

Abstract
Background and Aims
Targeting costimulatory receptors with agonistic antibodies is a promising cancer immunotherapy option. We aimed to investigate costimulatory receptor expression, particularly 4‐1BB (CD137 or tumor necrosis factor receptor superfamily member 9), on tumor‐infiltrating CD8+ T cells (CD8+ tumor‐infiltrating lymphocytes [TILs]) and its association with distinct T‐cell activation features among exhausted CD8+ TILs in hepatocellular carcinoma (HCC).
Approach and Results
Tumor tissues, adjacent nontumor tissues, and peripheral blood were collected from HCC patients undergoing surgical resection (n = 79). Lymphocytes were isolated and used for multicolor flow cytometry, RNA‐sequencing, and in vitro functional restoration assays. Among the examined costimulatory receptors, 4‐1BB was most prominently expressed on CD8+ TILs. 4‐1BB expression was almost exclusively detected on CD8+ T cells in the tumor—especially on programmed death 1 (PD‐1)high cells and not PD‐1int and PD‐1neg cells. Compared to PD‐1int and 4‐1BBnegPD‐1high CD8+ TILs, 4‐1BBposPD‐1high CD8+ TILs exhibited higher levels of tumor reactivity and T‐cell activation markers and significant enrichment for T‐cell activation gene signatures. Per‐patient analysis revealed positive correlations between percentages of 4‐1BBpos cells among CD8+ TILs and levels of parameters of tumor reactivity and T‐cell activation. Among highly exhausted PD‐1high CD8+ TILs, 4‐1BBpos cells harbored higher proportions of cells with proliferative and reinvigoration potential. Our 4‐1BB–related gene signature predicted survival outcomes of HCC patients in the The Cancer Genome Atlas cohort. 4‐1BB agonistic antibodies enhanced the function of CD8+ TILs and further enhanced the anti‐PD‐1–mediated reinvigoration of CD8+ TILs, especially in cases showing high levels of T‐cell activation.
Conclusion
4‐1BB expression on CD8+ TILs represents a distinct activation state among highly exhausted CD8+ T cells in HCC. 4‐1BB costimulation with agonistic antibodies may be a promising strategy for treating HCCs exhibiting prominent T‐cell activation.
Abbreviations
- CD8+ TILs
tumor‐infiltrating CD8+ T cells
- CTV
CellTrace Violet
- DEGs
differentially expressed genes
- DR3
death receptor 3
- FACS
fluorescence‐activated cell sorting
- GITR
glucocorticoid‐induced tumor necrosis factor receptor–related protein
- GSEA
gene set enrichment analysis
- GSVA
gene set variation analysis
- HCC
hepatocellular carcinoma
- ICI
immune checkpoint inhibitor
- IFN‐γ
interferon‐gamma
- IHL
intrahepatic lymphocyte
- HLA
human leukocyte antigen
- HVEM
herpesvirus entry mediator
- PBMC
peripheral blood mononuclear cell
- PD‐1
programmed cell death protein 1
- RNA‐seq
RNA‐sequencing
- SI
stimulation index
- TCF‐1
T‐cell factor 1
- TCGA
The Cancer Genome Atlas
- TCR
T‐cell receptor
- TIL
tumor‐infiltrating lymphocyte
- TME
tumor microenvironment
- TNF‐α
tumor necrosis factor alpha
- TNFR2
tumor necrosis factor receptor 2
- TNFRSF
tumor necrosis factor receptor superfamily member
Immune checkpoint inhibitors (ICIs) have revolutionized the treatment of various cancer types, and several agents targeting the programmed death 1 (PD‐1)/programmed death‐ligand 1 and cytotoxic T‐lymphocyte–associated protein 4 pathways are currently available for clinical use.1 Recent clinical trials of anti–PD‐1 therapy in patients with advanced hepatocellular carcinoma (HCC) show objective response rates of 16%‐20%,2, 3 prompting U.S. Food and Drug Administration approval of the anti–PD‐1 monoclonal antibodies, nivolumab and pembrolizumab, for use in HCC. However, the majority of HCC patients receiving anti–PD‐1 therapy still do not derive clinical benefit, highlighting the urgent need for immunotherapeutic strategies with improved therapeutic efficacy. To this end, research groups are investigating the use of various ICI‐based therapeutic strategies in combination with targeted agents, locoregional therapy, and other forms of immunotherapy.4
One promising therapeutic approach involves targeting costimulatory receptors, such as 4‐1BB, glucocorticoid‐induced tumor necrosis factor receptor–related protein (GITR), and OX‐40, with agonistic antibodies.1, 5, 6, 7 In addition to T‐cell receptor (TCR) signaling, costimulatory signaling is critical for full T‐cell activation and positively regulates T‐cell differentiation, effector function, survival, and memory formation.8, 9 Agonistic antibodies to costimulatory receptors may be used to potentiate these functional responses against tumors.1, 5, 6, 7 Among costimulatory receptors, 4‐1BB (tumor necrosis factor receptor superfamily member [TNFRSF] 9 or CD137) is considered one of the most compelling targets because of its capacity to activate exhausted T cells5, 10, 11, 12 and its potent antitumor efficacy shown in preclinical models.5, 11, 13, 14 Several clinical trials are evaluating the efficacy of 4‐1BB agonists combined with other immunotherapeutic strategies in multiple cancer types.5 However, little is known about the expression patterns of costimulatory receptors such as 4‐1BB on tumor‐infiltrating T cells or about the immunological and clinical implications of costimulatory receptor expression in HCC patients. Given the vital role of CD8+ T cells in eliciting antitumor functional responses15, 16, 17 and their substantial heterogeneity among HCCs,18, 19, 20 the rational development of therapies targeting costimulatory receptors will require investigation of the expression patterns of costimulatory receptors on tumor‐infiltrating CD8+ T cells (CD8+ tumor‐infiltrating lymphocytes [TILs]).
Many costimulatory receptors exhibit activation‐induced expression on T cells,8, 9 suggesting that their expression levels may represent the degree of T‐cell activation, and therapeutic costimulation conceptually targets T cells that have already been activated in the tumor microenvironment (TME). Therefore, delineation of the T‐cell activation features associated with costimulatory receptor expression will provide insights regarding how to maximize anti‐HCC T‐cell activation to improve the therapeutic efficacy of ICIs, as well as help identify additional targets involved in T‐cell activation in the TME. In particular, identification of a distinct T‐cell activation state among heterogeneously exhausted T cells could guide the development of T‐cell–activating approaches specifically targeting CD8+ TIL populations that have rigorously engaged in antitumor responses and subsequently acquired exhausted phenotypes. However, the heterogeneity of exhausted CD8+ TILs in the context of T‐cell activation in HCC remains largely unknown.
In this study, we aimed to comprehensively investigate the expression of costimulatory receptors on CD8+ TILs and its association with distinct features of T‐cell activation among exhausted CD8+ TILs in HCC. We focused on costimulatory receptor 4‐1BB because of its prominent expression on CD8+ TILs in HCC patients. We further examined the impact of 4‐1BB costimulation with agonistic antibodies on the functional responses of exhausted CD8+ TILs. Our results show that 4‐1BB expression identifies a distinctly activated population of exhausted CD8+ TILs and suggest that 4‐1BB costimulation may be a promising strategy for HCC patients with prominent T‐cell activation.
Materials and Methods
Study Patients and Clinical Samples
Clinical samples were obtained from a prospective cohort of 79 patients with pathologically confirmed HCC who underwent surgical resection at Asan Medical Center (Seoul, Korea) between April 2016 and April 2019. Fresh tumor tissue, adjacent nontumor liver tissue, and whole‐blood samples were collected. Table 1 summarizes the clinical characteristics of the study patients. We also obtained fresh tumor tissues from patients with other cancer types, and the corresponding data are summarized in Supporting Table S1. All included patients provided written informed consent, and this study was approved by the institutional review board.
Table 1.
Clinical Characteristics of the Study Patients With HCC
| Variable | n = 79 |
|---|---|
| Age (years) | 60.1 ± 9.9 |
| Male sex | 61 (77.9%) |
| Etiology | |
| HBV | 58 (73.4%) |
| HCV | 6 (7.6%) |
| NBNC | 15 (18.9%) |
| ALT (IU/mL) | 26 (18‐50) |
| INR | 1.06 ± 0.09 |
| Total bilirubin (mg/dL) | 0.6 ± 0.3 |
| Albumin (g/dL) | 3.8 ± 0.4 |
| AFP (ng/mL) | 9.8 (3.6‐231.0) |
| Liver cirrhosis | 26 (32.9%) |
| Microvascular invasion | 37 (46.8%) |
| Tumor diameter (cm) | 4.3 (3.1‐6.7) |
| Edmondson‐Steiner grade 3/4 (worst grade) | 58 (74.7%) |
Data are presented as no. (%) or mean ± SD or median (interquartile range).
Abbreviations: HBV, hepatitis B virus; HCV, hepatitis C virus; NBNC, non‐B non‐C; ALT, alanine aminotransferase; INR, international normalized ratio; AFP, alpha‐fetoprotein.
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by standard Ficoll‐Paque (GE Healthcare, Uppsala, Sweden) density gradient centrifugation. Intrahepatic lymphocytes (IHLs) and TILs were isolated from tissue samples as described.18 Briefly, tissue samples were cut into small pieces (2‐4 mm) and then transferred to a gentle MACS C‐Tube containing a mixture of enzymes (Milteny Biotec, Bergisch Gladbach, Germany). Next, samples were mechanically homogenized and enzymatically digested using the gentle MACS Octo Dissociator (Milteny Biotec). The resulting cell suspension was passed through a 70‐μm cell strainer, and then the cells were washed and cryopreserved.
Flow Cytometry and Immunophenotyping
Cells were stained using the LIVE/DEAD fixable red dead cell stain kit (Invitrogen, Carlsbad, CA) to gate out dead cells. Cells were washed once and then stained with fluorochrome‐conjugated antibodies against surface markers for 30 minutes at 4°C, followed by a second wash. For intracellular staining, cells were then fixed and permeabilized using a forkhead box protein P3 staining buffer kit (eBioscience, San Diego, CA) and then stained for intracellular proteins. Flow cytometry was performed using an LSR II instrument and FACSDiva software (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software (Treestar, San Carlos, CA).
RNA‐Sequencing and Data Analysis
RNA‐sequencing (RNA‐seq) was performed on PD‐1int, 4‐1BBnegPD‐1high, and 4‐1BBposPD‐1high CD8+ TILs (n = 5) that were sorted using a FACS Aria II cell sorter (BD Biosciences), as well as on total CD8+ TILs sorted using CD8 Microbeads (n = 10; Miltenyi Biotec). Each sorted cells sample had a purity of >90%. Sorted cells were cryopreserved in TRIzol reagent (Invitrogen), and RNA extraction was performed following the manufacturer's instructions.
Total RNA quality was assessed using the Agilent 2100 Bioanalyzer System. Extracted RNA samples were processed using the QuantSeq 3′ mRNA‐Seq Library Prep Kit (Lexogen, Vienna, Austria) and sequenced on an Illumina NextSeq 500. Read counts were normalized for effective library size, and differentially expressed genes (DEGs) were analyzed using DESeq2.21 DEGs were defined by a P value of <0.05 and an absolute fold change of >2. Based on P values, we determined the top 50 genes that were significantly up‐ or down‐regulated between 4‐1BBnegPD‐1high and 4‐1BBposPD‐1high CD8+ TILs. Gene set enrichment analysis (GSEA) was utilized to assess the enrichment of specific gene sets.22 For the RNA‐seq data, the Gene Expression Omnibus accession numbers are GSE132810 (for PD‐1int, 4‐1BBnegPD‐1high, and 4‐1BBposPD‐1high CD8+ TILs) and GSE132812 (for total CD8+ TILs).
T‐Cell Activation Gene Signatures from the Literature
The “Activation‐specific module” was directly obtained from the “ACT module” identified by Singer et al.23 The “CD8 in vivo activation” gene set was defined as the intersection of the DEGs between effector and naïve CD8+ T cells and of the DEGs between effector and memory CD8+ T cells in the article by Sankar et al.24 The “Effector and exhausted gene” set was defined as the sum of “cluster 2” and “cluster 6” as identified by Wherry et al.,25 which represents the genes commonly up‐regulated in effector and exhausted T cells. The “Acutely stimulated CD8+ T cell” gene set comprised DEGs that were up‐regulated in activated T cells compared to naïve T cells in the study by Giordano et al.26
The Cancer Genome Atlas Data Analysis
RNA‐seq data from the 370 HCC patients in The Cancer Genome Atlas (TCGA) were obtained from the GDC Data Portal (National Cancer Institute, Rockville, MD). Gene set variation analysis (GSVA) was performed using the R package, GSVA. Then, the enrichment score for the gene signatures was calculated by GSVA.27 Gene expression and survival data of an independent HCC cohort were obtained from GSE 7642728 and analyzed in the same manner. To conceptually compare the survival outcomes of the subcohorts, we used the enrichment score cutoff determined by the maximal chi‐square method using the R package, Maxstat, for each cohort.29
In Vitro Functional Assay
To represent costimulation under in vivo conditions, TILs were incubated at 37°C for 30 minutes with 10 μg/mL of agonistic antibodies for 4‐1BB (provided by ABL Bio, Seongnam, South Korea), GITR (79053‐2; BPS Bioscience, San Diego, CA), tumor necrosis factor receptor 2 (TNFR2; HM2007; Hycult Biotech, Uden, The Netherlands), or CD30 (81337; R&D Systems, Minneapolis, MN) or with the isotype controls, QA16A15 (Bio‐Legend), QA16A12 (Bio‐Legend), MOPC‐21 (Bio‐Legend), or 20116 (R&D Systems). Cells were then transferred to wells containing soluble anti‐CD3 antibody (1 ng/mL; OKT‐3; eBioscience) and either 10 μg/mL of blocking antibodies for PD‐1 (EH12.2H7; Bio‐Legend) or isotype control (MOPC‐21; Bio‐Legend). To measure CD8+ TIL proliferation, cells were labeled with CellTrace Violet (CTV; Invitrogen) and cultured for 72 hours. The mitotic index was calculated according to mitotic events, using the absolute number of precursor cells and the number of cells in each mitotic division. To standardize the baseline difference in proliferative capacity, we determined the stimulation index (SI) by dividing the mitotic index of the samples treated with anti–PD‐1 and/or anti–4‐1BB by that of the isotype control‐treated samples. We assessed interferon‐gamma (IFN‐γ) and tumor necrosis factor alpha (TNF‐α) production after 36 hours of culture. Brefeldin A and monensin (BD Biosciences) were added for the last 12 hours of incubation.
Results
4‐1BB is Prominently Expressed on CD8+ TILs, Especially on PD‐1high Cells
We first examined the expression levels of various activation‐induced costimulatory receptors of the TNFRSF9 on CD8+ TILs from HCC patients. Among the examined costimulatory receptors, 4‐1BB was more prominently expressed on CD8+ TILs compared to GITR, TNFR2, death receptor 3 (DR3), OX‐40, herpesvirus entry mediator (HVEM), and CD30 (Fig. 1A). To further delineate HCC‐specific aspects of costimulatory receptor expression, we also analyzed costimulatory receptor expression on CD8+ TILs from other cancer types (Supporting Table S1). Multicancer analysis of 4‐1BB expression revealed that the percentage of 4‐1BBpos CD8+ TILs was higher in HCC than in the other examined cancer types, although the difference between HCC and ovarian cancer was not statistically significant (Fig. 1B). In contrast, the percentages of CD8+ TILs expressing other costimulatory receptors were not higher, or were even lower, in HCC compared to in other cancer types (Supporting Fig. S1). Furthermore, among the examined TNFRSF costimulatory receptors, 4‐1BB was not the one most highly expressed in other examined cancer types (Supporting Fig. S2). These data indicate that prominent 4‐1BB expression on CD8+ TILs may be a notable feature of HCC; thus, we focused on 4‐1BB expression in our further investigations of the activation‐related features of CD8+ TILs and of its potential as an immunotherapeutic target for HCC patients.
Figure 1.
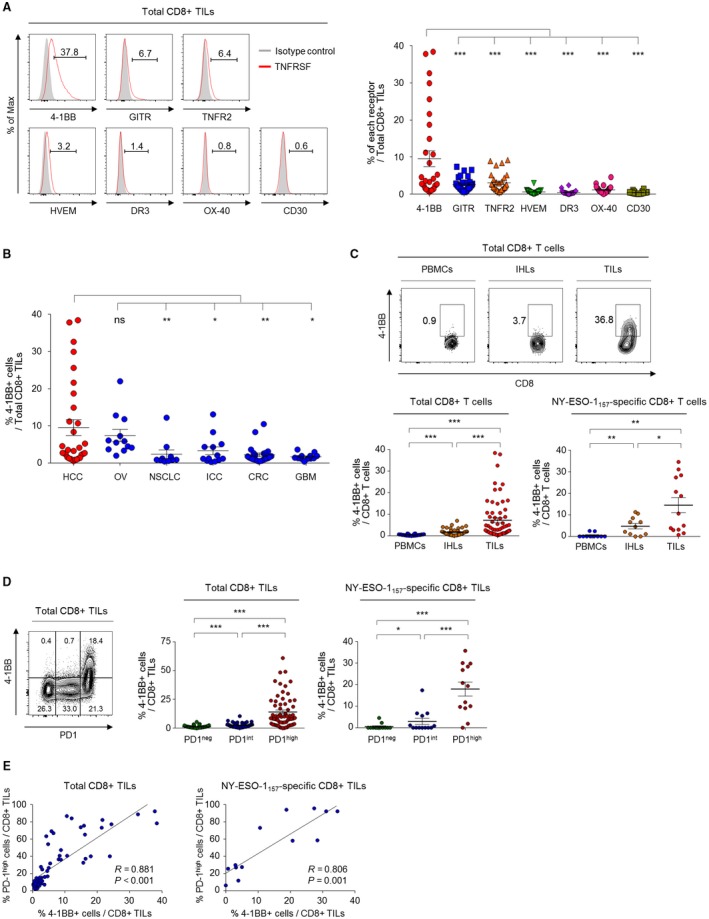
Compared to other costimulatory receptors, 4‐1BB is more prominently expressed on CD8+ TILs, especially on PD‐1high cells. (A) The percentage of CD8+ TILs that expressed various activation‐induced costimulatory receptors belonging to the TNFRSF. Left panel shows representative flow cytometry histograms. (B) Percentages of 4‐1BBpos CD8+ TILs in various cancer types: OV, ovarian cancer; NSCLC, non‐small‐cell lung cancer; ICC, intrahepatic cholangiocellular carcinoma; CRC, colorectal cancer; and GBM, glioblastoma multiforme. (C) 4‐1BB expression pattern across different CD8+ T‐cell fractions. The percentage of 4‐1BBpos cells was compared among the different T‐cell fractions of total and NY‐ESO‐1157‐165–specific CD8+ T cells. Left panel shows representative flow cytometry plots. (D) 4‐1BB expression according to differential PD‐1 expression levels. The percentage of 4‐1BBpos cells was compared among PD‐1high, PD‐1int and PD‐1neg subpopulations of total and NY‐ESO‐1157‐165–specific CD8+ TILs. (E) Correlations between the percentage of 4‐1BBpos CD8+ TILs and PD‐1high CD8+ TILs among the total CD8+ TILs and NY‐ESO‐1157–specific CD8+ TILs. Upper panel shows representative flow cytometry plots. Data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: ns, not significant.
Next, we examined 4‐1BB expression across different cellular fractions (i.e., PBMCs, IHLs, and TILs) and immunophenotypes of tumor‐antigen–specific CD8+ T cells using a human leukocyte antigen (HLA)‐A*0201 dextramer specific to NY‐ESO‐1157‐165. Total CD8+ T cells as well as NY‐ESO‐1157‐165–specific CD8+ T cells in the TIL fraction included significantly higher percentages of 4‐1BBpos cells compared to the IHL and PBMC fractions (Fig. 1C). The percentage of 4‐1BBpos cells among CD8+ TILs did not differ among HCCs with different etiologies (Supporting Fig. S3).
We recently demonstrated that PD‐1+ CD8+ TILs from HCC patients can be subdivided into PD‐1int and PD‐1high CD8+ TILs and that the latter group shows features of more‐severe T‐cell exhaustion.18 Another recent study found that PD‐1high CD8+ TILs exhibit high tumor reactivity.30 Therefore, we next examined 4‐1BB expression on CD8+ TILs according to differential PD‐1 expression, to investigate the role of 4‐1BB in the context of T‐cell exhaustion and tumor reactivity. Interestingly, 4‐1BB was almost exclusively expressed on PD‐1high CD8+ TILs (Fig. 1D). Most PD‐1int and PD‐1neg CD8+ TILs showed no 4‐1BB expression (Fig. 1D). A similar pattern was observed for NY‐ESO‐1157–specific CD8+ TILs (Fig. 1C, right panel). Frequencies of 4‐1BBpos cells among total and NY‐ESO‐1157–specific CD8+ TILs were positively correlated with frequencies of PD‐1high total and NY‐ESO‐1157–specific CD8+ TILs (Fig. 1E).
4‐1BB–Expressing PD‐1high CD8+ TILs Feature Immunophenotypically and Transcriptionally Prominent T‐Cell Activation
Exhausted CD8+ T cells are considered a primary target for therapeutic interventions involving ICIs.15, 16, 17, 30 Therefore, the delineation of distinct features of exhausted CD8+ TILs based on T‐cell activation status could help direct therapeutic strategies to maximize CD8+ TIL activation and assess the degree of preexisting T‐cell activation that can be targeted by immunotherapy. To further examine the immunological role of 4‐1BB as a T‐cell–activating receptor in phenotypically exhausted CD8+ TILs, we next investigated parameters of tumor reactivity (i.e., CD39 and CD103) and T‐cell activation (i.e., CD38 and HLA‐DR) among three distinct subpopulations of PD‐1+ CD8+ TILs determined by differential 4‐1BB and PD‐1 expression levels: PD‐1int, 4‐1BBnegPD‐1high, and 4‐1BBposPD‐1high subpopulations (Fig. 2A).
Figure 2.
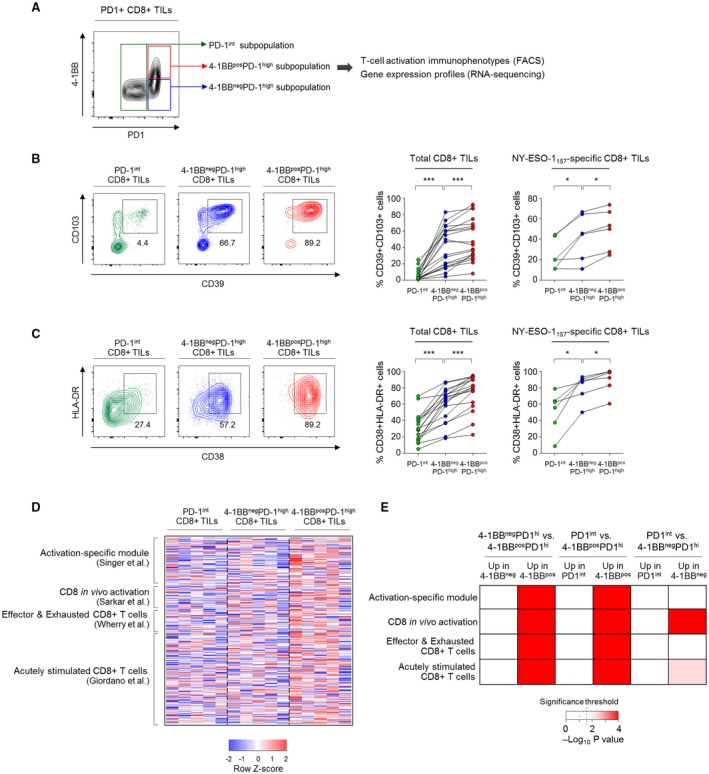
4‐1BB–expressing PD‐1high CD8+ TILs feature immunophenotypically and transcriptionally prominent T‐cell activation. (A) Representative flow cytometry plot showing subpopulations of PD‐1+ CD8+ TILs according to differential expression of PD‐1 and 4‐1BB. (B,C) Percentages of CD39+CD103+ cells (B) and CD38+HLA‐DR+ cells (C) among the PD‐1int, 4‐1BBnegPD‐1high, and 4‐1BBposPD‐1high subpopulations of total and NY‐ESO‐1157–specific CD8+ TILs. Left panels show representative flow cytometry plots (B,C). (D,E) RNA‐seq data comparing expression of genes related to T‐cell activation among FACS‐sorted PD‐1int, 4‐1BBnegPD‐1high and 4‐1BBposPD‐1high CD8+ TILs. (D) Heatmap showing expression levels of T‐cell activation gene signatures among the PD‐1int, 4‐1BBnegPD‐1high, and 4‐1BBposPD‐1high subpopulations. (E) Relative enrichment of T‐cell activation gene signatures among PD‐1int, 4‐1BBnegPD‐1high, and 4‐1BBposPD‐1high CD8+ TILs. Dashed line marks the significance threshold (P = 0.05). *P < 0.05; ***P < 0.001.
We first examined the proportion of a CD39+CD103+ CD8+ TIL subset representing tumor reactivity.31 The 4‐1BBposPD‐1high subpopulation showed the highest percentages of CD39+CD103+ cells among both total and NY‐ESO‐1157‐165–specific CD8+ TILs, followed by the 4‐1BBnegPD‐1high and PD‐1int subpopulations (Fig. 2B). The percentage of activated T cells (CD38+HLA‐DR+ cells) was higher among 4‐1BBposPD‐1high CD8+ TILs than in 4‐1BBnegPD‐1high CD8+ TILs, with the lowest frequency among PD‐1int CD8+ TILs (Fig. 2C). Additionally, expression levels of the TCR‐responsive transcription factors, interferon regulatory factor 4 (IRF4), basic leucine zipper transcription factor ATF‐like (BATF) and nuclear factor of activated T cells c1 (NFATc1), were higher among 4‐1BBposPD‐1high CD8+ TILs than among 4‐1BBnegPD‐1high and PD‐1int CD8+ TILs (Supporting Fig. S4).
To confirm the distinct activation‐related features in 4‐1BB–expressing CD8+ TILs, we performed RNA‐seq of fluorescence‐activated cell sorting (FACS)‐sorted PD‐1int, 4‐1BBnegPD‐1high, and 4‐1BBposPD‐1high CD8+ TILs. These subpopulations were compared with regard to the expression levels of four gene signatures that have been shown to represent T‐cell activation23, 24, 25, 26 (Fig. 2D). Notably, 4‐1BBposPD‐1high CD8+ TILs showed substantial enrichment of all four T‐cell activation gene signatures compared to PD‐1int and 4‐1BBnegPD‐1high CD8+ TILs (Fig. 2E). Only two of four gene sets were enriched in the 4‐1BBnegPD‐1high subpopulation compared to the PD‐1int subpopulation (Fig. 2E).
Overall, these results suggest that 4‐1BBposPD‐1high CD8+ TILs display immunophenotypes indicating higher degrees of T‐cell activation and tumor reactivity, as well as distinct enrichment of genes related to T‐cell activation. This highlights the heterogeneous activation status of exhausted CD8+ TILs according to 4‐1BB expression.
Percentage of 4‐1BBpos CD8+ TILs is Positively Correlated with Degree of CD8+ TIL Activation
To assess the implications of 4‐1BB expression on CD8+ TILs on a per‐patient basis, we examined the relationship between the percentage of 4‐1BBpos CD8+ TILs and activation‐related features of CD8+ TILs. The percentage of 4‐1BBpos cells was positively correlated with the percentages of CD39+CD103+ cells and CD38+HLA‐DR+ cells among total CD8+ TILs (Fig. 3A, upper panels), as well as among NY‐ESO‐1157‐165–specific CD8+ TILs (Fig. 3A, lower panels). On the other hand, the prominent correlations between the percentage of 4‐1BBpos CD8+ TILs and the percentages of CD39+CD103+ and CD38+HLA‐DR+ cells in HCC were not universally observed across other cancer types (Supporting Fig. S5), possibly attributed to the lower percentages of 4‐1BBpos CD8+ TILs in other cancer types.
Figure 3.
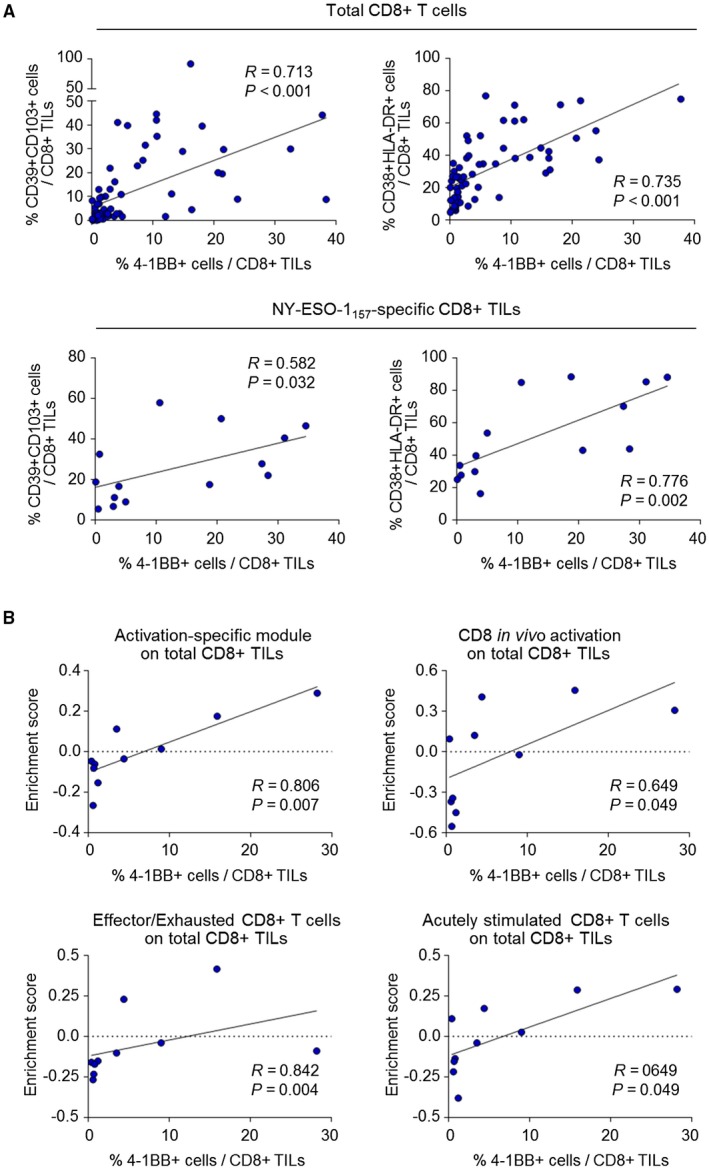
The percentage of 4‐1BBposCD8+ TILs is positively correlated with the degree of overall activation of CD8+ TILs. (A) Correlations between the percentage of 4‐1BBpos CD8+ TILs and the percentages of CD39+CD103+ cells and CD38+HLA‐DR+ cells, among total CD8+ TILs (left panel) and NY‐ESO‐1157‐165–specific CD8+ TILs (right panel). (B) RNA‐seq data from sorted total CD8+ TILs, analyzing the expression levels of T‐cell activation gene signatures, represented as the enrichment score obtained by GSVA. Correlations between the percentage of 4‐1BBpos CD8+ TILs and enrichment scores of T‐cell activation gene signatures were subsequently analyzed.
RNA‐seq data from sorted total CD8+ TILs revealed that expression levels of T‐cell activation gene signatures were positively correlated with the percentage of 4‐1BBpos cells among CD8+ TILs (Fig. 3B). This suggests that the frequency of 4‐1BBpos CD8+ TILs indicate the degree of overall T‐cell activation in the HCC microenvironment.
4‐1BBposPD‐1high CD8+ TILs Display Higher Levels of Parameters Indicating T‐Cell Proliferation and Reinvigoration than 4‐1BBnegPD‐1high CD8+ TILs
Next, we further investigated the immunological implications of 4‐1BB expression specifically in highly exhausted CD8+ T cells in the HCC microenvironment. To this end, we directly compared 4‐1BBpos and 4‐1BBneg cells among PD‐1high CD8+ TILs with regard to differential gene expression and factors related to T‐cell reinvigoration potential.
Examination of DEGs revealed that a total of 205 genes were up‐regulated and 364 genes were down‐regulated in 4‐1BBposPD‐1high CD8+ TILs compared to 4‐1BBnegPD‐1high CD8+ TILs (Fig. 4A). The genes that were up‐regulated in 4‐1BBposPD‐1high CD8+ TILs compared to 4‐1BBnegPD‐1high CD8+ TILs included several genes encoding other costimulatory receptors (i.e., TNFRSF8, TNFRSF4, TNFRSF18, and class I MHC‐restricted T‐cell–associated molecule gene [CRTAM]; Fig. 4A). Gene ontology term analysis indicated enrichment of cell‐cycle pathway genes among those that were up‐regulated in 4‐1BBposPD‐1high CD8+ TILs compared to 4‐1BBnegPD‐1high CD8+ TILs (Fig. 4B). This was further supported by the higher percentage of cells positive for the proliferation marker, Ki‐67, within the 4‐1BBposPD‐1high subpopulation compared to the 4‐1BBnegPD‐1high subpopulation, among both total and NY‐ESO‐1157‐165–specific CD8+ TILs (Fig. 4C and Supporting Fig. S6A). In contrast, genes related to cell‐death pathways were down‐regulated in 4‐1BBposPD‐1high CD8+ TILs compared to 4‐1BBnegPD‐1high CD8+ TILs (Supporting Fig. S6B).
Figure 4.
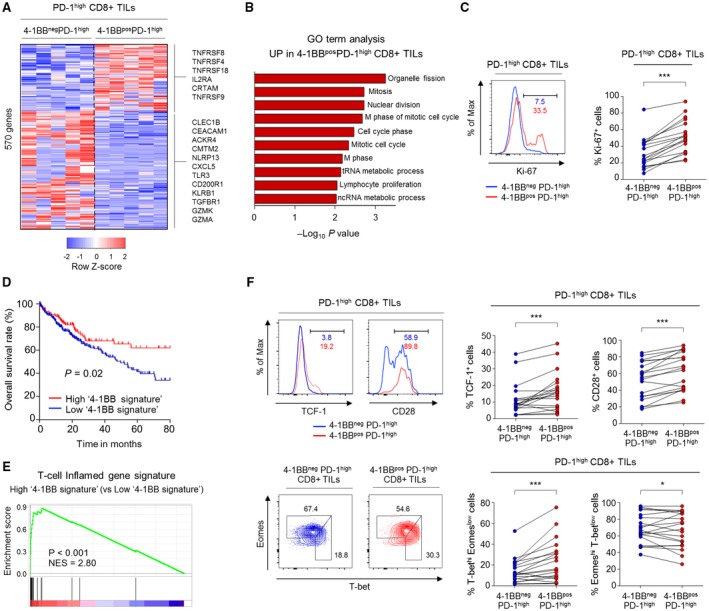
4‐1BBposPD‐1high CD8+ TILs exhibited higher levels of markers indicating T‐cell proliferation and reinvigoration compared to 4‐1BBnegPD‐1high CD8+ TILs. (A) Heatmap showing expression levels of 569 DEGs between 4‐1BBnegPD‐1high and 4‐1BBposPD‐1high CD8+ TILs. (B) Gene Ontology analysis for the DEGs that were up‐regulated in 4‐1BBposPD‐1high CD8+ TILs compared to in 4‐1BBnegPD‐1high CD8+ TILs. (C) Percentage of Ki‐67+ cells compared between 4‐1BBposPD‐1high and 4‐1BBnegPD‐1high CD8+ TILs. Left panel shows representative flow cytometry plot. (D) Kaplan‐Meier curve comparing survival outcomes among subgroups (i.e., high and low 4‐1BB signature groups) of HCC patients in the TCGA cohort, according to expression of the 4‐1BB signature. Cutoff was determined by the maximal chi‐square method. (E) GSEA was performed to compare the enrichment of a T‐cell–inflamed gene signature between the high and low 4‐1BB signature groups. (F) Expression levels of parameters involved in T‐cell reinvigoration (i.e., TCF‐1, CD28, and T‐bet/Eomes) were compared between 4‐1BBposPD‐1high and 4‐1BBnegPD‐1high CD8+ TILs. Left panel shows representative flow cytometry plots. *P < 0.05; ***P < 0.001. Abbreviations: ACKR4, atypical chemokine receptor 4; CEACAM1, carcinoembryonic antigen‐related cell adhesion molecule 1; CLEC1B, C‐type lectin domain family 1 member B; CMTM2, CKLF‐like MARVEL transmembrane domain‐containing 2; CXCL5, C‐X‐C motif chemokine ligand 5; Eomes, eomesodermin; GO, Gene Ontology; GZMA, granzyme 1, cytotoxic T‐lymphocyte–associated serine esterase 3; GZMK, granzyme K (serine protease, granzyme 3; tryptase II); IL2RA, interleukin‐2 receptor agonist; KLRB1, killer cell lectin‐like receptor B1; Max, maximum; ncRNA, noncoding RNA; NES, normalized enrichment score; NLRP13, NOD‐like receptor family pyrin domain containing 13; T‐bet, T‐box–containing protein expressed in T cells; TGFBR1, transforming growth factor beta receptor 1; tRNA, total RNA.
The top 50 genes that were significantly up‐ or down‐regulated in 4‐1BBposPD‐1high CD8+ TILs compared to 4‐1BBnegPD‐1high CD8+ TILs are summarized in Supporting Table S2. The top 50 genes up‐regulated in 4‐1BBposPD‐1high CD8+ TILs were defined as the “4‐1BB signature.” Mean level of 4‐1BB signature expression, as indicated by enrichment score, was highest in HCC compared to the other cancer types in the TCGA cohorts (Supporting Fig. S7A). We next examined the clinical implications of this 4‐1BB signature using data from the TCGA HCC cohort and found that tumors with high 4‐1BB signature were associated with better survival outcomes compared to those with low 4‐1BB signature (Fig. 4D). Moreover, compared to HCCs with low levels of 4‐1BB signature, those with high levels of 4‐1BB signature exhibited significant enrichment for a T‐cell–inflamed gene module representing active IFN‐γ response, cytotoxic effector function, and T‐cell–activating cytokines that is reportedly associated with response to anti–PD‐1 therapy32, 33 (Fig. 4E). In an independent HCC cohort, we also identified superior survival outcomes and enrichment of the T‐cell inflamed gene signature among the HCCs exhibiting high 4‐1BB signature expression (Supporting Fig. S7B,C).
We further examined several parameters associated with T‐cell reinvigoration potential by ICIs, including T‐cell factor 1 (TCF‐1),34, 35 CD28,36 and T‐bet/Eomes.37, 38 Compared to 4‐1BBnegPD‐1high CD8+ TILs, 4‐1BBposPD‐1high CD8+ TILs had higher percentages of TCF‐1+, CD28+, and T‐bethigh/Eomeslow cells, which represent subpopulations potentially reinvigorated by ICIs (Fig. 4F and Supporting Fig. S8). On the other hand, the proportion of terminally differentiated Eomeshigh/T‐betlow cells was lower in 4‐1BBposPD‐1high CD8+ TILs compared to 4‐1BBnegPD‐1high CD8+ TILs (Fig. 4F and Supporting Fig. S8).
Collectively, our results indicate that the 4‐1BB–related gene expression profile was associated with active antitumor response and that the subpopulation of 4‐1BB–expressing CD8+ TILs may retain higher proliferative and reinvigoration potential among highly exhausted CD8+ TILs.
4‐1BB Costimulation Further Enhances Function of CD8+ TILs and Anti‐PD‐1–Mediated CD8+ TIL Reinvigoration
Finally, we investigated whether 4‐1BB costimulation with agonistic antibodies against 4‐1BB could enhance the function of CD8+ TILs. In vitro functional assays revealed that 4‐1BB costimulation significantly enhanced CD8+ TIL proliferation, as indicated by SI (Fig. 5A) as well as IFN‐γ and TNF‐α production by CD8+ TILs (Supporting Fig. S9). To further characterize CD8+ TILs that favorably responded to anti–4‐1BB treatment, we subdivided CD8+ TIL samples into two subgroups based on their degree of functional response to anti–4‐1BB treatment, using median SI value as the cutoff (≥ the median SI vs. < the median SI). In accord with the proliferation assay results, compared to CD8+ TILs with lower SI values, CD8+ TILs with higher SI values exhibited greater relative increases of IFN‐γ and TNF‐α production by CD8+ TILs with anti–4‐1BB treatment (Fig. 5B). More important, compared to those with lower SI values, CD8+ TILs with higher SI values exhibited significantly larger percentages of 4‐1BB+, CD39+CD103+, CD38+HLA‐DR+, and Ki‐67+ cells (Fig. 5C). We also identified a robust correlation between functional anti–4‐1BB responses and percentage of 4‐1BBpos CD8+ TILs (Fig. 5D). Area under the curve analysis revealed that the percentage of 4‐1BBpos CD8+ TILs reliably distinguished the subgroup with a higher SI value by anti–4‐1BB treatment (i.e., ≥median SI; Fig. 5E).
Figure 5.
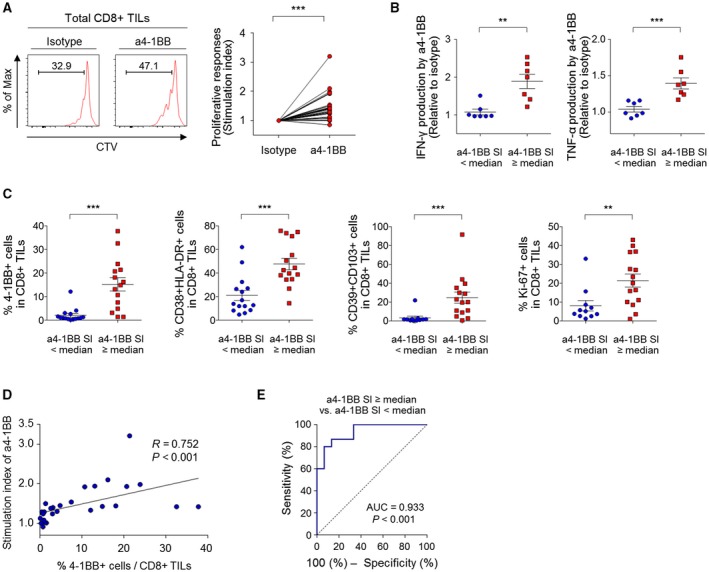
4‐1BB costimulation enhances the function of CD8+ TILs, especially in HCCs exhibiting prominent T‐cell activation. (A) Investigation of the effect of 4‐1BB costimulation on the proliferative response of CD8+ TILs, as indicated by SI. (B,C,E) Study patients were subdivided into two subgroups using the median SI by anti–4‐1BB as the cutoff. (B) Comparison between the two subgroups of the relative increases of IFN‐γ and TNF‐α production by CD8+ TILs with 4‐1BB costimulation. (C) Comparison between the two subgroups of the percentages of 4‐1BB+, CD39+CD103+, CD38+HLA‐DR+, and Ki‐67+ cells among CD8+ TILs. (D) Correlation between the percentage of 4‐1BBpos CD8+ TILs and the SI by 4‐1BB costimulation. (E) Receiver operating characteristic curve estimating the performance of the percentages of 4‐1BBpos CD8+ TILs with regard to distinguishing the two subgroups. Data are presented as mean ± SEM. (B,C) ** P < 0.01; ***P < 0.001. Abbreviation: Max, maximum.
Recent clinical trials of anti–PD‐1 therapy in HCC patients suggest a need to improve its therapeutic efficacy.2, 3 Thus, we next examined whether 4‐1BB costimulation could further enhance anti‐PD‐1–mediated T‐cell reinvigoration. While single PD‐1 blockade restored the abilities of CD8+ TILs to proliferate and produce IFN‐γ and TNF‐α, the combination of 4‐1BB costimulation and PD‐1 blockade further enhanced the abilities of CD8+ TILs to proliferate (Fig. 6A) and produce IFN‐γ and TNF‐α (Fig. 6B). Again, the percentage of 4‐1BBpos CD8+ TILs was positively correlated with the SI of combined 4‐1BB costimulation and PD‐1 blockade (Supporting Fig. S10A) and predicted the patient subgroup that exhibited more‐pronounced functional CD8+ TIL responses to this combined treatment (i.e., ≥median SI by anti–4‐1BB plus anti–PD‐1; Supporting Fig. S10B).
Figure 6.

4‐1BB costimulation further enhances anti‐PD‐1–mediated CD8+ TIL reinvigoration. (A,B) Efficacy of the combination of 4‐1BB costimulation and PD‐1 blockade compared to the efficacy of PD‐1 blockade or isotype control in terms of proliferative response (A) and cytokine production (B). (C) Comparison of the proliferative functional response of CD8+ TILs with various combinations of PD‐1 blockade and costimulatory agonists (i.e., anti–4‐1BB, anti‐GITR, anti‐TNFR2, and anti‐CD30). (D) The percentage of 4‐1BBpos cells was compared between samples treated with PD‐1 blocking antibodies versus isotype control upon anti‐CD3 stimulation (10 ng/mL) for 48 hours. In (A) and (C), data are presented as the SI of CD8+ TILs. In (B), data are presented as the relative ratio of the percentage of CD8+ TILs that produce IFN‐γ and TNF‐α by combined 4‐1BB costimulation and PD‐1 blockade or PD‐1 blockade alone, compared to that by isotype control. **P < 0.01; ***P < 0.001. Abbreviation: Max, maximum.
We then compared the efficacies of various combinations of PD‐1 blockade and costimulatory agonists, revealing that the combination of anti–4‐1BB plus anti–PD‐1 led to more pronounced functional CD8+ TIL responses compared to other combinations (i.e., anti–PD‐1 plus anti‐GITR, anti‐TNFR2, or anti‐CD30; Fig. 6C). PD‐1 blockade‐mediated T‐cell reinvigoration was accompanied by up‐regulation of 4‐1BB on CD8+ TILs (Fig. 6D), indicating that PD‐1 blockade may have beneficial effects on CD8+ TILs as part of immunotherapies targeting 4‐1BB.
Discussion
The therapeutic potential of targeting costimulatory receptors is currently under early clinical evaluation; however, much remains unknown about the biological and clinical implications of these receptors, which are essential for the rational design of immunotherapies. In this study, we investigated costimulatory receptor expression patterns on CD8+ TILs as well as the heterogeneity of T‐cell activation among exhausted CD8+ TILs in HCC. Compared to other examined activation‐induced costimulatory receptors, we found that 4‐1BB was most prominently expressed on CD8+ TILs, with expression almost exclusively on highly exhausted PD‐1high CD8+ TILs. 4‐1BBposPD‐1high CD8+ TILs displayed immunophenotypic and transcriptomic features of prominent T‐cell activation and higher levels of tumor reactivity markers. Among PD‐1high CD8+ TILs, 4‐1BB–expressing cells exhibited higher levels of proliferation and reinvigoration of potential markers. Finally, we showed that 4‐1BB costimulation with agonistic antibodies enhanced CD8+ TIL function and further enhanced anti‐PD‐1–mediated CD8+ TIL reinvigoration, especially in HCCs exhibiting prominent T‐cell activation.
This was a study to comprehensively characterize costimulatory receptor expression on CD8+ TILs and the associated T‐cell activation features in HCC patients. Previous studies describe the expression of 4‐1BB and other costimulatory receptors on CD8+ T cells, but earlier investigations have been limited in that they either used CD8+ T cells from peripheral blood39 or simply confirmed costimulatory receptor expression on tumor‐infiltrating T cells in HCC.19, 20 In HCC, 4‐1BB expression was most prominent among the tested activation‐induced costimulatory receptors in CD8+ TILs. However, this was not the case for the other examined cancer types. Additionally, compared to the other cancer types tested, HCC had a higher percentage of 4‐1BBpos CD8+ TILs, but had similar or even lower percentages of CD8+ TILs expressing other costimulatory receptors. Thus, the substantial differences in these features among various cancer types necessitate the detailed delineation of 4‐1BB–related T‐cell activation in HCC. Together with high levels of the 4‐1BB–related gene signature in HCC, these highlight the prominent 4‐1BB expression on CD8+ TILs and distinct 4‐1BB–related features as notable aspects of HCC that are not universally observed in other cancer types.
Another interesting finding is that 4‐1BB was almost exclusively expressed on PD‐1high CD8+ TILs, which reportedly represent highly exhausted and tumor‐reactive CD8+ TILs.18, 30 We recently demonstrated that PD‐1high CD8+ TILs exhibit features of more progressed T‐cell exhaustion compared to PD‐1int and PD‐1neg CD8+ TILs and that HCCs with higher proportions of PD‐1high CD8+ TILs are enriched for the T‐cell–inflamed gene signature.18 Similarly, Thommen et al. examined patients with non‐small‐cell lung cancer displaying typical phenotypic features of T‐cell exhaustion and reported that PD‐1high CD8+ TILs were associated with higher tumor recognition capacity and response to anti–PD‐1 therapy.30 These findings suggest that PD‐1high CD8+ TILs indicate active engagement against tumors and resultant exhausted phenotypes. Thus, expression of 4‐1BB on PD‐1high CD8+ TILs suggests that 4‐1BB may deliver costimulatory signals specifically to highly exhausted CD8+ TILs that have rigorously engaged in antitumor responses.
T‐cell exhaustion arises because of excessive and prolonged T‐cell activation to prevent immunopathology; therefore, these two states are closely interconnected and share features related to the cell‐cycle pathway, T‐cell migration, and cytotoxic effector function.23, 25, 26, 40, 41 Indeed, recent single‐cell transcriptomic analyses indicate that a proportion of CD8+ TILs that were previously identified as exhausted are indeed highly proliferating and undergoing a dynamic differentiation process.42, 43 This highlights the importance of distinguishing the subset of exhausted T cells in a viable T‐cell activation state. Compared to PD‐1int and 4‐1BBnegPD‐1high CD8+ TILs, we found that 4‐1BBposPD‐1high CD8+ TILs showed the highest levels of T‐cell activation markers and TCR‐responsive transcription factors as well as significant enrichment for T‐cell‐activation–related gene signatures. Importantly, a T‐cell activation gene signature that is uncoupled from dysfunction23 was specifically enriched in the 4‐1BBposPD‐1high subpopulation, but not in the 4‐1BBnegPD‐1high and PD‐1int subpopulations. 4‐1BBposPD‐1high CD8+ TILs also displayed the highest proportion of the tumor‐reactive CD39+CD103+ subset, indicating that the prominent T‐cell activation likely resulted from increased tumor reactivity of 4‐1BBpos cells. On the other hand, the percentage of 4‐1BBpos CD8+ TILs was positively correlated with the parameters of T‐cell activation analyzed on an individual basis, indicating that 4‐1BB may be a useful indicator of the overall activation status of exhausted CD8+ TILs.
Direct comparison between 4‐1BBpos and 4‐1BBneg cells among highly exhausted PD‐1high CD8+ TILs revealed substantial differences in the gene expression profiles and in parameters of T‐cell proliferation and reinvigoration. Indeed, genes up‐regulated in 4‐1BBposPD‐1high CD8+ TILs were associated with the cell‐cycle pathway and these cells displayed higher percentages of proliferation markers. Additionally, among PD‐1high CD8+ TILs, 4‐1BBpos cells retained a higher proportion of cells involved in T‐cell reinvigoration than 4‐1BBneg cells. Whereas 4‐1BB expression was previously shown to represent the tumor reactivity of CD8+ TILs,44 our study highlights immunological implications of 4‐1BB expression in exhausted CD8+ TILs: (1) The T‐cell‐activating receptor, 4‐1BB, is almost exclusively expressed on highly exhausted PD‐1high CD8+ TILs; (2) 4‐1BBpos CD8+ TILs harbor the highest degree of T‐cell activation, as well as proliferative and reinvigoration potential among highly exhausted CD8+ TILs; and (3) 4‐1BB expression represents the overall activation status of CD8+ TILs. Together, our findings indicate that 4‐1BB expression identifies a highly activated and immunologically viable population of phenotypically exhausted CD8+ TILs in HCC.
Another strength of our study is the identification of a T‐cell activation gene set that could help find targets to maximize T‐cell activation signals in HCC microenvironments. The 4‐1BB gene signature, derived from differential 4‐1BB expression among PD‐1high CD8+ TILs, was validated as predicting survival outcomes of patients in the TCGA cohort and in an independent HCC cohort. This suggests that the higher degree of T‐cell activation associated with 4‐1BB expression may reflect a robust antitumor response that leads to improved survival outcomes in HCC patients. Furthermore, HCCs with higher 4‐1BB signature expression showed enrichment of the T‐cell–inflamed gene signature, suggesting that 4‐1BB–related T‐cell activation directs active IFN‐γ and cytotoxic antitumor T‐cell responses, which are also associated with anti–PD‐1 response.32, 33 This gene set will be an important resource for mining additional key genes and pathways involved in T‐cell activation against HCC. It will also be interesting to validate whether this gene signature may be useful as a predictive biomarker for ICIs.
Using the in vitro functional assay system, we provide a demonstration that 4‐1BB costimulation promoted the functional enhancement of CD8+ TILs from HCC patients. Importantly, CD8+ TILs exhibiting higher degrees of T‐cell activation, tumor reactivity, and proliferation were associated with a better functional response to anti–4‐1BB treatment compared to those with low levels of preexisting T‐cell activation. These findings suggest that anti–4‐1BB therapy may be more effective in HCC patients exhibiting prominent T‐cell activation, which is linked to higher 4‐1BB expression on CD8+ TILs. Moreover, 4‐1BB expression reliably distinguished cases with a favorable CD8+ TIL response to anti–4‐1BB treatment. Therefore, 4‐1BB expression on CD8+ TILs may be useful for guiding the selection of patients who are likely to favorably respond to anti–4‐1BB treatment. These insights support the potential development of a clinical‐grade biomarker based on 4‐1BB expression such as immunohistochemical staining of 4‐1BB for HCC patients receiving anti–4‐1BB therapy.
We also found that 4‐1BB costimulation could further enhance PD‐1 blockade‐mediated T‐cell reinvigoration. The objective response rates of anti–PD‐1 therapy are reportedly around 20% in HCC patients2, 3; thus, there is an urgent need to improve the therapeutic efficacy of immunotherapy involving anti–PD‐1 therapy. Our findings indicate that 4‐1BB costimulation may be a potent therapeutic option in conjunction with anti–PD‐1 therapy. Our finding that PD‐1 blockade‐mediated T‐cell reinvigoration was accompanied by increased 4‐1BB expression implies that the combination of 4‐1BB costimulation and PD‐1 blockade can act synergistically. Together, our results provide insights that may guide the design of immunotherapy involving 4‐1BB costimulation. Although an issue has been raised regarding the hepatotoxicity of the 4‐1BB agonist, urelumab, it is generally considered that this problem can be overcome with urelumab dose adjustment,45 use of the other 4‐1BB agonist, utolimumab,46 and with agents having tumor‐specific properties5, 11, 47, 48 or that exhibit balanced agonistic strength with Fc gamma receptor (FcγR) affinity.14, 49 Future research may be required to further study the effects of 4‐1BB costimulation on other immune subsets, such as regulatory T cells,14 and the interactions with FcγRs of the antibodies14, 49 to support the optimal application of anti–4‐1BB therapy.
In conclusion, we demonstrated a distinct 4‐1BB–associated T‐cell activation state among exhausted CD8+ TILs in HCC. Our results provide rationale and evidence for the logical design of immunotherapies targeting costimulatory receptor 4‐1BB, as well as delineate immunological and clinical implications of T‐cell activation status in the HCC microenvironment. Treatment strategies involving 4‐1BB costimulation may be a promising therapeutic option for enhancing T‐cell activation against HCC. Future clinical studies are warranted to evaluate the efficacy of 4‐1BB agonists in HCC patients.
Author Contributions
H.D.K. and S.H.P. contributed to the conceptual design of the study and writing the manuscript. H.D.K., S.P., S.J., Y.J.L., H.L., S.M.H., J.Y.L., S.K., H.K.K., B.S.M., J.H.C., G.W.S., Y.S.J., E.C.S., S.H., and S.H.P. were involved in data acquisition. H.D.K., S.P., S.J., Y.S.J., E.C.S., and S.H.P. were involved in data analysis and interpretation.
Supporting information
Supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT; NRF‐2019R1A2C2005176; to S.H.P.) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI15C2859; to S.H.P.). This work was also supported by the KAIST Future Systems Healthcare Project from the Ministry of Science and ICT (to S.H.P.), Young Medical Scientist Research Grant through the Deawoong Foundation (DF‐201906‐0000003; to H.D.K.), and the Global Ph.D. Fellowship from the National Research Foundation of Korea (NRF‐2016H1A2A1906766; to H.D.K.).
Potential conflict of interest: Nothing to report.
Contributor Information
Shin Hwang, Email: shwang@amc.seoul.kr.
Su‐Hyung Park, Email: park3@kaist.ac.kr.
References
Author names in bold designate shared co‐first authorship.
- 1. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018;8:1069‐1086. [DOI] [PubMed] [Google Scholar]
- 2. El‐Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE‐224): a non‐randomised, open‐label phase 2 trial. Lancet Oncol 2018;19:940‐952. [DOI] [PubMed] [Google Scholar]
- 4. Greten TF, Lai CW, Li G, Staveley‐O’Carroll KF. Targeted and immune‐based therapies for hepatocellular carcinoma. Gastroenterology 2019;156:510‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov 2018;17:509‐527. [DOI] [PubMed] [Google Scholar]
- 6. Schaer DA, Hirschhorn‐Cymerman D, Wolchok JD. Targeting tumor‐necrosis factor receptor pathways for tumor immunotherapy. J Immunother Cancer 2014;2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sanmamed MF, Pastor F, Rodriguez A, Perez‐Gracia JL, Rodriguez‐Ruiz ME, Jure‐Kunkel M, et al. Agonists of co‐stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol 2015;42:640‐655. [DOI] [PubMed] [Google Scholar]
- 8. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013;13:227‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ward‐Kavanagh LK, Lin WW, Sedy JR, Ware CF. The TNF receptor superfamily in co‐stimulating and co‐inhibitory responses. Immunity 2016;44:1005‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015;21:581‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4–1BB: mechanistic rationale, clinical results, and future strategies. Blood 2018;131:49‐57. [DOI] [PubMed] [Google Scholar]
- 12. Vezys V, Penaloza‐MacMaster P, Barber DL, Ha SJ, Konieczny B, Freeman GJ, et al. 4–1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J Immunol 2011;187:1634‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, et al. Monoclonal antibodies against the 4–1BB T‐cell activation molecule eradicate established tumors. Nat Med 1997;3:682‐685. [DOI] [PubMed] [Google Scholar]
- 14. Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to costimulatory receptor 4–1BB enhance anti‐tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity 2018;49:958‐970.e7. [DOI] [PubMed] [Google Scholar]
- 15. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T‐cell invigoration to tumour burden ratio associated with anti‐PD‐1 response. Nature 2017;545:60‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daud AI, Loo K, Pauli ML, Sanchez‐Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti‐PD‐1 therapy in human melanoma. J Clin Invest 2016;126:3447‐3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim HD, Song GW, Park S, Jung MK, Kim MH, Kang HJ, et al. Association between expression level of PD1 by tumor‐infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology 2018;155:1936‐1950.e17. [DOI] [PubMed] [Google Scholar]
- 19. Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus‐related hepatocellular carcinoma. Gut 2019;68:916‐927. [DOI] [PubMed] [Google Scholar]
- 20. Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high‐dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci U S A 2017;114:E5900‐E5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Love M, Anders S, Huber W. Differential analysis of count data—the DESeq2 package. Genome Biol 2014;15(10):1186. [Google Scholar]
- 22. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, et al. A distinct gene module for dysfunction uncoupled from activation in tumor‐infiltrating T cells. Cell 2016;166:1500‐1511.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med 2008;205:625‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007;27:670‐684. [DOI] [PubMed] [Google Scholar]
- 26. Giordano M, Henin C, Maurizio J, Imbratta C, Bourdely P, Buferne M, et al. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J 2015;34:2042‐2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grinchuk OV, Yenamandra SP, Iyer R, Singh M, Lee HK, Lim KH, et al. Tumor‐adjacent tissue co‐expression profile analysis reveals pro‐oncogenic ribosomal gene signature for prognosis of resectable hepatocellular carcinoma. Mol Oncol 2018;12:89‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller R, Siegmund D. Maximally selected chi square statistics. Biometrics 1982;38:1011‐1016. [Google Scholar]
- 30. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD‐1(+) CD8(+) T cell pool with predictive potential in non‐small‐cell lung cancer treated with PD‐1 blockade. Nat Med 2018;24:994‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co‐expression of CD39 and CD103 identifies tumor‐reactive CD8 T cells in human solid tumors. Nat Commun 2018;9:2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN‐gamma‐related mRNA profile predicts clinical response to PD‐1 blockade. J Clin Invest 2017;127:2930‐2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan‐tumor genomic biomarkers for PD‐1 checkpoint blockade‐based immunotherapy. Science 2018;362:eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon‐Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)PD‐1(+)CD8(+) T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019;50:195‐211.e10. [DOI] [PubMed] [Google Scholar]
- 35. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon‐Copete S, et al. T cell factor 1‐expressing memory‐like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 2016;45:415‐427. [DOI] [PubMed] [Google Scholar]
- 36. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD‐1‐targeted therapies is CD28‐dependent. Science 2017;355:1423‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD‐L1 blockade. Proc Natl Acad Sci U S A 2008;105:15016‐15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012;338:1220‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inada Y, Mizukoshi E, Seike T, Tamai T, Iida N, Kitahara M, et al. Characteristics of immune response to tumor‐associated antigens and immune cell profile in patients with hepatocellular carcinoma. Hepatology 2019;69:653‐665. [DOI] [PubMed] [Google Scholar]
- 40. Fuertes Marraco SA, Neubert NJ, Verdeil G, Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front Immunol 2015;6:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science 2016;352:189‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard‐Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2019;176:775‐789.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018;564:268‐272. [DOI] [PubMed] [Google Scholar]
- 44. Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, et al. CD137 accurately identifies and enriches for naturally occurring tumor‐reactive T cells in tumor. Clin Cancer Res 2014;20:44‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist anti‐CD137 monoclonal antibody. Clin Cancer Res 2017;23:1929‐1936. [DOI] [PubMed] [Google Scholar]
- 46. Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, et al. Phase I study of single‐agent utomilumab (PF‐05082566), a 4‐1BB/CD137 agonist, in patients with advanced cancer. Clin Cancer Res 2018;24:1816‐1823. [DOI] [PubMed] [Google Scholar]
- 47. Compte M, Harwood SL, Munoz IG, Navarro R, Zonca M, Perez‐Chacon G, et al. A tumor‐targeted trimeric 4–1BB‐agonistic antibody induces potent anti‐tumor immunity without systemic toxicity. Nat Commun 2018;9:4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor‐targeted 4–1BB agonists for combination with T cell bispecific antibodies as off‐the‐shelf therapy. Sci Transl Med 2019;11:eaav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, et al. Optimization of 4–1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity. Nat Commun 2019;10:2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials