

Abstract
The removal of 5‐methyl‐deoxycytidine (mdC) from promoter elements is associated with reactivation of the silenced corresponding genes. It takes place through an active demethylation process involving the oxidation of mdC to 5‐hydroxymethyl‐deoxycytidine (hmdC) and further on to 5‐formyl‐deoxycytidine (fdC) and 5‐carboxy‐deoxycytidine (cadC) with the help of α‐ketoglutarate‐dependent Tet oxygenases. The next step can occur through the action of a glycosylase (TDG), which cleaves fdC out of the genome for replacement by dC. A second pathway is proposed to involve C−C bond cleavage that converts fdC directly into dC. A 6‐aza‐5‐formyl‐deoxycytidine (a‐fdC) probe molecule was synthesized and fed to various somatic cell lines and induced mouse embryonic stem cells, together with a 2′‐fluorinated fdC analogue (F‐fdC). While deformylation of F‐fdC was clearly observed in vivo, it did not occur with a‐fdC, thus suggesting that the C−C bond‐cleaving deformylation is initiated by nucleophilic activation.
Keywords: demethylation, DNA modifications, epigenetics, formylcytidine
Please release Me, let Me go: The demethylation process of 5‐methyl‐deoxycytidine (mdC), which is associated with the reactivation of silenced genes, was investigated. In the course of active demethylation in stem cells, mdC is oxidized to 5‐formyl‐deoxycytidine (fdC), which is then deformylated, potentially after addition of a nucleophile at the C6‐position.
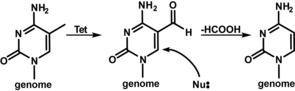
The nucleobase modification 5‐formyl‐deoxycytidine (fdC, 1) is found in stem cells during early development and in the brain.1, 2, 3, 4, 5 These tissues are particularly rich in 5‐hydroxymethyl‐deoxycytidine (hmdC) from which fdC (1) is produced.6, 7 The formation of hmdC and fdC requires oxidation reactions that are performed by α‐ketoglutarate‐dependent Tet enzymes, with 5‐methyl‐deoxycytidine (mdC) being the initial starting molecule.8, 9, 10 This cascade of oxidation reactions is a part of an active demethylation process, in which mdC as a silencer of transcription is replaced by unmodified dC.11 The central molecule that is removed seems to be fdC.12, 13 It can be cleaved out of the genome by a dedicated DNA glycosylase, which creates an abasic site that is further processed, leading to the insertion of an unmodified dC.14 Because abasic sites are harmful DNA‐repair intermediates that can cause genome instability, it was suggested early on that fdC might be directly deformylated to dC by C−C bond cleavage.15, 16 Evidence for the existence of such a direct deformylation process was recently reported.17 Model studies showed that direct deformylation of fdC and potentially also decarboxylation of 5‐carboxy‐deoxycytidine (cadC) are indeed possible.15 Nevertheless, it requires activation of the nucleobase by a nucleophilic addition to the C6 position. For fdC, an additional hydrate formation on the formyl group seems to be necessary, as depicted in Figure 1 A. Although activation with a helper nucleophile is well known as the central mechanistic process during the methylation of dC to mdC by DNA methyltransferases (Dnmts),18, 19 it remains to be confirmed whether such activation occurs in vivo as well. In this work, we investigated this hypothesis with two probe molecules, 2′‐fluorinated‐fdC (F‐fdC, 2) and 6‐aza‐fdC (a‐fdC, 3). The two compounds were simultaneously fed to different cell types, including primed stem cells. This led to random incorporation of these bases at the “C” sites in the respective genomes. Furthermore, it led to the presence of F‐fdC and a‐fdC not only at CpG sites. Ultrasensitive UHPLC‐QQQ‐MS2 was subsequently used to interrogate the chemical processes that occur at F‐fdC and a‐fdC in the genomes. The data show that while F‐fdC is efficiently deformylated, this does not occur for a‐fdC. The only difference between the two nucleobases is the presence of an in‐ring nitrogen atom (6‐aza atom), which features a lone pair that prohibits nucleophilic addition. These results thus provide strong evidence that nucleophilic activation is the central governing mechanistic event that is required for C−C bond cleavage in vivo.
Figure 1.
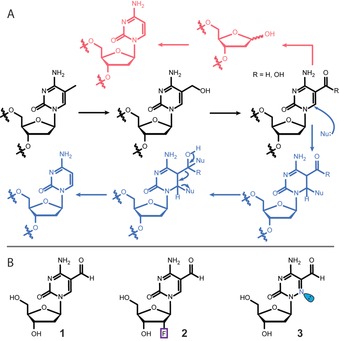
A) The mdC removal pathways that involve oxidation to hmdC, fdC, and cadC followed by either base‐excision repair (magenta) or C−C bond cleavage (blue). B) Structures of fdC (1) and the two probe molecules 2 and 3 used for this study.
The fluorinated nucleoside F‐fdC (2) was recently introduced by us as a deformylation probe.17 Compound 2 is an antimetabolite that is effectively incorporated into the genomes of growing cells. The 2′‐fluoro group is required to block all types of glycosylases, so that base‐excision repair is efficiently inhibited. This ensures high levels of F‐fdC (2) in the genome, as required to observe potential deformylation processes.
The synthesis of the novel nucleoside a‐fdC (3) is depicted in Scheme 1. The synthesis was started with bromo pyruvic acid (4), which we first converted into the semicarbazone 5, followed by conversion into the acid chloride, subsequent cyclization, and hydrolysis to give hydroxymethylated 6‐aza‐uracil (6).20 Vorbrüggen nucleosidation with Hoffers’ chlorosugar subsequently provided the nucleoside 7 as a mixture of the α‐ and β‐anomers, which could be separated by recrystallization. Next, we acetyl‐protected the hydroxymethyl group to give 8, and then used a standard procedure to convert the U base 8 into the C‐derived base 9 by amination of the 4‐triazole intermediate with ammonium hydroxide. This led to the concomitant cleavage of the acetyl protecting group. Dess–Martin oxidation of 9 to 10 and final removal of the toluoyl groups furnished the 6‐aza‐5‐formyl‐deoxycytidine nucleoside (a‐fdC) 3 in a good total yield of 22 % with respect to 6 (Supporting Information).
Scheme 1.
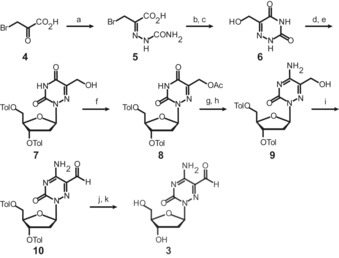
Synthesis of the probe molecule a‐fdC (3). a) semicarbazide⋅HCl, NaOAc, HOAc, H2O, 0 °C to r.t., 2.5 h, 49 %. b) pyridine, SOCl2, 80 °C, 75 min. c) H2O, 110 °C, 17 h, 74 % over 2 steps. d) TMSCl, HMDS, 135 °C, 75 min, then e) Hoffer's chlorosugar, CHCl3, r.t., 17 h, 56 % over 2 steps. f) Ac2O, pyridine, r.t., 22 h, 96 %. g) 1,2,4‐triazole, POCl3, NEt3, MeCN, 0 °C to r.t., 18 h, then h) NH4OH, 1,4‐dioxane, 40 °C, 5 h, 84 %. i) Dess–Martin periodinane, CH2Cl2, −15 °C to r.t., 1 h, 89 %. j) NaOMe, MeOH, benzene, r.t., 1.5 h, then k) reversed‐phase HPLC, 54 %.
Compound 3 features a nitrogen atom instead of a carbon atom at the 6‐position, which possesses a lone pair that blocks any nucleophilic addition to this position. Compound 3 is consequently a perfect model system to investigate whether such a nucleophilic activation is required for the deformylation, as mechanistically postulated (Figure 1 A).
The nucleosides 2 and 3 were subsequently added at a concentration of 350 μm to the media of Neuro‐2a, RBL‐2H3, CHO‐K1 cells for 72 hours (see the Supporting Information). During this time, the nucleosides are converted in vivo into the corresponding triphosphates and then incorporated into the genome of the dividing cells. Initial studies in which we fed the nucleosides individually allowed us to determine that neither compound decreases cell viability up to a concentration of 400 μm, thus the experiments were conducted below the toxicity level. In addition, we tested 2 and 3 at 350 μm on E14 TDG +/− and −/− mouse embryonic stem cells (mESCs) under a three‐day priming process with C/R media. This system allowed us to exclude the BER pathway, leading to a detectable and quantifiable accumulation of natural fdC (see the Supporting Information). After three days, the cells were harvested and lysed, and the genomic DNA was extracted using an optimised protocol (see the Supporting Information). This was followed by an enzymatic digestion of gDNA to single nucleosides and analysed according to a method that we reported recently in detail.21 The obtained nucleoside mixture containing mostly the canonical nucleosides dA, dC, dG, and dT, plus the non‐canonical nucleosides mdC, hmdC, and fdC, as well as the incorporated molecules F‐fdC and a‐fdC and their potential downstream products (F‐dC, F‐mdC, a‐dC, a‐mdC). Nucleosides were separated by ultra‐HPL chromatography and characterized by coupling of the UHPLC system to a triple‐quadrupole mass spectrometer. For exact quantification of the nucleosides by isotope dilution, isotopically labelled standards of F‐fdC and of the product F‐dC were spiked into the analysis mixture as internal standards (see the Supporting Information). To enable exact quantification, calibration curves using these standards were determined (see the Supporting Information). Quantification was performed in the linear region.
During the analysis, we noted that an unusually low amount of a‐fdC (3) was detected because it showed a broad elution profile with very low intensity (Figure 2 B). All attempts to sharpen the elution profile in order to gain sensitivity failed. NMR analysis of compound 3 showed the reason for broad elution profile (see the Supporting Information). Due to the additional electron‐withdrawing in‐ring nitrogen atom, compound 3 exists partially as its hydrate in aqueous solution (20 %, see the Supporting Information). Although the ease of hydrate formation may foster deformylation, the hydrate/carbonyl equilibrium makes efficient detection of compound 3 basically impossible. In order to circumvent the problem, we started to derivatize a‐fdC (3) before analysis with methoxyamine. Addition of CH3ONH2 (150 mm) to the digestion solution indeed provided the methoxyoxime of a‐fdC in quantitative yield after just 15 min at 25 °C and pH 10. The naturally present fdC (1) and the probe molecule F‐fdC (2), however, react as well, but unfortunately not quantitatively. To reduce impurities during MS measurements, we decided against using a catalyst for oxime formation. We therefore decided to analyse the digested DNA in two batches. The first one contained the digested untreated DNA to quantify all bases other than a‐fdC. In the second batch, we treated the digested DNA with methoxyamine for a‐fdC quantification. For quantification of the derivatized a‐fdC, we constructed an external calibration curve (see the Supporting Information).
Figure 2.
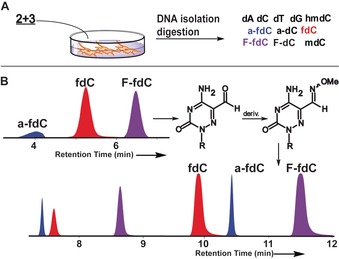
A) Overview of the experimental steps with the feeding and analysis. B) Analysis scheme and the reaction of a‐fdC with methoxyamine to block hydrate formation and of a typical UHPL‐chromatogram before (C‐8 column) and after derivatization (C‐18 column) for exact quantification. Peak splitting is due isomerization (blue peaks: a‐fdC, red peaks: fdC, and purple peaks: F‐fdC).
With this method in hand, we next quantified all nucleosides present in the genome of the cells treated with a mixture of 2+3.
Figure 3 A shows that we indeed detected the fluorinated F‐dC (2), thus confirming very efficient deformylation activity. We tested different cell types and found different levels of deformylation activity. But in all cases, the conversion of F‐fdC into F‐dC was clearly detectable. Most interesting is that we observed the highest deformylation activity in cells associated with neuronal properties. This is in line with neurons featuring the highest levels of hmdC and fdC. In contrast, Figure 3 B shows that for a‐fdC (3), we were unable to detect any formation of the deformylated compound a‐dC despite the high propensity of 3 to exist in the hydrated form, which is one prerequisite for efficient C−C bond cleavage. This result suggests that the ability to react with a nucleophile at the 6‐position is also required in vivo for efficient deformylation.
Figure 3.
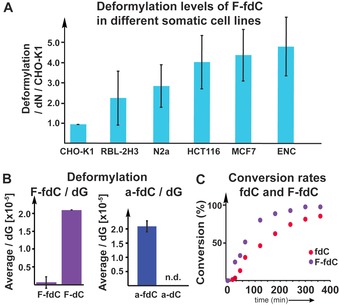
A) Deformylation data for F‐fdC in different cell types, showing that F‐fdC is deformylated in very different cells. Deformylation rate was calculated by the F‐dC+F‐mdC/dN per F‐fdC/dN, then the values were normalized to the cell line with the lowest deformylation level (CHO‐K1=1). B) The deformylation of F‐fdC/dG and a‐fdC/dG, showing the induced differences due to C6‐carbon‐to‐nitrogen exchange. C) The bisulfite data show that the deformylation of fdC and F‐fdC is comparable, thus showing that the 2′‐F substitution has only a small accelerating effect, whereas the reaction of a‐fdC could not be detected.
In order to substantiate this result, we next performed in vitro studies with bisulfite. Bisulfite is a strong nucleophile that has been reported to cause deformylation of fdC by first attacking the C6 position, followed by conversion of the C5, C6‐saturated fdC adduct into the bisulfite adduct, which then undergoes deformylation.22 The deformylated product dC is then further converted into dU by the well‐known bisulfite‐induced deamination reaction of dC (see the Supporting Information). Indeed, when we reacted fdC with bisulfite, we observed efficient deformylation and deamination to dU. We then studied to what extent the reaction is influenced by the 2′‐F atom present in F‐fdC, in order to estimate whether the in vivo deformylation could be just the result of the 2′‐F atom. Treatment of F‐fdC with bisulfite also led to deformylation and deamination to F‐dU, and indeed the reaction is a little faster compared to fdC (see Figure 3 C). Although the difference is measurable, it is in total rather small. With these data in hand, we can conclude that we may overestimate the amount of deformylation that can occur with fdC lacking the 2′‐F atom. We can certainly exclude that deformylation in vivo occurs only with F‐fdC. It is unfortunate that we are unable to measure the direct deformylation of fdC because of the presence of efficient BER processes. A TDG −/− cell line showed a huge increase in fdC compared to the TDG +/−, whereas a‐fdC and F‐fdC stay constant, thus showing that these compounds are indeed not repaired by the TDG protein (see the Supporting Information). The bisulfite studies, however, show that the F‐fdC compound is not a perfect but sufficient reporter of this C−C bond cleavage. Treatment of a‐fdC (3) with bisulfite did not provide the deformylated product a‐dC under any circumstances, showing that the inability to react with a nucleophile at the 6‐position totally blocks the C−C bond cleavage. We can therefore conclude that the deformylation of fdC during active demethylation requires oxidation of mdC to fdC. fdC can undergo a direct C−C bond cleavage to dC, but this reaction requires a helper nucleophile to attack the C6‐position, which is blocked in the case of a‐fdC by the lone pair introduced by the C6‐carbon‐to‐nitrogen exchange. While the chemistry that allows the transformation of fdC into dC is now elucidated, we next need to find the nucleophiles that perform the reaction in vivo.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) GRK2338 (Project ID 321812289), SFB1309 (PID 325871075), SFB1361 (PID 393547839) and SPP1784 (PID 255344185). This project has received additional funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement n° EPiR 741912) and through a H2020 Marie Skłodowska‐Curie Action (LightDyNAmics, 765866).
A. Schön, E. Kaminska, F. Schelter, E. Ponkkonen, E. Korytiaková, S. Schiffers, T. Carell, Angew. Chem. Int. Ed. 2020, 59, 5591.
Dedicated to Dr. Klaus Römer on the occasion of his 80th birthday
Contributor Information
M. Sc. Alexander Schön, https://www.carellgroup.de
Prof. Dr. Thomas Carell, Email: thomas.carell@lmu.de.
References
- 1. Wagner M., Steinbacher J., Kraus T. F. J., Michalakis S., Hackner B., Pfaffeneder T., Perera A., Müller M., Giese A., Kretzschmar H. A., Carell T., Angew. Chem. Int. Ed. 2015, 54, 12511–12514; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12691–12695. [Google Scholar]
- 2. Liu S., Wang J., Su Y., Guerrero C., Zeng Y., Mitra D., Brooks P. J., Fisher D. E., Song H., Wang Y., Nucleic Acids Res. 2013, 41, 6421–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Song C. X., Szulwach K. E., Dai Q., Fu Y., Mao S. Q., Lin L., Street C., Li Y., Poidevin M., Wu H., Gao J., Liu P., Li L., Xu G. L., Jin P., He C., Cell 2013, 153, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pfaffeneder T., Hackner B., Truss M., Münzel M., Müller M., Deiml C. A., Hagemeier C., Carell T., Angew. Chem. Int. Ed. 2011, 50, 7008–7012; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7146–7150. [Google Scholar]
- 5. Bachman M., Uribe-Lewis S., Yang X., Burgess H. E., Iurlaro M., Reik W., Murrell A., Balasubramanian S., Nat. Chem. Biol. 2015, 11, 555–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kriaucionis S., Heintz N., Science 2009, 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi D. Q., Ali I., Tang J., Yang W. C., Front. Genet. 2017, 8, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Münzel M., Globisch D., Carell T., Angew. Chem. Int. Ed. 2011, 50, 6460–6468; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6588–6596. [Google Scholar]
- 9. Spruijt C. G., Gnerlich F., Smits A. H., Pfaffeneder T., Jansen P. W. T. C., Bauer C., Münzel M., Wagner M., Müller M., Khan F., Eberl H. C., Mensinga A., Brinkman A. B., Lephikov K., Müller U., Walter J., Boelens R., Van Ingen H., Leonhardt H., Carell T., Vermeulen M., Cell 2013, 152, 1146–1159. [DOI] [PubMed] [Google Scholar]
- 10. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., Zhang Y., Science 2011, 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neri F., Incarnato D., Krepelova A., Rapelli S., Anselmi F., Parlato C., Medana C., DalBello F., Oliviero S., Cell Rep. 2015, 10, 674–683. [DOI] [PubMed] [Google Scholar]
- 12. Su M., Kirchner A., Stazzoni S., Müller M., Wagner M., Schröder A., Carell T., Angew. Chem. Int. Ed. 2016, 55, 11797–11800; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11974–11978. [Google Scholar]
- 13. Fu T., Liu L., Yang Q. L., Wang Y., Xu P., Zhang L., Liu S., Dai Q., Ji Q., Xu G. L., He C., Luo C., Zhang L., Chem. Sci. 2019, 10, 7407–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rahimoff R., Kosmatchev O., Kirchner A., Pfaffeneder T., Spada F., Brantl V., Müller M., Carell T., J. Am. Chem. Soc. 2017, 139, 10359–10364. [DOI] [PubMed] [Google Scholar]
- 15. Schiesser S., Pfaffeneder T., Sadeghian K., Hackner B., Steigenberger B., Schröder A. S., Steinbacher J., Kashiwazaki G., Höfner G., Wanner K. T., Ochsenfeld C., Carell T., J. Am. Chem. Soc. 2013, 135, 14593–14599. [DOI] [PubMed] [Google Scholar]
- 16. Schiesser S., Hackner B., Pfaffeneder T., Müller M., Hagemeier C., Truss M., Carell T., Angew. Chem. Int. Ed. 2012, 51, 6516–6520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6622–6626. [Google Scholar]
- 17. Iwan K., Rahimoff R., Kirchner A., Spada F., Schröder A. S., Kosmatchev O., Ferizaj S., Steinbacher J., Parsa E., Müller M., Carell T., Nat. Chem. Biol. 2018, 14, 72–78. [DOI] [PubMed] [Google Scholar]
- 18. Jeltsch A., ChemBioChem 2002, 3, 274–293. [DOI] [PubMed] [Google Scholar]
- 19. Du Q., Wang Z., Schramm V. L., Proc. Natl. Acad. Sci. USA 2016, 113, 2916–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alekseeva I. V., Shalamai A. S., Shalamai V. S., Chernetski V. P., Ukr. Khim. Zh. (Ukr. Ed.) 1976, 42, 398–401. [Google Scholar]
- 21. Traube F. R., Schiffers S., Iwan K., Kellner S., Spada F., Müller M., Carell T., Nat. Protoc. 2019, 14, 283–312. [DOI] [PubMed] [Google Scholar]
- 22. Kriukienė E., Liutkevičiūtėa Z., Klimašauskas S., Chem. Soc. Rev. 2012, 41, 6916–6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary