

Abstract
Background and Aims
Sustained virologic response (SVR) to interferon (IFN)‐free therapies ameliorates portal hypertension (PH); however, it remains unclear whether a decrease in hepatic venous pressure gradient (HVPG) after cure of hepatitis C translates into a clinical benefit. We assessed the impact of pretreatment HVPG, changes in HVPG, and posttreatment HVPG on the development of hepatic decompensation in patients with PH who achieved SVR to IFN‐free therapy. Moreover, we evaluated transient elastography (TE) and von Willebrand factor to platelet count ratio (VITRO) as noninvasive methods for monitoring the evolution of PH.
Approach and Results
The study comprised 90 patients with HVPG ≥ 6 mm Hg who underwent paired HVPG, TE, and VITRO assessments before (baseline [BL]) and after (follow‐up [FU]) IFN‐free therapy. FU HVPG but not BL HVPG predicted hepatic decompensation (per mm Hg, hazard ratio, 1.18; 95% confidence interval, 1.08‐1.28; P < 0.001). Patients with BL HVPG ≤ 9 mm Hg or patients who resolved clinically significant PH (CSPH) were protected from hepatic decompensation. In patients with CSPH, an HVPG decrease ≥ 10% was similarly protective (36 months, 2.5% vs. 40.5%; P < 0.001) but was observed in a substantially higher proportion of patients (60% vs. 24%; P < 0.001). Importantly, the performance of noninvasive methods such as TE/VITRO for diagnosing an HVPG reduction ≥ 10% was inadequate for clinical use (area under the receiver operating characteristic curve [AUROC], < 0.8), emphasizing the need for HVPG measurements. However, TE/VITRO were able to rule in or rule out FU CSPH (AUROC, 0.86‐0.92) in most patients, especially if assessed in a sequential manner.
Conclusions
Reassessment of HVPG after SVR improved prognostication in patients with pretreatment CSPH. An “immediate” HVPG decrease ≥ 10% was observed in the majority of these patients and was associated with a clinical benefit, as it prevented hepatic decompensation. These results support the use of HVPG as a surrogate endpoint for interventions that lower portal pressure by decreasing intrahepatic resistance.
Abbreviations
- ACLD
advanced chronic liver disease
- AUROC
area under the receiver operating characteristic curve
- BL
baseline
- cACLD
compensated advanced chronic liver disease
- CHC
chronic hepatitis C
- CI
confidence interval
- CSPH
clinically significant portal hypertension
- CTP
Child‐Turcotte‐Pugh
- FU
follow‐up
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HE
hepatic encephalopathy
- HR
hazard ratio
- HVPG
hepatic venous pressure gradient
- IFN
interferon
- IL‐6
interleukin 6
- LBP
lipopolysaccharide‐binding protein
- MELD
Model for End‐Stage Liver Disease
- NASH
nonalcoholic steatohepatitis
- NSBB
nonselective beta‐blockers
- PLT
platelet count
- PH
portal hypertension
- SVR
sustained virologic response
- TE
transient elastography
- VITRO
von Willebrand factor to platelet count ratio
- VWF
von Willebrand factor
Interferon (IFN)‐free therapies have revolutionized the treatment of patients with chronic hepatitis C (CHC). Even in previous difficult‐to‐cure patient populations, such as patients with human immunodeficiency virus coinfection1, 2 or compensated advanced chronic liver disease (cACLD) and PH,3, 4 rates of sustained virologic response (SVR) increased to nearly 100%. Accordingly, the focus of attention has shifted to the regression of liver disease and risk stratification concepts for personalized follow‐up (FU) of patients who had advanced chronic liver disease (ACLD) before treatment and achieved SVR.5, 6, 7
Four studies have investigated the impact of viral suppression by SVR to IFN‐free treatments on the evolution of PH,8, 9, 10, 11 as assessed by hepatic venous pressure gradient (HVPG). Although patients with pretreatment subclinical PH (i.e., HVPG 6‐9 mm Hg) did not progress to clinically significant portal hypertension (CSPH; HVPG ≥ 10 mm Hg12, 13, 14), less than one fourth of patients who had CSPH before treatment resolved CSPH.8, 10, 11 Accordingly, the vast majority of patients remained at risk for hepatic decompensation despite the cure of hepatitis C virus (HCV) infection. However, nearly two thirds of patients with CSPH before antiviral therapy had an HVPG decrease ≥ 10% after achieving SVR to IFN‐free regimens,8, 10 which denotes a clinically meaningful decrease according to the Baveno VI chapter on the impact of etiological therapy.13 This recommendation is primarily based on studies evaluating the HVPG response to nonselective beta‐blockers (NSBB).15 However, there is also limited evidence from patients with HVPG ≥ 12 mm Hg due to alcoholic liver disease (ALD) who were advised to abstain from alcohol (HVPG decrease cutoff ≥ 15%),16 or patients with compensated cirrhosis due to nonalcoholic steatohepatitis (NASH) treated with simtuzumab or placebo (HVPG decrease cutoff ≥ 20%).17 Besides variceal (re‐)bleeding, HVPG response to conventional NSBB ± vasoactive drugs has been shown to decrease the risks of occurrence/worsening of ascites and its complications.15 In addition, some studies even reported (trends toward) a decrease in hepatic encephalopathy (HE), a decompensating event that is less closely linked to PH.15 However, conventional NSBB have completely different modes of action, as compared with etiological therapies or other treatments that primarily act by decreasing intrahepatic resistance.18, 19 HVPG is currently not accepted as a surrogate endpoint for the accelerated approval of medical therapies for PH by regulatory authorities, because there is limited evidence supporting its use as a surrogate endpoint for other treatments than NSBB.20 This is particularly problematic in patients with cACLD, in whom the incidence of hepatic decompensation is comparatively low, resulting in tremendously larger trials and longer study periods, if clinical endpoints (e.g., hepatic decompensation) are assessed. Because resources are limited, only a minority of the emerging treatment approaches are tested in phase 3 clinical trials assessing direct endpoints, which greatly inhibits scientific progress in the field of PH.21
Thus, we aimed to assess the impact of pretreatment HVPG, changes in HVPG, and posttreatment values on the development of hepatic decompensation in patients with PH who achieved SVR to IFN‐free therapy. Moreover, we investigated the use of transient elastography (TE) and von Willebrand factor (VWF) for monitoring the evolution of PH and the impact of HCV cure on markers of bacterial translocation and inflammation.22
Patients and Methods
Study Design and Population
Ninety patients with PH (HVPG ≥ 6 mm Hg12, 13, 14) who underwent HVPG and TE before (baseline [BL], October 2013 to April 2018) and after IFN‐free therapy at the Medical University of Vienna were prospectively characterized and followed for the development of clinical events after the end of treatment. An explorative analysis on changes in markers of bacterial translocation and inflammation (lipopolysaccharide‐binding protein [LBP] and interleukin 6 [IL‐6]) was performed in a subgroup of 73 patients.
Of note, a considerable proportion of our study cohort has been included in a previous study investigating the impact of SVR to IFN‐free therapies on HVPG (and TE)8; however, this previous study did not evaluate the association between hemodynamic changes and clinical outcomes (i.e., the primary objective of the present study) or the impact of HCV cure on markers of bacterial translocation and inflammation.
Clinical and Laboratory Parameters
VWF/platelet count (PLT) ratio (VITRO) was calculated by dividing VWF antigen levels (%) over PLT (G × L−1), as previously described.23, 24
See Supporting Information for further information.
HCV Therapy
All patients were treated with IFN‐free therapies. The choice of the regimen was at the physicians’ discretion and depended on the availability of early‐access programs, reimbursement policies, and national25, 26 and international27, 28, 29 clinical practice guidelines at the time of treatment initiation. Treatment durations ranged from 8 to 24 weeks.
HVPG and Liver Stiffness Measurement
HVPG measurements were performed by the Vienna Hepatic Hemodynamic Lab at the Medical University of Vienna in accordance with a standardized operating procedure30 and in the absence of NSBB and nitrates. In patients on NSBB, treatment was paused 5 days before HVPG measurements. Liver stiffness was measured using TE (FibroScan; Echosens, Paris, France).
In patients with CSPH (HVPG ≥ 10 mm Hg12, 13, 14), a clinically meaningful change in HVPG was defined by a decrease of ≥10%, as recommended by the Baveno VI consensus for etiologic therapies.13 Subclinical and pronounced PH were defined by an HVPG of 6‐9 mm Hg and ≥16 mm Hg, respectively.12, 13, 14
Clinical Events
In patients with cACLD, hepatic decompensation was defined by variceal bleeding, incident ascites, or incident HE, whereas in patients with decompensated ACLD, further hepatic decompensation was defined by variceal (re‐)bleeding, requirement of paracentesis, admission for grade 3/4 HE, or development of grade 3/4 HE during admission.
Although new onset of jaundice is commonly referred to as a decompensating event in natural history studies,31 we did not incorporate jaundice in the definition of hepatic decompensation, because this term is poorly defined.32 This is in line with previous studies investigating risk factors for hepatic decompensation.32, 33, 34
Statistical Analysis
Ethics
This study was conducted in accordance with the Declaration of Helsinki and approved by the local ethics committee of the Medical University of Vienna. Patients were consented for HVPG measurements.
Results
Baseline Characteristics
The majority of patients were male (73%), with a mean age of 54.5 ± 1 years (Supporting Table S1). Overall, 22% of patients had a history of hepatic decompensation and 28% were Child‐Turcotte‐Pugh (CTP) score stage B, whereas no CTP score stage C patients were included. The median BL Model for End‐Stage Liver Disease (MELD) score was 9 (3) points. All patients with varices and/or previous variceal bleeding (n = 34) were on NSBB treatment, except for 4 patients in primary prophylaxis who were intolerant to NSBB. Moreover, 8 additional patients received carvedilol for arterial hypertension. Accordingly, 38 (42%) patients were receiving NSBB treatment. Median BL HVPG, which was assessed 1.71 (10.3) months before treatment initiation, was 14 (8) mm Hg. Twenty‐three (26%) patients had subclinical PH (i.e., HVPG 5‐9 mm Hg; median HVPG, 8 [3] mm Hg) at BL, whereas HVPG values of 10‐15 mm Hg (median HVPG, 12 [4] mm Hg) and ≥16 mm Hg (median HVPG, 19 [4] mm Hg) were observed in 29 (32%) and 38 (42%) patients, respectively. Accordingly, the prevalence of CSPH at BL was 74% (67/90).
Baseline characteristics of the subgroup of patients with pretreatment CSPH are shown in Table 1.
Table 1.
Comparison of BL Characteristics, Changes During Antiviral Therapy, and Characteristics at FU HVPG Measurement Between Patients With CSPH at BL Who Had or Did Not Have an HVPG Decrease ≥ 10%
| Patient Characteristics | All, n = 67 | HVPG Decrease ≥ 10%, n = 40 | No HVPG Decrease ≥ 10%, n = 27 | P Value |
|---|---|---|---|---|
| Age, years | 54.5 ± 1.18 | 56.1 ± 1.4 | 52.2 ± 2 | 0.101 |
| Sex | ||||
| Male | 45 (67%) | 25 (63%) | 20 (74%) | 0.428 |
| Female | 22 (33%) | 15 (38%) | 7 (26%) | |
| BL BMI, kg × m−2 | 25.8 ± 0.6 | 25.8 ± 0.7 | 25.8 ± 1 | 0.966 |
| ≥25 kg × m−2 | 35 (52%) | 20 (50%) | 15 (56%) | 0.655 |
| ≥30 kg × m−2 | 12 (18%) | 7 (18%) | 5 (19%) | 1 |
| Δ BMI, kg × m−2 | 0.211 (2.366) | 0.106 (2.236) | 0.316 (2.564) | 0.691 |
| FU BMI, kg × m−2 | 26 ± 0.6 | 26 ± 0.75 | 26 ± 0.91 | 0.976 |
| ≥25 kg × m−2 | 38 (57%) | 23 (58%) | 15 (56%) | 0.875 |
| ≥30 kg × m−2 | 11 (16%) | 6 (15%) | 5 (19%) | 0.745 |
| Alcohol consumption | ||||
| Abstinent | 57 (85%) | 34 (85%) | 23 (85%) | 0.517 |
| Nonabstinent but below threshold* | 5 (7%) | 4 (10%) | 1 (4%) | |
| Above threshold* | 5 (7%) | 2 (5%) | 3 (11%) | |
| HCV genotype | ||||
| 1 | 45 (67%) | 25 (63%) | 20 (74%) | |
| 3 | 15 (22%) | 11 (28%) | 4 (27%) | 0.545 |
| 4 | 7 (10%) | 4 (10%) | 3 (11%) | |
| History of hepatic decompensation | 13 (19%) | 7 (18%) | 6 (22%) | 0.632 |
| BL CTP score, points | 6 (2) | 6 (2) | 7 (2) | 0.075 |
| Stage A | 43 (64%) | 30 (75%) | 13 (48%) | 0.037 |
| Stage B | 24 (36%) | 10 (25%) | 14 (52%) | |
| Δ CTP score, points | 0 (0) | 0 (0) | 0 (0) | 0.577 |
| FU CTP score, points | 6 (2) | 6 (1) | 6 (3) | 0.066 |
| BL MELD score, points | 9 (3) | 9 (3) | 10 (4) | 0.009 |
| Δ MELD score, points | 0 (2) | 0 (1) | −1 (2) | 0.521 |
| FU MELD score, points | 9 (2) | 8 (3) | 10 (4) | 0.027 |
| Varices | 33 (49%) | 17 (43%) | 16 (59%) | 0.178 |
| Small | 17 (52%) | 10 (59%) | 7 (44%) | 0.387 |
| Large | 16 (48%) | 7 (41%) | 9 (56%) | |
| NSBB treatment | 35 (52%) | 19 (48%) | 16 (59%) | 0.345 |
| BL HVPG, mm Hg | 16.2 ±0.5 | 15.8 ±0.6 | 16.7 ±0.9 | 0.395 |
| ≥16 mm Hg | 38 (57%) | 21 (53%) | 17 (63%) | 0.397 |
| Absolute Δ HVPG, mm Hg | −2.64 ± 0.46 | −5.1 ± 0.34 | 0.93 ± 0.49 | <0.001 |
| Relative Δ HVPG, % | −17.8 ± 2.9 | −33 ± 2.3 | 4.56 ± 2.61 | <0.001 |
| FU HVPG, mm Hg | 12 (7) | 10.5 (5) | 17 (11) | <0.001 |
| ≤5 mm Hg | 4 (6%) | 4 (10%) | 0 (0%) | <0.001 |
| 6‐9 mm Hg | 12 (18%) | 12 (30%) | 0 (0%) | |
| 10‐15 mm Hg | 29 (43%) | 19 (48%) | 10 (37%) | |
| ≥16 mm Hg | 22 (33%) | 5 (13%) | 17 (63%) | |
| BL liver stiffness, kPa† | 27.7 (25.4) | 27 (19.7) | 29.1 (24.1) | 0.269 |
| Absolute Δ liver stiffness, kPa† | −4.6 (11.9) | −6.8 (11) | −3.6 (19.7) | 0.004 |
| Relative Δ liver stiffness, %† | −18.1 (43.1) | −31.9 (35.9) | −8.22 (52.8) | <0.001 |
| FU liver stiffness, kPa† | 23 (26) | 19.1 (13.5) | 32.4 (27) | 0.002 |
| BL PLT, G × L−1 | 83 (49) | 100.5 (43) | 68 (49) | 0.004 |
| Absolute Δ PLT, G × L−1 | 7.16 ± 2.95 | 8.73 ± 3.99 | 4.85 ± 4.36 | 0.523 |
| Relative Δ PLT, G × L−1 | 8.11 (27.89) | 8.6 (24.79) | 3.03 (26.28) | 0.51 |
| FU PLT, G × L−1 | 89 (64) | 101 (62) | 77 (59) | 0.015 |
| BL VWF, %‡ | 293 (151) | 283 ± 15.7 | 311 ± 18 | 0.267 |
| Absolute Δ VWF, %‡ | −58 (90) | −83.3 ± 10 | −26.6 ± 8.8 | <0.001 |
| Relative Δ VWF, %‡ | −19.1 (26.4) | −30.1 (23.5) | −9.7 (19.9) | <0.001 |
| FU VWF, %‡ | 202 (141) | 179 (88) | 275 (198) | 0.003 |
| BL VITRO, %‡ | 3.38 (3.42) | 2.98 (2.41) | 4.64 (3.56) | 0.014 |
| Absolute Δ VITRO‡ | −0.791 (1.176) | −0.979 (1.442) | −0.382 (1.532) | 0.002 |
| Relative Δ VITRO, %‡ | −25.5 (38.6) | −34.1 (28.9) | −11.2 (43.7) | 0.013 |
| FU VITRO, %‡ | 2.4 (2.64) | 1.96 (1.96) | 3.31 (4.56) | 0.002 |
>30 g/day and >20 g/day for males and females, respectively.48
Information available in 64 patients.
Information available in 65 patients.
Abbreviation: BMI, body mass index.
Baseline Characteristics According to the Severity of PH
Change in Hepatic Venous Pressure Gradient After Treatment
The FU HVPG measurement was performed 8.79 (6.67) months after the treatment initiation, which was 4.15 (6.58) months after the end of IFN‐free therapy.
Absolute and relative changes in HVPG in this series of 90 patients are depicted in Supporting Fig. S1 and were consistent with our previous report on 60 patients, who were also included in this study.8 No patient with subclinical PH at BL progressed to CSPH (i.e., FU HVPG ≥ 10 mm Hg); however, 57% (13/23) of these patients 38/90 resolved PH (i.e., FU HVPG ≤ 5 mm Hg). Among patients with CSPH at BL, 6% (4/67) of patients had an FU HVPG of ≤ 5 mm Hg, 18% (12/67) of patients regressed to subclinical PH (i.e., HVPG ≥ 5‐9 mm Hg), and CSPH persisted in 76% (51/67). Of note, the proportion of patients with profound PH (i.e., HVPG ≥ 16 mm Hg) decreased from 57% (38/67) at BL to 33% (22/67) at FU.
Importantly, in patients with CSPH at BL, an HVPG decrease ≥ 10% was substantially more common (60% [40/67]) than a regression to HVPG values ≤ 9 mm Hg after antiviral therapy (24% [16/67]; P < 0.001).
Severity of PH and Clinical Events
During a median follow‐up of 35.3 (21.8) months, 11 patients developed hepatic decompensation (n = 7 first hepatic decompensation; n = 4 further hepatic decompensation): n = 5 ascites, n = 5 HE, and n = 1 variceal bleeding. Three patients underwent liver transplantation. The death of 1 patient with further hepatic decompensation was considered liver related, whereas 1 compensated patient died of a non‐liver‐related (cardiac) cause.
Although there was only a trend toward an increased risk of posttreatment hepatic decompensation with increasing BL HVPG (per mm Hg, hazard ratio [HR], 1.13; 95% confidence interval [CI], 0.94‐1.28; P = 0.069), FU HVPG showed a strong association with hepatic decompensation (per mm Hg; HR, 1.18; 95% CI, 1.08‐1.28; P < 0.001). Accordingly, although BL HVPG stratified patients into subgroups with different risks of hepatic decompensation after treatment (log‐rank: P = 0.04; Fig. 1), its discriminatory ability seemed to improve if HVPG was assessed during FU: Whereas none of the patients with FU HVPG ≤ 9 mm Hg (0/39) had hepatic decompensation, the Kaplan‐Meier estimates for hepatic decompensation at 36 months in patients with FU HVPG values of 10‐15 mm Hg and ≥16 mm Hg were 11.1% and 40.1%, respectively.
Figure 1.
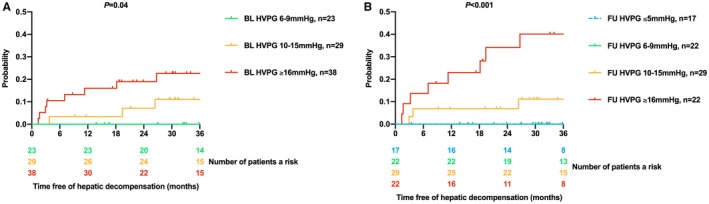
Cumulative incidence of hepatic decompensation after treatment according to (A) BL and (B) FU HVPG strata.
Correlation Between the Change in Hepatic Venous Pressure Gradient After Treatment and Clinical Events
In the population at risk for hepatic decompensation (i.e., patients with CSPH at BL), FU HVPG (area under the receiver operating characteristic curve [AUROC], 0.819; 95% CI, 0.687‐0.951; P = 0.001) as well as the absolute (AUROC, 0.872; 95% CI, 0.74‐1; P < 0.001) and relative (AUROC, 0.877; 95% CI, 0.749‐1; P < 0.001) changes in HVPG were associated with hepatic decompensation. In contrast, BL HVPG was not predictive (AUROC, 0.657; 95% CI, 0.468‐0.951; P = 0.101).
Although the highest Youden’s index was observed at a relative HVPG decrease of 5% (sensitivity, 90.9%/specificity, 80.4%), we used the similarly sensitive but slightly less specific 10% cutoff (sensitivity, 90.9%/specificity, 69.6%) for further analyses, as recommended by the Baveno VI chapter on the impact of etiological treatment.13
Patients with CSPH at BL who achieved an HVPG decrease ≥ 10% had a substantially lower risk of hepatic decompensation after treatment (2.5% vs. 40.5% at 36 months; P < 0.001; Fig. 2). The results remained basically unchanged, if only events occurring after the FU HVPG‐measurement were considered (2.6% vs. 30.7% at 36 months; P = 0.003). When stratifying patients according to the presence or absence of previous hepatic decompensation, patients with compensated CSPH at BL (n = 54) were completely protected from hepatic decompensation, if HVPG decreased by ≥ 10% (0% vs. 36.4% at 36 months; P < 0.001). In the limited number of patients with decompensated CSPH included in our study (n = 13), the difference in the risk of further hepatic decompensation (14.3% vs. 55.6% at 36 months; P = 0.22) did not attain statistical significance.
Figure 2.
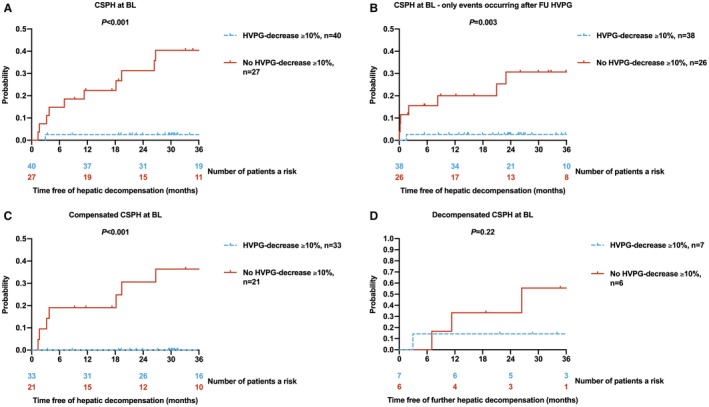
Cumulative incidence of hepatic decompensation after treatment according to HVPG decrease ≥ 10% from BL to FU. (A) All patients with CSPH at BL, (B) all patients with CSPH at BL, only considering events that occurred after the FU HVPG measurement, and subgroups of patients with (C) compensated CSPH and (D) decompensated CSPH at BL.
Importantly, achieving an HVPG decrease ≥ 10% was associated with a substantially decreased risk of hepatic decompensation (adjusted HR, 0.102; 95% CI, 0.012‐0.863; P = 0.036), even when accounting for history of hepatic decompensation as well as treatment‐induced changes in CTP and MELD score, or their posttreatment values (Table 2).
Table 2.
Univariate and Multivariate Cox Regression Analyses on Determinants of Hepatic Decompensation After Treatment
| Parameter | Univariate Analysis, n = 67 | Multivariate Analysis, n = 67 | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | aHR | 95% CI | P Value | |
| History of hepatic decompensation | 2.5 | 0.732‐8.56 | 0.166 | — | — | — |
| Change in CTP score, per point | 1.55 | 0.84‐2.87 | 0.179 | — | — | — |
| FU CTP score, per point | 2.11 | 1.45‐3.06 | < 0.001 | 1.484 | 1.01‐2.17 | 0.043 |
| Change in MELD score, per point | 1.26 | 0.9‐1.76 | 0.176 | — | — | — |
| FU MELD score, per point | 1.25 | 1.13‐1.38 | 0.001 | 1.15 | 1.01‐1.32 | 0.042 |
| HVPG decrease ≥ 10% | 0.056 | 0.007‐0.483 | 0.006 | 0.102 | 0.012‐0.863 | 0.036 |
Abbreviation: aHR, adjusted hazard ratio.
Comparison With Other Potential Definitions of HVPG Response
Using more restrictive definitions of HVPG response, such as an HVPG decrease ≥ 20%, HVPG decrease ≥ 20% or to ≤9 mm Hg, or an HVPG decrease ≥ 20% or to ≤ 12 mm Hg (in the subgroup of patients with BL HVPG > 12 mm Hg), did not increase the sensitivity for hepatic decompensation during FU, because 1 decompensated patient with HVPG response developed further hepatic decompensation, regardless of the definition used (Supporting Table S2). However, a lower proportion of patients (49%‐51%) achieved HVPG response according to these definitions, and thus, additional patients without hepatic decompensation were assigned to the HVPG nonresponse group. Accordingly, the probability of hepatic decompensation in patients with HVPG nonresponse tended to decrease, suggesting an inferior discriminatory ability of these more restrictive definitions of HVPG response.
PH and De Novo Hepatocellular Carcinoma
Six patients were diagnosed with de novo hepatocellular carcinoma (HCC), who all had CSPH at BL. However, HCC seemed to develop independently of the evolution of PH during treatment: Two patients who developed FU HCC showed an HVPG decrease to ≤ 9 mm Hg at FU. Moreover, 83% (5/6) had an HVPG decrease ≥ 10%.
Factors Associated With an HVPG Decrease ≥ 10% in Patients With Pretreatment CSPH
Please see Table 1 and the Supporting Information.
Noninvasive Diagnosis of CSPH at FU and HVPG Decrease ≥ 10%
In the overall study population, liver stiffness (AUROC, 0.92; 95% CI, 0.863‐0.978; P < 0.001), platelet count (AUROC, 0.816; i.e., 1 minus 0.184, due to indirect association; 95% CI, 0.724‐0.908; P < 0.001), VWF (AUROC, 0.807; 95% CI, 0.714‐0.9; P < 0.001), and VITRO (AUROC, 0.877; 95% CI, 0.802‐0.952; P < 0.001) showed a high diagnostic accuracy for CSPH at FU (Supporting Table S3; Fig. 3).
Figure 3.
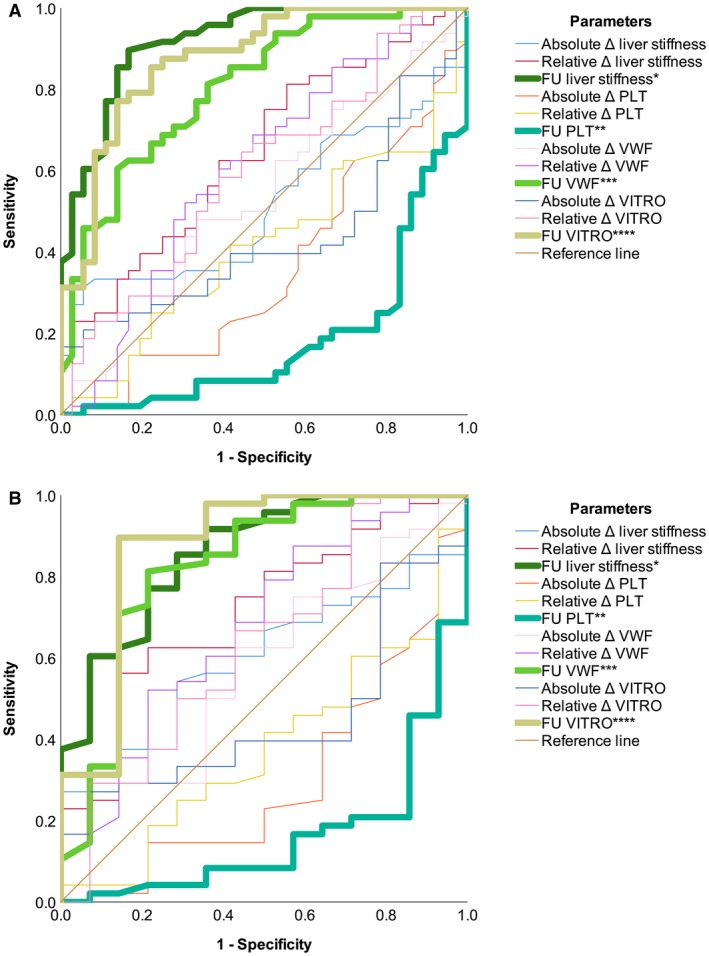
Performance of noninvasive markers for diagnosing CSPH at FU. Bold lines indicate parameters statistically significantly associated with the condition. (A) All patients (AUROC: *0.92; **0.816, i.e., 1 minus 0.184, due to indirect association; ***0.807; ****0.877). (B) Subgroup of patients with CSPH at BL (AUROC: *0.86; **0.836, i.e., 1 minus 0.164, due to indirect association; ***0.822; ****0.876).
The diagnostic indices of the previously defined FU liver stiffness cutoffs for ruling out (12.4 kPa; sensitivity, 97.9%) and ruling in FU CSPH (25.3 kPa; specificity, 94.4%)8 remained unchanged despite adding additional patients, with 39.3% (33/84) of patients being unclassifiable. The false‐negative patient had an FU liver stiffness of 11.8 kPa and an FU HVPG value of 17 mm Hg and did not achieve an HVPG response ≥ 10%; however, this patient did not develop hepatic decompensation.
The newly defined FU VITRO cutoffs for ruling out and ruling in FU CSPH were 0.95 (sensitivity, 95.8%) and 3.3 (specificity, 94.4%), with 53.6% (45/84) of patients in the diagnostic gray zone. The false‐negative patient had an FU VITRO 0.92, an FU liver stiffness of 17.1 kPa, and an FU HVPG value of 17 mm Hg and did not achieve an HVPG response ≥ 10%; however, this patient did not develop hepatic decompensation.
Using these tests sequentially (i.e., assessing FU VITRO in patients who were unclassifiable based on FU liver stiffness and vice versa) statistically significantly decreased the proportion of patients in the diagnostic gray zone to 25% (21/84; P = 0.047 and P < 0.001 compared with FU liver stiffness and VITRO alone, respectively), while maintaining a high sensitivity (liver stiffness first, 93.8%; VITRO first, 93.8%) and specificity (liver stiffness first, 87.1%; VITRO first, 90.3%) for ruling out and ruling in FU CSPH, respectively.
Interestingly, the diagnostic performance of liver stiffness tended to worsen if the regression of CSPH was assessed, i.e., when the analysis was restricted to patients with CSPH at BL (AUROC, 0.86; 95% CI, 0.751‐0.969; P < 0.001). In contrast, the AUROC of PLT and VWF numerically increased in this subgroup of patients, with VITRO showing the highest diagnostic accuracy (AUROC, 0.876; 95% CI, 0.749‐1; P < 0.001) of all assessed parameters.
Although liver stiffness, platelet count, VWF, and VITRO, as well as their relative/absolute changes during treatment, showed statistically significant associations with HVPG decrease ≥ 10%, all AUROC values were below 0.8.
Evolution of HVPG and Hepatic Decompensation During Long‐Term FU
Thirteen (19%) out of 67 patients with CSPH at BL underwent an additional HVPG measurement (“last”) after the first assessment of FU HVPG. These HVPG measurements were performed 31.2 ± 3.4 months after treatment initiation, which was 26.8 ± 3.36 months after the end of IFN‐free therapy and 20.1 ± 2.8 months after the first posttreatment HVPG measurement.
In these selected patients, HVPG decreased from 15.2 ± 1.7 to 11.8 ± 1.8 mm Hg, which corresponds to an absolute change of −3.38 ± 0.68 mm Hg (P < 0.001) and a relative change of −24.4 ± 4.1% (Supporting Fig. S2). The proportion of patients who resolved CSPH increased from 15% (2/13) at the second (“FU”) assessment to 46% (6/13) at the last HVPG measurement. Seven (54%) out of 13 patients had achieved an HVPG decrease ≥ 10% at the second (“FU”) HVPG assessment, which was maintained in all patients. Moreover, 4 additional patients achieved an HVPG decrease ≥ 10% at the last measurement, resulting in an overall rate of long‐term HVPG decrease ≥ 10% of 85% (11/13) in this series of patients.
Interestingly, we observed a close correlation between hemodynamic changes and the patients’ clinical course: Whereas all (11/11) patients who maintained/achieved an HVPG decrease ≥ 10% at last HVPG measurement had an uneventful posttreatment FU period (including 5 patients with a history of hepatic decompensation), both patients without a decrease ≥ 10% at last HVPG had hepatic decompensation (Fig. 4).
Figure 4.
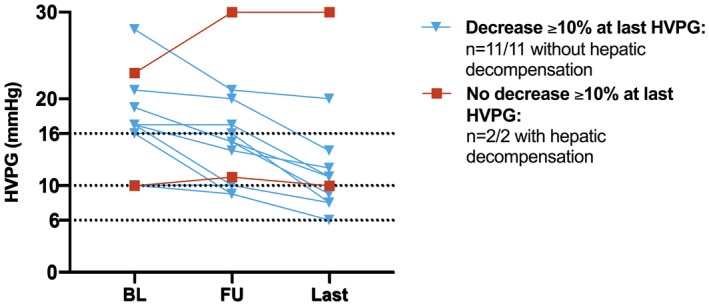
Evolution of HVPG in 13 patients who underwent an additional (“last”) HVPG measurement after the first FU measurement and correlation with hepatic decompensation after treatment.
Exploratory Analysis on Changes in Markers of Bacterial Translocation and Inflammation
Discussion
The approval of highly effective IFN‐free therapies has ushered in a new era in the treatment of CHC.2, 4 Because even in patients with PH, SVR is almost universal, CHC is seen as an excellent model for the study of liver disease regression after the cure of the primary etiologic factor.
Although viral suppression in chronic hepatitis B may have comparable effects,35 its effect on HVPG has only been studied in a small series of patients and was not correlated with direct endpoints. Moreover, despite the high prevalence of ALD, information on the impact of alcohol abstinence on HVPG and its predictive value for clinical events is limited.16 Finally, the prognostic impact of HVPG decreases due to pharmacological therapies for NASH is currently being investigated. The results of a recent randomized controlled trial on simtuzumab, a monoclonal antibody against lysyl oxidase‐like 2, in patients with compensated cirrhosis suggest that an HVPG decrease ≥ 20% is associated with a lower risk of hepatic decompensation.17 However, simtuzumab has been found to be ineffective in this setting, and one third of patients included in this analysis have been assigned to the placebo group.36 Thus, this study confirms the prognostic value of HVPG and its changes over time in compensated NASH cirrhosis rather than providing strong evidence for the use of HVPG response as a surrogate outcome for the clinical effectiveness of an etiological treatment.17 Accordingly, insights from patients with CHC are of great importance for advancing the understanding of the association between changes in HVPG due to therapies targeting intrahepatic resistance (i.e., etiological treatments) and clinical outcomes. Evidence from such studies is essential for promoting HVPG as a surrogate endpoint accepted by regulatory authorities.
In a previous study investigating hemodynamic changes during antiviral therapy with pegylated interferon and ribavirin, pretreatment HVPG/CSPH was predictive of hepatic decompensation during follow‐up.37 However, because of the low efficacy of this regimen in patients with cirrhosis, only a minority achieved SVR, and thus, the prognostic impact of changes in HVPG and posttreatment values was not assessed. Although pretreatment HVPG strata were predictive of clinical events in our study, the discriminatory ability of these HVPG strata improved substantially when posttreatment HVPG values were considered. This may be explained by patients with pretreatment CSPH being reclassified as having no or only subclinical PH (i.e., HVPG ≤ 9 mm Hg) after treatment, which was fully protective of hepatic decompensation. In addition, the proportion of patients with profound PH (HVPG ≥ 16 mm Hg, i.e., patients at high risk for hepatic decompensation) decreased after treatment, which may have enriched this subgroup with patients who have already passed a potential point of no return in the natural history of liver disease, resulting in a particularly bad prognosis. Accordingly, posttreatment but not pretreatment HVPG was predictive of hepatic decompensation in AUROC analysis if only patients at risk (i.e., patients with pretreatment CSPH) were considered. This is in line with the recent observation that a dynamic model based on serial HVPG measurements is closely linked with the risk of hepatic decompensation and/or death.38
However, the concept of using HVPG cutoffs for risk stratification has an important limitation, which has also been observed in studies assessing the HVPG response to NSBB.15 Although a decrease below a certain cutoff (i.e., HVPG ≤ 12 mm Hg) was protective and thus highly sensitive for (recurrent) variceal bleeding, such a decrease was only achieved by a small proportion of patients. Therefore, this cutoff was combined with the more commonly obtained HVPG reduction by ≥ 20% to increase the specificity of HVPG response.15 In order to further increase the positive predictive value (PPV) of this definition in patients at a lower risk of variceal bleeding, the Baveno VI faculty recently adopted the more specific 10% cutoff for primary prophylaxis.13 Interestingly, the Youden’s index‐derived optimal cutoff would have been ≥5% in our study; however, considering the imprecision of the measurement, using this cutoff may have substantially limited the reproducibility of HVPG response assessment in patients with HVPG < 20 mm Hg. Accordingly, we used the slightly less specific 10% cutoff, which is also recommended by the Baveno VI chapter on the impact of etiological treatment.13 On the one hand, it cannot be ruled out that treatments targeting intrahepatic resistance (e.g., statins) have beneficial effects despite achieving less pronounced but consistent reductions in HVPG.20 Especially in patients undergoing etiological therapy, even small changes may denote a trend in the “right” direction (i.e., disease regression). However, on the other hand, the required HVPG reduction could also be higher,39 because of the absence of the nonhemodynamic effects of NSBB.18, 19 In our study, more restrictive definitions of HVPG response (e.g., HVPG decrease ≥ 20%) seemed to worsen its discriminatory ability, which argues against the latter hypothesis.
In patients with pretreatment CSPH, an HVPG reduction ≥ 10% translated into a clinical benefit, especially in patients with cACLD who were completely protected from hepatic decompensation. In contrast, the trend toward a lower risk of further hepatic decompensation in patients with an HVPG decrease ≥ 10% did not attain statistical significance, which may be explained by the low number (n = 13) of patients with decompensated CSPH included in our study. Data obtained in a series of 13 patients with posttreatment CSPH undergoing a third HVPG measurement further strengthened the link between relative changes in HVPG and hepatic decompensation. Importantly, the use of relative changes in HVPG, rather than posttreatment HVPG, substantially increased the proportion of patients correctly identified as being at low risk (i.e., specificity/PPV of HVPG response). This is in line with the findings of the abovementioned studies, which defined HVPG response to NSBB treatment.
In line with previous reports,8, 10 patients with more advanced liver disease were less likely to achieve HVPG response, possibly because of the perpetuation of mechanisms promoting PH in some of these patients. Although pretreatment values of HVPG and most noninvasive markers of PH were comparable between patients with an HVPG reduction ≥ 10%, or without, we observed profound differences in the absolute and relative changes, as well as posttreatment values of liver stiffness, VWF, and VITRO score, indicating that these markers of PH may have utility for the noninvasive monitoring of the evolution of PH after SVR.
Although HVPG measurement is generally safe and well tolerated,40 its clinical use is limited by its invasiveness, and its availability is mostly restricted to academic centers. Accordingly, the development/validation of noninvasive methods is essential for promoting personalized medicine in patients with ACLD who achieved SVR. Posttreatment liver stiffness and VITRO showed an excellent diagnostic performance for FU CSPH. Nevertheless, when restricting the analysis to patients with pretreatment CSPH in order to evaluate the regression of CSPH, the AUROC of TE numerically decreased, whereas the AUROC of VITRO increased. This might be a consequence of liver stiffness being less strongly correlated with HVPG at values ≥ 10‐12 mm Hg in patients without NSBB treatment,41 and VWF may show a better correlation with HVPG in this patient population.22, 42
We confirmed the diagnostic value of our previously defined liver stiffness cutoffs (sensitivity/specificity of about 95%) for ruling in (25.3 kPa) and ruling out (12.4 kPa) posttreatment CSPH; however, a high proportion of patients were unclassifiable. Applying VITRO cutoffs chosen to obtain similar sensitivity and specificity (ruling out, 0.95/ruling in, 3.3), the proportion of patients within the diagnostic gray zone was even higher. However, performing these tests in a sequential manner substantially reduced the proportion of unclassifiable patients and thus increased their potential value for clinical practice. In addition, Thabut and colleagues43 recently validated the Baveno VI criteria for screening for high‐risk varices in patients with SVR/viral suppression. Accordingly, there is an increasing number of noninvasive tools that may guide clinical decision making in this setting. Still, these noninvasive methods do not confer the same information as HVPG response, as they are incapable of monitoring the dynamics of PH with adequate accuracy. It would have been interesting to assess the diagnostic performance of spleen stiffness in this setting, because changes in spleen stiffness have recently shown some promise as a noninvasive marker of HVPG response to carvedilol.44 Future studies assessing clinical endpoints in larger cohorts should aim to refine the use of combinations and/or serial assessments of noninvasive tests for risk stratification.
SVR to IFN‐free therapy was associated with a decrease in LBP and tended to lower IL‐6 levels. However, changes were highly heterogenous and independent of the evolution of PH. Interestingly, a similar pattern has been observed in a study investigating the impact of NSBB treatment on measures of gastrointestinal permeability and LBP/IL‐6 levels.45 Although NSBB treatment lowered HVPG and decreased all of these parameters, the changes were not interrelated. This may suggest that PH and bacterial translocation as well as inflammation are not as closely linked as assumed by some authors. Moreover, a recent study demonstrated substantial interindividual variability in measures of inflammation (i.e., peaks of IL‐6 that resolved spontaneously),46 which may have interfered with the analysis of repeated assessments.
Of note, de novo HCC occurred only in patients with pretreatment CSPH, which is in line with the previous observations that patients with CSPH bear a 6‐fold increased risk of HCC47 as compared with patients with cirrhosis but without CSPH. However, incident HCC was not linked to the changes in PH, as it commonly occurred in patients who showed clear evidence of liver disease regression. This suggests that despite the amelioration of hepatic inflammation,9 previously acquired genetic alterations and residual morphological changes/changes in the hepatic microenvironment in patients with pretreatment CSPH may play a central role in hepatocarcinogenesis after SVR. Because death due to complications of PH was rare, we would like to highlight the importance of screening programs for the timely diagnosis of HCC in these patients, who otherwise mostly have a favorable prognosis.5, 7
We must acknowledge several limitations of our study. Because of limited sample size, we combined patients with cACLD and decompensated ACLD for the main analyses. However, we were able to confirm the prognostic value of an HVPG decrease ≥ 10% in an analysis restricted to the important subgroup of patients with cACLD. Finally, we abstained from performing competing risk analyses,31 as the incidence of competing risks (i.e., liver transplantation/death without previous hepatic decompensation) was very low. Furthermore, it cannot be assumed that the observations on long‐term changes in HVPG are generalizable, as only a minority of patients underwent an additional (“last”) HVPG measurement, which may have led to selection bias. However, the profound long‐term changes in HVPG raise questions about the optimal time point for the assessment of posttreatment HVPG. In our study, the time intervals between the end of treatment and posttreatment HVPG measurements were not standardized, although they were within a reasonable range in all patients. The median time span was close to the time to SVR assessment (i.e., 12 weeks). A later assessment may have resulted in more pronounced decreases in HVPG, which may have increased the rate of an HVPG reduction ≥ 10% and thus may have restratified more patients to lower‐risk HVPG strata. However, it would have also led to a higher number of episodes of hepatic decompensation occurring before the posttreatment HVPG measurement.
In conclusion, reassessment of HVPG after the cure of hepatitis C improved prognostication. Patients with subclinical PH before/after IFN‐free treatment are at negligible risk for hepatic decompensation, as we did not observe progression to CSPH. The cure of the primary etiologic factors induced an “immediate” HVPG decrease ≥ 10% in 60% of patients with pretreatment CSPH, which translated into a clinically meaningful benefit, as it prevented hepatic decompensation. This finding provides important evidence for the use of HVPG as a surrogate endpoint for interventions that lower portal pressure by decreasing intrahepatic resistance. Noninvasive methods such as TE and VITRO score cannot substitute HVPG measurement, as they are incapable of monitoring the dynamics of PH with adequate accuracy; however, they may be used for ruling in or ruling out posttreatment CSPH.
Supporting information
Supported by a grant from the Medical Scientific Fund of the Major of the City of Vienna (No. 17035) as well as the Andrew K. Burroughs short‐term training fellowship of the European Association for the Study of the Liver.
Potential conflict of interest: Dr. Chromy consults for, advises, is on the speakers’ bureau for, and/or received travel support from AbbVie and Gilead. He consults for, advises, and/or is on the speakers’ bureau for MSD. Dr. Ferenci consults for, advises, is on the speakers’ bureau for, and/or received grants from Gilead. He consults for, advises, and/or is on the speakers’ bureau for AbbVie, Bristol‐Myers Squibb, and MSD. Dr. Garcia‐Pagan consults for, advises, and/or is on the speakers’ bureau for Cook and W.L. Gore & Associates. He received grants from Conatus, Exalenz, Novartis, and Theravance. Dr. Hernandez‐Gea consults for, advises, and/or is on the speakers’ bureau for W.L Gore & Associates. Dr. Kozbial received travel support from AbbVie, Gilead, and Bristol‐Myers Squibb. Dr. Mandorfer consults for, advises, is on the speakers’ bureau for, and/or received travel support from AbbVie, Bristol‐Myers Squibb, and Gilead. He consults for, advises, and/or is on the speakers’ bureau for W.L. Gore & Associates. Dr. Steindl‐Munda consults for, advises, is on the speakers’ bureau for, and/or received travel support from AbbVie and Gilead. She consults for, advises, and/or is on the speakers’ bureau for Intercept and MSD. She received travel support from Falk. Dr. Peck‐Radosavljevic consults for, advises, is on the speakers’ bureau for, and/or received travel support from AbbVie and Gilead. He consults for, advises, and/or is on the speakers’ bureau for Bristol‐Myers Squibb and MSD. Dr. Pinter consults for, advises, is on the speakers’ bureau for, and/or received travel support from Bayer and Bristol‐Myers Squibb. He consults for, advises, and/or is on the speakers’ bureau for Eisai, Ipsen, Lilly, and MSD. Dr. Reiberger consults for, advises, is on the speakers’ bureau for, and/or received grants/travel support from AbbVie, Boehringer Ingelheim, Gilead, MSD, and W.L. Gore & Associates. He consults for, advises, and/or is on the speakers’ bureau for Intercept and Siemens. He received grants from Philips. Dr. Schwabl received travel support from AbbVie, Falk, and Gilead. Dr. Stättermayer consults for, advises, and/or is on the speakers’ bureau for Boehringer Ingelheim, Gilead, and MSD. Dr. Trauner consults for, advises, is on the speakers’ bureau for, and/or received grants/travel support from Albireo, Falk, Gilead, Intercept, and MSD. He consults for consults for, advises, and/or is on the speakers’ bureau for Bristol‐Myers Squibb, Novartis, Phenex, and Regulus. He received grants/travel support from AbbVie and Takeda. He is also co‐inventor of patents on the medical use of 24‐norursodeoxycholic acid.
References
Author names in bold designate shared co‐first authorship.
- 1. Mandorfer M, Schwabl P, Steiner S, Scheiner B, Chromy D, Bucsics T, et al. Interferon‐free treatment with sofosbuvir/daclatasvir achieves sustained virologic response in 100% of HIV/hepatitis C virus‐coinfected patients with advanced liver disease. AIDS 2016;30:1039‐1047. [DOI] [PubMed] [Google Scholar]
- 2. Mandorfer M, Schwabl P, Steiner S, Reiberger T, Peck‐Radosavljevic M. Advances in the management of HIV/HCV coinfection. Hepatol Int 2016;10:424‐435. [DOI] [PubMed] [Google Scholar]
- 3. Mandorfer M, Kozbial K, Freissmuth C, Schwabl P, Stattermayer AF, Reiberger T, et al. Interferon‐free regimens for chronic hepatitis C overcome the effects of portal hypertension on virological responses. Aliment Pharmacol Ther 2015;42:707‐718. [DOI] [PubMed] [Google Scholar]
- 4. Ferenci P, Kozbial K, Mandorfer M, Hofer H. HCV targeting of patients with cirrhosis. J Hepatol 2015;63:1015‐1022. [DOI] [PubMed] [Google Scholar]
- 5. Jacobson IM, Lim JK, Fried MW. American Gastroenterological Association Institute Clinical Practice Update‐Expert Review: care of patients who have achieved a sustained virologic response after antiviral therapy for chronic hepatitis C infection. Gastroenterology 2017;152:1578‐1587. [DOI] [PubMed] [Google Scholar]
- 6. Lens S, Berbel C, Forns X, García‐Pagán JC. Portal hypertension reverses following successful antiviral treatment for HCV: fact or fiction? Curr Hepatol Rep 2018;17:209‐217. [Google Scholar]
- 7. Ioannou GN, Feld JJ. What are the benefits of a sustained virologic response to direct‐acting antiviral therapy for hepatitis C virus infection? Gastroenterology 2019;156:446‐460. [DOI] [PubMed] [Google Scholar]
- 8. Mandorfer M, Kozbial K, Schwabl P, Freissmuth C, Schwarzer R, Stern R, et al. Sustained virologic response to interferon‐free therapies ameliorates HCV‐induced portal hypertension. J Hepatol 2016;65:692‐699. [DOI] [PubMed] [Google Scholar]
- 9. Schwabl P, Mandorfer M, Steiner S, Scheiner B, Chromy D, Herac M, et al. Interferon‐free regimens improve portal hypertension and histological necroinflammation in HIV/HCV patients with advanced liver disease. Aliment Pharmacol Ther 2017;45:139‐149. [DOI] [PubMed] [Google Scholar]
- 10. Lens S, Alvarado‐Tapias E, Marino Z, Londono MC, LLop E, Martinez J, et al. Effects of all‐oral anti‐viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus‐associated cirrhosis. Gastroenterology 2017;153:1273‐1283. [DOI] [PubMed] [Google Scholar]
- 11. Afdhal N, Everson GT, Calleja JL, McCaughan GW, Bosch J, Brainard DM, et al. Effect of viral suppression on hepatic venous pressure gradient in hepatitis C with cirrhosis and portal hypertension. J Viral Hepat 2017;24:823‐831. [DOI] [PubMed] [Google Scholar]
- 12. Bosch J, Abraldes JG, Berzigotti A, Garcia‐Pagan JC. The clinical use of HVPG measurements in chronic liver disease. Nat Rev Gastroenterol Hepatol 2009;6:573‐582. [DOI] [PubMed] [Google Scholar]
- 13. de Franchis R, Baveno VIF. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol 2015;63:743‐752. [DOI] [PubMed] [Google Scholar]
- 14. Reiberger T, Puspok A, Schoder M, Baumann‐Durchschein F, Bucsics T, Datz C, et al. Austrian consensus guidelines on the management and treatment of portal hypertension (Billroth III). Wien Klin Wochenschr 2017;129:135‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mandorfer M, Hernández‐Gea V, Reiberger T, Garca‐Pagan JC. Hepatic venous pressure gradient‐response in non‐selective beta‐blocker treatment – is it worth measuring? Curr Hepatol Rep 2019;18:174‐186. [Google Scholar]
- 16. Vorobioff J, Groszmann RJ, Picabea E, Gamen M, Villavicencio R, Bordato J, et al. Prognostic value of hepatic venous pressure gradient measurements in alcoholic cirrhosis: a 10‐year prospective study. Gastroenterology 1996;111:701‐709. [DOI] [PubMed] [Google Scholar]
- 17. Sanyal AJ, Harrison SA, Ratziu V, Abdelmalek MF, Diehl AM, Caldwell S, et al. The natural history of advanced fibrosis due to nonalcoholic steatohepatitis: data from the simtuzumab trials. Hepatology 2019. Apr 16. 10.1002/hep.30664. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18. Mandorfer M, Reiberger T. Beta blockers and cirrhosis, 2016. Dig Liver Dis 2017;49:3‐10. [DOI] [PubMed] [Google Scholar]
- 19. Reiberger T, Mandorfer M. Beta adrenergic blockade and decompensated cirrhosis. J Hepatol 2017;66:849‐859. [DOI] [PubMed] [Google Scholar]
- 20. Abraldes JG, Trebicka J, Chalasani N, D'Amico G, Rockey DC, Shah VH, et al. Prioritization of therapeutic targets and trial design in cirrhotic portal hypertension. Hepatology 2019;69:1287‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vilaseca M, Guixe‐Muntet S, Fernandez‐Iglesias A, Gracia‐Sancho J. Advances in therapeutic options for portal hypertension. Therap Adv Gastroenterol 2018;11:1756284818811294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mandorfer M, Schwabl P, Paternostro R, Pomej K, Bauer D, Thaler J, et al. Von Willebrand factor indicates bacterial translocation, inflammation, and procoagulant imbalance and predicts complications independently of portal hypertension severity. Aliment Pharmacol Ther 2018;47:980‐988. [DOI] [PubMed] [Google Scholar]
- 23. Maieron A, Salzl P, Peck‐Radosavljevic M, Trauner M, Hametner S, Schofl R, et al. Von Willebrand Factor as a new marker for non‐invasive assessment of liver fibrosis and cirrhosis in patients with chronic hepatitis C. Aliment Pharmacol Ther 2014;39:331‐338. [DOI] [PubMed] [Google Scholar]
- 24. Hametner S, Ferlitsch A, Ferlitsch M, Etschmaier A, Schofl R, Ziachehabi A, et al. The VITRO score (Von Willebrand factor antigen/thrombocyte ratio) as a new marker for clinically significant portal hypertension in comparison to other non‐invasive parameters of fibrosis including ELF test. PLoS One 2016;11:e0149230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sarrazin C, Berg T, Buggisch P, Dollinger MM, Hinrichsen H, Hofer H, et al. S3 guideline hepatitis C addendum [in German]. Z Gastroenterol 2015;53:320‐334. [DOI] [PubMed] [Google Scholar]
- 26. Sarrazin C, Zimmermann T, Berg T, Neumann UP, Schirmacher P, Schmidt H, et al. Prophylaxis, diagnosis and therapy of hepatitis‐C‐virus (HCV) infection: the German guidelines on the management of HCV infection ‐ AWMF‐Register‐No.: 021/012 [in German]. Z Gastroenterol 2018;56:756‐838. [DOI] [PubMed] [Google Scholar]
- 27. European Association for Study of Liver . EASL recommendations on treatment of hepatitis C 2015. J Hepatol 2015;63:199‐236. [DOI] [PubMed] [Google Scholar]
- 28. European Association for the Study of the Liver . EASL recommendations on treatment of hepatitis C 2016. J Hepatol 2017;66:153‐194. [DOI] [PubMed] [Google Scholar]
- 29. European Association for the Study of the Liver . EASL recommendations on treatment of hepatitis C 2018. J Hepatol 2018;69:461‐511. [DOI] [PubMed] [Google Scholar]
- 30. Reiberger T, Schwabl P, Trauner M, Peck‐Radosavljevic M, Mandorfer M. Measurement of the hepatic venous pressure gradient and transjugular liver biopsy. J Vis Exp. In press. [DOI] [PubMed] [Google Scholar]
- 31. D'Amico G, Morabito A, D'Amico M, Pasta L, Malizia G, Rebora P, et al. Clinical states of cirrhosis and competing risks. J Hepatol 2018;68:563‐576. [DOI] [PubMed] [Google Scholar]
- 32. Ripoll C, Groszmann R, Garcia‐Tsao G, Grace N, Burroughs A, Planas R, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology 2007;133:481‐488. [DOI] [PubMed] [Google Scholar]
- 33. Berzigotti A, Garcia‐Tsao G, Bosch J, Grace ND, Burroughs AK, Morillas R, et al. Obesity is an independent risk factor for clinical decompensation in patients with cirrhosis. Hepatology 2011;54:555‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scheiner B, Steininger L, Semmler G, Unger LW, Schwabl P, Bucsics T, et al. Controlled attenuation parameter does not predict hepatic decompensation in patients with advanced chronic liver disease. Liver Int 2019;39:127‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manolakopoulos S, Triantos C, Theodoropoulos J, Vlachogiannakos J, Kougioumtzan A, Papatheodoridis G, et al. Antiviral therapy reduces portal pressure in patients with cirrhosis due to HBeAg‐negative chronic hepatitis B and significant portal hypertension. J Hepatol 2009;51:468‐474. [DOI] [PubMed] [Google Scholar]
- 36. Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 2018;155:1140‐1153. [DOI] [PubMed] [Google Scholar]
- 37. Lens S, Rincon D, Garcia‐Retortillo M, Albillos A, Calleja JL, Banares R, et al. Association between severe portal hypertension and risk of liver decompensation in patients with hepatitis C, regardless of response to antiviral therapy. Clin Gastroenterol Hepatol 2015;13:1846‐1853. [DOI] [PubMed] [Google Scholar]
- 38. Abraldes JG, Garcia‐Tsao G, Ripoll C, Patch D, Grace ND, Bosch J, et al. Dynamic prediction of the risk of decompensation/death in patients with compensated cirrhosis based on serial hepatic venous pressure gradient (HVPG) measurements. Hepatology 2018;68:97A. [Google Scholar]
- 39. Delgado MG, Bosch J. HVPG measurements as a surrogate of clinical events in cirrhosis: experience from clinical trials. Curr Hepatol Rep 2019;18:164‐173. [Google Scholar]
- 40. Casu S, Berzigotti A, Abraldes JG, Baringo MA, Rocabert L, Hernandez‐Gea V, et al. A prospective observational study on tolerance and satisfaction to hepatic haemodynamic procedures. Liver Int 2015;35:695‐703. [DOI] [PubMed] [Google Scholar]
- 41. Reiberger T, Ferlitsch A, Payer BA, Pinter M, Homoncik M, Peck‐Radosavljevic M; Vienna Hepatic Hemodynamic Lab . Non‐selective beta‐blockers improve the correlation of liver stiffness and portal pressure in advanced cirrhosis. J Gastroenterol 2012;47:561‐568. [DOI] [PubMed] [Google Scholar]
- 42. La Mura V, Reverter JC, Flores‐Arroyo A, Raffa S, Reverter E, Seijo S, et al. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut 2011;60:1133‐1138. [DOI] [PubMed] [Google Scholar]
- 43. Thabut D, Bureau C, Layese R, Bourcier V, Hammouche M, Cagnot C, et al. Validation of Baveno VI criteria for screening and surveillance of esophageal varices in patients with compensated cirrhosis and a sustained response to antiviral therapy. Gastroenterology 2019;156:997‐1009. [DOI] [PubMed] [Google Scholar]
- 44. Kim HY, So YH, Kim W, Ahn DW, Jung YJ, Woo H, et al. Non‐invasive response prediction in prophylactic carvedilol therapy for cirrhotic patients with esophageal varices. J Hepatol 2018;70:412‐422. [DOI] [PubMed] [Google Scholar]
- 45. Reiberger T, Ferlitsch A, Payer BA, Mandorfer M, Heinisch BB, Hayden H, et al. Non‐selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL‐6 in patients with cirrhosis. J Hepatol 2013;58:911‐921. [DOI] [PubMed] [Google Scholar]
- 46. Fernandez J, Claria J, Amoros A, Aguilar F, Castro M, Casulleras M, et al. Effects of albumin treatment on systemic and portal hemodynamics and systemic inflammation in patients with decompensated cirrhosis. Gastroenterology 2019;157:149‐162. [DOI] [PubMed] [Google Scholar]
- 47. Ripoll C, Groszmann RJ, Garcia‐Tsao G, Bosch J, Grace N, Burroughs A, et al. Hepatic venous pressure gradient predicts development of hepatocellular carcinoma independently of severity of cirrhosis. J Hepatol 2009;50:923‐928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol 2016;64:1388‐1402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials