

Abstract
Alkynes are an important class of organic molecules due to their utility as versatile building blocks in synthesis. Although efforts have been devoted to the difunctionalization of alkynes, general and practical strategies for the direct hydroalkylation and alkylarylation of terminal alkynes under mild reaction conditions are less explored. Herein, we report a photoredox/nickel dual‐catalyzed anti‐Markovnikov‐type hydroalkylation of terminal alkynes as well as a one‐pot arylalkylation of alkynes with alkyl carboxylic acids and aryl bromides via a three‐component cross‐coupling. The results indicate that the transformations proceed via a new mechanism involving a single‐electron transfer with subsequent energy‐transfer activation pathways. Moreover, steady‐state and time‐resolved fluorescence‐spectroscopy measurements, density functional theory (DFT) calculations, and wavefunction analysis have been performed to give an insight into the catalytic cycle.
Keywords: alkynes, arylalkylation, DFT calculations, hydroalkylation, nickel, photoredox reactions
Meditation on alkylation: A photoredox/nickel dual‐catalyzed anti‐Markovnikov‐type hydroalkylation of terminal alkynes as well as a one‐pot alkylarylation of alkynes with alkyl carboxylic acids and aryl bromides via a three‐component cross‐coupling are presented. The reactions take place efficiently, with excellent regioselectivity and moderate to excellent stereoselectivity under mild conditions.
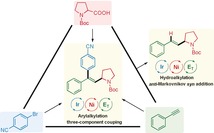
Introduction
The development of improved methods for the efficient construction of multiple carbon–carbon and/or carbon–heteroatom bonds in one synthetic operation is of great interest. In this context, the potential of multi‐component reactions (MCRs) as a tool to increase efficiency, productivity, reduce costs, and decrease the environmental impact has been long recognized.1 Furthermore, multi‐component reactions are highly attractive as they allow the assembly of complex structures starting from simple starting materials. The feasibility of MCRs in the functionalization of carbon–carbon triple bonds, which are ubiquitous in organic compounds, has been confirmed especially in the synthesis of heterocycles.2 In contrast, the application of MCRs to the dicarbofunctionalization of readily accessible terminal alkynes, affording structurally defined multi‐substituted alkenes, has been rarely realized.3 Moreover, alternative protocols require the use of organo‐metal species and multistep transformations.4 Therefore, it is desirable to develop general, efficient, and practical protocols for the dicarbofunctionalization of terminal alkynes.5
In addition to dicarbofunctionalization, the hydrocarbofunctionalization of terminal alkynes is a fundamental transformation leading to valuable substituted alkene products. While the hydroarylation of terminal alkynes has been established,6 hydroalkylation is rare.7 Thus, the development of general, efficient, and practical protocols for the hydroalkylation of terminal alkynes would be an important advancement.
Recently, photo‐mediated transition‐metal catalysis has emerged as a powerful method in organic synthesis.8 In particular, photoredox/nickel dual catalysis shows advantages and important products have been afforded by using this strategy.9 Notably, photoredox/nickel dual catalysis has recently found application in the hydroalkylation of terminal and internal alkynes.10, 11 Considerable progress has been made by the Wu group to realize the hydroalkylation of internal alkynes with ether and amide C(sp3)−H bonds with high selectivity.10 During the course of this work, a decarboxylative hydroalkylation of aliphatic alkynes with alkyl carboxylic acids was achieved, giving the corresponding product in Markovnikov regioselectivity.11 However, protocols for the hydroalkylation of alkynes, especially with the opposite regioselectivity, are still lacking.
Despite the significant progress made in photoredox and transition‐metal dual‐catalyzed two‐component cross‐coupling reactions, less is known about challenging multi‐component cross‐couplings via this dual catalysis. Considering the great significance of the one‐pot difunctionalization of alkynes, we herein report a photoredox/nickel dual‐catalyzed anti‐Markovnikov‐type hydroalkylation of alkynes as well as the one‐pot arylalkylation of alkynes with alkyl carboxylic acids and aryl bromides via three‐component cross‐coupling under mild conditions (Scheme 1). A wide range of disubstituted and trisubstituted olefin products have been accessed in a syn‐addition manner, which is difficult to achieve using traditional methods. This protocol features the utilization of feedstock starting materials, mild reaction conditions, high functional‐group tolerance, complex molecular applications, successful scale‐up, and a novel mechanism involving single‐electron transfer (SET) and energy‐transfer (E T) activation pathways.
Scheme 1.
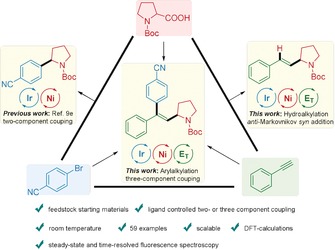
Photoredox/nickel dual‐catalyzed transformations involving aryl bromides, carboxylic acids, and alkynes.
Results and Discussion
We initiated our study by finding suitable conditions for the catalytic hydroalkylation of alkynes using ethynylbenzene 1 a and Boc‐Pro‐OH 2 a as model substrates, Cs2CO3 as base, Pc1 as photocatalyst, NiCl2⋅glyme as the nickel source, and DMF as solvent, which afforded the corresponding anti‐Markovnikov‐type hydroalkylation product 3 a in 31 % yield only (Table 1, entry 1). Various solvents were evaluated next, and acetonitrile was found to give a slightly higher yield and stereoselectivity (entries 2–4). When other bases were used, the reaction did not occur or took place with lower yields (entries 5–7). The photocatalyst screening revealed that Pc4 gives the product with higher stereoselectivity without decreasing the yield (entries 8–12). Several commonly used nickel catalysts were evaluated next; however, no further improvement was observed (entries 13–16). Using 10 equiv of H2O as an additive decreased the yield (entry 17). Finally, the best yield (72 %) was obtained after adjusting the amount of radical precursor and base, and performing the reaction in a mixture of solvents for a prolonged reaction time (entries 18 to 20). Using 10 mol % dtbbpy as ligand decreased the yield and the stereoselectivity (entry 21). Reducing the amount of carboxylic acid 2 a to 1 equiv slightly decreased the yield (57 % yield, entry 22), while adding 2 equiv of H2O was beneficial for the formation of the desired product (65 % yield, entry 23). Control experiments were conducted, and no product was obtained in the absence of the nickel catalyst and photocatalyst (entries 24 and 25).
Table 1.
Reaction conditions for the optimization of the photoredox/nickel dual‐catalyzed hydroalkylation of alkynes.

|
Entry[a] |
Pc |
[Ni] |
Base |
Solvent |
Yield (%)[b] |
|---|---|---|---|---|---|
|
1 |
Pc1 |
NiCl2⋅glyme |
Cs2CO3 |
DMF |
31 (60:40) |
|
2 |
Pc1 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
32 (66:34) |
|
3 |
Pc1 |
NiCl2⋅glyme |
Cs2CO3 |
DMSO |
28 (60:40) |
|
4 |
Pc1 |
NiCl2⋅glyme |
Cs2CO3 |
acetone |
31 (63:37) |
|
5 |
Pc1 |
NiCl2⋅glyme |
K2CO3 |
CH3CN |
31 (63:37) |
|
6 |
Pc1 |
NiCl2⋅glyme |
Na2CO3 |
CH3CN |
16 (60:40) |
|
7 |
Pc1 |
NiCl2⋅glyme |
Li2CO3 |
CH3CN |
0 |
|
8 |
Pc2 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
9 (71:29) |
|
9 |
Pc3 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
15 (69:31) |
|
10 |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
32 (73:27) |
|
11 |
Pc5 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
10 (63:37) |
|
12 |
Pc6 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
9 (60:40) |
|
13 |
Pc4 |
Ni(cod)2 |
Cs2CO3 |
CH3CN |
24 (73:27) |
|
14 |
Pc4 |
NiBr2 |
Cs2CO3 |
CH3CN |
23 (72:28) |
|
15 |
Pc4 |
Ni(acac)2 |
Cs2CO3 |
CH3CN |
12 (75:15) |
|
16 |
Pc4 |
Ni(OAc)2⋅4 H2O |
Cs2CO3 |
CH3CN |
18 (74:26) |
|
17[c] |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
20 (73:27) |
|
18[d] |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN |
45 (73:27) |
|
19[e] |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN+DMF |
54 (73:27) |
|
20[f] |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN+DMF |
72 (73:27) |
|
21[f,g] |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN+DMF |
15 (68:32) |
|
22[h] |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN+DMF |
57 (73:27) |
|
23[h,i] |
Pc4 |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN+DMF |
65 (73:27) |
|
24[e] |
Pc4 |
– |
Cs2CO3 |
CH3CN+DMF |
0 |
|
25[e] |
– |
NiCl2⋅glyme |
Cs2CO3 |
CH3CN+DMF |
0 |
[a] Reaction conditions: ethynylbenzene (0.1 mmol), Boc‐Pro‐OH (0.2 mmol), photocatalyst (0.001 mmol), [Ni] (0.01 mmol), base (0.2 mmol) in solvent (2.0 mL) at room temperature under irradiation with a 34 W blue LED for 12 h. [b] GC yield and calibrated Z/E‐ratio using (E)‐stilbene as internal standard. [c] H2O (10 equiv) was added. [d] Boc‐Pro‐OH (5 equiv) and Cs2CO3 (5 equiv) in 5 mL CH3CN, 24 h. [e] Boc‐Pro‐OH (5 equiv) and Cs2CO3 (5 equiv) in 2.5 mL CH3CN and 2.5 mL DMF, 24 h. [f] Boc‐Pro‐OH (3 equiv) and Cs2CO3 (1 equiv) in 2.5 mL CH3CN and 2.5 mL DMF, 48 h. [g] With 10 mol % dtbbpy as ligand. [h] Boc‐Pro‐OH (1 equiv) and Cs2CO3 (1 equiv) in 2.5 mL CH3CN and 2.5 mL DMF, 48 h. [i] H2O (2 equiv) was added. Pc=photocatalyst. 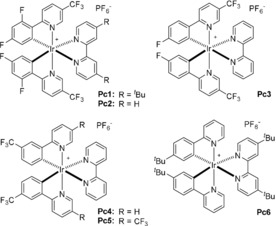
With the optimized reaction conditions in hand, the scope of the alkyne hydroalkylation with respect to both reaction partners was evaluated (Table 2). A series of electronically and sterically diverse terminal alkynes could be converted into the corresponding disubstituted alkenes in moderate to high yields. Although ethynylbenzenes bearing a methyl group in the para‐ and meta‐position underwent this transformation in good yields, ortho‐methyl‐substituted ethynylbenzene gave the corresponding product in moderate yield only, indicating that the process can be hampered by steric hindrance (3 b–3 d). Alkynes possessing electron‐donating and electron‐withdrawing functional groups such as methoxy, methylthio, fluoro, trifluoromethyl, cyano, and ester groups were all suitable substrates for this transformation (3 f–3 h and 3 j–3 q). Importantly, some potentially sensitive groups such as borate ester, chloride, and even bromide were well tolerated under our reaction conditions, allowing further functionalization of the generated products 3 r–3 u. Although substrates bearing electron‐withdrawing functional groups reacted with Boc‐Pro‐OH in only moderate yields (3 n, 3 p, and 3 r), good to high yields and better stereoselectivity were obtained when they reacted with a tertiary amino acid (3 o, 3 q, and 3 s). Also, product 3 v bearing a bicyclic residue was obtained in moderate yield. Moreover, this protocol could be readily extended to estrone‐ and heterocycle‐derived alkynes. Notably, chloride and trifluoromethyl functional groups in ortho‐position on the phenyl residue of ethynylbenzene improved the stereoselectivity of the transformation significantly, giving pure Z‐isomers of the corresponding products (3 y and 3 z). Notably, 3 z was obtained as Z‐isomer in a good yield of 71 % when the reaction was conducted on a 2.0 mmol scale, showing the scalability and practicability of this newly developed mild hydroalkylation protocol.
Table 2.
Substrate scope for the photoredox/nickel dual‐catalyzed hydroalkylation of alkynes.[a]

|
|
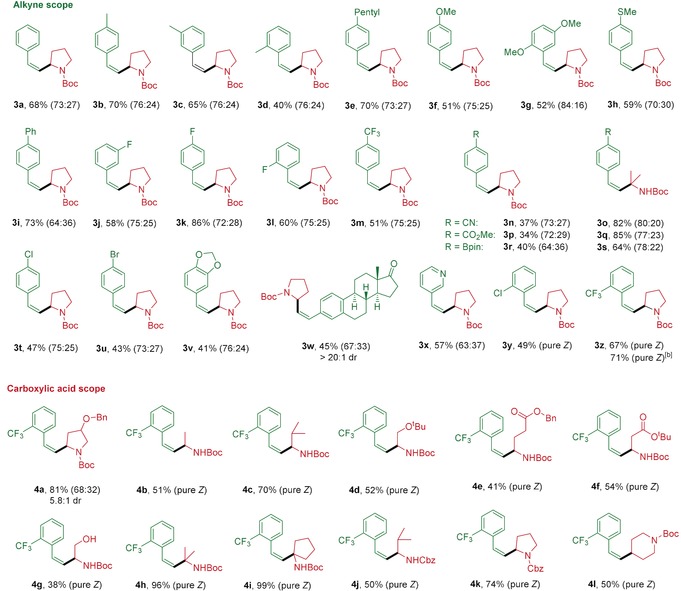
[a] Reaction conditions: alkyne (0.1 mmol), alkyl carboxylic acid (0.3 mmol), Pc4 (0.001 mmol), NiCl2⋅dme (0.01 mmol), Cs2CO3 (0.1 mmol) in DMF (2.5 mL) and CH3CN (2.5 mL) at room temperature under irradiation with a 34 W blue LED for 48 h. The values in parenthesis represent the Z/E‐ratio of the product determined by 1H NMR analysis. [b] The reaction was performed on a 2.0 mmol scale.
Next, the scope of alkyl carboxylic acids was explored. A wide range of Boc‐protected cyclic and acyclic secondary amino acids bearing alkyl, ether, ester, and even unprotected hydroxy functionalities underwent the reaction in moderate to high yields (4 a–4 g) with excellent stereoselectivity. Boc‐protected tertiary amino acids could improve the reaction efficiency significantly, giving the corresponding products 4 h and 4 i with excellent yields and stereoselectivities. Also, Cbz‐protected amino acids and common alkyl carboxylic acid were suitable substrates for this reaction.
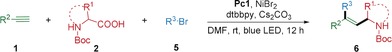
Finally, we addressed the more challenging arylalkylation of alkynes via a photoredox/nickel dual‐catalyzed three‐component cross‐coupling under mild reaction conditions. After evaluating various reaction parameters, the optimal reaction conditions were assigned as follows: Pc1 as photocatalyst, inexpensive NiBr2 as nickel catalyst, dtbbpy as ligand, and Cs2CO3 as base in DMF at room temperature for 12 h. We next examined the scope with respect to the three coupling partners (Table 3). Aryl bromides bearing electron‐withdrawing functional groups such as ester, ketone, and cyano underwent this three‐component cross‐coupling smoothly (6 a–6 c). Bicyclic and difunctionalized aryl bromides gave the corresponding trisubstituted alkenes in good yields (6 d and 6 e). Strikingly, the protocol was also applied to pharmaceutically related heterocyclic and natural‐product‐derived aryl bromides, giving the corresponding products in good yields (6 f–6 h).
Table 3.
Substrate scope for the photoredox/nickel dual‐catalyzed arylalkylation of alkynes.[a]
|
|
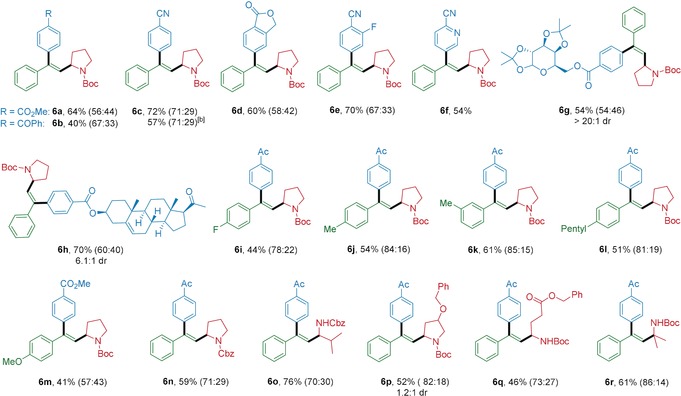
[a] Reaction conditions: aryl bromide (0.2 mmol), alkyl carboxylic acid (0.4 mmol), alkyne (0.6 mmol), Pc1 (0.002 mmol), NiBr2 (0.02 mmol), Cs2CO3 (0.4 mmol) in DMF (2.0 mL) at room temperature under irradiation with a 34 W blue LED for 12 h. The values in parenthesss represent the ratio of syn‐addition product to anti‐addition product determined by 1H NMR and NOESY analysis. [b] The reaction was performed on a 2.0 mmol scale.
Regarding the scope of alkynes, electron‐rich and electron‐poor terminal alkynes were suitable for this transformation (6 i–6 m). Likewise, various Boc‐ and Cbz‐ protected cyclic and acyclic secondary amino acids, as well as a tertiary amino acid, underwent this reaction in moderate to good yields (6 m–6 r). Also, 6 c was obtained in 57 % yield with 71:29 stereoselectivity when the reaction was performed on a 2.0 mmol scale.
To get an insight into these newly developed hydroalkylation and arylalkylation protocols, control experiments were conducted. When the reaction was conducted in deuterated acetonitrile and DMF, no deuterated product was observed (Scheme 2 a). When 2 equiv D2O was added to the reaction, the product was generated with 80:20 H/D ratio, which may be due to the formation of CsDCO3 via direct or indirect H/D exchange with D2O (Scheme 2 b,c). These results show that the hydrogen atom originates from carboxylic acids via the generation of CsHCO3. The isomerization efficiency for the pure E‐isomer in the presence of Pc4 is comparable to the model hydroalkylation reaction, indicating the crucial role of the photocatalyst in the generation of the Z‐isomer via an energy‐transfer process (Scheme 2 d).12 To gain more insight into the SET process, we first performed cyclic‐voltammetry (CV) measurements of Pc4 used in the hydroalkylation reactions (Scheme 2 e). The reduction potential of the triplet excited‐state photocatalyst 3*Pc4 [E 1/2(Ir*III/IrII)=+1.37 V vs. SCE in CH3CN]13 is thermodynamically favored to induce the single‐electron oxidation of deprotonated α‐amino acids (Boc‐Pro‐OCs (E 1/2=+0.95 V vs. SCE in CH3CN).9e Next, steady‐state Stern–Volmer luminescence‐quenching experiments were conducted by the addition of various concentrations of substrates (2 a or 1 a) to Pc4. The results show that the photocatalyst Pc4 was not quenched (Scheme 2 h). However, the deprotonated 2 a (2 a+Cs2CO3) quenched the photocatalyst Pc4 (Scheme 2 f) and the Stern–Volmer plot displayed a linear correlation (Scheme 2 h), showing the importance of the base in the generation of the alkyl radical. Additionally, a lifetime measurement of 3*Pc4 using time‐resolved emission spectroscopy was also carried out. Here, a relatively stable and long‐lived 3*Pc4 was observed (1127 ns, Figure S11 in the Supporting Information) which decreased monotonously with the increase in quencher concentration (2 a+Cs2CO3; Scheme 2 g). Furthermore, the slopes of the steady‐state and time‐resolved Stern–Volmer plots are very similar (Scheme 2 h), indicating that the major quenching process is the dynamic quenching of excited‐state 3*Pc4 by Boc‐Pro‐OCs.14 Also, the electron‐transfer (ET) rate constant k ET was determined by plotting difference between the observed rate constant (k obs) and the ground‐state recovery rate (k GSR) vs. different concentrations of quencher (2 a+Cs2CO3). A slope of (3.12±0.12)×109 L mol−1 s−1 was observed, which correlates with the ET rate constant, k ET, for 2 a and Pc4, while the intercept represents the backward reaction rate (Scheme 2 i), which is negligible, showing that this SET process is irreversible.15
Scheme 2.
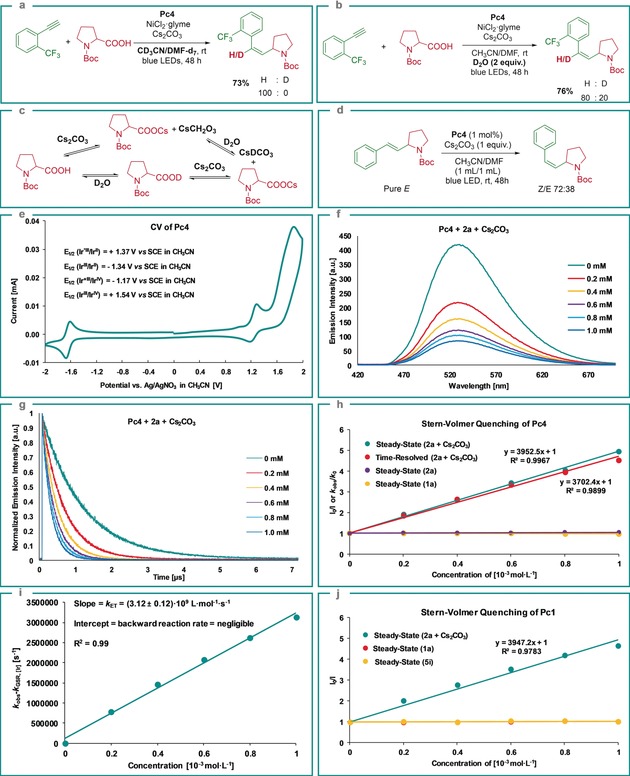
Mechanistic investigation. a) Reaction conducted in CD3CN and DMF‐d6. b) 2 equiv D2O was added. c) Formation of CsDCO3 via direct or indirect H/D exchange with D2O. d) Z/E‐isomerization via energy transfer using photocatalyst Pc4. e) CV measurement of Pc4. f) Steady‐state Stern–Volmer experiment of Pc4 and Boc‐Pro‐OCs (2 a+Cs2CO3). g) Phosphorescence lifetimes of 3*Pc4 at different concentrations of quencher (2 a+Cs2CO3). h) Combined quenching data of steady‐state and time‐resolved studies of Pc4. i) Stern–Volmer analysis yielded a rate constant, k ET, of (3.12±0.12)×109 L mol−1 s−1 by the SET between 3*Pc4 and Boc‐Pro‐OCs (2 a+Cs2CO3). The backward reaction rate is negligible. j) Steady‐state Stern–Volmer quenching analysis of Pc1 in the presence of Boc‐Pro‐OCs (2 a+Cs2CO3), phenylacetylene 1 a or p‐bromoacetophenone 5 i.
Furthermore, a steady‐state quenching experiment showed that the photocatalyst Pc1 used in arylalkylation reactions was quenched by Boc‐Pro‐OCs, while it was not quenched by phenylacetylene 1 a and p‐bromoacetophenone 5 i (Scheme 2 j). Based on these results, mechanisms for the photoredox/nickel dual‐catalyzed hydroalkylation and arylalkylation of alkynes are proposed (Scheme 3 a,b). First, the IrIII complex absorbs visible light and forms a long‐lived triplet excited state *IrIII complex.8b Oxidation of the carboxylic acid by the *IrIII complex and subsequent CO2 extrusion generates the alkyl radical intermediate 7,9e which is trapped by the NiI complex 8 to afford NiII intermediate 9. Reduction of 9 by the IrII reductant gives NiI intermediate 10, which undergoes 1,2‐migratory alkyne insertion to form NiI intermediate 11.11, 16 For the hydroalkylation process, protonation of 11 via intermediate 12 generates the E‐isomer 13 (concerted protonation demetallation, CPD) along with the formation of NiI complex 8.17 Next, the Z‐isomer 15 is obtained via intermediate 14 from the E‐isomer 13 through an energy‐transfer pathway (Scheme 3 a).3c Likewise, for arylalkylation, the generated NiI intermediate 19 undergoes the oxidative addition of an aryl halide to give intermediate 20, followed by reductive elimination to deliver the anti‐addition three‐component coupling product 21. At last, the syn‐addition three‐component coupling product 22 is obtained via the energy‐transfer pathway (Scheme 3 b).3c
Scheme 3.

Proposed mechanism for the photoredox/nickel dual‐catalyzed a) hydroalkylation and b) arylalkylation of alkynes.
Furthermore, DFT calculations for the migratory‐insertion and the concerted protonation–demetallation steps were conducted to rationalize and support the proposed mechanism (Scheme 4 a). As a model reaction, we investigated the cross‐coupling between phenylacetylene 1 a and Boc‐Pro‐OH 2 a. The migratory insertion step commenced with the coordination of phenylacetylene to the active alkyl‐NiI catalyst A, forming the intermediate complex B. Next, the alkyl group at the nickel center migrates to the terminal carbon atom of the alkyne to generate the NiI intermediate C via the transition state B‐TS with an energy barrier of only 6.3 kcal mol−1. In the next step, CsHCO3 coordinates to the formed NiI intermediate C to give NiI complex D, followed by the concerted protonation–demetallation to form the NiI intermediate E via the transition state D‐TS. Finally, the product is liberated along with the formation of the NiI complex F with a free energy gain of 10.4 kcal mol−1. Alternatively, the pathway in which the alkyl group at the nickel center migrates to the internal carbon atom of the alkyne was also calculated. The energy barrier of this pathway is 16.8 kcal mol−1 (B‐TS′), which is highly unfavorable compared the 6.3 kcal mol−1 of B‐TS. Additionally, the migratory insertion of the alkyne at the NiII center was calculated, which also shows anti‐Markovnikov regioselectivity; however, the energy barrier is considerably higher (Figure S13). To further explain the regioselectivity of the migratory insertion step, wavefunction analysis was conducted to calculate the ADCH (atomic‐dipole‐moment‐corrected Hirshfeld) atomic charge18 of the C(sp)‐carbon atoms C1 and C2 in the transition states B‐TS and B‐TS′ (Scheme 4 b).19 The results show that in the transition state B‐TS, the atomic charges for C1 and C2 are −0.75 and +0.25 a.u., respectively, while in the transition state B‐TS′, C2 and C1 have atomic charges of −0.47 and +0.22 a.u., respectively. Thus, the alkyl group is prone to migrate to C2 in B‐TS as it is more electrophilic than C1 in B‐TS′, and at the same time, C1 in B‐TS is considerably more nucleophilic than C2 in B‐TS′ and thus easier to bind to the nickel center.
Scheme 4.
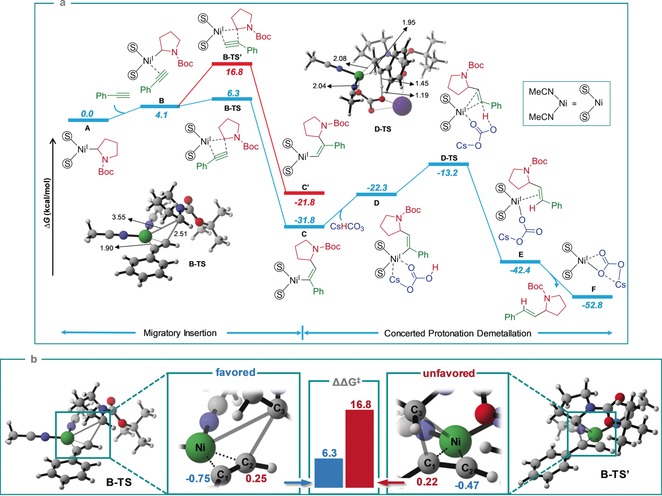
a) DFT‐computed energy profiles for the migratory insertion and concerted protonation–demetallation steps. Free energies in solution (in kcal mol−1) calculated at SMD(acetonitrile)‐M06/Def2‐TZVPP//PBE/Def2‐TZVP(Ni)/Def2‐SVP (other atoms) Selected optimized geometries are shown. Bond lengths in Å. b) Energy barrier for migratory insertion with different regioselectivities and the ADCH atomic charges of carbon atoms C1 and C2 at the transition states.
Conclusion
In conclusion, the hydroalkylation and arylalkylation of alkynes presented herein is an efficient approach for the synthesis of a series of highly valuable disubstituted and trisubstituted alkenes via photoredox/nickel dual catalysis which takes place under mild reaction conditions. Both protocols possess a broad substrate scope including complex starting materials and good functional‐group tolerance. Notably, the reactions take place efficiently, with excellent regioselectivity and moderate‐to‐excellent stereoselectivity (syn‐addition manner). Steady‐state and time‐resolved fluorescence‐spectroscopy measurements were conducted, confirming the dynamic quenching between the photocatalyst and deprotonated amino acid. Additionally, DFT calculations as well as wavefunction analysis were performed to reveal the regioselectivity of the migratory insertion step. Owing to the broad substrate scope and successful scale‐up, these protocols should find application in the synthesis of highly valuable disubstituted and trisubstituted alkenes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
C.Z. acknowledges the King Abdullah University of Science and Technology (KAUST) for support and the KAUST Supercomputing Laboratory for providing computational resources of the supercomputer Shaheen II. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. 617044 (SunCatChem).
H. Yue, C. Zhu, R. Kancherla, F. Liu, M. Rueping, Angew. Chem. Int. Ed. 2020, 59, 5738.
References
- 1.
- 1a. Tietze L. F., Modi A., Med. Res. Rev. 2000, 20, 304–322; [DOI] [PubMed] [Google Scholar]
- 1b. Masson G., Neuville L., Bughin C., Fayol A., Zhu J., Top. Heterocycl. Chem. 2010, 1–24;20871791 [Google Scholar]
- 1c. Dömling A., Wang W., Wang K., Chem. Rev. 2012, 112, 3083–3135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Zhu J., Wang Q., Wang M., Multicomponent reactions in organic synthesis, Wiley-VCH, Weinheim, 2015; [Google Scholar]
- 1e. Levi L., Mueller T. J., Chem. Soc. Rev. 2016, 45, 2825–2846. [DOI] [PubMed] [Google Scholar]
- 2. Müller T. J. J., Deilhof K., in Multicomponent Reactions in Organic Synthesis (Eds.: J. Zhu, Q. Wang, M.-X. Wang), Wiley-VCH, Weinheim, 2015, pp. 333–378. [Google Scholar]
- 3.
- 3a. Li Z., García-Domínguez A., Nevado C., J. Am. Chem. Soc. 2015, 137, 11610–11613; [DOI] [PubMed] [Google Scholar]
- 3b. Li Z., García-Domínguez A., Nevado C., Angew. Chem. Int. Ed. 2016, 55, 6938–6941; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7052–7055; [Google Scholar]
- 3c. Guo L., Song F., Zhu S., Li H., Chu L., Nat. Commun. 2018, 9, 4543; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Zhu C., Yue H., Maity B., Atodiresei I., Cavallo L., Rueping M., Nat. Catal. 2019, 2, 678–687. [Google Scholar]
- 4.
- 4a. Wipf P., Waller D. L., Reeves J. T., J. Org. Chem. 2005, 70, 8096–8102; [DOI] [PubMed] [Google Scholar]
- 4b. Cheung C. W., Hu X., Chem. Eur. J. 2015, 21, 18439–18444. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Chinchilla R., Najera C., Chem. Rev. 2014, 114, 1783–1826; [DOI] [PubMed] [Google Scholar]
- 5b. Trost B. M., Li C.-J., Modern alkyne chemistry: Catalytic and atom-economic transformations, Wiley-VCH, Weinheim, 2014. [Google Scholar]
- 6.
- 6a. Kitamura T., Eur. J. Org. Chem. 2009, 1111–1125; [Google Scholar]
- 6b. Yamamoto Y., Chem. Soc. Rev. 2014, 43, 1575–1600; [DOI] [PubMed] [Google Scholar]
- 6c. Boyarskiy V. P., Ryabukhin D. S., Bokach N. A., Vasilyev A. V., Chem. Rev. 2016, 116, 5894–5986. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Cheung C. W., Zhurkin F. E., Hu X., J. Am. Chem. Soc. 2015, 137, 4932–4935; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Li J., Zhang J., Tan H., Wang D. Z., Org. Lett. 2015, 17, 2522–2525; [DOI] [PubMed] [Google Scholar]
- 7c. Suess A. M., Uehling M. R., Kaminsky W., Lalic G., J. Am. Chem. Soc. 2015, 137, 7747–7753; [DOI] [PubMed] [Google Scholar]
- 7d. Uehling M. R., Suess A. M., Lalic G., J. Am. Chem. Soc. 2015, 137, 1424–1427; [DOI] [PubMed] [Google Scholar]
- 7e. Li Y., Ge L., Qian B., Babu K. R., Bao H., Tetrahedron Lett. 2016, 57, 5677–5680; [Google Scholar]
- 7f. Lu X.-Y., Liu J.-H., Lu X., Zhang Z.-Q., Gong T.-J., Xiao B., Fu Y., Chem. Commun. 2016, 52, 5324–5327; [DOI] [PubMed] [Google Scholar]
- 7g. Ouyang X. H., Song R. J., Liu B., Li J. H., Adv. Synth. Catal. 2016, 358, 1903–1909; [Google Scholar]
- 7h. Nakamura K., Nishikata T., ACS Catal. 2017, 7, 1049–1052. [Google Scholar]
- 8.
- 8a. Parasram M., Gevorgyan V., Chem. Soc. Rev. 2017, 46, 6227–6240; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Twilton J., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W., Nat. Rev. Chem. 2017, 1, 0052; [Google Scholar]
- 8c. Huo H., Shen X., Wang C., Zhang L., Röse P., Chen L.-A., Harms K., Marsch M., Hilt G., Meggers E., Nature 2014, 515, 100–103; [DOI] [PubMed] [Google Scholar]
- 8d. Meng Q., Schirmer T., Katou K., König B., Angew. Chem. Int. Ed. 2019, 58, 5723–5728; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5779–5784; [Google Scholar]
- 8e. Zhang L., Si X., Yang Y., Zimmer M., Witzel S., Sekine K., Rudolph M., Hashmi A. S. K., Angew. Chem. Int. Ed. 2019, 58, 1823–1827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1837–1841. [Google Scholar]
- 9.Examples:
- 9a. Cavalcanti L. N., Molander G. A., Top. Curr. Chem. 2016, 362, 1–23; [Google Scholar]
- 9b. Tellis J. C., Kelly C. B., Primer D. N., Jouffroy M., Patel N. R., Molander G. A., Acc. Chem. Res. 2016, 49, 1429–1439; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Milligan J. A., Phelan J. P., Badir S. O., Molander G. A., Angew. Chem. Int. Ed. 2019, 58, 6152–6163; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6212–6224; [Google Scholar]
- 9d. Tellis J. C., Primer D. N., Molander G. A., Science 2014, 345, 433–436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Zuo Z., Ahneman D. T., Chu L., Terrett J. A., Doyle A. G., MacMillan D. W., Science 2014, 345, 437–440; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9f. Zheng S., Gutiérrez-Bonet Á., Molander G. A., Chem 2019, 5, 339–352; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9g. Matsui J. K., Gutiérrez-Bonet Á., Rotella M., Alam R., Gutierrez O., Molander G. A., Angew. Chem. Int. Ed. 2018, 57, 15847–15851; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16073–16077; [Google Scholar]
- 9h. Dumoulin A., Matsui J. K., Gutiérrez-Bonet Á., Molander G. A., Angew. Chem. Int. Ed. 2018, 57, 6614–6618; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6724–6728; [Google Scholar]
- 9i. Li J., Luo Y., Cheo H. W., Lan Y., Wu J., Chem 2019, 5, 192–203; [Google Scholar]
- 9j. Corcé V., Chamoreau L. M., Derat E., Goddard J. P., Ollivier C., Fensterbank L., Angew. Chem. Int. Ed. 2015, 54, 11414–11418; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11576–11580; [Google Scholar]
- 9k. Oderinde M. S., Jones N. H., Juneau A., Frenette M., Aquila B., Tentarelli S., Robbins D. W., Johannes J. W., Angew. Chem. Int. Ed. 2016, 55, 13219–13223; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13413–13417; [Google Scholar]
- 9l. Oderinde M. S., Frenette M., Robbins D. W., Aquila B., Johannes J. W., J. Am. Chem. Soc. 2016, 138, 1760–1763; [DOI] [PubMed] [Google Scholar]
- 9m. Fan L., Jia J., Hou H., Lefebvre Q., Rueping M., Chem. Eur. J. 2016, 22, 16437–16440; [DOI] [PubMed] [Google Scholar]
- 9n. Yue H., Zhu C., Rueping M., Angew. Chem. Int. Ed. 2018, 57, 1371–1375; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1385–1389; [Google Scholar]
- 9o. Huang L., Rueping M., Angew. Chem. Int. Ed. 2018, 57, 10333–10337; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10490–10494; [Google Scholar]
- 9p. Kancherla R., Muralirajan K., Arunachalam S., Rueping M., Trends Chem. 2019, 1, 510–523; [Google Scholar]
- 9q. Dewanji A., Krach P. E., Rueping M., Angew. Chem. Int. Ed. 2019, 58, 3566–3570; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3604–3608; [Google Scholar]
- 9r. Huang L., Zhu C., Yi L., Yue H., Kancherla R., Rueping M., Angew. Chem. Int. Ed. 2020, 59, 457–464; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 465–472. [Google Scholar]
- 10. Deng H.-P., Fan X.-Z., Chen Z.-H., Xu Q.-H., Wu J., J. Am. Chem. Soc. 2017, 139, 13579–13584. [DOI] [PubMed] [Google Scholar]
- 11. Till N. A., Smith R. T., MacMillan D. W., J. Am. Chem. Soc. 2018, 140, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh K., Staig S. J., Weaver J. D., J. Am. Chem. Soc. 2014, 136, 5275–5278. [DOI] [PubMed] [Google Scholar]
- 13.The reduction potential of triplet excited state photocatalyst 3*Pc4 was obtained by converting the ground-state redox potential to the excited-state redox pontential; see the Supporting Information for more details.
- 14. Arias-Rotondo D. M., McCusker J. K., Chem. Soc. Rev. 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Nuhant P., Oderinde M. S., Genovino J., Juneau A., Gagné Y., Allais C., Chinigo G. M., Choi C., Sach N. W., Bernier L., Fobian Y. M., Bundesmann M. W., Khunte B., Frenette M., Fadeyi O. O., Angew. Chem. Int. Ed. 2017, 56, 15309–15313; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15511–15515; [Google Scholar]
- 15b. Teders M., Henkel C., Anhäuser L., Strieth-Kalthoff F., Gómez-Suárez A., Kleinmans R., Kahnt A., Rentmeister A., Guidi D., Glorius F., Nat. Chem. 2018, 10, 981–988. [DOI] [PubMed] [Google Scholar]
- 16. Huggins J. M., Bergman R. G., J. Am. Chem. Soc. 1981, 103, 3002–3011. [Google Scholar]
- 17. Maity B., Koley D., ChemCatChem 2018, 10, 566–580. [Google Scholar]
- 18.
- 18a. Lu T., Chen F., J. Theor. Comput. Chem. 2012, 11, 163–183; [Google Scholar]
- 18b. Lu T., Chen F., Acta Phys. Chim. Sin. 2012, 28, 1–18. [Google Scholar]
- 19.ADCH atomic charges were calculated by Multiwfn 3.7. see: Lu T., Chen F., J. Comput. Chem. 2012, 33, 580–592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary