

Abstract
ACTB encodes β‐cytoplasmic actin, an essential component of the cytoskeleton. Based on chromosome 7p22.1 deletions that include the ACTB locus and on rare truncating ACTB variants, a phenotype resulting from ACTB haploinsufficiency was recently proposed. We report putative ACTB loss‐of‐function variants in four patients. To the best of our knowledge, we report the first 7p22.1 microdeletion confined to ACTB and the second ACTB frameshifting mutation that predicts mRNA decay. A de‐novo ACTB p.(Gly302Ala) mutation affects β‐cytoplasmic actin distribution. All four patients share a facial gestalt that is distinct from that of individuals with dominant‐negative ACTB variants in Baraitser‐Winter cerebrofrontofacial syndrome. Two of our patients had strikingly thin and sparse scalp hair. One patient had sagittal craniosynostosis and hypospadias. All three affected male children have attention deficits and mild global developmental delay. Mild intellectual disability was present in only one patient. Heterozygous ACTB deletion can allow for normal psychomotor function.
Keywords: ACTB, intellectual disability, loss‐of‐function, sparse scalp hair, β‐cytoplasmic actin
ACTB encodes β‐cytoplasmic actin, an essential component of the cytoskeleton. Based on rare chromosome 7p22.1 deletions that include the ACTB locus and on rare truncating ACTB variants, a phenotype resulting from ACTB haploinsufficiency was recently proposed. We report the first 7p22.1 microdeletion confined to ACTB, and the second ACTB frameshifting mutation that predicts mRNA decay. Mild intellectual disability was present in one patient. Attention deficits and mild global developmental delay were present in three patients. Normal psychomotor function was seen in one patient with a heterozygous ACTB deletion. Heterozygous ACTB deletion can allow for normal psychomotor function.
1. INTRODUCTION
Distinct phenotypes have been reported with distinct classes of heterozygous variants in ACTB, which encodes β‐actin, an essential component of the cytoskeleton (a) the majority of reported pathogenic variants in ACTB are de‐novo missense changes in exons 2‐4 and were identified in patients with Baraitser‐Winter cerebrofrontofacial syndrome (BWCFF). These mutations are causing the disorder by gain‐of‐function or dominant‐negative mechanisms (Di Donato et al., 2014; Riviere et al., 2012; Verloes et al., 2015). Among patients with BWCF, seven have been identified with juvenile‐onset dystonia and the ACTB variant p.Arg183Trp (Procaccio et al., 2006; Skogseid et al., 2018). Whether dystonia represents a rare nonspecific complication of BWCFF or a manifestation specific to the ACTB variant p.Arg183Trp remains to be resolved; (b) deletion of ACTB was considered responsible for the clinical features observed in patients with rare, nonrecurrent 7p22.1 microdeletions (Cuvertino et al., 2017; Shimojima et al., 2016). Such microdeletions of ≤2 Mb in size were identified in patients with developmental delay, intellectual disability, short stature and microcephaly (7p22.1 microdeletion syndrome); a recent compilation of 23 microdeletions had identified a common deleted region of 0.37 Mb involving the FBLX18, ACTB, FSCN1, RNF216, and ZNF815P genes in such patients (Cuvertino et al., 2017). Recently, a de‐novo 60 kb microdeletion encompassing only FBXL18 and ACTB was identified in a 23‐month‐old child displaying symptoms of 7p22.1 microdeletion syndrome and distinctive facial features (Palumbo et al., 2018); (c) most recently, heterozygous variants in exons 5 and 6 of ACTB were reported in six patients with thrombocytopenia, minor facial dysmorphism, and microcephaly, with and without mild intellectual disability (Latham et al., 2018).
We report here four patients with putative ACTB loss‐of‐function variants, with intellectual disability being mild or absent, and with three male patients sharing attention deficits and behavioral problems. Strikingly thin scalp hair is present in two patients, and craniosynostosis and hypospadias in one patient. The sharing of patient‐related data was facilitated by the GeneMatcher tool (Sobreira, Schiettecatte, Valle, & Hamosh, 2015).
The human subjects in this study were tested for diagnostic purposes after obtaining written informed consent. Patient 1 is an 11‐year‐old male with mild developmental delay and attention deficit. He attends a mainstream school with 1 year of delay and performance in the lower range. Two episodes of generalized seizures occurred at an age of 5 years, and since then he remained seizure‐free without anticonvulsive treatment. From the age of 3 years, he developed an axonal neuropathy that progresses slowly. He has high‐arched feet and requires ankle‐foot orthoses for ambulation; there is no sensory involvement at age 11 years. The neuropathy is paternally inherited, and present also in a younger brother; there is mild facial dysmorphism (Figure 1a). Routine blood count at age 6 years was normal (no thrombocytopenia). Patient 2 is the 35‐year‐old mother of patient 1. She completed lower secondary education. She has strikingly thin and sparse scalp hair (Figure 1b). Facial gestalt shows wavy interrupted eyebrows, dense eyelashes, wide nose, wide mouth, prominent cheeks, and chin. She has hyperopia. Several routine blood counts were normal. Patient 3 is a 4‐year‐old male with slow weight gain in the neonatal period. There is a global developmental delay which improves with speech and language therapy. His language skills at the age of 4 years are at the level of 2.5–3 years old. Autism spectrum disorder was diagnosed by Autism Diagnostic Observation Schedule. He does demonstrate a social interest in other children but lacks the skills to engage with them appropriately. He has starring spells, but EEGs have been normal. Cranial magnetic resonance imaging (MRI) was normal, but a CT of the skull showed sagittal craniosynostosis at an age of 2 years. He had hypospadias with surgical repair, phimosis, penile torsion, and congenital chordee. There is facial dysmorphism with deeply set eyes with prominent brow ridge, a broad nasal tip, large prominent ears and pointed chin, and sparse scalp hair (Figure 1c). He has long toes with 2nd and 5th toe clinodactyly. He has a small patent ductus arteriosus at an age of 4 years, and spells of tachycardia. In several blood counts, no thrombocytopenia has ever been noted. Patient 4 is a 6‐year‐old male. Attention deficit disorder was diagnosed. He attends a normal school with support. His comprehension is good but speech is delayed. He has thin scalp hair, deep set eyes, wide nose, deep‐set columella, strabismus, and myopia (Figure 1d). Anthropometric patient data are compiled in Table 1.
Figure 1.
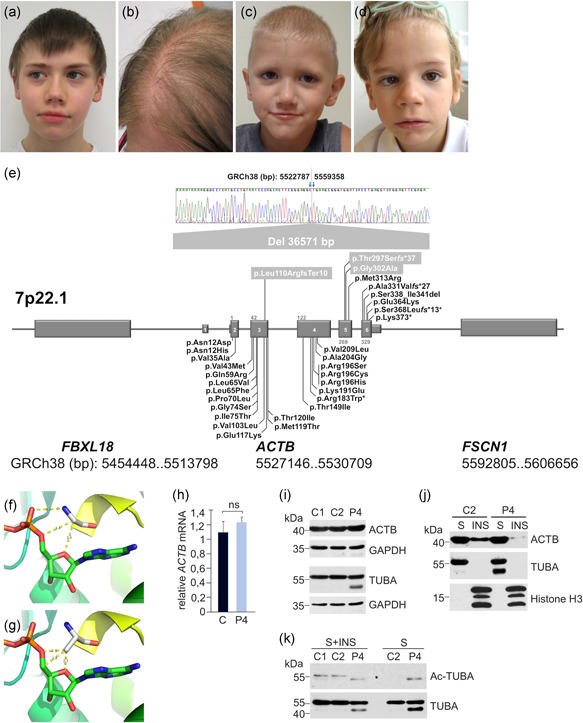
Clinical findings, ACTB variant compilation, and effects of the ACTB p.(Gly302Ala) variant. (a) Patient 1 at the age of 11 years. (b) Patient 2 at the age of 35 years. (c) Patient 3 at the age of 6 years. (d) Patient 4 at the age of 4 years. Shared facial dysmorphism consists of wavy interrupted eyebrows, dense eyelashes, wide nose, wide mouth, prominent cheeks, and chin. Sparse scalp hair in patients 2 and 4. (e) Compilation of novel and reported ACTB variants associated with BWCFF, putative ACTB loss‐of‐function and ACTB‐AST (chromosomal order of genes and exons not drawn to scale). Modeling of ACTB residue Gly‐302 (f) and variant Ala‐302 (g). (h) The relative ACTB messenger RNA expression in a healthy donor (C) and Patient 4 (P4) was assessed by quantitative real‐time polymerase chain reaction using the method. Values were normalized to the amount of human ribosomal protein L32 mRNA. Immunoblot analysis of ACTB and α‐tubulin (TUBA) in total lysates, NP‐40 soluble (S) and insoluble (INS) fractions of blood mononuclear cells from healthy individuals (C1 and C2) and Patient 4 (P4). GAPDH and histone H3 were used as loading controls (i,j). (k) TUBA acetylation was analyzed in total lysates (S + INS) and NP‐40 soluble (S) fraction obtained from control and Patient 4 blood mononuclear cells using anti‐Ac‐TUBA and anti‐TUBA antibodies. BWCFF, Baraitser‐Winter cerebrofrontofacial syndrome
Table 1.
Clinical history and findings in four patients with putative ATCB loss‐of‐function variants
| Patient | Sex | Age (years) | Prenatal and neonatal history, birth measures | Postnatal growth retardation | Microcephaly | Motor delay | Speech delay | Intellectual disability | Behavioral, psychiatric and neurological features | Facial gestalt | Malformation and physical anomalies | ACTB Variant | Inheritance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 11 | Normal 40 weeks 2,900 g 48 cm 33 cm | N (3rd‐10th centile) | N (10th‐25th centile) | Y | Y | None | Attention deficit, seizures at age 5 years, additional paternally inherited axonal neuropathy | Wavy interrupted eyebrows, dense eyelashes, wide nose, wide mouth, prominent cheeks and chin | Anisometropia, unilateral high‐grade hyperopia and astigmatism | Complete heterozygous ACTB gene deletion | Maternal (son of patient 2) |
| 2 | F | 35 | SGA 40 weeks 2,120 g | N (25th centile) | N (25th centile) | N | N | None | None | Sparse scalp hair, wavy interrupted eyebrows, dense eyelashes, wide nose, wide mouth, prominent cheeks and chin | Hyperopia | Complete heterozygous ACTB gene deletion | Unknown |
| 3 | M | 4 | Normal At term 3,350 g 54 cm Slow neonatal weight gain | N (10th‐25th centile) | N (10th‐25th centile) | Y | Y | None | Autism, staring spells with normal EEG, possible attention deficit and impulse control disorder | Sparse scalp hair, deeply set eyes with prominent brow ridge, broad nasal tip, pointed chin, large prominent ears | Long toes with 2nd and 5th toe clinodactyly, sagittal craniosynostosis, hypospadias, patent ductus arteriosus | Heterozygous c.890_891delCA; p.Thr297Serfs*37 | De novo? – no access to paternal sample |
| 4 | M | 6 | Normal 40 weeks 2950 g 49 cm | N (25th‐50th centile) | N (3rd‐10th centile) | Y | Y | Mild, WISC: FSIQ 66 | Attention deficit, visuospatial deficits, speech delay | Thin hair, deep set eyes, wide nose, deep set columella | Strabismus, myopia |
Heterozygous c.905G>C; p.(Gly302Ala) MutationTaster: pathogenic (0.999; range 0–1) CADD score: pathogenic (24.0; range 0–100) PolyPhen2: benign (0.281; range 0–1) Provean: deleterious (−4.177, range ‐40–12.5, cutoff:<−2.5) |
De novo |
Abbreviations: CADD, combined annotation dependent depletion; F, female; FSIQ, full scale intelligence quotient; ID, intellectual disability; M, male; N, no or none known; SGA, small for gestational age; U, unknown; WISC, Wechsler intelligence scale for children; Y, yes; y, years.
Whole‐exome sequencing (WES) was performed in all patients (Baumann, Steichen‐Gersdorf, Krabichler, Muller, & Janecke, 2017) as no specific clinical diagnosis was suggested. WES variation data were filtered for low allele frequencies corresponding to a rare monogenic disease, using data from two population cohorts (Exome Sequencing Project [ESP] and the Exome Aggregation Consortium, ExAC; http://exac.broadinstitute.org/). We identified three distinct ACTB variants in these families (submitted to the Leiden open variation database, https://grenada.lumc.nl/LSDB_list/lsdbs/ACTB), that are all predicted to cause ACTB loss‐of‐function; these variants are shown in Figure 1e, together with a compilation of reported ACTB variants and their classification.
Patient 1 and his mother (patient 2) harbor an intrachromosomal deletion of 36.6 kb (NC_000007.14:g.5522788_5559357del) on their shared chromosome 7, which completely removes the ACTB gene, and leaves approximately 9 and 36 kb of intergenic sequences intact with respect to its neighboring genes, FBXL18 and FSCN1. To the best of our knowledge, this represents the first 7p22.1 microdeletion that is confined to ACTB. The boundaries of this intrachromosomal 7p22.1 deletion, which was detected in WES data with the panelcn.MOPS software (Povysil et al., 2017), were narrowed down by quantitative polymerase chain reaction (qPCR) to ultimately facilitate breakpoint sequencing in patients 1 and 2. ACTB PCR, qPCR, and sequencing primer sequences were based on the NCBI reference sequence for mRNA (NM_001101.4), and genomic DNA (NG_007992.1). Primer sequences and PCR conditions are available from the authors upon request. Nucleotide numbering reflects complementary DNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence. Patient 3 carries a heterozygous ACTB c.890_891delCA frameshift variant in exon 5 (p.Thr297Serfs*37), which is predicted to trigger nonsense‐mediated mRNA decay (NMD; Lykke‐Andersen & Jensen, 2015). Patient 4 carries a de‐novo p.(Gly302Ala) variant in ACTB; the introduction of the larger alanine for glycine is predicted to bring the alanine residue in hydrogen‐bonding distance to ADP as modeled on pdb structure 6FM2 (Kotila et al., 2018; Figure 1f,g). The effect of this missense variant was predicted to be deleterious by three of four different in silico prediction programs, Provean (Choi, Sims, Murphy, Miller, & Chan, 2012), MutationTaster (Schwarz, Rodelsperger, Schuelke, & Seelow, 2010), and CADD (Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019) and to be benign by Polyphen2 (Adzhubei et al., 2010; Table 1). The expression of ACTB was analyzed in mononuclear cells isolated from patient 4 and healthy donor blood samples; we found that that the p.(Gly302Ala) mutation neither affects ACTB mRNA nor total ACTB levels (Figure 1h,i). Fractionation of cell lysates into detergent‐soluble (s) and insoluble (ins) fractions, however, revealed a substantial decrease in the amount of cytoskeleton‐associated ACTB in P4 lymphocytes (Figure 1j). We also detected an elevated level of an α‐tubulin form with a lower molecular weight in the patient (Figure 1i,j). This form showed a high degree of acetylation in the soluble fraction and was not detected in control samples (Figure 1k). Our findings indicate that the expression of ACTB p.(Gly302Ala) variant is likely to affect the lymphocyte cytoskeleton organization by changing the cellular distribution and properties of microfilament and microtubule specific proteins, β‐actin and α‐tubulin, respectively.
We performed WES in three unrelated boys with mildly delayed motor and language development, and deficits in attention span and social communication skills. These patients share the facial features with patients described by Latham et al. (2018), which, however, might not be sufficiently specific to clinically diagnose a disease entity. The identified ACTB deletion in patients 1 and 2, and the frameshift mutation in patient 3 are supposed to abrogate ACTB protein production from these alleles; unfortunately, no material was available from these patients to study their ACTB transcript and protein levels, and address the question of ACTB haploinsufficiency. A recent study found no consistent differences in β‐cytoplasmic actin levels in cells from four individuals with ACTB microdeletions versus controls; nevertheless, these authors proposed that ACTB haploinsufficiency causes the symptoms of 7p22.1 microdeletion syndrome, where the core features were intellectual disability, developmental delay and mild facial dysmorphism (Cuvertino et al., 2017). In that study, 30 of 33 individuals had microdeletions in 7p22.1 with at least 3 further genes deleted in addition to ACTB. The sizes of the different intrachromosomal deletions were very variable, as were the degree of intellectual disability and developmental problems in these patients (Cuvertino et al., 2017). Three patients in that study had heterozygous truncating ACTB variants of which only one was likely to trigger NMD of ACTB; this patient had mild developmental delay and slight social inhibition, but had no other features of 7p22.1 microdeletion syndrome; this patient also had compound‐heterozygous variants in DPYD (Cuvertino et al., 2017). Our study thus emphasizes that the deletion of one ACTB copy is not associated with intellectual disability per se, and can go without developmental delay and behavioral problems as seen in our patient 2. However, mild developmental delay and attention deficits with behavioral problems might emerge as typical although inconstant features of putative ACTB loss‐of‐function variants. Most recently, six individuals from four unrelated families carrying de‐novo or cosegregating heterozygous variants in exons 5 and 6 of ACTB were described. Common features amongst this cohort of patients with 3′‐ACTB variants included developmental delay, microcephaly, and transient or permanent thrombocytopenia with platelet anisotropy and enlarged platelets. Five of six patients presented with microcephaly and two of six patients had a mild intellectual disability. The authors named this condition ACTB‐associated syndromic thrombocytopenia (ACTB‐AST; Latham et al., 2018). Two late‐truncating variants identified in ACTB‐AST, p.(Ala331Val_fs*27) and p.(Ser368Leu_fs*13), are not expected to trigger NMD. The p.(Ser368Leu_fs*13) was identified in a 5‐year‐old female with a history of recurrent thrombocytopenia during the first year of life, microcephaly, periventricular nodular heterotopias, and a low normal IQ. The same variant had been reported before in a patient with feeding difficulties, moderate intellectual disability, microcephaly, hyperactivity, dystonia and tracheoesophageal fistula, esophageal atresia, overlapping toes, a short foot, and tapered fingers and pectus excavatum (Cuvertino et al., 2017). This represents an observation of variable involvement in patients with the same ACTB variant, and other observations are those of patient 1 and 2 in our study here, and patients 1 and 2 in Latham et al. (2018), where patient 1 presented with mild developmental delay, incomplete cleft lip, heart defect, microcephaly, and thrombocytopenia. His father, patient 2, only displayed thrombocytopenia. Father and child carried a p.(Met313Arg) variant in exon 5 (Latham et al., 2018), a rare instance of an ACTB missense variant not associated with BWCFF. The child's phenotype resembles the first patient described with an ACTB variant, a de novo p.(Glu364Lys) variant in exon 6 associated with moderate intellectual disability, abnormal white blood cell counts, and thrombocytopenia (Nunoi et al., 1999).
In the absence of functional and mechanistic studies, we hypothesize that the patients in our report, together with those described by Latham et al. (2018) represent a phenotypic spectrum with mild ID, facial dysmorphism, and variable effects on platelet morphology and function. This phenotypic spectrum is, however, widely different from BWCFF syndrome. BWCFF is a well‐defined syndrome with recognizable facial features, developmental disability, neuronal migration defects, hearing loss, ocular colobomas, heart and renal defects, and progressive muscle wasting (Verloes et al., 2015) due to heterozygous missense variants in ACTB, that exert a gain‐of‐function effect.
Two of our patients had strikingly thin and sparse scalp hair, which was also seen in a number of patients reported with 7p22.1 microdeletion syndrome, which might argue for the presence of some degree of ACTB haploinsufficiency. The same holds for urogenital malformations, which were seen frequently in the cohort of patients with larger 7p22.1 deletions, including an instance of hypospadias and an instance of a micropenis. Hypospadias was present in our patient 3, suggesting that ACTB haploinsufficiency might contribute to this malformation. In contrast, craniosynostosis was not reported in patients with 7p22.1 deletions, suggesting that the craniosynostosis seen in patient 3 might be unrelated to ACTB haploinsufficiency. Among ten individuals with 7p22.1 microdeletion syndrome investigated, eight were found to have some structural abnormality detected by brain MRI. In our study, a single patient undergoing brain MRI had normal results, showing that structural brain defects are not a mandatory feature associated with putative ACTB loss‐of‐function variants or haploinsufficiency.
Coexamination of actin and microtubule cytoskeleton constituents in megakaryocytes and thrombocytes from patients with 3′ ACTB variants indicated that these variants inhibited the final stages of platelet maturation by compromising microtubule organization, and variably brought about qualitative changes to β‐actin filaments (Latham et al., 2018). In fibroblasts from patients with 7p22.1 microdeletion syndrome, altered cell shape, and migration, reduced cell proliferation, altered expression of cell‐cycle genes, and decreased amounts of nuclear, but not β‐cytoplasmic actin were observed. Reduced cell attachment surface area, volume and migratory capacity of ACTB‐AST fibroblasts were also seen; variants affected polarization, movement, and morphology at the single‐cell level, whilst the wild‐type phenotype was rescued with cell–cell contact formation (Latham et al., 2018). ACTB‐AST variants also caused altered expression ratios of different actins and the highly disordered nature of β‐tubulin in patient‐derived platelets was demonstrated. Along these lines, we hypothesize that the novel p.(Gly302Ala) variant disturbs the fine balance between actin and microtubule cytoskeletal organization.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENT
This work was supported by grant no. 16678 from Jubiläumsfonds der Österreichischen Nationalbank.
Baumann M, Beaver EM, Palomares‐Bralo M, et al. Further delineation of putative ACTB loss‐of‐function variants: A 4‐patient series. Human Mutation. 2020;41:753–758. 10.1002/humu.23970
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7, 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, M. , Steichen‐Gersdorf, E. , Krabichler, B. , Muller, T. , & Janecke, A. R. (2017). A recognizable type of syndromic short stature with arthrogryposis caused by bi‐allelic SEMA3A loss‐of‐function variants. Clinical Genetics, 92, 86–90. 10.1111/cge.12967 [DOI] [PubMed] [Google Scholar]
- Choi, Y. , Sims, G. E. , Murphy, S. , Miller, J. R. , & Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One, 7, e46688 10.1371/journal.pone.0046688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvertino, S. , Stuart, H. M. , Chandler, K. E. , Roberts, N. A. , Armstrong, R. , Bernardini, L. , … Banka, S. (2017). ACTB loss‐of‐function mutations result in a pleiotropic developmental disorder. American Journal of Human Genetics, 101, 1021–1033. 10.1016/j.ajhg.2017.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donato, N. , Rump, A. , Koenig, R. , Der Kaloustian, V. M. , Halal, F. , Sonntag, K. , … Verloes, A. (2014). Severe forms of Baraitser‐Winter syndrome are caused by ACTB mutations rather than ACTG1 mutations. European Journal of Human Genetics, 22, 179–183. 10.1038/ejhg.2013.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotila, T. , Kogan, K. , Enkavi, G. , Guo, S. , Vattulainen, I. , Goode, B. L. , & Lappalainen, P. (2018). Structural basis of actin monomer re‐charging by cyclase‐associated protein. Nature Communications, 9, 1892 10.1038/s41467-018-04231-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham, S. L. , Ehmke, N. , Reinke, P. Y. A. , Taft, M. H. , Eicke, D. , Reindl, T. , … Di Donato, N. (2018). Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nature Communications, 9, 4250 10.1038/s41467-018-06713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke‐Andersen, S. , & Jensen, T. H. (2015). Nonsense‐mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nature Reviews Molecular Cell Biology, 16, 665–677. 10.1038/nrm4063 [DOI] [PubMed] [Google Scholar]
- Nunoi, H. , Yamazaki, T. , Tsuchiya, H. , Kato, S. , Malech, H. L. , Matsuda, I. , & Kanegasaki, S. (1999). A heterozygous mutation of beta‐actin associated with neutrophil dysfunction and recurrent infection. Proceedings of the National Academy of Sciences of the United States of America, 96, 8693–8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo, O. , Accadia, M. , Palumbo, P. , Leone, M. P. , Scorrano, A. , Palladino, T. , … Carella, M. (2018). Refinement of the critical 7p22.1 deletion region: Haploinsufficiency of ACTB is the cause of the 7p22.1 microdeletion‐related developmental disorders. European Journal of Medical Genetics, 61, 248–252. 10.1016/j.ejmg.2017.12.008 [DOI] [PubMed] [Google Scholar]
- Povysil, G. , Tzika, A. , Vogt, J. , Haunschmid, V. , Messiaen, L. , Zschocke, J. , … Wimmer, K. (2017). panelcn.MOPS: Copy‐number detection in targeted NGS panel data for clinical diagnostics. Human Mutation, 38, 889–897. 10.1002/humu.23237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccio, V. , Salazar, G. , Ono, S. , Styers, M. L. , Gearing, M. , Davila, A. , … Wainer, B. H. (2006). A mutation of beta ‐actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. American Journal of Human Genetics, 78, 947–960. 10.1086/504271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M. (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47, D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere, J. B. , van Bon, B. W. , Hoischen, A. , Kholmanskikh, S. S. , O'Roak, B. J. , Gilissen, C. , … Dobyns, W. B. (2012). De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser‐Winter syndrome. Nature Genetics, 44, 440–444. 10.1038/ng.1091. S441‐442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Rodelsperger, C. , Schuelke, M. , & Seelow, D. (2010). MutationTaster evaluates disease‐causing potential of sequence alterations. Nature Methods, 7, 575–576. 10.1038/nmeth0810-575 [DOI] [PubMed] [Google Scholar]
- Shimojima, K. , Narai, S. , Togawa, M. , Doumoto, T. , Sangu, N. , Vanakker, O. M. , … Yamamoto, T. (2016). 7p22.1 microdeletions involving ACTB associated with developmental delay, short stature, and microcephaly. European Journal of Medical Genetics, 59, 502–506. 10.1016/j.ejmg.2016.09.008 [DOI] [PubMed] [Google Scholar]
- Skogseid, I. M. , Rosby, O. , Konglund, A. , Connelly, J. P. , Nedregaard, B. , Jablonski, G. E. , … Glover, J. C. (2018). Dystonia‐deafness syndrome caused by ACTB p.Arg183Trp heterozygosity shows striatal dopaminergic dysfunction and response to pallidal stimulation. Journal of Neurodevelopmental Disorders, 10, 17 10.1186/s11689-018-9235-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira, N. , Schiettecatte, F. , Valle, D. , & Hamosh, A. (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36, 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verloes, A. , Di Donato, N. , Masliah‐Planchon, J. , Jongmans, M. , Abdul‐Raman, O. A. , Albrecht, B. , … Pilz, D. T. (2015). Baraitser‐Winter cerebrofrontofacial syndrome: Delineation of the spectrum in 42 cases. European Journal of Human Genetics, 23, 292–301. 10.1038/ejhg.2014.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.