

Abstract
Scar formation after injury of the brain or spinal cord is a common event. While glial scar formation by astrocytes has been extensively studied, much less is known about the fibrotic scar, in particular after stroke. Platelet‐derived growth factor receptor ß‐expressing (PDGFRß+) pericytes have been suggested as a source of the fibrotic scar depositing fibrous extracellular matrix (ECM) proteins after detaching from the vessel wall. However, to what extent these parenchymal PDGFRß+ cells contribute to the fibrotic scar and whether targeting these cells affects fibrotic scar formation in stroke is still unclear. Here, we utilize male transgenic mice that after a permanent middle cerebral artery occlusion stroke model have a shift from a parenchymal to a perivascular location of PDGFRß+ cells due to the loss of regulator of G‐protein signaling 5 in pericytes. We find that only a small fraction of parenchymal PDGFRß+ cells co‐label with type I collagen and fibronectin. Consequently, a reduction in parenchymal PDGFRß+ cells by ca. 50% did not affect the overall type I collagen or fibronectin deposition after stroke. The redistribution of PDGFRß+ cells to a perivascular location, however, resulted in a reduced thickening of the vascular basement membrane and changed the temporal dynamics of glial scar maturation after stroke. We demonstrate that parenchymal PDGFRß+ cells are not the main contributor to the fibrotic ECM, and therefore targeting these cells might not impact on fibrotic scar formation after stroke.
Keywords: collagen, extracellular matrix, fibronectin, fibrotic scar, glial scar, pericytes, RRID:AB_2082660, RRID:AB_2105706, RRID:AB_2162497, RRID:AB_217595, RRID:AB_2298772, RRID:AB_298179, RRID:AB_305808, RRID:AB_354858, RRID:AB_393571, RRID:AB_467492, RRID:SCR_002798, RRID:SCR_003070, RRID:SCR_010279, stroke
After stroke, a fibrotic scar is formed, where the injured tissue is replaced with extracellular matrix. Parenchymal pericytes are not the main contributor to this deposition, and targeting pericytes after stroke does not affect scar formation.
Significance.
The scar that forms after an injury to the brain is needed to replace injured tissue and to limit the spreading of inflammation to uninjured tissue. However, scar formation also limits functional recovery. Little is known about which cells are contributing to scar formation, potentially providing a target to modulate this healing process. Pericytes, which detach from their normal position around blood vessels, have been suggested to be the source of scar‐forming cells. Here we provide evidence that detaching pericytes are not the major contributor to the fibrotic scar and hence, targeting these cells did not change the fibrotic scar.
1. INTRODUCTION
Scar formation is a common response to tissue injury in different organs, including the central nervous system (CNS). It is the result of a repair mechanism that replaces damaged tissue with extracellular matrix (ECM). After injury to the CNS, different cell types participate to encapsulate the affected tissue in order to prevent extensive inflammation and further neurodegeneration (Kawano et al., 2012). Scar formation in the CNS occurs in response to traumatic injuries, lesions caused by autoimmunity or infections, and after stroke (Dias & Göritz, 2018). The current view distinguishes between the well‐studied glial scar and the less‐characterized fibrotic scar in the CNS (Fernández‐Klett & Priller, 2014).
In the chronic phase of stroke, a glial scar is formed by resident‐reactive astroglia that assemble around the lesion core in the penumbra to seal off the intact tissue from the damaged ischemic core (Wanner et al., 2013). Enveloped by this glial scar, a dense fibrotic scar develops within the ischemic core. In this stiff fibrotic scar, fibrous ECM proteins such as type I collagen and fibronectin are produced by different cell types participating in fibrotic scar formation (Fernández‐Klett & Priller, 2014). In comparison to other tissues, the ECM within the CNS contains small amounts of fibrous ECM proteins, and is composed by a network of proteoglycans, hyaluronans, tenascins, and link‐proteins (Lau, Cua, Keough, Haylock‐Jacobs, & Yong, 2013). Therefore, it is not only providing mechanical support, but serves as a substrate for the compartmentalization of the extracellular space and functions as a scaffold during development and adult neurogenesis (Dityatev, Seidenbecher, & Schachner, 2010; Kwok, Dick, Wang, & Fawcett, 2011). The increased deposition of fibrous ECM proteins after stroke resulting in a stiff fibrotic scar is suggested to impede the anatomical plasticity within the CNS and therefore impact negatively on learning and memory (Fernández‐Klett & Priller, 2014). One of the cell types recently suggested to be associated with a fibrotic response in the CNS are platelet‐derived growth factor receptor ß‐expressing (PDGFRß+) pericytes (Fernandez‐Klett et al., 2012; Göritz et al., 2011; Hesp et al., 2017; Reeves, Jardim, Sisodiya, Thom, & Liu, 2019; Zehendner et al., 2015).
Pericytes are perivascular cells that are embedded in the vascular basement membrane and lining capillaries (Armulik, Genové, & Betsholtz, 2011). They show multiple responses to acute ischemic stroke (Cai et al., 2016; Kamouchi, Ago, Kuroda, & Kitazono, 2011), but only few studies so far have examined the role of pericytes in the chronic phase of stroke. These studies suggest that pericytes play an active role in tissue remodeling after stroke (Fernandez‐Klett et al., 2012; Makihara et al., 2014; Roth et al., 2019). In particular, it has been suggested that PDGFRß+ pericytes detach from the vessel wall and migrate into the injured parenchyma where they form a dense network. Here they become “scar‐forming pericytes” producing ECM and thereby shaping the fibrotic scar (Fernandez‐Klett et al., 2012; Makihara et al., 2014; Shen et al., 2012). In particular, their migration away from the perivascular location into the parenchyma has been associated with this phenotypic switch toward ECM‐producing myofibroblasts (Fernandez‐Klett et al., 2012). Pericytes have also been suggested to shape the formation of the glial scar as their absence has been shown to distort the polarization of astrocytes toward the infarct core (Shen et al., 2012).
However, particularly in stroke, it is unclear to what extent the parenchymal PDGFRß+ pericytes contribute to the fibrotic scar. It is also currently not known if migration into the parenchyma is essential for pericytes to produce ECM.
We have previously shown that loss of regulator of G‐protein signaling 5 (RGS5) in brain pericytes leads to a shift in the fate choice between the vascular and parenchymal location of pericytes after ischemic stroke (Roth et al., 2019). In RGS5‐knockout (KO) mice, we found a ca 50% reduction in parenchymal PDGFRß+ density and an increased number of perivascular pericytes during the chronic phase after stroke. The loss of RGS5 in pericytes and the associated preservation of perivascular pericytes improved the integrity of blood–brain barrier (BBB) and vessel remodeling (Roth et al., 2019). However, whether the spatial redistribution of PDGFRß+ cells in these RGS5‐KO mice has an impact on the fibrotic and glial scar formation after stroke remains unknown.
Utilizing RGS5‐KO mice in an experimental stroke model, we investigated the impact of reduced numbers of parenchymal PDGFRß+ pericytes on the properties of the fibrotic and glial scar formation.
We confirm previous findings that RGS5‐KO mice have reduced numbers of parenchymal PDGFRß+ cells in the infarct core at 7 days after stroke. However, only a small fraction of these parenchymal PDGFRß+ cells are participating in ECM production and, therefore, the spatial redistribution of pericytes in RGS5‐KO does not affect type I collagen or fibronectin deposition. However, wild‐type (WT) mice showed pronounced vascular basement membrane thickening after stroke, this response was significantly reduced in RGS5‐KO mice. In addition, the temporal dynamics of glial scar maturation differed between RGS5‐KO and WT mice.
In this study, we show that parenchymal PDGFRß+ pericytes are not the main contributor to ECM production in the fibrotic scar and reduction in their numbers does not have an impact on the fibrotic scar in stroke.
2. MATERIAL AND METHODS
2.1. Animals
A knockout–knockin reporter mouse strain, where green fluorescent protein (GFP) is expressed under the RGS5 promotor was used in this study (Nisancioglu et al., 2008). We used 8‐ to 12‐week‐old Rgs5gfp/gfp mice (referred to as RGS5‐KO mice, n = 10). In RGS5‐KO mice, both alleles of RGS5 are replaced with GFP, resulting in only GFP expression upon RGS5 promotor activation, without the production of RGS5 protein. WT mice (Rgs5+/+, n = 10) were used as a control. About 20 male mice were used in this study. Only male mice were included to allow for direct comparison with previous studies and to reduce biological variability.
RGS5‐KO mice have previously been characterized. RGS5‐KO mice are viable, fertile, and develop without observable defects. Under physiological conditions, pericyte numbers and the vascular density are comparable to WT controls (Nisancioglu et al., 2008; Özen et al., 2018).
Animals were housed under standard conditions with a 12 hours light/dark cycle and with access to food and water ad libitum. Four to five mice were kept in the same cage, and their cages where enriched with nesting material, a ladder, and wooden chewing sticks. All experimental procedures were approved by the Ethics committee of Lund University.
2.2. Permanent middle cerebral artery occlusion
The distal part of the left middle cerebral artery (MCA) was permanently occluded to induce a focal cerebral ischemia as previously described (Llovera, Roth, Plesnila, Veltkamp, & Liesz, 2014). In brief, animals were anesthetized with 1.5% isoflurane and an incision was made between the left lateral part of the orbit and the left ear. The parotid gland and the temporal muscle were moved, and a small craniotomy was made above the anterior distal branch of the MCA. After exposure of the MCA, it was permanently occluded by electrocoagulation using an electrosurgical unit (ICC50; Erbe, Germany). Marcain was locally applied and the wound was sutured. No animals were excluded from analysis. Sham‐operated animals underwent the same procedure except occlusion of the MCA which did not result in fibrotic or glial scar formation (Supporting Information Figure S1).
2.3. Tissue processing
The mice were killed at 7 and 14 days after permanent middle cerebral artery occlusion (pMCAO) by injecting 150 μl Pentobarbital vet (60 mg/ml, APL) and transcardially perfusing with phosphate‐buffered saline (PBS) followed by 4% paraformaldehyde (PFA). After the removal of brains, they were placed in 4% PFA for postfixation overnight, before they were then placed in 30% sucrose in PBS and sectioned in 12 series with a microtome (Leica SM 2010R) in coronal sections of 40 µm.
2.4. Immunohistochemistry
For fluorescent immunohistochemistry, the brain sections were washed three times in PBS for 5 min and then blocked for 30 min in 5% normal donkey (Biowest, catalog number S2170‐100) or goat serum (Biowest, catalog number S2000‐100) in 0.25% Triton‐X100 (Alfa Aesar) in PBS (PBS‐TX). For PDGFRß detection, sections were pretreated with citrate buffer for 20 min at 80°C. Sections were incubated with primary antibodies (Table 1) overnight at room temperature in 3% serum in PBS‐TX. The following antibodies were used: rabbit anti‐type I collagen (1:400, Rockland, RRID:AB_217595), rat anti‐PDGFRß (1:200, eBioSience, RRID:AB_467492), rabbit anti‐PDGFRß (1:200, Cell Signaling, RRID:AB_2162497), mouse anti‐fibronectin (1:100, BD Biosciences, RRID:AB_2105706), anti rabbit‐type IV collagen (1:400, AbD Serotec, RRID:AB_2082660), rabbit anti‐laminin (1:400, Abcam, RRID:AB_298179), rabbit anti‐GFAP (1:400, GFAP, RRID:AB_305808), rat anti‐CD31 (1:400, BD Pharmingen, RRID:AB_393571), mouse anti‐NeuN (1:400, BD Pharmingen, RRID:AB_2298772), and goat anti‐Podocalyxin (1:400, R&D Systems, RRID:AB_354858). After washing, the staining was visualized using fluorophore‐conjugated secondary antibodies (Invitrogen). Specificity of secondary antibodies was determined by following the same staining protocol, but without adding primary antibodies (Supporting Information Figure S1).
Table 1.
List of antibodies used in this study
| Name | Company | Species | Cat. number | RRID | Conc. used |
|---|---|---|---|---|---|
| Type I collagen | Rockland | Rabbit; pAb | 600‐401‐103‐0.5 | RRID: AB_217595 | 1:400 |
| PDGFRß | eBioScience | Rat; mAB | 14‐1402‐81 | RRID: AB_467492 | 1:200 |
| PDGFRß | Cell signaling | Rabbit, mAb | 3169S | RRID: AB_2162497 | 1:200 |
| Fibronectin | BD biosciences | Mouse; mAb | 610077 | RRID:AB_2105706 | 1:100 |
| Type IV collagen | AbD Serotec | Rabbit; pAb | 2150‐1470 | RRID: AB_2082660 | 1:500 |
| Laminin | Abcam | Rabbit; pAb | ab11575 | RRID: AB_298179 | 1:400 |
| Glial fibrillary acidic protein (GFAP) | Abcam | Rabbit; pAb | ab7260 | RRID: AB_305808 | 1:400 |
| CD31 | BD pharmingen | Rat; mAb | 550274 | RRID: AB_393571 | 1:400 |
| NeuN | Millipore | Mouse; mAb | MAB377 | RRID: AB_2298772 | 1:400 |
| Podocalyxin | R&D systems | Goat; pAB | AF1556‐SP | RRID: AB_354858 | 1:400 |
Abbreviations: mAb, monoclonal antibody; pAb, polyclonal antibody.
2.5. Image processing and cell counting
Fluorescent immunostainings were visualized using a Leica SP8 confocal microscope. Three consecutive sections spanning the infarct core were analyzed (between +1.2 mm to −1.0 mm from bregma). Three images were taken with a 63× objective within the infarct core and the peri‐infarct area, respectively. Images were analyzed using ImageJ (NIH, RRID:SCR_003070). To quantify the cell numbers, cells were counted manually throughout the entire Z‐stack (15 um) using the ImageJ counting tool in a blinded manner. The numbers of cells were determined per section and three sections per animal were averaged. Cell numbers are presented as per mm2. Parenchymal and perivascular PDGFRß+ cells were distinguished by their morphology and location in relation to capillaries (Roth et al., 2019) (illustrated in Figure 1c). PDGFRß+ cells with a clear cell soma and processes around capillaries were classified as perivascular PDGFRß+ cells, while PDGFRß+ cells located distant from the vessel with an amoeboid‐like morphology and multipolar irregular cell projections were classified as parenchymal PDGFRß+ cells (Reeves et al., 2019). For the double‐labeling with type I collagen or fibronectin, PDGFRß+ cells were classified according to their morphology. The location of parenchymal and perivascular PDGFRß+ cells was confirmed in WT mice as shown in Supporting Information Figure S3. The density of the basement membrane was assessed by staining the basement membrane for type IV collagen and laminin and using the ImageJ area measurement tool. The density was expressed as the percentage of the area positive for a basement membrane marker of the total area analyzed. The thickness of the vascular basement membrane was measured using the vascular markers CD31 and podocalyxin in combination with type IV collagen and laminin staining, respectively, and high‐resolution confocal images were acquired. The thickness was measured on a single z‐stack as the distance between the podocalyxin+ or CD31+ capillary wall to the outer edge of the basement membrane using the distance tool in ImageJ. This was repeated along capillaries with 5 µm distance between each measurement. The glial scar thickness was defined as the distance between the border of the infarct core and outer border of the peri‐infarct area delineated by hypertrophic GFAP+ cells (indicated with dotted line in Figure 6a). DAPI was used as a nuclear counterstain to identify single cells for all analysis. For infarct volume assessment, whole series were stained with the neuronal marker NeuN and scanned with a high‐resolution scanner. The infarcted area, defined as the area depleted of NeuN cells, and the area of the contralateral and ipsilateral hemisphere was measured, and the volume of the infarct was calculated subsequently, taking the thickness of the sections and the numbers of series into account. The percentage of the infarcted volume was calculated as 100 × ((V contralateral hemisphere−V ipsilateral hemisphere w/o infarct)/V contralateral hemisphere).
Figure 1.

Loss of RGS5 in pericytes results in decreased numbers of parenchymal platelet‐derived growth factor receptor ß‐positive (PDGFRß+) cells in the fibrotic scar after stroke. (a) Representative confocal images showing PDGFRß+ cells (red) in the fibrotic scar within the infarct core surrounded by a glial fibrillary acidic protein (GFAP+) glial scar (green) at 7 and 14 days in wild‐type (WT) and RGS5‐knockout (KO) mice. (b) Representative confocal images of the infarct core, illustrating the spatial distribution of PDGFRß+ cells (red) in relation to blood vessels (podocalyxin, cyan) of RGS5‐KO and WT mice at 7 and 14 days after stroke. (c) 3D representations of a PDGFRß+ cell (red) in the parenchyma distance to a vessel (cyan) (upper panel) and a perivascular PDGFRß+ cell contacting the vessel. (d) Quantification of the density of PDGFRß+ area in RGS5‐KO and WT mice at 7. (e) Quantification of number of parenchymal PDGFRß+ cells in RGS5‐KO and WT mice at 7 days. (f) Quantification of number of perivascular PDGFRß+ cells in RGS5‐KO and WT mice at 7 days. (g) Quantification of density of PDGFRß+ area in RGS5‐KO and WT mice at 14 days. (h) Quantification of number of parenchymal PDGFRß+ cells in RGS5‐KO and WT mice at 14 days. (i) Quantification of number of perivascular PDGFRß+ cells in RGS5‐KO and WT mice at 14 days. IC, infarct core; PI, peri‐infarct area; Pdclx, podocalyxin. N = 5; data are represented in box and whiskers plots (median, lower, and upper quartiles, minimum and maximal value, and “+” indicates mean). **p < 0.01, ***p < 0.001, Student's t‐test. Scale bars 200, 20, 5 µm
Figure 6.

RGS5‐KO mice show earlier polarization of glial scar after stroke. (a) Confocal images of glial fibrillary acidic protein (GFAP) (red) and 4',6'‐Diamidin‐2‐phenylindol (DAPI) (white) at 7 and 14 days. First column shows an overview of the glial scar, with the infarct core outlined. The box indicates where the picture on the second column was taken. (b) GFAP+ cell outside the peri‐infarct area, taken as indicated with asterisk in panel a. (c) Quantification of GFAP+ cell numbers at 7 days in peri‐infarct area showing decreased numbers in RGS5‐KO mice. (d) Quantification of GFAP density in peri‐infarct area at 7 days. (e) Quantification of the thickness of the glial scar (as highlighted in overview picture) at 7 days. (f) Quantification of GFAP+ cell numbers at 14 days in the peri‐infarct area. (g) Quantification of GFAP density in the peri‐infarct area at 14 days. (h) Quantification of the thickness of the glial scar at 14 days. IC: infarct core. N = 5. Data shown in box and whiskers plots (median, lower and upper quartiles, minimum and maximal value, and “+” indicates mean). **p < 0.05, ***p < 0.001. Student's t‐test. Scale bars: 200, 20, 10 µm
2.6. Figure composition
Figures were composed in Adobe Illustrator CC2017 (Adobe, RRID:SCR_010279). For representative confocal images, maximum projections of 15 z‐stacks were generated with ImageJ. Three‐dimensional orthogonal views were generated in ImageJ, and the center image shows a single z‐stack surrounded by the orthogonal view throughout 15 z‐stacks.
2.7. Statistics
GraphPad Prism version 7.0c (Graph Pad, RRID:SCR_002789) was used for statistical analysis of the data. All data were normally distributed (Shapiro–Wilk test). Data are reported as mean ± SD, 95% confidence interval (CI) and illustrated in box and whiskers plots. Sample size was defined according to previous studies (Özen et al., 2018; Roth et al., 2019). For two‐group comparisons, Student t‐test or multiple t tests were performed, as indicated in figure legends. Statistical significance for multiple t tests was determined using the Bonferroni method. Significance was set at p < 0.05.
3. RESULTS
3.1. Reduced numbers of parenchymal PDGFRß+ cells in RGS5‐KO mice after stroke
First we confirmed that after ischemic stroke, a fibrotic scar developed within the infarct core that was densely packed with PDGFRß+ cells (Fernandez‐Klett et al., 2012; Makihara et al., 2014; Renner et al., 2003; Roth et al., 2019). This fibrotic scar was clearly demarcated by a GFAP‐immunoreactive glial scar (Figure 1a).
We have also previously demonstrated that loss of RGS5 in pericytes results in a phenotypic shift of pericytes with reduced density of the PDGFRß+ immunoreactive area and increased numbers of perivascular PDGFRß+ cells at 7 days after stroke (Roth et al., 2019). Here, we confirm these previous results and verify that the density of the PDGFRß+ area was reduced by ca. 50% in RGS5‐KO mice compared to WT mice at 7 days after stroke (RGS5‐KO: 13.7 ± 3.6%, 95% CI [9.0, 18.3]; WT: 26.0 ± 6.4%, 95% CI [17.6, 34.3]; t(8) = 4.106, p = 0.006) (Figure 1b,d). This was consistent with a ca. 50% reduction in the number of parenchymal PDGFRß+ cells in RGS5‐KO mice versus WT mice (RGS5‐KO: 259 ± 41 cells/mm2, 95% CI [208, 311]; WT: 496 ± 81 cells/mm2 , 95% CI [350, 597]; t(8) = 5.786, p < 0.001) (Figure 1b,c,e), whereas the number of perivascular PDGFRß+ cells was increased by ca. twofold in RGS5‐KO mice compared to WT mice (RGS5‐KO: 528 ± 92 cells/mm2, 95% CI [413, 642]; WT: 281 ± 35 cells/mm2 95% CI [237, 325]; t(8)=5.580, p < 0.001) (Figure 1b,c,f). We confirmed that perivascular PDGFRß+ cells were embedded within the vascular basement membrane, while parenchymal PDGFRß+ cells were not embedded (Supporting Information Figure S2).
After 14 days, the total area occupied by PDGFRß+ cells decreased, reflecting the contraction of the ischemic lesion (Figure 1a). The above differences between RGS5‐KO and WT mice remained as a trend but did not reach statistical significance (Figure 1g–i).
3.2. Few parenchymal PDGFRß+ cells participate in type I collagen deposition in the infarct core after stroke
PDGFRß+ cells in the parenchyma have been described to contribute to the production of the ECM proteins type I collagen and fibronectin (Fernandez‐Klett et al., 2012; Makihara et al., 2014). Therefore, we next investigated whether the significant reduction in parenchymal PDGFRß+ cells in RGS5‐KO mice at 7 days had an impact on type I collagen production after stroke.
Overall, the type I collagen expression was highly increased in response to stroke within the infarct area in RGS5‐KO and WT mice at both time points (Figure 2a, left column). The expression of type I collagen was distributed around the vascular structures, as well as in the parenchyma (Figure 2a, second panel; Supporting Information Figure S3). Even though RGS5‐KO mice had significantly less parenchymal PDGFRß+ cells at 7 days after stroke, the overall density of type I collagen within the infarct core was not changed in RGS5‐KO mice in comparison to WT mice (RGS5‐KO: 6.7 ± 0.9%, 95% CI [5.5, 7.9]; WT: 6.9 ± 1.9%, 95% CI [4.4, 9.3]; t(8) = 0.124, p = 0.934) (Figure 2b). We therefore examined the contribution of parenchymal PDGFRß+ cells to the production of type I collagen. Interestingly, only around 20% of parenchymal PDGFRß+ cells colocalized with type I collagen at 7 days after stroke (Figure 2a,c). The number of parenchymal PDGFRß+ cells expressing type I collagen was not significantly different in RGS5‐KO mice in comparison to WT mice (RGS5‐KO: 26 ± 10 cells/mm2, 95% CI [8, 42]; WT: 80 ± 35 cells/mm2, 95% CI [23, 137]; t(16) = 1.400, p = 0.373) (Figure 2c). Notably, in both genotypes the majority of type I collagen was located around vascular structures rather than in the parenchyma (Figure 2a). As nearly all perivascular PDGFRß+ cells were co‐labeling with type I collagen, the number of type I collagen +/PDGFRß+ perivascular cells was significantly higher in RGS5‐KO mice in comparison to WT mice (RGS5‐KO: 508 ± 73 cells/mm2, 95% CI [391, 625]; WT: 280 ± 15 cells/mm2, 95% CI [145, 303]; t(16)=6.203, p < 0.001) (Figure 2d).
Figure 2.

Majority of type I collagen deposition occurs around perivascular but not parenchymal platelet‐derived growth factor receptor ß‐positive (PDGFRß+) cells. (a) Confocal images of type I collagen (cyan), PDGFRß (red), and DAPI (blue) at 7 and 14 days. Left column shows an increase in type I collagen within the infarct core after stroke (outlined with dotted lines) in wild‐type (WT) and RGS5‐KO mice at both time points. Box indicates that second column images were taken within the infarct core. Second column shows distribution of type I collagen in relation to PDGFRß staining, with respective single stainings on the right. White arrow indicating that majority of parenchymal PDGFRß+ cells are negative for type I collagen. Yellow arrows indicate the rare presence of parenchymal PDGFRß+/ type I collagen cells. Orthogonal view of higher magnifications illustrate examples of parenchymal PDGFRß‐positive cells negative for type I collagen (′), parenchymal PDGFRß‐positive/type I collagen‐positive cells (″) and perivascular PDGFRß‐positive/type I collagen‐positive cells (‴). (b) Quantification of type I collagen density at 7 days. (c) Quantification of total number of parenchymal PDGFRß cells that are either positive or negative for type I collagen at 7 days. (d) Quantification of number of total perivascular PDGFRß cells, that are either positive or negative for type I collagen at 7 days. (e) Quantification of type I collagen density at 14 days. (f) Quantification of the total number of parenchymal PDGFRß cells, that are either positive or negative for type I collagen at 14 days. (g) Quantification of number of total perivascular PDGFRß cells, that are either positive or negative for type I collagen at 14 days. IC, infarct core, Coll I, type I collagen. N = 5. Data shown in box and whiskers plots (median, lower, and upper quartiles, minimum and maximal value, and “+” indicates mean). **p < 0.01, ***p < 0.001. Student's t‐test (b, e) and multiple t‐tests with Bonferroni post hoc analysis (c, d, f, g). Scale bars: 200, 20, 10 µm
After 14 days, the density of type I collagen did not show any difference between RGS5‐KO and WT (Figure 2e). Furthermore, the percentage of parenchymal PDGFRß+ cells colocalizing with type I collagen decreased to approximately 15%, and their numbers did not show a significant difference between RGS5‐KO and WT mice (RGS5‐KO: 72 ± 9 cells/mm2, 95% CI [57, 87]; WT: 89 ± 32 cells/mm2, 95% CI [37, 140]; t(16)=0.465, p > 0.999) (Figure 2f). The number of perivascular PDGFRß+ cells colocalizing with type I collagen remained higher in RGS5‐KO mice in comparison to WT mice, but did not reach statistical significance (RGS5‐KO: 451 ± 59 cells/mm2; RGS5‐KO: 325 ± 67 cells/mm2; t(12) = 2.955; p = 0.061) (Figure 2g).
3.3. PDGFRß+ cells are not the main contributor to fibronectin production after ischemic stroke
We next investigated if parenchymal PDGFRß+ cells contribute to fibronectin deposition. Similar to type I collagen, fibronectin expression was highly increased within the infarct core (Figure 3a, left column). After 7 days, the density of fibronectin was similar between RGS5‐KO and WT mice (RGS5‐KO: 22.8 ± 3.9%, 95% CI [18.0, 27.6]; WT: 18.1 ± 7.6%, 95% CI [8.3, 28.1], t(8) = 1.365, p = 0.277) (Figure 3b), suggesting that RGS5 does not regulate fibronectin disposition by pericytes after stroke. Overall, only around 10% of parenchymal PDGFRß+ cells colocalized with fibronectin and this was not significantly affected by the genotype (RGS5‐KO: 28 ± 9 cells/mm2, 95% CI [12, 42]; WT: 55 ± 22 cells/mm2, 95% CI [20, 90]; t(24) = 0.387, p > 0.999) (Figure 3c). Despite the increased number of total perivascular PDGFRß+ cells in RGS5‐KO mice, there was no difference in the number of perivascular PDGFRß+ cells colocalizing with fibronectin (RGS5‐KO: 53 ± 13 cells/mm2, 95% CI [33, 73]; WT: 61 ± 32 cells/mm2, 95% CI [8, 112]; t(8) = 0.417, p > 0.999) (Figure 3d). The majority of fibronectin+ cells did not co‐label with PDGFRß and were located in the parenchyma, and the total number of parenchymal fibronectin+ cells was similar between RGS5‐KO and WT mice (RGS5‐KO: 656 ± 73 cells/mm2, 95% CI [539, 773]; WT: 594 ± 153 cells/mm2, 95% CI [350, 838]; t(24) = 0.999, p = 0.492) (Figure 3c).
Figure 3.
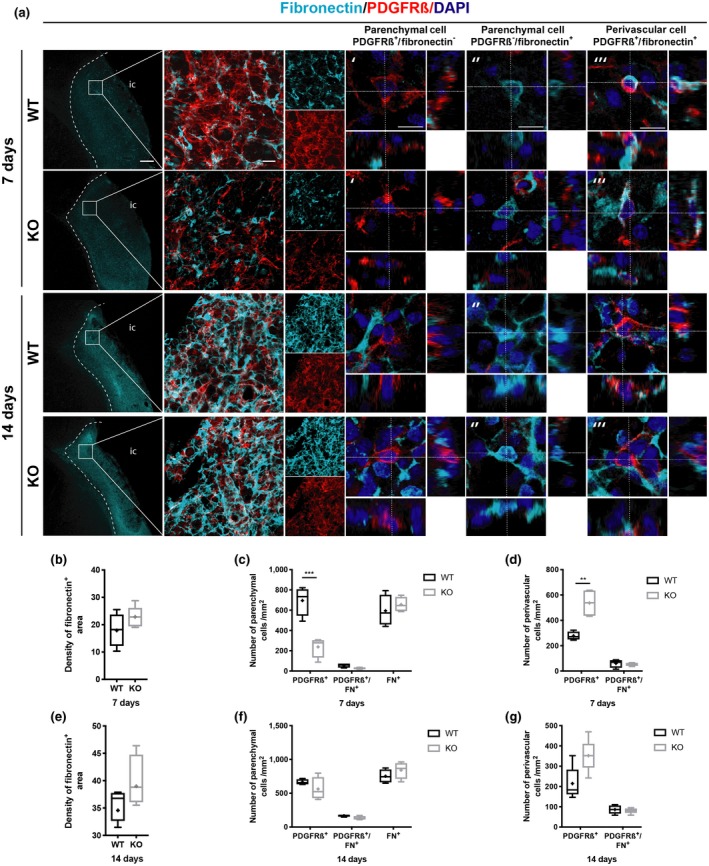
Only very few platelet‐derived growth factor receptor ß (PDGFRß+) cells contribute to fibronectin deposition. (a) Confocal images of fibronectin (cyan), PDGFRß (red), and DAPI (blue) at 7 and 14 days. First column shows increase in fibronectin deposition within the infarct core after stroke (outlined with dotted lines) in wild‐type (WT) and RGS5‐KO mice at both time points. Boxes indicate that second column images were taken within the infarct core. Second column shows the distribution of fibronectin in relation to PDGFRß staining, with respective single staining on the right. Higher magnifications show orthogonal view of a parenchymal PDGFRß+ cell that is negative for fibronectin (′), parenchymal fibronectin+ cell that is PDGFRß‐negative (″), and perivascular PDGFRß+/fibronectin+ cell (‴). (b) Quantification of fibronectin density at 7 days. (c) Quantification of total number of parenchymal cells that are either PDGFRß+ or FN+, and number of double positive cells at 7 days after stroke. (d) Quantification of total number perivascular PDGFRß+ cells and perivascular PDGFRß+/fibronectin+ cells at 7 days after stroke. (e) Quantification of fibronectin density at 14 days. (f) Quantification of total parenchymal cells that are either PDGFRß+ or fibronectin+, and number of double positive cells at 14 days after stroke. (g) Quantification of total number of perivascular PDGFRß+ cells and perivascular PDGFRß+/fibronectin+ cells at 14 days after stroke. FN, fibronectin; IC, infarct core. N = 5. Data shown in box and whiskers plots (median, lower and upper quartiles, minimum and maximal value, and “+” indicates mean). **p < 0.01. Student's t‐test (b, e) and multiple t‐tests with Bonferroni post‐hoc analysis (c, d, f, g). Scale bars: 200, 20, 10 µm
After 14 days, the density of the fibronectin+ area further increased in comparison to 7 days but did not show any difference between RGS5‐KO and WT mice (RGS5‐KO: 39.9 ± 5.7%, 95% CI [32.4, 47.35]; WT: 35.7 ± 2.9%, 95% CI [31.1, 40.4]; t(8) = 1.501, p = 0.260) (Figure 3e). Around 20% of parenchymal PDGFRß+ cells colocalized with fibronectin after 14 days, with similar numbers between RGS5‐KO and WT mice (RGS5‐KO: 133 ± 33 cells/mm2, 95% CI [89, 176]; WT: 167 ± 14 cells/mm2, 95% CI [144, 189]; t(24) = 0.495, p = 0.627) (Figure 3f). Similarly, there was no difference in the number of perivascular PDGFRß+ cells colocalizing with fibronectin (RGS5‐KO:80 ± 19 cells/mm2, 95% CI [55, 105]; WT: 86 ± 24 cells/mm2, 95% CI [48, 125]; t(16) = 0.150, p > 0.999) (Figure 3g). Further, the total number of fibronectin+ cells remained unaffected by the genotype (RGS5‐KO: 842 ± 153 cells/mm2, 95% CI [643, 1041]; WT: 752 ± 100 cells/mm2, 95% CI [593, 911]; t(24) = 1.314, p = 0.615) (Figure 3f).
3.4. RGS5‐KO mice have a reduced thickening of the vascular basement membrane in response to stroke
We then examined whether the increased number of perivascular PDGFRß+ cells in RGS5‐KO mice after 7 days affected the pathological thickening of the vascular basement membrane after stroke (Figure 4). The vascular basement membrane has been reported to thicken after injury (Gonul et al., 2002; Nehls, Schuchardt, & Drenckhahn, 1994), and accordingly the expression of type IV collagen was increased around vessels in the infarct core (Figure 4a, left panel). There was no significant difference in the density of type IV collagen between RGS5‐KO and WT mice at both time points (7 days RGS5‐KO: 8.1 ± 2.6%, 95% CI [1.5, 14.6]; WT: 4.9 ± 1.7%, 95% CI [0.5; 9.3]; t(8) = 1.748, p = 0.155; 14 days RGS5‐KO: 4.5 ± 0.9%, 95% CI [3.3, 5.7]; WT:4.5 ± 1.3%, 95% CI [2.7, 6.28]; t(8) = 0.000, p > 0.999) (Figure 4b,d). However, the thickness of type IV collagen around the vessels was significantly reduced in RGS5‐KO mice compared to WT mice at 7 and this also remained significantly different at 14 days after stroke (7 days RGS5‐KO: 0.7 ± 0.006 µm, 95% CI [0.719, 0.747]; WT: 0.9 ± 0.04 µm, 95% CI [0.776, 1.004]; t(8) = 5.875, p = 0.004; 14 days RGS5‐KO: 0.7 ± 0.09 µm, 95% CI [0.437, 0.910]; WT: 1.3 ± 0.2 µm, 95% CI [0.766, 1.774], t(8) = 4.611, p = 0.009) (Figure 4c,e).
Figure 4.
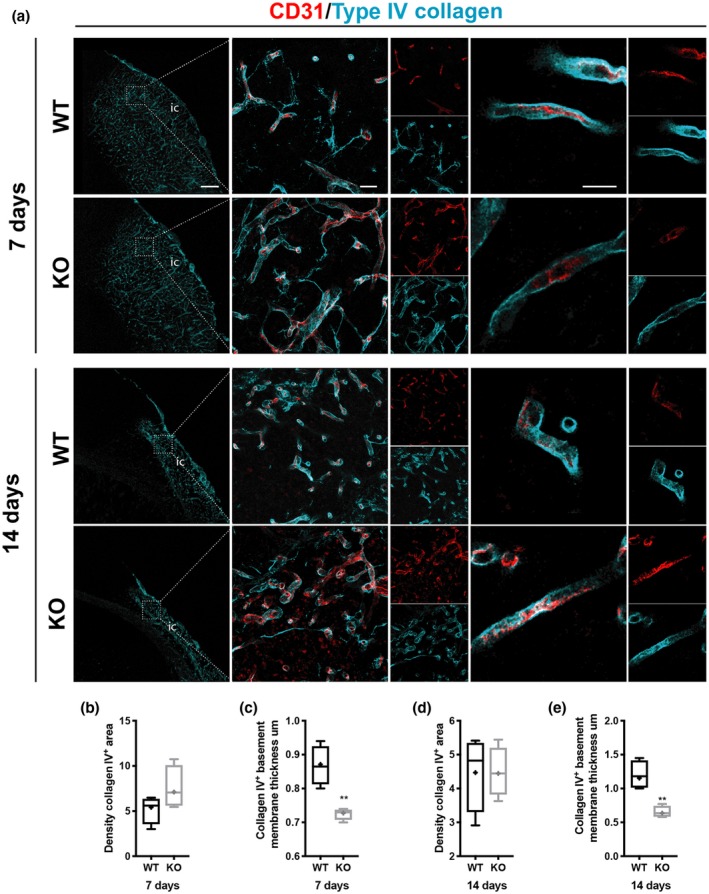
RGS5‐KO mice have reduced thickness of type IV collagen+ vascular basement membrane after stroke. (a) Confocal images of type IV collagen (cyan) and the blood vessel marker CD31 (red) at 7 and 14 days. Left column shows type IV collagen distribution in wild‐type (WT) and RGS5‐KO mice at both time points. The box indicates where the higher magnification picture in middle column has been taken from. Right column shows a high magnification of single z‐stack through a blood vessel to illustrate the thickness of the vascular basement membrane that is reduced in RGS5‐KO mice. (b) Quantification of type IV Collagen area density at 7 days. (c) Quantification of thickness of type IV collagen+ vascular basement membrane at 7 days. (d) Quantification of type IV collagen area density at 14 days. (e) Quantification of thickness of type IV collagen+ vascular basement membrane at 14 days. IC: infarct core. N = 5. Data shown in box and whiskers plots (median, lower and upper quartiles, minimum and maximal value, and “+” indicates mean). **p < 0.01. Student's t‐test. Scale bars: 200, 20, 10 µm
We also investigated if the reduced thickening was applicable for other vascular basement membrane proteins and therefore examined a second vascular basement membrane component, laminin (Figure 5). Similar to type IV collagen, the expression of laminin was increased around blood vessels within the infarct core (Figure 5a, left panel). There was no significant difference between genotypes in the overall density of laminin at 7 and 14 days after stroke (Figure 5b,d). However, similar to type IV collagen, the pathological thickening was reduced in RGS5‐KO mice at both time points (7 days RGS5‐KO: 0.8 ± 0.1 µm, 95% CI [0.658, 0.961]; WT: 1.1 ± 0.3 µm, 95% CI [0.795, 1.413]; t(8) = 2.426, p = 0.038; 14 days RGS5‐KO: 0.6 ± 0.05 µm, 95% CI [0.559, 0.706]; WT: 0.8 ± 0.1 µm, 95% CI [0.672, 1.013]; t(8) = 3.604, p = 0.011) (Figure 5c,e).
Figure 5.
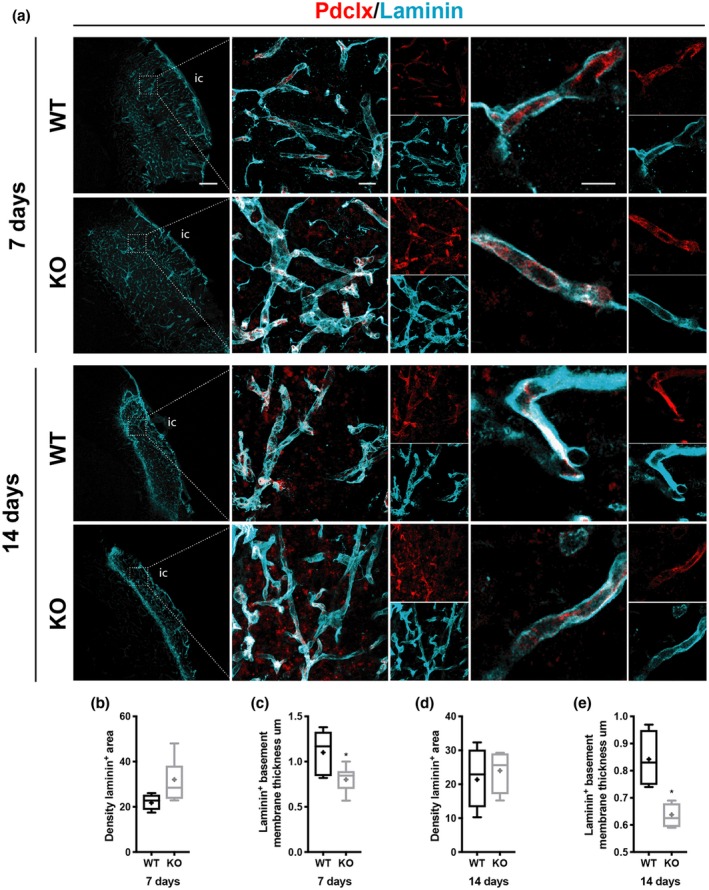
RGS5‐KO mice have reduced thickness of laminin+ vascular basement membrane after stroke. (a) Confocal images of laminin (cyan) and blood vessel marker Pdclx (red) at 7 and 14 days. The box indicates where the higher magnification picture in middle column has been taken from. Right column shows high magnification of single z‐stack through a blood vessel to illustrate the reduced thickness of the vascular basement membrane in RGS5‐KO mice. (b) Quantification of laminin+ area density at 7 days. (c) Quantification of the thickness of laminin+ vascular basement membrane at 7 days. (d) Quantification of laminin area density at 14 days. (e) Quantification of the thickness of laminin+ vascular basement membrane at 14 days. IC, infarct core. Pdclx, podocalyxin. N = 5. Data shown in box and whiskers plots (median, lower and upper quartiles, minimum and maximal value, and “+” indicates mean). *p < 0.05. Student's t‐test. Scale bars: 200, 20, 10 µm
3.5. RGS5‐KO mice show an earlier polarization of the glial scar after stroke
The fibrotic scar is demarcated by a GFAP‐immunoreactive glial scar (Figure 1a). The cross talk of astrocytes and pericytes is important in the regulation of tissue survival (Bonkowski, Katyshev, Balabanov, Borisov, & Dore Duffy, 2011); therefore we next investigated if the RGS5‐mediated shift in pericyte localization affected astrocytes and the formation of the glial scar by analyzing morphological changes in astrocytes (Figure 6).
At 7 days after stroke, there were no GFAP+ cells within the infarct core of RGS5‐KO and WT mice (Figure 6a). In the peri‐infarct area, however, GFAP expression was highly increased. GFAP+ cells in the peri‐infarct area had a hypertrophic cell soma in both RGS5‐KO and WT mice compared to astrocytes outside the peri‐infarct area (Figure 6a). In the peri‐infarct area of WT mice, GFAP+ cells primarily had a stellate morphology and the peri‐infarct area was densely packed with GFAP+ cells (Figure 6a). In contrast, in the peri‐infarct area of RGS5‐KO mice, GFAP+ cells had a different morphology with polarized processes toward the infarct core. In comparison to WT mice, the number and density of GFAP+ cells were significantly reduced in RGS5‐KO mice (RGS5‐KO: 546 ± 21 cells/mm2, 95% CI [512, 580], 19.1 ± 1.3%, 95% CI [16.9, 21.1]; WT: 801 ± 59 cells/mm2, 95% CI [705, 896], 30.7 ± 0.9%, 95% CI [29.3, 32.2]; t(8) = 8.008, p < 0.001 and t(8) = 14.55, p < 0.001, respectively) (Figure 6b,c). Furthermore, there was a significant reduction in thickness of the glial scar in RGS5‐KO mice (RGS5‐KO: 154.3 ± 24 µm, 95% CI [129.5, 179.1]; WT: 226.2 ± 18.5 µm, 95% CI [196.8, 255.6]; t(8) = 5.099, p = 0.001) (Figure 6a,d, indicated between dotted lines).
After 14 days, GFAP+ cells in the peri‐infarct area of both genotypes were hypertrophic and showed a polarized morphology with elongated processes toward the infarct core. There was no difference in the number and the density of GFAP+ cells between RGS5‐KO and WT mice at 14 days after stroke (RGS5‐KO: 1,098 ± 302 cells/mm2, 95% CI [617, 1579], 31.6 ± 14.4%, 95% CI [8.6, 54.5]; WT: 842 ± 102 cells/mm2, 95% CI [494, 1041], 19.2.7 ± 7.9%, 95% CI [6.5,31.7]; t(8)=1.901, p = 0.227 and t(8) = 1.514, p = 0.181, respectively) (Figure 6e,f). However, the glial scar thickness was now significantly increased in RGS5‐KO mice in comparison to WT mice (RGS5‐KO: 353.0 ± 28.4 µm, 95% CI [316.1, 389.9]; WT: 220.0 ± 38.9 µm, 95% CI [169.4, 270.7]; t(8) = 6.751, p = 0.009) (Figure 6g).
We have previously reported that RGS5‐KO mice have similar infarct volumes compared to WT mice at 7 days after stroke (Roth et al., 2019). To investigate whether the redistribution of PDGFRß+ cells had an impact on the infarct volume at 14 days, NeuN staining was performed (Figure 7). No difference in infarct volume was observed between RGS5‐KO and WT mice at 14 days after stroke (RGS5‐KO: 5.3 ± 1.4 mm2, 95% CI [3.9, 6.6]; WT: 4.0 ± 3.1 [0.1, 9.9], t(8) = 0.520, p = 0.378).
Figure 7.
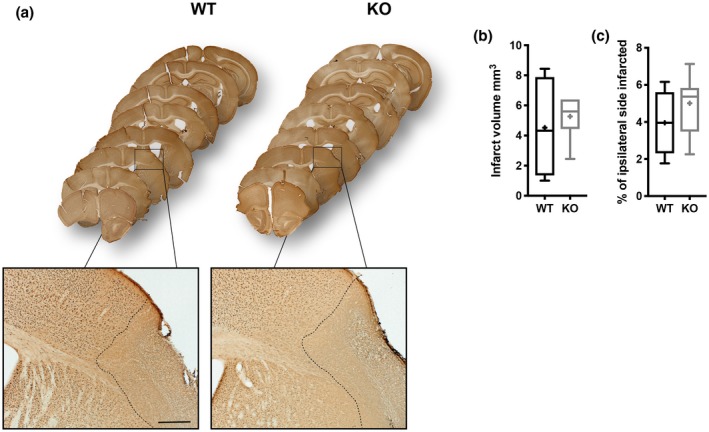
Loss of RGS5 in pericytes does not affect stroke size at 14 days after stroke. (a) Whole brain series of wild‐type (WT) and RGS5‐KO mice 14 days after stroke stained with NeuN. Box indicates where higher magnification was taken. Dotted lines indicate area that is depleted of NeuN+ cells. (b) Quantification of the infarct volume. (c) Quantification of the relative infarcted area. N = 5. Data shown in box and whiskers plots (median, lower and upper quartiles, minimum and maximal value, and “+” indicates mean), Student's t‐test. Scale bar: 200 µm
4. DISCUSSION
Here we examined the role of parenchymal PDGFRß+ cells in the formation of the fibrotic scar after stroke. We demonstrate that a 50% reduction in parenchymal PDGFRß+ cells in the ischemic core does not translate into changes in the ECM deposition in the ischemic core as most of the ECM is produced by cells negative for PDGFRß. The redistribution of PDGFRß+ cells toward a perivascular location, however, lead to a reduced pathological thickening of the vascular basement membrane consisting of type IV collagen and laminin and was associated with an earlier polarization of the glial scar in RGS5‐KO mice.
Pericytes detach from the vessel wall in response to CNS injury within hours after the insult and migrate into the adjacent parenchyma (Dore‐Duffy et al., 2000; Duz, Oztas, Erginay, Erdogan, & Gonul, 2007; Gonul et al., 2002) where they form a dense network of PDGFRß+ cells. These PDGFRß+ cells have been interpreted as a fibrotic scar responsible for the deposition of the ECM proteins type I collagen and fibronectin (Fernandez‐Klett et al., 2012; Makihara et al., 2014). Here, we have taken advantage of a transgenic mouse model that leads to a shift from a parenchymal to a perivascular location of PDGFRß+ cells due to the loss of RGS5 in pericytes (Nisancioglu et al., 2008; Roth et al., 2019). The resulting decrease in parenchymal PDGFRß+ cells after stroke allows us to address their proposed contribution to fibrotic scar formation.
Surprisingly, only a small fraction of the parenchymal PDGFRß+ cells colocalized with type I collagen and fibronectin. Our findings were determined after careful confocal analysis distinguishing parenchymal and perivascular PDGFRß+ cells. Other studies have reported higher co‐labeling of PDGFRß+ cells with at least fibronectin, although the authors did not distinguish their specific location (Fernandez‐Klett et al., 2012) or quantify cell numbers (Makihara et al., 2014). In addition, we utilize a permanent MCAO model, that combines a high reproducibility with a defined cortical stroke (Llovera et al., 2014), whereas others have used a reperfusion or photothrombotic stroke model (Fernandez‐Klett et al., 2012; Makihara et al., 2014) making it difficult to directly compare results.
Our data suggest that the contribution of parenchymal pericytes to ECM production in the fibrotic scar after stroke is rather negligible. Consequently, a 50% reduction in these cells did not have any significant impact on the overall ECM density in the ischemic core. This implicates that pericyte detachment and migration might not be as crucial for fibrosis after stroke as previously suggested (Fernandez‐Klett et al., 2012; Fernández‐Klett & Priller, 2014).
The origin of scar‐forming cells is still debated. In particular, fibroblasts have been shown to co‐label with type I collagen after stroke and other CNS injuries (Kelly et al., 2016; Komuta et al., 2009; Riew, Choi, Kim, Jin, & Lee, 2018; Soderblom et al., 2013). To date, the only linage‐tracing study that suggests a pericyte origin of scar‐forming cells was performed in spinal cord injury (Göritz et al., 2011). Using a GLAST promotor, Göritz et al. showed that a specific subtype of pericytes is contributing to scar formation after spinal cord injury, confirming our results that only a small fraction of parenchymal PDGFRß+ cells are producing type I collagen and fibronectin. Whether all parenchymal PDGFRß+ cells originate from pericytes after stroke can only be fully addressed using lineage tracing. However, our study shows that targeting a pericyte‐specific gene results in a redistribution of the location of PDGFRß+ cells, supporting their pericyte origin. Determining the origin of scar‐forming cells could be of great clinical interest. Accordingly, reducing pericyte‐derived fibrotic scar tissue promotes functional recovery after spinal cord injury (Dias et al., 2018).
Our data further indicate that loss of RGS5 in pericytes does not directly affect type I collagen and fibronectin production after stroke, as the overall deposition of these ECM proteins was not changed. This suggests that the described role of RGS5 in ECM production in other organs might not be applicable to brain pericytes after stroke (Bahrami et al., 2014; Holobotovskyy et al., 2013; Li et al., 2010). RGS proteins are conserved, and the fact that that loss of RGS5 does not directly affect type I collagen and fibronectin could be due to compensatory upregulation of other RGS proteins (Ganss, 2015). However, a previous study has shown that loss of RGS5 did not result in compensatory upregulation of RGS4 and RGS16 (Nisancioglu et al., 2008).
The redistribution of PDGFRß+ cell toward a perivascular location in RGS5‐KO mice, however, had implications on the pathological thickening of the vascular basement membrane after stroke. The ischemic insult results in a modulation of the vascular basement membrane proteins, by both increased degradation and thickening of the vascular basement membrane (Fukuda et al., 2004; Gonul et al., 2002; Nehls et al., 1994). We show that the vascular basement membrane components, type IV collagen and laminin, are increased around blood vessels within the infarct core during the chronic phase after stroke, which is in line with the previous data from mouse and human stroke samples (Fernandez‐Klett et al., 2012). The reduced thickening of the vascular basement membrane after stroke in RGS5‐KO mice may at least partially be due to the reduced BBB breakdown we have previously reported in RGS5‐KO mice during the acute and chronic phase after stroke (Özen et al., 2018; Roth et al., 2019). BBB breakdown results in the migration of monocytes, neutrophils, and macrophages into the infarcted area, contributing to the inflammatory process that modulates the basement membrane proteins (Rosell et al., 2008; Sixt et al., 2001). Additionally, it has been shown that the changes in the vascular basement membrane can be associated with pericyte expression of proteases (Dore‐Duffy et al., 2000; Nehls et al., 1994), and accordingly the higher numbers of perivascular PDGFRß+ cells in RGS5‐KO mice could result in a higher production of proteases. The decreased thickening of the vascular basement membrane might have implications for the vascular remodeling occurring in this phase after stroke, as especially type IV collagen has been shown to contribute to inhibition of angiogenesis (Colorado et al., 2000; Mundel & Kalluri, 2007). Angiogenesis and vascular remodeling are important factors in functional recovery, and increased vessel density in stroke patients correlates with survival time (Krupinski et al., 1994). Hence, reduced thickening of the vascular basement membrane in RGS5‐KO mice could potentially have effects on stroke outcome through angiogenesis and vascular remodeling. Indeed, we have previously reported that RGS5‐KO mice have a more preserved vasculature with increased vascular density within the infarct core (Roth et al., 2019), which might also explain why the overall density of the vascular basement membrane was not changed. The assessment of the vascular basement membrane was conducted using confocal analysis. Compared to electron microscopy, this method has a lower resolution and provides less structural information which limits the conclusions that can be drawn.
We show that in the chronic phase of stroke, the fibrotic scar is surrounded by a glial scar, in line with previous observations (Fernandez‐Klett et al., 2012). We demonstrate that the redistribution of PDGFRß+ cells in RGS5‐KO mice results in an earlier polarization of the glial scar, suggesting a potential cross talk between pericytes and astrocytes after stroke. Pericytes have been shown to influence the polarization of astrocytes under physiological conditions (Armulik et al., 2010; Gundersen, Vindedal, Skare, & Nagelhus, 2014), and the cell–cell communication between astrocytes and pericytes is important in BBB maintenance (Bertossi, Girolamo, Errede, Virgintino, & Roncali, 2003; Bonkowski et al., 2011). Upon stroke, the direct cell–cell contact within the ischemic core is lost, contributing to the breakdown of the BBB. A study in GFAP‐KO mice suggests that pericytes proliferate and increase their coverage around blood vessels to compensate for the lack of astrocyte‐end feet connections (Bonkowski et al., 2011). Interestingly, it has been shown that conditional deletion of PDGFRß during stroke resulted in a disrupted glial scar formation (Shen et al., 2012). The underlying mechanisms of how PDGFRß+ cells contribute to the segregation of the glial and fibrotic scar, however, remain unclear. The increase in ECM deposition in the infarct core has been implicated to induce astrocyte activation (Ellison et al., 1998; Hashimoto et al., 2005; Summers, Kangwantas, Nguyen, Kielty, & Pinteaux, 2010); however, our data suggest that at least type I collagen and fibronectin are not the main drivers in astrocyte activation.
However, the observed changes in morphology and reduced numbers of GFAP+ cells in RGS5‐KO mice might also be due to secondary effects, as the formation of the glial scar is closely coordinated with the inflammatory response after stroke (Fitch & Silver, 2008; Sofroniew, 2015). Accordingly, the reduced numbers of GFAP+ astrocytes in RGS5‐KO after 7 days might be due to the decreased or delayed proliferation of astrocytes (Li et al., 2014). Several factors have been described to result in astrocyte proliferation and morphological changes, including BBB disruption (Schachtrup et al., 2010; Sofroniew, 2009), suggesting that the reduced BBB breakdown in RGS5‐KO mice leads to a delayed proliferation (Özen et al., 2018; Roth et al., 2019) supported by the fact that the number of GFAP+ cells does not show a difference between the genotypes at 14 days anymore. A delayed proliferation might also explain the thicker glial scar seen in RGS5‐KO mice after stroke.
In several CNS disorders, including stroke, chronic neuroinflammation, traumatic brain injury, and brain tumors, a glial scar with a dual role forms around the injury side (Fitch & Silver, 2008). On one hand, the glial scar limits the spread of inflammation but on the other hand, it inhibits the functional recovery, suggesting that modulating the glial scar might improve functional recovery (Kawano et al., 2012; McKeon, Schreiber, Rudge, & Silver, 1991; Sofroniew, 2009).
The observed changes in the distribution of PDGFRß+ cells, however, did not have an impact on the final outcome assessed as stroke size at 14 days after stroke, suggesting that targeting parenchymal PDGFRß+ cells in the fibrotic scar after stroke do not constitute a therapeutic target in this model. A limitation of the study is the usage of only male mice. Further studies would be needed to address a possible impact of the gender on scar formation after ischemic stroke.
Scar formation after stroke is important for tissue remodeling, while also representing a major barrier to regeneration. Here we show that targeting PDGFRß+ cell distribution does not affect fibrotic scar formation after stroke. Our study shows that especially parenchymal PDGFRß+ cells do only minimally contribute to the fibrotic scar formation in ischemic stroke. Consequently, a shift from parenchymal to perivascular PDGFRß+ cells does not affect ECM deposition after stroke, while resulting in a decreased pathological thickening of the vascular basement membrane and an earlier polarization of the glial scar. While a shift of pericytes toward a perivascular phenotype does not affect fibrotic scar formation, it however remains an important target for vascular remolding after stroke.
DECLARATION OF TRANSPARENCY
The authors, reviewers and editors affirm that in accordance to the policies set by the Journal of Neuroscience Research, this manuscript presents an accurate and transparent account of the study being reported and that all critical details describing the methods and results are present.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, M.R. and G.P.; Methodology, M.R. and G.P.; Investigation, M.R., A.E., C.A., R.C., and G.P; Formal Analysis, M.R., A.E., R.C., and G.P; Resources, G.G.; Writing – Original Draft, M.R., A.E., R.C., and G.P; Writing – Review & Editing, M.R., A.E., R.C., and G.P.; Visualization, M.R.; Supervision, G.P.; Project Administration, G.P.; Funding Acquisition, G.P.
Supporting information
Figure S1. No fibrosis occurs in sham‐operated animals. (a) Confocal images of WT and KO mice 7 days after sham operation showing that sham operation does not induce changes PDGFRß+ cells, deposition of ECM or astrocyte reactivity. (b) Specificity of the secondary antibody was tested at 7 days after stroke, showing that secondary antibodies did not bind unspecifically in the infarct core. Pdclx: Podocalyxin, Scale bars: 20 μm.
Figure S2. Perivascular and parenchymal PDGFRß+. (a) Three‐dimensional confocal representation showing examples of perivascular (top) and parenchymal (bottom) PDGFRß+ cells (red). Perivascular PDGFRß+ cell (red) is in contract with a capillary (podocalyxin, red) and is covered by laminin (blue). Parenchymal PDGFRß+ cell is located in parenchyma without contact to capillaries and is not embedded in laminin. Images were taken in the infarct core of WT mice to allow to co‐staining of four markers. (b) Schematic representation of the location of perivascular and parenchymal PDGFRß+ cells in relation to a capillary and the vascular basement membrane. Scale bar 5 μm.
Figure S3. Type I collagen distribution within the infarct core at 7 days after stroke. (a) Confocal images of type I collagen (cyan) around vasculature (CD31, red) within the infract core of WT and RGS5‐KO mice. (b) Confocal images of PDGFRß (red), type I collagen (cyan), vasculature (podocalyxin, green), and nuclear marker DAPI (grey) in infarct core of WT mice. First row shows maximal projection of four z‐stacks, with individual stainings in second row. Third row shows orthogonal view with a single x‐stack. (′) parenchymal PDGFRß+/type I collagen- cell, (″) parenchymal PDGFRß+/type I collagen+ cell, (″′) perivascular PDGFRß+/type I collagen+ cell. Scale bar: 20 μm (in a) and 5 μm (in b).
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
ACKNOWLEDGMENTS
We thank Alicja Flasch for excellent technical help with Immunohistochemistry and Ilknur Özen for valuable discussions.
Roth M, Enström A, Aghabeick C, Carlsson R, Genové G, Paul G. Parenchymal pericytes are not the major contributor of extracellular matrix in the fibrotic scar after stroke in male mice. J Neuro Sci. 2020;98:826–842. 10.1002/jnr.24557
Edited by Eric M. Prager. Statistics Editor: David McArthur. Reviewed by Byung Kim and Helia Tenza.
The peer review history for this article is available at https://publons.com/publon/10.1002/jnr.24557.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Armulik, A. , Genové, G. , & Betsholtz, C. (2011). Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Developmental Cell, 21(2), 193–215. 10.1016/j.devcel.2011.07.001 [DOI] [PubMed] [Google Scholar]
- Armulik, A. , Genové, G. , Mäe, M. , Nisancioglu, M. H. , Wallgard, E. , Niaudet, C. , … Betsholtz, C. (2010). Pericytes regulate the blood‐brain barrier. Nature, 468(7323), 557–561. 10.1038/nature09522 [DOI] [PubMed] [Google Scholar]
- Bahrami, A. J. , Gunaje, J. J. , Hayes, B. J. , Riehle, K. J. , Kenerson, H. L. , Yeung, R. S. , … Mahoney, W. M., Jr . (2014). Regulator of G‐protein signaling‐5 is a marker of hepatic stellate cells and expression mediates response to liver injury. PLoS One, 9(10), e108505 10.1371/journal.pone.0108505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertossi, M. , Girolamo, F. , Errede, M. , Virgintino, D. , & Roncali, L. (2003). Effects of 6‐aminonicotinamide gliotoxin on blood‐brain barrier differentiation in the chick embryo cerebellum. Anatomy and Embryology, 207(3), 209–219. 10.1007/s00429-003-0335-4 [DOI] [PubMed] [Google Scholar]
- Bonkowski, D. , Katyshev, V. , Balabanov, R. D. , Borisov, A. , & Dore Duffy, P. (2011). The CNS microvascular pericyte: Pericyte‐astrocyte crosstalk in the regulation of tissue survival. Fluids and Barriers of the CNS, 8(1), 8 10.1186/2045-8118-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, W. , Liu, H. , Zhao, J. , Chen, L. Y. , Chen, J. , Lu, Z. , & Hu, X. (2016). Pericytes in brain injury and repair after ischemic stroke. Translational Stroke Research, 8(2), 1–15. 10.1007/s12975-016-0504-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colorado, P. C. , Torre, A. , Kamphaus, G. , Maeshima, Y. , Hopfer, H. , Takahashi, K. , … Kalluri, R. (2000). Anti‐angiogenic cues from vascular basement membrane collagen. Cancer Research, 60(9), 2520–2526. 5918959B-84B4-4F47-8452-8C01E2589708 [PubMed] [Google Scholar]
- Dias, D. O. , & Göritz, C . (2018). Fibrotic scarring following lesions to the central nervous system. Matrix Biology, 68–69, 561–570. 10.1016/j.matbio.2018.02.009 [DOI] [PubMed] [Google Scholar]
- Dias, D. O. , Kim, H. , Holl, D. , Solnestam, B. W. , Lundeberg, J. , Carlén, M. , … Frisén, J. (2018). Reducing pericyte‐derived scarring promotes recovery after spinal cord injury. Cell, 173(1), 153–165. 10.1016/j.cell.2018.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev, A. , Seidenbecher, C. I. , & Schachner, M. (2010). Compartmentalization from the outside: The extracellular matrix and functional microdomains in the brain. Trends in Neurosciences, 33(11), 503–512. 10.1016/j.tins.2010.08.003 [DOI] [PubMed] [Google Scholar]
- Dore‐Duffy, P. , Owen, C. , Balabanov, R. , Murphy, S. , Beaumont, T. , & Rafols, J. A. (2000). Pericyte migration from the vascular wall in response to traumatic brain injury. Microvascular Research, 60(1), 55–69. 10.1006/mvre.2000.2244 [DOI] [PubMed] [Google Scholar]
- Duz, B. , Oztas, E. , Erginay, T. , Erdogan, E. , & Gonul, E. (2007). The effect of moderate hypothermia in acute ischemic stroke on pericyte migration: An ultrastructural study. Cryobiology, 55(3), 279–284. 10.1016/j.cryobiol.2007.08.009 [DOI] [PubMed] [Google Scholar]
- Ellison, J. A. , Velier, J. J. , Spera, P. , Jonak, Z. L. , Wang, X. , Barone, F. C. , & Feuerstein, G. Z . (1998). Osteopontin and its integrin receptor alpha(v)beta3 are upregulated during formation of the glial scar after focal stroke. Stroke, 29(8), 1698–1706; discussion 1707. DBBC27D7‐9C30‐41F2‐817B‐4689A6EF78F5 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Klett, F. , Potas, J. R. , Hilpert, D. , Blazej, K. , Radke, J. , Huck, J. , … Priller, J. (2012). Early loss of pericytes and perivascular stromal cell‐induced scar formation after stroke. Journal of Cerebral Blood Flow and Metabolism, 33(3), 153–165. 10.1038/jcbfm.2012.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Klett, F. , & Priller, J. (2014). The fibrotic scar in neurological disorders. Brain Pathology, 24(4), 404–413. 10.1111/bpa.12162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch, M. T. , & Silver, J. (2008). CNS injury, glial scars, and inflammation: Inhibitory extracellular matrices and regeneration failure. Experimental Neurology, 209(2), 294–301. 10.1016/j.expneurol.2007.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, S. , Fini, C. A. , Mabuchi, T. , Koziol, J. A. , Eggleston, L. L. , & del Zoppo, G. J. (2004). Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke, 35(4), 998–1004. 10.1161/01.STR.0000119383.76447.05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganss, R. (2015). Keeping the balance right: Regulator of G protein signaling 5 in vascular physiology and pathology. Progress in Molecular Biology and Translational Science, 133, 93–121. 10.1016/bs.pmbts.2015.02.003 [DOI] [PubMed] [Google Scholar]
- Gonul, E. , Duz, B. , Kahraman, S. , Kayali, H. , Kubar, A. , & Timurkaynak, E. (2002). Early pericyte response to brain hypoxia in cats: An ultrastructural study. Microvascular Research, 64(1), 116–119. 10.1006/mvre.2002.2413 [DOI] [PubMed] [Google Scholar]
- Göritz, C. , Dias, D. O. , Tomilin, N. , Barbacid, M. , Shupliakov, O. , & Frisén, J. (2011). A pericyte origin of spinal cord scar tissue. Science, 333(6039), 238–242. 10.1126/science.1203165 [DOI] [PubMed] [Google Scholar]
- Gundersen, G. A. , Vindedal, G. F. , Skare, O. , & Nagelhus, E. A. (2014). Evidence that pericytes regulate aquaporin‐4 polarization in mouse cortical astrocytes. Brain Structure and Function, 219(6), 2181–2186. 10.1007/s00429-013-0629-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, M. , Koda, M. , Ino, H. , Yoshinaga, K. , Murata, A. , Yamazaki, M. , … Moriya, H. (2005). Gene expression profiling of cathepsin D, metallothioneins‐1 and ‐2, osteopontin, and tenascin‐C in a mouse spinal cord injury model by cDNA microarray analysis. Acta Neuropathologica, 109(2), 165–180. 10.1007/s00401-004-0926-z [DOI] [PubMed] [Google Scholar]
- Hesp, Z. C. , Yoseph, R. Y. , Suzuki, R. , Wilson, C. , Nishiyama, A. , & McTigue, D. M. (2017). Proliferating NG2 cell‐dependent angiogenesis and scar formation alter axon growth and functional recovery after spinal cord injury in mice. Journal of Neuroscience, 38(6), 3953–3916. 10.1523/JNEUROSCI.3953-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holobotovskyy, V. , Manzur, M. , Tare, M. , Burchell, J. , Bolitho, E. , Viola, H. , … Ganss, R. (2013). Regulator of G‐protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circulation Research, 112(5), 781–791. 10.1161/CIRCRESAHA.111.300142 [DOI] [PubMed] [Google Scholar]
- Kamouchi, M. , Ago, T. , Kuroda, J. , & Kitazono, T. (2011). The possible roles of brain pericytes in brain ischemia and stroke. Cellular and Molecular Neurobiology, 32(2), 159–165. 10.1007/s10571-011-9747-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano, H. , Kimura‐Kuroda, J. , Komuta, Y. , Yoshioka, N. , Li, H. P. , Kawamura, K. , … Raisman, G. (2012). Role of the lesion scar in the response to damage and repair of the central nervous system. Cell and Tissue Research, 349(1), 169–180. 10.1007/s00441-012-1336-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, K. K. , MacPherson, A. M. , Grewal, H. , Strnad, F. , Jones, J. W. , Yu, J. , … Siegenthaler, J. A. (2016). Col1a1+ perivascular cells in the brain are a source of retinoic acid following stroke. BMC Neuroscience, 17(1), 49 10.1186/s12868-016-0284-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuta, Y. , Teng, X. , Yanagisawa, H. , Sango, K. , Kawamura, K. , & Kawano, H. (2009). Expression of transforming growth factor‐β receptors in meningeal fibroblasts of the injured mouse brain. Cellular and Molecular Neurobiology, 30(1), 101–111. 10.1007/s10571-009-9435-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupinski, J. , Kaluza, J. , Kumar, P. , Kumar, S. , Path, F. R. C. , & Wang, J. M . (1994). Role of angiogenesis in patients with cerebral ischemic stroke. Stroke, 25, 1794–1798. [DOI] [PubMed] [Google Scholar]
- Kwok, J. C. F. , Dick, G. , Wang, D. , & Fawcett, J. W. (2011). Extracellular matrix and perineuronal nets in CNS repair. Developmental Neurobiology, 71(11), 1073–1089. 10.1002/dneu.20974 [DOI] [PubMed] [Google Scholar]
- Lau, L. W. , Cua, R. , Keough, M. B. , Haylock‐Jacobs, S. , & Yong, V. W. (2013). Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nature Reviews Neuroscience, 14(10), 722–729. 10.1038/nrn3550 [DOI] [PubMed] [Google Scholar]
- Li, H. , He, C. , Feng, J. , Zhang, Y. , Tang, Q. , Bian, Z. , … Huang, C . (2010). Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proceedings of the National Academy of Sciences of the United States of America, 107(31), 13818–13823. 10.1073/pnas.1008397107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Zhang, N. , Lin, H.‐Y. , Yu, Y. , Cai, Q.‐Y. , Ma, L. , & Ding, S. (2014). Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis‐induced ischemia in adult mice. BMC Neuroscience, 15(1), 58 10.1186/1471-2202-15-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovera, G. , Roth, S. , Plesnila, N. , Veltkamp, R. , & Liesz, A. (2014). Modeling stroke in mice: Permanent coagulation of the distal middle cerebral artery. Journal of Visualized Experiments, 89, e51729 10.3791/51729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makihara, N. , Arimura, K. , Ago, T. , Tachibana, M. , Nishimura, A. , Nakamura, K. , … Kitazono, T. (2014). Involvement of platelet‐derived growth factor receptor β in fibrosis through extracellular matrix protein production after ischemic stroke. Experimental Neurology, 264C, 127–134. 10.1016/j.expneurol.2014.12.007 [DOI] [PubMed] [Google Scholar]
- McKeon, R. J. , Schreiber, R. C. , Rudge, J. S. , & Silver, J. (1991). Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. Journal of Neuroscience, 11(11), 3398–3411. 10.1523/JNEUROSCI.11-11-03398.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundel, T. M. , & Kalluri, R. (2007). Type IV collagen‐derived angiogenesis inhibitors. Microvascular Research, 74(2–3), 85–89. 10.1016/j.mvr.2007.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls, V. , Schuchardt, E. , & Drenckhahn, D. (1994). The effect of fibroblasts, vascular smooth muscle cells, and pericytes on sprout formation of endothelial cells in a fibrin gel angiogenesis system. Microvascular Research, 48(3), 349–363. 10.1006/mvre.1994.1061 [DOI] [PubMed] [Google Scholar]
- Nisancioglu, M. H. , Mahoney, W. M. Jr , Kimmel, D. D. , Schwartz, S. M. , Betsholtz, C. , & Genové, G. (2008). Generation and characterization of rgs5 mutant mice. Molecular and Cellular Biology, 28(7), 2324–2331. 10.1128/MCB.01252-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özen, I. , Roth, M. , Barbariga, M. , Gaceb, A. , Deierborg, T. , Genové, G. , & Paul, G. (2018). Loss of regulator of G‐protein signaling 5 leads to neurovascular protection in stroke. Stroke, 49, 2182–2190. 10.1161/STROKEAHA.118.020124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, C. , Jardim, A. P. , Sisodiya, S. M. , Thom, M. , & Liu, J. Y. W. (2019). Spatiotemporal dynamics of PDGFRβ expression in pericytes and glial scar formation in penetrating brain injuries in adults. Neuropathology and Applied Neurobiology, 45(6), 609–627. 10.1111/nan.12539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner, O. , Tsimpas, A. , Kostin, S. , Valable, S. , Petit, E. , Schaper, W. , & Marti, H. H. (2003). Time‐ and cell type‐specific induction of platelet‐derived growth factor receptor‐beta during cerebral ischemia. Molecular Brain Research, 113(1–2), 44–51. [DOI] [PubMed] [Google Scholar]
- Riew, T.‐R. , Choi, J.‐H. , Kim, H. L. , Jin, X. , & Lee, M.‐Y. (2018). PDGFR‐β‐positive perivascular adventitial cells expressing nestin contribute to fibrotic scar formation in the striatum of 3‐NP intoxicated rats. Frontiers in Molecular Neuroscience, 11, 1–21. 10.3389/fnmol.2018.00402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell, A. , Cuadrado, E. , Ortega‐Aznar, A. , Hernández‐Guillamon, M. , Lo, E. H. , & Montaner, J. (2008). MMP‐9‐positive neutrophil infiltration is associated to blood‐brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke, 39(4), 1121–1126. 10.1161/STROKEAHA.107.500868 [DOI] [PubMed] [Google Scholar]
- Roth, M. , Gaceb, A. , Enström, A. , Padel, T. , Genové, G. , Özen, I. , & Paul, G. (2019). Regulator of G‐protein signaling 5 regulates the shift from perivascular to parenchymal pericytes in the chronic phase after stroke. FASEB Journal, 33, 1–9. 10.1096/fj.201900153R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup, C. , Ryu, J. K. , Helmrick, M. J. , Vagena, E. , Galanakis, D. K. , Degen, J. L. , … Akassoglou, K. (2010). Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF‐beta after vascular damage. Journal of Neuroscience, 30(17), 5843–5854. 10.1523/JNEUROSCI.0137-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Ishii, Y. , Xu, G. , Dang, T. C. , Hamashima, T. , Matsushima, T. , … Sasahara, M. (2012). PDGFR‐β as a positive regulator of tissue repair in a mouse model of focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism, 32(2), 353–367. 10.1038/jcbfm.2011.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt, M. , Engelhardt, B. , Pausch, F. , Hallmann, R. , Wendler, O. , & Sorokin, L. M. (2001). Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood‐brain barrier in experimental autoimmune encephalomyelitis. Journal of Cell Biology, 153(5), 933–946. 10.1083/jcb.153.5.933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderblom, C. , Luo, X. , Blumenthal, E. , Bray, E. , Lyapichev, K. , Ramos, J. , … Lee, J. K. (2013). Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. Journal of Neuroscience, 33(34), 13882–13887. 10.1523/JNEUROSCI.2524-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends in Neurosciences, 32(12), 638–647. 10.1016/j.tins.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nature Reviews Neuroscience, 16(5), 249–263. 10.1038/nrn3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers, L. , Kangwantas, K. , Nguyen, L. , Kielty, C. , & Pinteaux, E. (2010). Adhesion to the extracellular matrix is required for interleukin‐1 beta actions leading to reactive phenotype in rat astrocytes. Molecular and Cellular Neurosciences, 44(3), 272–281. 10.1016/j.mcn.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner, I. B. , Anderson, M. A. , Song, B. , Levine, J. , Fernandez, A. , Gray‐Thompson, Z. , … Sofroniew, M. V. (2013). Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3‐dependent mechanisms after spinal cord injury. Journal of Neuroscience, 33(31), 12870–12886. 10.1523/JNEUROSCI.2121-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehendner, C. M. , Sebastiani, A. , Hugonnet, A. , Bischoff, F. , Luhmann, H. J. , & Thal, S. C. (2015). Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Scientific Reports, 5, 13497 10.1038/srep13497 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. No fibrosis occurs in sham‐operated animals. (a) Confocal images of WT and KO mice 7 days after sham operation showing that sham operation does not induce changes PDGFRß+ cells, deposition of ECM or astrocyte reactivity. (b) Specificity of the secondary antibody was tested at 7 days after stroke, showing that secondary antibodies did not bind unspecifically in the infarct core. Pdclx: Podocalyxin, Scale bars: 20 μm.
Figure S2. Perivascular and parenchymal PDGFRß+. (a) Three‐dimensional confocal representation showing examples of perivascular (top) and parenchymal (bottom) PDGFRß+ cells (red). Perivascular PDGFRß+ cell (red) is in contract with a capillary (podocalyxin, red) and is covered by laminin (blue). Parenchymal PDGFRß+ cell is located in parenchyma without contact to capillaries and is not embedded in laminin. Images were taken in the infarct core of WT mice to allow to co‐staining of four markers. (b) Schematic representation of the location of perivascular and parenchymal PDGFRß+ cells in relation to a capillary and the vascular basement membrane. Scale bar 5 μm.
Figure S3. Type I collagen distribution within the infarct core at 7 days after stroke. (a) Confocal images of type I collagen (cyan) around vasculature (CD31, red) within the infract core of WT and RGS5‐KO mice. (b) Confocal images of PDGFRß (red), type I collagen (cyan), vasculature (podocalyxin, green), and nuclear marker DAPI (grey) in infarct core of WT mice. First row shows maximal projection of four z‐stacks, with individual stainings in second row. Third row shows orthogonal view with a single x‐stack. (′) parenchymal PDGFRß+/type I collagen- cell, (″) parenchymal PDGFRß+/type I collagen+ cell, (″′) perivascular PDGFRß+/type I collagen+ cell. Scale bar: 20 μm (in a) and 5 μm (in b).
Transparent Science Questionnaire for Authors
Transparent Peer Review Report
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.