

Abstract
We introduce divalent 3D DNA origami cuboids as truly monodisperse colloids and harness their ability for precision functionalization with defined patches and defined numbers of supramolecular binding motifs. We demonstrate that even adamantane/β‐cyclodextrin host/guest inclusion complexes of moderate association strength can induce efficient supracolloidal fibrillization at high dilution of the 3D DNA Origami as a result of cooperative multivalency. We show details on the assembly of Janus and non‐Janus 3D DNA origami into supracolloidal homo‐ and heterofibrils with respect to multivalency effects, electrostatic screening, and stoichiometry. We believe that the merger of 3D DNA origami with colloidal self‐assembly and supramolecular motifs provides new synergies at the interface of these disciplines to better understand multivalency effects, to promote structural complexity, and add non‐DNA assembling and switching mechanisms to DNA nanoscience.
Keywords: 3D DNA Origami, multivalence, self-assembly, supracolloidal polymerization, supramolecular chemistry
Combined forces: Adamantane and β‐cyclodextrin functionalized 3D DNA origami Janus colloids are used for cooperative multivalent interaction enhancement of supramolecular host/guest interactions resulting in 1D‐fibril formation. The merger of DNA nanotechnology and supramolecular chemistry gives access to study multivalent effects at a colloidal level.
Self‐assembly in colloid science provides model systems to understand phase behavior and structure formation reminiscent of atomic crystals.1 In the recent decade, much effort has been devoted to patchy colloidal molecules with surface‐anisotropic or shape‐anisotropic interaction patterns to increase structural complexity.2 Janus particles with broken symmetry stick out as a unique particle class as they allow directionality to be encoded into self‐assemblies and to realize applications in interface stabilization, for self‐propelling objects or as highly precise biosensors.3 Concurrently, the assembly interactions have been diversified towards the inclusion of sophisticated supramolecular interactions as these allow advanced switchability or self‐sorting in multicomponent systems.4 Despite such progress, clear frontiers exist in understanding how molecule‐like interactions translate to larger systems and to which extent orientation, directionality and cooperativity in surface‐confined molecular interactions play a role for structure formation.5 Promoting a fundamental understanding of how supramolecular interactions and multivalency guide nanoparticle assembly relies on having monodisperse objects with the highest control over patch size and the density of functionalization.
Despite progress in the fabrication of anisotropic particles, such as block‐copolymer self‐assembly, microfluidics, lithography, or phase separation,3 the ultimate precision of patch generation with nanometer resolution and molecularly programmed multivalency still remains a considerable challenge. 3D DNA origami has been emerging as a method for the preparation of arbitrarily shaped, truly monodisperse and addressable all‐DNA nanoparticles.6 It has been employed for the generation of intricate single objects,7 plasmonic structures,8 enzyme complexes,9 and hierarchical self‐assembly into higher ordered structures.10 Reports of using 3D DNA origami as patchy nanoobjects for supracolloidal assembly11—a topic of current interest in the colloid and nanoparticle field4d, 12—mainly employ DNA‐mediated interactions13 to organize 3D DNA origami structures in a hierarchical fashion. Although DNA interactions are highly programmable, the implementation of non‐DNA interactions is important to broaden response profiles and to provide a means for preparing orthogonal switches.
We identify that at the interface of precision nanoparticle synthesis, supramolecular interactions, and colloidal self‐assembly, 3D DNA origami nanoparticles are ideal to understand how supramolecular interactions manifest their multivalent binding in colloidal self‐assembly, because the amount of interacting strands and the patch size are programmable through the structural design. Herein, we demonstrate the use of patchy 3D DNA origami structures as colloidal patchy nanoparticles with Janus character, and describe in detail how the precise arrangement of supramolecular host/guest interactions can be used to achieve supraparticular self‐assembly into 1D fibrils harnessing cooperative multivalency. This work thus builds a bridge and promotes mutual understanding for the fields of DNA nanoscience, colloid/nanoparticle science and supramolecular interactions.
As the monodisperse nanoparticle, we employ a bivalent 3D DNA origami cuboid.10c It is folded from a m13mp18 cyclic scaffold ssDNA and a 5‐fold excess of ssDNA staple strands in a temperature ramp and purified by membrane spin‐filtration to remove excess staple strands (Figure 1 a). The cuboid consists of 72 double helices aligned in a honeycomb lattice with the two patches bearing ssDNA docking strands, which can be functionalized with supramolecular units that are appended to ssDNA sequences. Both sides can be orthogonally addressed to lead to a Janus cuboid with divalent character, whereby one patch houses the guest units, while the other side houses the host units.
Figure 1.
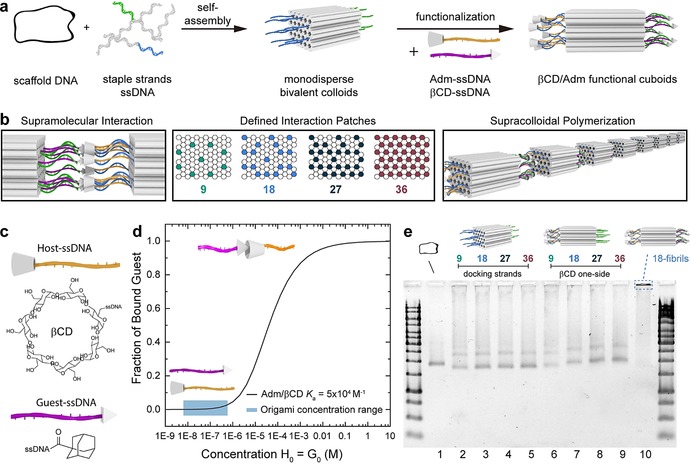
Overview of host/guest functionalized bivalent 3D DNA origami cuboids. a) Preparation of 3D DNA origami cuboids and subsequent functionalization by addition of adamantane (Adm) or beta‐cyclodextrin (βCD) ssDNA. b) Supramolecular interaction is facilitated by perfect alignment of patch geometry and four different densities are implemented. c) Adm and βCD functionalities are attached to ssDNA strands complementary to one patch on the cuboid. d) Concentration‐dependent binding of Adm/βCD inclusion complexes. Typical 3D DNA origami concentrations are indicated. e) Folding and functionalization of the cuboids visualized by agarose gel electrophoresis.
The host/guest interactions are located on both ends of the same duplexes running through the origami lattice, which allows for a perfect alignment of the interactions once two origami patches approach each other. We designed four cuboids with different numbers of docking strands (9, 18, 27, 36; Figure 1 b). This can be achieved by adding the appropriate docking strands during the folding, to assure that indeed only a certain valency is generated. The remaining duplexes of the origami were passivated with T9 overhangs to avoid unspecific aggregation. Later, we will also turn to supracolloidal copolymerization by functionalization of two batches of origami with the host and guest motifs located at both patches of the individual origami, resulting in a system wherein A2 and B2 assemble into A2B2 heterofibrils. The tunability in valency is a unique advantage of 3D DNA Origami structures over any other nanoparticle structure, as it allows to precisely engineer the amount of supramolecular units at both faces. Thereby details about multivalency effects are accessible. As the supramolecular, non‐DNA based‐interaction we chose adamantane (Adm) and beta‐cyclodextrin (βCD) inclusion complexation (Figure 1 c). Although this interaction is relatively strong with respect to most βCD interactions,14 it has not been considered so far in the DNA origami field due to a perceived mismatch of association constant, K a, and typical origami concentrations. The average K a of Adm/βCD is around 5×104 m −1.15 By calculating the binding isotherm, it can be estimated that a concentration in the millimolar range is needed for efficient 1:1 binding in solution, that is, having complex formation greater than 80 % (Figure 1 d).16 In contrast, typical 3D DNA origami nanoparticle concentrations are only in the nm to μm regime and we employ a concentration of 20 nm. Depending on the valency of the cuboid (9, 18, 27, 36), the total concentration of Adm and βCD units is still only at a maximum of around 1 μm, hence a factor of 103 lower than needed for a greater than 80 % binding. We however hypothesized that the multivalency, arising from placing the interacting motifs into close proximity at a patch may facilitate chelate cooperativity, greatly supporting robust supracolloidal fibril formation.17
The Adm and βCD end‐functionalized ssDNA strands (T m=45 °C) were prepared by amide coupling and copper‐free click reactions, respectively, and purified by HPLC. The functional ssDNA strands were usually employed in a two‐fold excess to ensure complete saturation of the docking strands on the patches. The successful preparation and functionalization of the cuboids can be confirmed by agarose gel electrophoresis (AGE). The more compact nature of the folded cuboid is visible by its increased electrophoretic mobility (Figure 1 e, lane 2) compared to the cyclic scaffold strand (Figure 1 e, lane 1). Only insignificant differences in migration are observed for the cuboids carrying different docking strand densities (Figure 1 e, lanes 3–6). However, once one patch is functionalized, for instance with ssDNA‐βCD, significantly lower electrophoretic mobility is observed with increasing degree of functionalization as a result of the lower charge‐to‐mass ratio (Figure 1 e, lane 7–9).
Next, we turn to the analysis of the supracolloidal polymerization. In the absence of Adm‐ssDNA and βCD‐ssDNA most of the docking‐strand‐modified cuboids are in monomeric form (Figure 2 a). Statistical analysis of TEM images gives an average degree of polymerization (X̄n) of 1.1–1.2 for the four different cuboids (9, 18, 27, 36), corresponding to a unimer fraction of greater than 80 %. Addition of the host and guest ssDNA strands triggers the supramolecular polymerization and the X̄n increases with the number of connector positions, hence the multivalency available on the cuboids (Figure 2 b). The distribution of species broadens at higher X̄n with a maximum X̄n of 12 for 36‐host/guest functionalized origami (36‐HG) after 1 h. Almost no monomeric cuboids are visible in TEM and longer chains are the predominant species. This corroborates the AGE (Figure 1 e, lane 10) and clearly shows that long nanofibrils with micrometer length can be formed. Hence, the multivalency at the patches leads to effective binding, even at a low total concentration of cuboids (Figure 1 d). Robust fibril formation (that is, the formation of fibrils occurs with the same precision (regarding time, artifacts, length of fibrils) also in the presence of competing binding partners in solution) also occurs in the presence of an excess of the functionalization strands versus the docking strands on the cuboid and the X̄n does not change significantly for all four connector numbers (Figure 2 c), which demonstrates the high cooperativity resulting from the spatially organized multivalent interactions. Only the addition of a high excess of a single partner, that is, the guest molecules, breaks the fibrils formed into monomeric species again (Figure 2 d).
Figure 2.
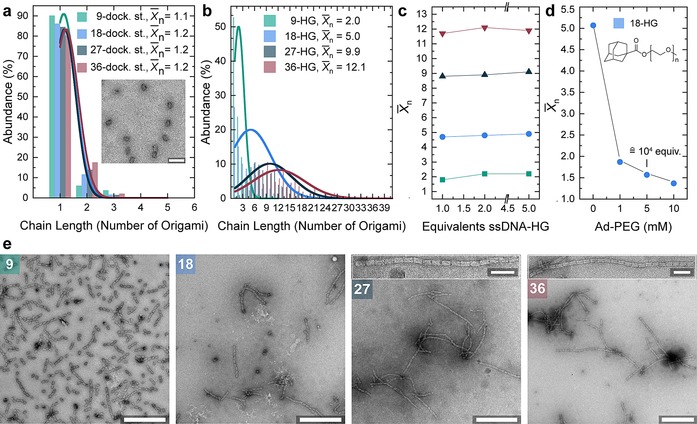
Supracolloidal polymerization based on multivalent host/guest binding. Origami concentration is 20 nm. a) Unfunctionalized docking‐strand modified cuboids are predominantly in monomeric form. Inset: 18‐docking‐strand cuboid. Scale bar 100 nm. b) Number fraction of species (abundance) in percent for the four HG densities. The X̄n of host/guest cuboid increases together with the number of HG‐units attached. Distribution of species obtained from statistical analysis of clearly visible species in negatively stained TEM. Curves show a normal distribution function. c) The X̄n as a function of the equivalents of ssDNA‐HG added to the solution. d) Addition of PEGylated‐adamantane in high concentration breaks fibrils into monomeric species. e) Negatively stained TEM images after 1 h assembly time. Scale bars 500 nm. Insets show neatly aligned origami fibrils. Inset scale bars 100 nm.
More insights into the cooperativity of the multivalent binding can be obtained by estimating the colloidal‐level binding constants K a between two origami structures as a function of the connector strand density using image analysis as introduced by Turberfield and co‐workers.18 Indeed, the K a constant scales non‐linearly as a function of the connector strand density and lies orders of magnitude above the monovalent, molecular‐level host/guest interactions present on the origami structures (ca. 5×104 m −1; Table 1, Figure S3). Interestingly, when recalculating the K a constant by the number of connectors, it becomes clear that the individual binding units are enhanced by 3–4 orders of magnitude and that the binding is over proportionally strengthened when increasing the number of connector strands.
Table 1.
K a between two origami structures estimated as a function of multivalency.
|
Origami valency |
K a [M−1] |
K a/connector number |
|---|---|---|
|
9 |
2.4×108 |
2.7×107 |
|
18 |
1.6×109 |
8.9×107 |
|
27 |
1.2×1010 |
4.4×108 |
|
36 |
2.6×1010 |
7.2×108 |
The dynamic supramolecular nature of the interaction also facilitates the assembly of neatly aligned cuboids (Figure 2 e). Owing to the intrinsic stoichiometry match in Janus cuboids bearing the host and guest motifs on opposite sides, there are no stoichiometry problems for the assembly, which after all is reminiscent of a step‐growth polymerization. Since the supracolloidal polymerization is triggered once both Adm‐ssDNA and βCD‐ssDNA are added to the cuboid solution, we also investigated the temporal development of the nanofibril length, X̄n for the four functionalization densities at four times (10 min, 1 h, 16 h, 48 h) by ex situ TEM. X̄n increases for all the systems with time, nearly doubling between 10 min to 2 days (Figure 3 a). Higher functionalization densities result in a faster assembly especially in the beginning. Already after 10 min the X̄n of the 36‐HG origami is almost four‐times higher than the one of 9‐HG origami and after two days supramolecular fibrils with a X̄n of 15 and up to 48 repeats form for the 36‐HG origami.
Figure 3.
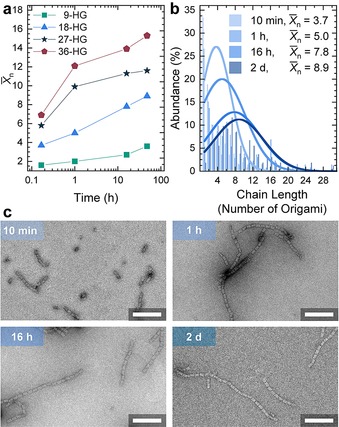
Temporal development of the fibril length depending on the functionalization degree. Origami concentration is 20 nm. a) Temporal evolution of X̄n for the 4 different host/guest origami. b) Number fraction of species (abundance) in percent for 18‐HG origami in dependence of the assembly time. c) TEM images for 18‐HG origami cuboids after the indicated times. Scale bars 250 nm.
Since the concentration of the cuboids is constant in all samples, the faster reaction kinetics indicates more effective collisions leading to assembly at high functionalization densities, while lower functionalization densities behave in a more dynamic way with less effective collisions and the ability for dissociation. Overall it is evident again that larger functionalization degrees lead to a faster assembly into long nanofibrils.
DNA assemblies are generally sensitive towards salinity because this influences the screening of the electrostatic repulsion of the negatively charged phosphate backbone. Especially in 3D DNA origami structures a certain minimum concentration of divalent Mg2+ is necessary for correct folding. The effect of charge screening is also important for hierarchical assembly or conformational changes of 3D DNA origami as demonstrated for DNA base‐stacking assemblies10b and DNA base‐pairing interactions.19 We therefore anticipated that also supramolecular host/guest‐mediated 3D DNA origami assemblies are highly cation dependent. Indeed, at lower [Mg2+] the X̄n decreases for all HG densities. Supracolloidal polymerization of 9 and 18‐HG origamis is completely absent at 1 mm Mg2+ (Figure 4 a) and can be modulated over a wide range of X̄n. In contrast, 36 connector origami structures are less sensitive to changes of the ionic strength and high X̄n result also at 1 mm Mg2+. In fact, for 27 and 36‐HG origamis, there is no lower cation concentration which prevents supracolloidal assembly since dissolution of colloids occurs at too low salinity before the suppression of assembly. The removal of salt from already formed 18‐HG origami fibrils leads to disassembly into short oligomers and monomers and with resupply of magnesium ions the initial X̄n can be restored (Figure 4 b). These results underscore important principles for designing switchable host/guest interactions in the future, as many switchable host/guest interactions do not switch fully between K a=0 and high K a, but rather exhibit a high K a and a low K a.20 We suggest that, in case of multivalent interactions, a balance between electrostatic repulsion and K a needs to be found to have efficient switching.
Figure 4.
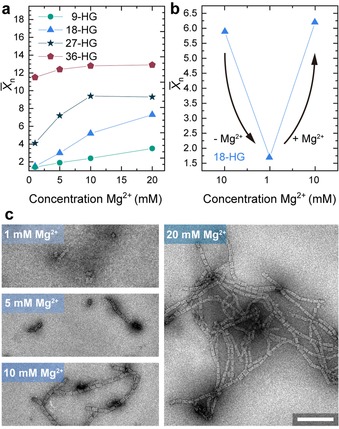
Influence of the Mg2+ concentration the nanofibril length. Origami concentration is 20 nm. a) X̄n for the four connector densities after 1 h of assembly time. The X̄n increases with increasing [Mg2+]. b) Reducing the [Mg2+] results in disassembly of 18‐HG origami fibrils. After resupply of [Mg2+] the initial X̄n can be restored. c) TEM images show fibril lengths for 18‐HG origami cuboids at different [Mg2+]. Scale bars 250 nm.
To demonstrate the programmable nature of 3D DNA origami structures as colloidal building blocks, we also investigated bi‐particulate supracolloidal copolymerization, again as function of the density of host/guest units (Figure 5). As known from step‐growth polymerization in molecular systems, it is important to achieve control over the stoichiometry to reach high degrees of “conversion of functional groups”. Therefore, we split the solution of docking‐strand‐modified cuboids in two batches and functionalized each batch on both sides either with Adm‐ssDNA or βCD‐ssDNA. This allows exactly the same cuboid concentration to be maintained in both batches. Subsequently, both batches can be mixed together in different ratios. As expected, the X̄n reaches a maximum in a mixture of 50:50 host:guest type cuboids, while the individual cuboids do not show any fibril formation. Similar to above, the 1D fibrils of cuboids with higher functionalization density show a higher X̄n. Unbalanced mixtures show reduced X̄n originating from integration of the species present in lower concentration and end capping of the growing nanofibrils by the cuboid being present in excess. The overall X̄n of the 50:50 supracolloidal copolymerizations is lower than the AB Janus system originating presumably from a reduced probability of collision of two fitting origami.
Figure 5.
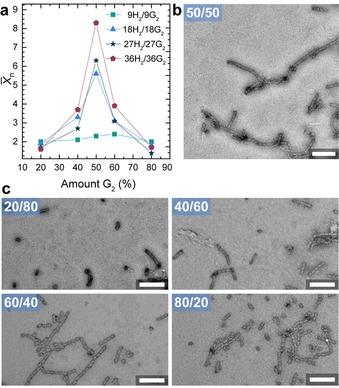
Supracolloidal copolymerization of H2 and G2 origami. a) Influence of the ratio of Host (H2)‐origami versus Guest (G2)‐origami on X̄n as a function of multivalency. b) Negatively stained TEM image represent origami fibrils consisting of 18‐H2 and 18‐G2 origami in a 50:50 mixture. c) Negatively stained TEM image of origami fibrils consisting of 18‐H2 and 18‐G2 origami in the indicated ratios. Assembly time 1 h and [Mg2+]=10 mm. Total origami concentration 20 nm. Scale bar 250 nm.
In conclusion we have shown for the first time that 3D DNA origami structures can be assembled using relatively weak host/guest supramolecular interactions (with respect to the operating particle concentration) by exploiting multivalency effects due to geometric confinement of the interacting species in patch volumes giving rise to chelate cooperativity. The highly defined and localized interaction patches support multivalent binding and greatly enhance the total binding strength of the supramolecular interactions even at low origami concentrations. In our system, the degree of polymerization of the resulting supracolloidal fibrils can be tuned by the number of connector pairs on the cuboids, the assembly time, and the electrostatic repulsion. Formed assemblies can be reversibly disassembled by modulating the ionic strength or can be disassembled by addition of high concentrations of competing guest molecules. Supracolloidal polymerizations can be conducted from a single Janus‐type building blocks (A–B) leading to perfect stoichiometry and the longest fibrils, or by copolymerization of two building blocks (A2/B2), in which the X̄n is highly dependent on the ratio of origami structures. We believe that 3D DNA origami offers fascinating possibilities for nanometer‐precise patch generation on nanoparticles enabling further in‐depth studies of cooperative binding events of different supramolecular binding motifs.
The principles should be widely applicable to other types of supramolecular interactions allowing for a cross‐fertilization at the interface of supramolecular chemistry, 3D DNA origami, and colloid science. Given the large amount of switchable interactions known for cyclodextrin‐based inclusion complexes (or other type of supramolecular interactions), new switching methods can for instance be encoded for DNA origami that may not be accessible easily using classical DNA‐based methods.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge funding through the ERC Starting Grant “TimeProSAMat” (ID: 677960). S.L. acknowledges a scholarship of the Fonds der Chemischen Industrie. This work made use of the microscopy facilities provided through the Core Facility “Imaging of Materials Systems” at the FIT.
S. Loescher, A. Walther, Angew. Chem. Int. Ed. 2020, 59, 5515.
References
- 1.
- 1a. Macfarlane R. J., Lee B., Jones M. R., Harris N., Schatz G. C., Mirkin C. A., Science 2011, 334, 204–208; [DOI] [PubMed] [Google Scholar]
- 1b. Auyeung E., Li T. I., Senesi A. J., Schmucker A. L., Pals B. C., de la Cruz M. O., Mirkin C. A., Nature 2014, 505, 73–77. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Glotzer S. C., Solomon M. J., Nat. Mater. 2007, 6, 557–562; [DOI] [PubMed] [Google Scholar]
- 2b. Liu B., Wu Y., Zhao S., Chem. Eur. J. 2018, 24, 10562–10570; [DOI] [PubMed] [Google Scholar]
- 2c. Gong Z., Hueckel T., Yi G. R., Sacanna S., Nature 2017, 550, 234–238. [DOI] [PubMed] [Google Scholar]
- 3. Walther A., Mueller A. H., Chem. Rev. 2013, 113, 5194–5261. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Wang Y., Wang Y., Breed D. R., Manoharan V. N., Feng L., Hollingsworth A. D., Weck M., Pine D. J., Nature 2012, 491, 51–55; [DOI] [PubMed] [Google Scholar]
- 4b. de Feijter I., Albertazzi L., Palmans A. R., Voets I. K., Langmuir 2015, 31, 57–64; [DOI] [PubMed] [Google Scholar]
- 4c. Stricker L., Fritz E. C., Peterlechner M., Doltsinis N. L., Ravoo B. J., J. Am. Chem. Soc. 2016, 138, 4547–4554; [DOI] [PubMed] [Google Scholar]
- 4d. Han K., Go D., Tigges T., Rahimi K., Kuehne A. J., Walther A., Angew. Chem. Int. Ed. 2017, 56, 2176–2182; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2208–2214; [Google Scholar]
- 4e. Han K., Go D., Hoenders D., Kuehne A. J. C., Walther A., ACS Macro Lett. 2017, 6, 310–314. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Whitesides G., Mathias J., Seto C., Science 1991, 254, 1312–1319; [DOI] [PubMed] [Google Scholar]
- 5b. Whitesides G. M., Grzybowski B., Science 2002, 295, 2418–2421. [DOI] [PubMed] [Google Scholar]
- 6. Loescher S., Groeer S., Walther A., Angew. Chem. Int. Ed. 2018, 57, 10436–10448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10594–10607. [Google Scholar]
- 7.
- 7a. Dietz H., Douglas S. M., Shih W. M., Science 2009, 325, 725–730; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Douglas S. M., Dietz H., Liedl T., Hogberg B., Graf F., Shih W. M., Nature 2009, 459, 414–418; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Langecker M., Arnaut V., Martin T. G., List J., Renner S., Mayer M., Dietz H., Simmel F. C., Science 2012, 338, 932–936; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Funke J. J., Ketterer P., Lieleg C., Korber P., Dietz H., Nano Lett. 2016, 16, 7891–7898. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Kuzyk A., Schreiber R., Zhang H., Govorov A. O., Liedl T., Liu N., Nat. Mater. 2014, 13, 862–866; [DOI] [PubMed] [Google Scholar]
- 8b. Zhou C., Duan X., Liu N., Nat. Commun. 2015, 6, 8102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Zhang T., Hartl C., Frank K., Heuer-Jungemann A., Fischer S., Nickels P. C., Nickel B., Liedl T., Adv. Mater. 2018, 30, 1800273. [DOI] [PubMed] [Google Scholar]
- 9. Zhao Z., Fu J., Dhakal S., Johnson-Buck A., Liu M., Zhang T., Woodbury N. W., Liu Y., Walter N. G., Yan H., Nat. Commun. 2016, 7, 10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Amoako G., Zhou M., Ye R., Zhuang L., Yang X., Shen Z., Chin. Sci. Bull. 2013, 58, 3019–3022; [Google Scholar]
- 10b. Gerling T., Wagenbauer K. F., Neuner A. M., Dietz H., Science 2015, 347, 1446–1452; [DOI] [PubMed] [Google Scholar]
- 10c. Tigges T., Heuser T., Tiwari R., Walther A., Nano Lett. 2016, 16, 7870–7874; [DOI] [PubMed] [Google Scholar]
- 10d. Wagenbauer K. F., Sigl C., Dietz H., Nature 2017, 552, 78–83; [DOI] [PubMed] [Google Scholar]
- 10e. Hong F., Zhang F., Liu Y., Yan H., Chem. Rev. 2017, 117, 12584–12640. [DOI] [PubMed] [Google Scholar]
- 11. Yonamine Y., Cervantes-Salguero K., Minami K., Kawamata I., Nakanishi W., Hill J. P., Murata S., Ariga K., Phys. Chem. Chem. Phys. 2016, 18, 12576–12581. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Liu K., Nie Z., Zhao N., Li W., Rubinstein M., Kumacheva E., Science 2010, 329, 197–200; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Groeschel A. H., Schacher F. H., Schmalz H., Borisov O. V., Zhulina E. B., Walther A., Mueller A. H., Nat. Commun. 2012, 3, 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Jungmann R., Scheible M., Kuzyk A., Pardatscher G., Castro C. E., Simmel F. C., Nanotechnology 2011, 22, 275301; [DOI] [PubMed] [Google Scholar]
- 13b. Woo S., Rothemund P. W., Nat. Chem. 2011, 3, 620–627; [DOI] [PubMed] [Google Scholar]
- 13c. Bayrak T., Helmi S., Ye J., Kauert D., Kelling J., Schonherr T., Weichelt R., Erbe A., Seidel R., Nano Lett. 2018, 18, 2116–2123; [DOI] [PubMed] [Google Scholar]
- 13d. Pfeifer W., Lill P., Gatsogiannis C., Sacca B., ACS Nano 2018, 12, 44–55. [DOI] [PubMed] [Google Scholar]
- 14. Rekharsky M. V., Inoue Y., Chem. Rev. 2014, 114, 10940–10975.25247843 [Google Scholar]
- 15.
- 15a. Cromwell W. C., Bystrom K., Eftink M. R., J. Phys. Chem. 1985, 89, 326–332; [Google Scholar]
- 15b. Eftink M. R., Andy M. L., Bystrom K., Perlmutter H. D., Kristol D. S., J. Am. Chem. Soc. 1989, 111, 6765–6772. [Google Scholar]
- 16. Thordarson P., Chem. Soc. Rev. 2011, 40, 1305–1323. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Mulder A., Huskens J., Reinhoudt D. N., Org. Biomol. Chem. 2004, 2, 3409–3424; [DOI] [PubMed] [Google Scholar]
- 17b. Hunter C. A., Anderson H. L., Angew. Chem. Int. Ed. 2009, 48, 7488–7499; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7624–7636; [Google Scholar]
- 17c. Dubel N., Liese S., Scherz F., Seitz O., Angew. Chem. Int. Ed. 2019, 58, 907–911; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 918–923. [Google Scholar]
- 18. Jin J., Baker E. G., Wood C. W., Bath J., Woolfson D. N., Turberfield A. J., ACS Nano 2019, 13, 9927–9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marras A. E., Shi Z., M. G. Lindell, 3rd , Patton R. A., Huang C. M., Zhou L., Su H. J., Arya G., Castro C. E., ACS Nano 2018, 12, 9484–9494. [DOI] [PubMed] [Google Scholar]
- 20. Yang Y. W., Sun Y. L., Song N., Acc. Chem. Res. 2014, 47, 1950–1960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary