adv. 2014, 4, 58826–58851. [Google Scholar]
- 11.
- 11a. Butkevich A. N., Sednev M. V., Shojaei H., Belov V. N., Hell S. W., Org. Lett. 2018, 20, 1261–1264; [DOI] [PubMed] [Google Scholar]
- 11b. Wold F., Ballou C. E., J. Biol. Chem. 1957, 227, 301–312. [PubMed] [Google Scholar]
- 12.Hydrolysis with aq. Et3N/H2CO3 buffer (after removal of POCl3 and trimethyl phosphate) transforms O-alkyldichlorophosphates to primary alkyl phosphates and cleaves the N–P bond formed upon phosphorylation of the weakly nucleophilic arylamine.
- 13. Li T., Giasson R., J. Am. Chem. Soc. 1994, 116, 9890–9893. [Google Scholar]
- 14.
- 14a. Itoh T., Mase T., Org. Lett. 2004, 6, 4587–4590; [DOI] [PubMed] [Google Scholar]
- 14b. Mispelaere-Canivet C., Spindler J.-F., Perrio S., Beslin P., Tetrahedron 2005, 61, 5253–5259. [Google Scholar]
- 15. Schultz H. S., Freyermuth H. B., Buc S. R., J. Org. Chem. 1963, 28, 1140–1142. [Google Scholar]
- 16.
- 16a. Evangelista R. A., Guttman A., Chen F.-T. A., Electrophoresis 1996, 17, 347–351; [DOI] [PubMed] [Google Scholar]
- 16b. Ruhaak L. R., Steenvoorden E., Koeleman C. A. M., Deelder A. M., Wuhrer M., Proteomics 2010, 10, 2330–2336; [DOI] [PubMed] [Google Scholar]
- 16c. Chen F.-T. A., Dobashi T. S., Evangelista R. A., Glycobiology 1998, 8, 1045–1052. [DOI] [PubMed] [Google Scholar]
- 17.Yields were determined by integration of HPLC peak areas for the free dyes and their conjugates at isosbestic points; see Table S1 in the Supporting Information.
- 18. 6-H+G moves slower than APTS+G (Figure S5).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary