

Abstract
Most phenotypic screens aiming to discover new antimalarial chemotypes begin with low cost, high-throughput tests against the asexual blood stage (ABS) of the malaria parasite life cycle. Compounds active against the ABS are then sequentially tested in more difficult assays that predict whether a compound has other beneficial attributes. Although applying this strategy to new chemical libraries may yield new leads, repeated iterations may lead to diminishing returns and the rediscovery of chemotypes hitting well-known targets. Here, we adopted a different strategy to find starting points, testing ∼70,000 open source small molecules from the Global Health Chemical Diversity Library for activity against the liver stage, mature sexual stage, and asexual blood stage malaria parasites in parallel. In addition, instead of using an asexual assay that measures accumulated parasite DNA in the presence of compound (SYBR green), a real time luciferase-dependent parasite viability assay was used that distinguishes slow-acting (delayed death) from fast-acting compounds. Among 382 scaffolds with the activity confirmed by dose response (<10 μM), we discovered 68 novel delayed-death, 84 liver stage, and 68 stage V gametocyte inhibitors as well. Although 89% of the evaluated compounds had activity in only a single life cycle stage, we discovered six potent (half-maximal inhibitory concentration of <1 μM) multistage scaffolds, including a novel cytochrome bc1 chemotype. Our data further show the luciferase-based assays have higher sensitivity. Chemoinformatic analysis of positive and negative compounds identified scaffold families with a strong enrichment for activity against specific or multiple stages.
Keywords: malaria, multistage antimalarials, drug discovery, chemoinformatic analysis, whole genome sequencing (WGS)
Malaria parasites remain a lethal threat for nearly half the world’s population and claimed an estimated 435,000 lives in 2017.1 Efforts to prevent malaria come primarily through vector control measures, such as the use of insecticide-treated bednets2,3 with symptomatic relief provided by small molecule therapeutics. Although worldwide efforts have led to substantial declines in morbidity and mortality over the past 20 years, progress has recently stalled, with the WHO malaria reports now noting increases in cases after years of decline.1 Although there are many possible reasons, including population growth, insecticide resistance, political conflict, and parasite resistance to artemisinin-based combination therapies,4 the fact remains that current treatment options are suboptimal. Importantly, current therapies that are widely used either allow continued disease transmission (artemisinin derivatives and 4-aminoquinolines) or are associated with high rates of resistance (artemisinin derivatives, 4-aminoquinolines, antifolates, and cytochrome bc1 inhibitors),5 limiting their widespread use.
In addition to resistance liabilities, few commonly used therapies prevent malaria. Malaria infections starts in the liver via the delivery of about 100 sporozoites by the bite of a female Anopheles mosquito.6,7 The parasites migrate to the liver where the exoerythrocytic forms (EEF) develop; disease symptoms only occur when asexual blood stage (ABS) parasites emerge from the liver and begin replicating in erythrocytes, 1 to 2 weeks later.8,9 The prevention of malaria infection by killing EEFs is ideal because most malaria deaths come from complications that arise during the subsequent ABS. Furthermore, as parasite numbers are very low (in contrast to an ABS infection, which may involve billions of parasites), resistance is less likely to emerge. Artemisinin combination therapies, the most widely used treatments, do not prevent malaria, and while antifolates (pyrimethamine) can prevent infection, the widespread and undisciplined use has rendered them largely ineffective (although they are still used for seasonal prophylaxis). Mitochondrial inhibitors, such as atovaquone, also have resistance issues and are expensive.
Even fewer therapies block malaria transmission. During asexual replication, 1–5% of ABS parasites will differentiate into gametocytes (GAMs), the cells that can immediately differentiate into male and female gametes once the cells sense that they have left the vertebrate host. Mature gametocytes (stage V) are the only parasite forms able to survive in the mosquito midgut and are responsible for disease transmission.10−12 Patients who take a drug that only targets the ABS may continue to transmit malaria parasites to their neighbors. Primaquine and now tafenoquine are the only drugs used to block transmission, but both are toxic to patients with glucose-6 phosphate deficiencies.13
Armed with our knowledge of the parasite’s life cycle, the field has come to recognize that better drugs are possible.14 This has spurred a search for potent scaffolds that act throughout the parasite’s complex life cycle,15 primarily using phenotypic screening with cell culture models that predict activity against different parasite life cycle stages, such as the exoerythrocytic (liver) stage, the blood stage, and stage V gametocytes.16 Multistage active compounds have been identified by iteratively screening libraries through successive life cycle stages,17−19 and promising antimalarial drug candidates are currently progressing through clinical trials following the chemotype’s discovery with such screening strategies.20−23 However, stepwise filtering fails to identify all multistage active scaffolds, since hits from only one stage are carried forward. Furthermore, many stage-specific compounds, which could be formulated into combination therapies for a multistage drug, are ignored by these methods.
Previous large-scale ABS phenotypic screens have typically relied on readouts that measure parasite abundance rather than parasite viability. For example, the DNA intercalating dye, SYBR Green I, indicates decreases in the amounts of parasite DNA after 72 h of compound exposure relative to untreated control cultures.24−26 This method is simple and cost-effective with a minimum number of liquid transfer steps but is theoretically less sensitive than other methods as it detects the accumulation of total DNA: after 72 h only, a 10- to 20-fold difference between treated and untreated wells might be observed for a fast-acting compound like artemisinin, but slow-acting compounds may only give a 50% reduction in signal. Alternatively, Gamo et al. used a lactate dehydrogenase assay that gives a readout that is proportional to parasite numbers.27 Both of these methods could fail to identify antimalarial compounds, which act more slowly or are less potent, including so-called, delayed-death inhibitors. Modified ABS assays could theoretically detect new antimalarial chemotypes that might have been overlooked in previous screens.
Here, we describe the life cycle-wide antimalarial screen of the Global Health Chemical Diversity Library (GHCDL).28 This library, consisting of ∼70,000 compounds, was screened in parallel against P. falciparum (Pf) ABS, P. falciparum stage V GAMs, and P. berghei liver stage (PbLuc). In addition, for the ABS stage, we explore the use of different incubation times and more sensitive luciferase assays, allowing the discovery of compounds that act more slowly against the ABS parasites and that might have been overlooked in previous screens that assessed total DNA synthesis in the parasite after inhibitor treatment.27 Our findings highlight a new chemical space to fill the current void of delayed death, transmission blocking, and causal prophylactic antimalarials.
Results
The Global Health Chemical Diversity Library Is Enriched for Druglike Chemotypes with No Known Antimalarial Activity
The GHCDL (Figure 1, Table S1) is comprised of 68,614 commercially available compounds that were selected for their diversity and adherence to lead-like physiochemical properties. All but 30 compounds obey the standard Lipinski Rule of Five,29 with an average molecular weight of 320.7 Da (184.20 to 492.23 Da), mean logP of 2.0 (−3.27 to 6.35), mean hydrogen bond donor count of 0.9 (0 to 5), and mean hydrogen bond acceptor count of 5.6 (1 to 10; see Table S2). In addition, because ideal antimalarials should be orally bioavailable, the library was constructed to give a topological polar surface area (TPSA) average of 64.2 Å2 (16.13 to 160.88 Å2), with an average rotatable bond count of 4.5 (0 to 10).30 The degree of carbon saturation as a surrogate for successful clinical testing outcome has been previously described31 and was emphasized in the GHCDL with an average fraction of sp3 hybridized carbon count (Fsp3) of 0.48 (0 to 1.0). Care was taken to maximize the library’s value by excluding scaffolds that were similar to those molecules in the Medicine for Malaria Venture’s drug development portfolio. This same library has been screened against other pathogens including Cryptosporidium, of which 31 compounds were shown to inhibit C. parvum development in HCT-8 cells.32 It has also been used to look for inhibitors of gamete formation in malaria parasites.33
Figure 1.
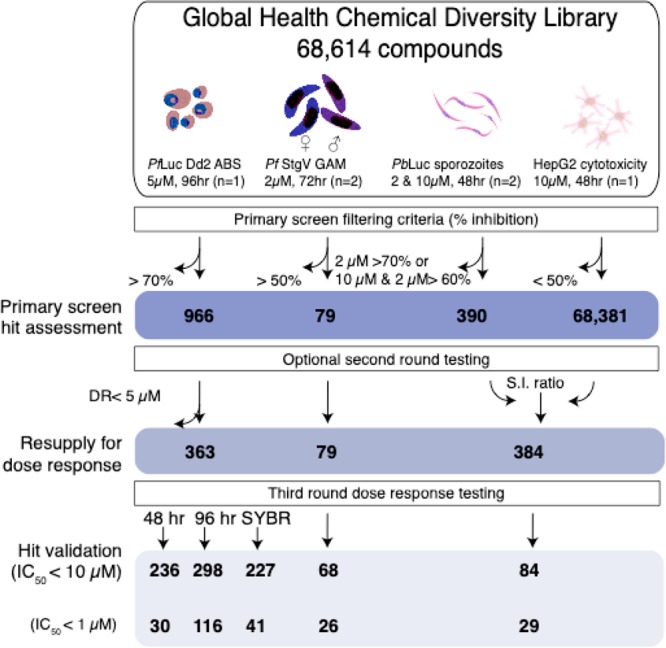
Global Health Chemical Diversity Library screening workflow. A set of 68,614 small molecules was sequentially screened across the Plasmodium life cycle. Horizontal arrows indicate the removal of unqualified compounds at a given criterion while vertical arrows track the progress of desirable ones. Primary screening data were filtered by inhibition to generate a list of reconfirmation worthy hits. Commercially available compounds for each stage were resupplied and reconfirmed in dose response to round off a series of potent leads. Total PbLuc active compounds includes recombinant luciferase inhibitors.
To assess the structural diversity of the library, we clustered the entire GHCDL by scaffold similarity using ECFP4 fingerprints and a Dice similarity coefficient (DSC) of 0.7. This generated an extensive similarity network, with scaffold families segregating into 11,194 discrete clusters with an average size of 5.19 members (stdev = 26.3). In the analysis, 10,411 compounds were considered singletons. The largest cluster consisted of 2131 compounds.
To demonstrate diversity, the compounds were coclustered with known antimalarials and compounds with confirmed activity (Table S3), which were discovered in past high-throughput efforts (24,595).17,27,34−36 Of these, only 2 compounds were identical to compounds in the GHCDL, and 60 showed a similarity of >75%. One compound, DDD01037968, bears the diaminopyrimidine scaffold found in antifolates such as pyrimethamine as well as 13 antimalarial chemotypes in the query list. As described below, DDD01037968 was active in ABS screens and would be expected to inhibit dihydrofolate reductase on the basis of past studies.24
A Luciferase ABS Screen Shows Increased Sensitivity for Slow-Acting Compounds
To interrogate the library, we first performed a screen for activity against the ABS. To improve the low signal-to-noise ratio inherent with methods that measure overall parasite growth, we used an alternative method that relies on a P. falciparum Dd2 (PfLuc) line expressing firefly luciferase37 under the control of the pfhrp3 (5′ UTR)/pfhrp2 (3′ UTR) promoter. Studies have shown that, in the presence of luciferin, these parasites emit light in proportion to the infection rate per well in real time.37 In order to capture even more compounds, our primary screen further used a 96 h incubation time to identify slow-acting compounds, also known as delayed-death inhibitors. These compounds typically act against the parasite’s apicoplast and have been commonly overlooked during previous large-scale screens that have used a 72 h incubation period.27 Interference with canonical apicoplast function is thought to cause the latent killing of the untreated daughter-generation parasites, resulting in a delayed-death phenotype.38 Known antimalarials, such as doxycycline, work by this mechanism and are used as prophylactic treatments.
The primary assay was run at a compound concentration of 5 μM. Briefly, 8 μL of an ABS parasite mixture (∼10,000 parasites) was dispensed into 1536-well plates previously spotted with 64,811 GHCDL compounds (the remaining 3642 library compounds arrived later and were evaluated separately). After a 96 h incubation, 2 μL of luciferin (BrightGlo) was added, and the luminescent signal was measured. Bioluminescence was normalized to DMSO (negative control) and artemisinin-treated (positive control) wells for the single point screens. For this primary screen, assay fidelity was determined on a per plate basis using both the Z′-factor and the potency variance of the positive (artemisinin) and negative (DMSO) controls. The mean Z′ value was 0.60 (range of 0.43–0.75), with only one plate falling below our ideal 0.5 threshold (Figure S1). Both controls were consistent, with a mean inhibition for artemisinin and DMSO equaling 99.6 ± 1.03% and −3.91 ± 29.2%, respectively.
The ABS primary screen yielded 950 compounds with >70% parasitic bioluminescence inhibition, relative to the controls (Table S1). Because single replicate screening will have more false positives and negatives (especially in low volume HTS formats where instrument tolerance impacts the accuracy of compound and culture dispensing), reconfirmation of the available 924 single point hits was performed against ABS parasites in single point triplicate at 1.25 μM. Even at this reduced concentration, 355 compounds averaged ≥50% inhibition and 175 compounds maintained ≥70% inhibition across three replicates (Table S4). As a final step, hits were retested (from DMSO stocks) in an 8-point (5–0.00232 μM; 96 h) dose response series against PfLuc Dd2, yielding 363 with potent activity (<3 μM). Following the primary screen and subsequent reconfirmation assays, another 3642 library compounds arrived from the library supplier (University of Dundee) (Table S4) and were funneled directly into single point triplicate testing at 1.25 μM. Another 16 compounds, which averaged >50% inhibition across all three replicates, were advanced to 11-point dose response testing, along with the hits from Set 1.
From these (Sets 1 and 2), compounds were resupplied as fresh powders and evaluated in 11-point dose response tests (Pfluc; n = 2) for the two time points. For additional dose response testing, we also sought to distinguish potential delayed-death inhibitors, such as clindamycin, from compounds that work quickly, like artesunate,39 by using two different incubation times, 48 and 96 h. Altogether, 298 compounds were reconfirmed with an IC50 of less than 10 μM at 96 h (PfLuc). Of these, 29 compounds showed a 48 h PfLuc IC50 of ≤1 μM with 116 others showing the same level of activity (<1 μM) in the 96 h PfLuc assay.
To further validate these compounds, we counterscreened the compounds in a 96 h SYBR green proliferation assay (Table S4). Interestingly, only 41 were active (IC50 < 1 μM) in the 96 h SYBR green assay. While some of the Pfluc hits could be biochemical firefly luciferase inhibitors, most (70 of 87) were not active (<1 μM) in the 48 h PfLuc assay or in the P. berghei luciferase liver assay described below. Interestingly, we observed that the average potency of our delayed-death control, clindamycin,38 at 96 h was 5.20 ± 1.25 nM (n = 4) and >10 μM (n = 4) in the luciferase and SYBR green assays, respectively (Figure 2a,b). Doxycycline, another delayed-death control, behaved similarly at 96 h (IC50 of 0.55 ± 0.20 μM) but showed some activity (7.18 ± 1.59 μM) in the 48 h PfLuc assay. These data show that, even at 96 h, the SYBR green 1 DNA replication assay lacks the sensitivity to robustly detect delayed-death inhibitors. In fact, the majority of the compounds that were detected with our 96 h Pfluc assay were missed in an ABS lactate dehydrogenase screen of GHCDL.33
Figure 2.

Delayed-death inhibitors. (a) Screening plate view. Heatmaps from the PfLuc dose response assays were superimposed to show preferential compound efficacy at the 48 h (light yellow) or 96 h (red) incubation times. Compounds were titrated in dose response and in identical patterns across both representative plates shown. Artemisinin controls are positive for parasite death at all time points, while delayed-death controls including clindamycin and doxycycline are only active at 96 h. The highest potency ABS inhibitor, DDD01057375, shows a similar trend to delayed-death controls. (b) Dose response curves for clindamycin for ABS and liver stage. (c) From the 68 delayed-death inhibitors, 22 belonged to scaffold families that were enriched for delayed-death inhibitors, relative to the entire library. Active delayed-death members are shown as dark red diamonds while inactive members are shown in pink. Additional data as well as error measurements are available in Tables S1 and S4. Data can be interactively explored at http://www.ndexbio.org/#/networkset/1ac81391-eebd-11e9-bb65-0ac135e8bacf?accesskey=8f0a93f855278c5ffdb1e8e52c3ca7b1983ac8452eb3b8409f99cc39fd783082.
For classification purposes, delayed-death inhibitors were those that showed a 48 to 96 h PfLuc IC50 ratio of >5. Using this criterion, 68 compounds (Figure 2c) were identified that could be grouped in 10 clusters with more than 2 hits in the delayed-death data set (probability of enrichment by chance = 1.11 × 10–8 to 5.95 × 10–5). This repeated, independent discovery of near identical chemotypes suggests that the screening method is effective (Figure 2). These 68 inhibitors preserve no structural similarity (Dice similarity coefficient of >70%) to 5 previously identified delayed-death inhibitors.38,40 However, a noteworthy quinoline substructure is present in DDD01057375, exhibiting the highest potency (7.14 × 10–3 μM; 95% CI of 0.004–0.01 μM) in the 96 h assay format against ABS parasites. These data highlight that delayed-death inhibitors were likely omitted from previous, large-scale screens focused on the ABS.
A Causal Prophylaxis Screen Identifies New Liver Stage Chemotypes
To identify compounds with potential prophylactic activity, we used a P. berghei rodent malaria liver stage model41 and HepG2-A16-CD81 hepatocytes.42 The P. berghei parasites express a firefly luciferase transgene reporter under the control of the eukaryotic elongation factor 1A (pbeef1aa) promoter43 that is active during the liver stage. The advantage of this rodent model is that it has a 48 h liver stage developmental time, which prevents hepatoma cell overgrowth. In addition, the murine species model represents a more abundant and safer alternative to human-infecting species while the use of hepatoma cells provides day to day consistency, making them ideal for high-throughput screening. We simultaneously conducted a library-wide cytotoxicity counter screen against uninfected hepatocytes to ensure that a loss of signal was not from host cell death. The luciferase-expressing sporozoites allow the assay to be run in 1536-well format and to have a low cost.19
For the screen, 5 μL of HepG2-A16-CD81 hepatocytes containing 3 × 103 cells was seeded into 1536-well plates, which had been prespotted with 10 nL of library compound (2 μM final assay concentration (Figure 1)) and incubated overnight. For PbLuc infected plates, approximately 750 purified sporozoites in 5 μL of screening media were added to each well and incubated for 48 h before measuring luciferase activity. Compounds were tested for toxicity in parallel (see Methods) by measuring uninfected hepatocyte viability using CellTiter-Glo. In total, 328 compounds inhibited parasite bioluminescence by >70% at 2 μM, while only 67 compounds inhibited uninfected hepatocyte growth by more than 50%. This hit rate was slightly lower than expected for an agnostic library, which prompted us to rescreen the entire library at 10 μM. The PbLuc and cytotoxicity assay plates for this second replicate would be stamped in-house with 50 nL of library compound per well, using the same compound plates as the sexual stage screen described below. All the assay parameters were kept similar, except the change in concentration and shortening the time the compounds spent in each well prior to infection. This yielded 1440 compounds with >75% PbLuc inhibition, of which 103 were found to be cytotoxic (>50% HepG2 inhibition). All potential false positive effectors from the CellTiter-Glo assay were filtered from subsequent studies (Table S1). Naturally, there were more primary hits from the 10 μM than the 2 μM screen due to the higher testing concentration.
For reconfirmation, we sourced all the hits that inhibited PbLuc at >70% and mean HepG2 at <50% in the 2 μM screen or PbLuc at >60% in both 2 and 10 μM screens and mean HepG2 at <50%. The resulting compounds were acquired as DMSO stocks and retested in the dose response (50–2.82 × 10–4 μM or 25–141.13 × 10–6 μM) against PbLuc and uninfected hepatocytes (Table S5). Liver stage activity was reconfirmed for 84 molecules with an IC50 < 10 μM, 29 of which averaged submicromolar efficacy across two or more replicates (Table S3). The majority of these compounds were weak inhibitors, although 18 submicromolar hits were found (Table S5).
Subsequent cluster analysis of PbLuc inhibitors validated their data: of the 84 compounds, 21 were members of 10 distinct clusters (111 total nodes with 3 to 54 nodes per cluster enriched for liver stage activity (Figure 3, green)). Cluster 402 contains members that are similar to triclocarban, and similar dual stage molecules (e.g., MMV665852) have been noted in previous publications.44 Resistance is associated with amplifications of pfatp2. All alkyl derivatives of cluster 756 qualified for dose response testing, with two cluster representatives, DDD01027733 and DDD01027481 (p-valueEEF of 8.94 × 10–7), yielding an IC50’s of 0.096 and 0.138 μM, respectively. An analog of the aforementioned substructure has shown inhibition of the human dihydroorotate dehydrogenase (DHODH), the Plasmodium homologue being a well characterized target.45 Importantly, cytotoxicity in both DDD01027733 and DDD01027481 was >25 μM in HepG2 hepatocytes.
Figure 3.

Liver active compounds. (a) 111 compounds belonging to one of 10 clusters enriched for gametocyte activity are shown. Gray diamonds show an activity of less than 10 μM, and green circles show compounds that were not selected for dose response testing or were inactive. Probability values were calculated using the hypergeometric mean function. (b) Activity tests against P. vivax patient isolates for three independent measurements and two compounds from cluster 756 (highlighted in green in (a)) as well as the controls. Parasite exoerythrocytic forms were stained with an HSP70 antibody and were counted on day 7. Three replicates were performed, and the data shows the mean and standard deviation. The number of events (schizonts and hypnozoites) were counted by reading 200 fields per well. Additional data for all compounds can be found in Table S5. Data can be explored at http://www.ndexbio.org/#/networkset/1ac81391-eebd-11e9-bb65-0ac135e8bacf?accesskey=8f0a93f855278c5ffdb1e8e52c3ca7b1983ac8452eb3b8409f99cc39fd783082.
Because compounds identified with the P. berghei model might have species specific activity, several compounds (DDD01027733 and DDD01027481) were tested against human P. vivax parasites derived from patients along with controls atovaquone and GNF179. The treatment of HC04 cells with the compounds for 24 h prior to the addition of P. vivax sporozoites resulted in no detectable hypnozoites or schizonts after treatment with nanomolar concentrations (Figure 3b). These data confirm that the P. berghei liver stage model is relevant to human infections.
Biochemical Luciferase Inhibitors May Have Antimalarial Activity
While bioluminescent reporter systems allow for efficient high-throughput screens, biochemical luciferase inhibitors could theoretically give false positive hits.46 To investigate this possibility, potent dose response hits were tested against recombinant firefly luciferase (rLuci) (Photinus pyralis) in the presence of luciferin (Table S5). These data identified 8 previously unreported luciferase inhibitors with submicromolar IC50 values against rLuc in two independent replicates (Figure S2). Interestingly, four analogs, including the compound DDD01061024, were enriched for luciferase activity (p-valuerLUCi = 4.16 × 10–12). The remaining 4 compounds were structurally distinct from one another; however, DDD01252313 closely resembles PTC124 (Ataluren), a drug used to treat Duchenne muscular dystrophy. The 3,5-aryl-oxadiazole core of these analogs can reversibly inhibit firefly luciferase but not Renilla reniformis luciferase.47
Although DDD01061024 potently inhibited rLuc, we noted that it showed an IC50 of 0.344 μM against P. falciparum Dd2 using a luciferase-independent 96 h SYBR green I-based ABS assay (see Methods), suggesting antiparasitic activity in addition to recombinant luciferase activity. These blood stage results were further validated by using a lactate dehydrogenase (LDH) ABS assay (IC50 of 0.521 μM, Figure S3b) and a liver stage high content imaging assay (IC50 of 0.375 μM, Figure S3d). In vitro evolution and whole genome analysis revealed that three parasite clones that had acquired resistance to DDD01061024 each bore an independently derived nonsynonymous mutation (all V259L) in cytochrome b (PfCYTB; mal_mito_3) in the Q0 binding pocket. Molecular docking studies against a Pf CYTB homology model suggest DDD01061024 occupies this quinone binding site in its lowest free energy state (Figure S3a) in the same relative position as atovaquone, which is also known to target the Q0 binding site of cytochrome bc1.48 Mutations within this site, and indeed at our V259L residue, were previously reported using other known cytochrome b inhibitors including ELQ40049 and RYL-552.50 As expected, the atovaquone IC50 value against wild-type parasites was 0.377 nM, which jumped 11-fold to 4.19 nM in DDD01061024 mutants (Figure S3c). These data overall show that recombinant luciferase activity may be misleading and confirm that the electron transport chain is an important target for compounds that act against both the blood stage and the liver stage.
Fewer Compounds Are Active against Stage V Gametocytes
Gametocytes are responsible for malaria transmission, and killing them is expected to block the spread of malaria. To identify possible transmission-blocking inhibitors, we screened the GHCDL against stage V gametocytes.51 Stage V gametocytes were specifically favored for screening due to their intrinsic chemical resilience over immature forms52 and their pharmacodynamically favorable circulation within the bloodstream. This strategy mimics in vivo conditions for the parasites, thus providing an ideal model for malaria sexual stage drug discovery.
A prolific gametocyte generating clone of P. falciparum (NF54-G3) was isolated in-house and used to culture mature sexual stages for these studies. To induce a sufficient quantity of mature gametocytes for screening, tightly synchronized asexual blood stages at 6–8% parasitemia were stressed for 24 h with 50% spent media. Morphology was tracked by daily blood smears. The addition of N-acetyl glucosamine (NAG) at 0–9 days postinduction prevented the reinfection of undifferentiated asexual stages. For the primary screen, 1536-well assay plates were stamped in duplicate with 10 nL of library compounds for a 2 μM final testing concentration. Infected cultures were added at 0.75% gametocytemia in 10 μL of screening media and 1.25% hematocrit. Viability of the gametocytes was measured after 72 h of compound treatment using the dye MitoTracker Red CMXRos, which selectively labels parasites with intact mitochondrial membrane potential.53 This yielded 79 hits with an average gametocyte count reduction of >50% relative to the DMSO negative control. To reconfirm these hits, all 79 compounds were retested against stage V gametocytes in triplicate 10-point dose response (10–5.08 × 10–4 μM). A high reconfirmation rate was obtained, with 68 compounds giving an IC50 value of less than 10 μM and 26 compounds with submicromolar IC50 values (Table S6). Many of these compounds were not active against blood stages, highlighting the physiological differences between the two stages.
As with the delayed-death inhibitors, cluster analysis supported the robustness of our approach. Nine clusters were identified with hit numbers ranging in size from 2 to 5 compounds (Figure 4) and cluster sizes ranging from 3 to 12 members. The probability of discovering five or more independent members of a cluster of 8 compounds with only 68 hits and a library size of 68,614 by chance is very low (p = 4.8 × 10–14). As described above, coclustering with stage V compounds that were discovered using sequential testing showed little overlap.
Figure 4.

Stage V gametocyte active network. The diagram shows nine clusters (51 compounds) enriched for gametocyte activity (24 active compounds). Dark purples diamonds show the activity of less than 10 μM, and light purple circles show compounds that were not selected for dose response testing or were inactive. Probability values were calculated using the hypergeometric mean function. The cluster identity of the inactive members is shown in Table S3. Data can be explored at http://www.ndexbio.org/#/networkset/1ac81391-eebd-11e9-bb65-0ac135e8bacf?accesskey=8f0a93f855278c5ffdb1e8e52c3ca7b1983ac8452eb3b8409f99cc39fd783082.
Scaffold Analysis Shows Enrichment of Novel Antimalarial Chemotypes with Dual Stage Activity
The advantage of our testing cascade is to reveal scaffold families with dual stage activity. To gain confidence in scaffold family members, we analyzed the complete set of 417 compounds with an IC50 value of less than 10 μM in one of the assays, of which 173 were members of 66 clusters with 2 or more active members. The majority (36) of these 66 were comprised of compounds active in only one life cycle stage. Of the remaining, 14 were ABS only active with one or more members having delayed-death activity on the basis of the criterion described above (96 h IC50 > 5× 48 h IC50). Altogether, there were 13 groups with multistage representation (Figure 5): seven clusters (8828, 8128, 4193, 3800, 3056, 2587 and 3063) with either ABS (DD or otherwise) or stage V GAM activity, five with ABS and liver representation (3452, 2025, 2748, 5349, and 7624), and one with stage V GAM and liver (2247).
Figure 5.

Potent multistage and stage-specific enriched clusters. (a) Classification and number of single stage and multistage confirmed hits (IC50 < 10 μM) belonging to enriched clusters (173 compounds). (b) Clusters that have a higher number of active compounds than that expected by chance. The hypergeometric mean was calculated for each cluster, and only those clusters enriched for bioactivity are shown (p < 0.005, 171 compounds). The central node of each cluster represents the maximum common substructure (MCS). Clusters are anchored to each other on the basis of their affinity for a given life cycle stage, while multistage active compounds are further differentiated by color.
The most enriched set of dual blood stage inhibitors (cluster ID 8128) contains a pyrimidoazepine substructure, previously described as a serotonin receptor antagonist but not known to have antimalarial activity.54 A thiophene heterocycle adjacent to the tertiary amine was potent under 1 μM in both blood stages (DDD01243664), with imidazole (DDD01254777) and thiazole/pyrazole (DDD01258014/DDD01255965; p = 6.11 × 10–10) substitutions favoring potency in ABS and GAMs, respectively. Another GAM inhibitory cluster with at least one potent ABS member (cluster ID 2587) boasts a pyridine-thiazole core, similar to previously known ABS potent scaffolds MMV007907, MMV001246, and MMV665909.55 However, the pyridine substitutions in our inhibitors lay opposite to those previously described. This linkage independent blood stage selectivity highlights the dual stage potential of this cluster’s core substructure. Cluster 3033 bears a similar substructure to the dual blood stage active DDD01057018 (cluster ID 2587). However, the nitrogen-rich substitutions in DDD01058843 (p = 3.86 × 10–5) seemingly reduce its ABS potency, while retaining potent gametocytocidal activity (Table S3).
The core hexahydrobenzo[4,5]thieno[2,3-d]pyrimidine of the G/L cluster 2247 has shown Plasmodium dihydrofolate reductase (PfDHFR) inhibition,56 although our library analogs contain a unique dihydropyrimidine substructure, which may account for the lack of ABS activity seen in other DHFR inhibitors. Alternatively, the lack of activity in the ABS stage may be due to dhfr mutations in our Dd2 parasite line.
The most significant cluster with combined asexual and liver stage potency was 3452. All aryl modifications were dual active in both primary screens, while a 5-(pyridine-3-yl)pyrimidine linkage (DDD01060831; 24.8% PbLuc inhibition at 10 μM) lost meaningful efficacy. One cluster representative, DDD01061024, was tested in dose response for both stages and yielded submicromolar potency in both (Figure 5b). This scaffold was evaluated for target identification studies, as described above, and is a cytochrome bc1 inhibitor (Figure S3). Cytochrome bc1 inhibitors are expected to have dual stage activity.34
Delayed-death inhibitors act against the malaria parasite apicoplast, which is thought to be important for the synthesis of type II fatty acid synthesis (FASII) at least in the ABS.57 Previous studies have shown that apicoplast-targeting antibiotics do not impact the liver stages but that the apicoplasts fail to segregate, resulting in poor blood stage infection.58 Of the 71 compounds with a delayed death 48 h/96 h IC50 ratio of >5, only one (DDD01250400) had confirmed liver stage activity. These data support the notion that the inhibition of type II fatty acid synthesis does not give a detectable cellular phenotype when inhibition occurs during the early liver stage.
Discussion
Previous efforts to identify multistage active antimalarials employed a stepwise screening approach whereby nonoverlapping hits from later-tested stages would be lost. This approach, while sometimes fruitful,59 fixates on the few inhibitors with life cycle spanning efficacy in a given library and ignores the value of potent, stage-specific scaffolds. Given the rarity of multistage Plasmodium inhibitors found in small molecule libraries,60 greater emphasis should be placed on combination therapies comprised of complementary inhibitors. This strategy is already used in the field with drugs like Malarone (atovaquone–proguanil) and several different artemisinin combination therapies (for example, artemether–lumefantrine). However, except in a few cases like primaquine−artemisinin and atovaquone–proguanil, clinically approved combination therapies lack a gametocytocidal or liver stage component. To this end, we generated a panoramic view of bioactivity in the Global Health Chemical Diversity Library, marking the largest life cycle-wide screening effort in Plasmodium to date.
This work provides dozens of potential new starting points for the development of symptomatic relief, transmission blocking, and prophylactic drugs, the latter two being critical components highlighted by the Medicine for Malaria Venture’s (MMV) call to new therapies.61 It remains unclear if the lack of pan-active inhibitors here is a function of the chemical space explored by the GHCDL or if the probability of finding such inhibitors is exceptionally low. With an abundance of hits throughout the parasitic life cycle, cheminformatic analysis allowed for a probabilistic determination of scaffold activity. Nearly half of these hits belonged to enriched bioactive clusters, thus establishing a structure activity profile that can be leveraged by groups in lead development. While we continue focusing on these enriched sets, we acknowledge that many potent singletons remain. Thus, all screening data will remain open access as tools for future investigation.
Few other groups have looked for delayed-death inhibitors. Delayed-death inhibitors, while potentially unsuitable for providing symptomatic relief, could be useful if employed as part of a long-acting chemoprophylaxis. Overall, our data suggest the advantage in throughput gained by using a luminescent reporter for ABS and liver stage screening outweighs the risk of detecting false positive luciferase inhibitors, despite reliance on counterscreening. This is evident by our consistent rediscovery of analogs to known antimalarials and emphasizes the predictive nature of our screening models.
The higher number of ABS inhibitors versus stage V GAMs is perhaps unsurprising given that relatively few essential pathways are known to exclusively disrupt sexual stage function (such as translation repression,62 autophagy,63 or meiosis64). Nonetheless, with conserved blood stage targets including protein translation (targeted by puromycin), protein secretion (KAF15665), or protein degradation (carmaphycin B and epoxomycin), a higher incidence of dual blood stage inhibitors (versus with either exoerythrocytic form) is more likely. Still, numerous structurally distinct scaffolds with concurrent liver and asexual blood stage activity have been linked to mitochondrial electron transport chain disruption.66 This is in-line with our findings of DDD01061024 and its proposed target, cytochrome bc1.
By exclusively testing against stage V GAMs in our sexual stage screens, we likely missed inhibitors that act on fully differentiated forms occurring within the mosquito midgut. However, compounds that affect mature intraerythrocytic sexual stages have shown a sterilizing effect in mosquitos by standard membrane feeding assays (SMFA).51 While SMFAs remain the gold standard for testing a compound’s transmission blocking capabilities, their overall throughput makes large-scale screening impractical. Furthermore, as stage V gametocytes persist in the host’s bloodstream for days, interventions there present a pharmacodynamic advantage versus the fertilization window that occurs over minutes within the mosquito midgut.
Despite potentially missing some human liver stage inhibitors by using a murine malaria species, the practical advantages of P. berghei as a surrogate of human liver stage infection are noteworthy. In addition to the safer handling characteristics of murine malaria, the acquisition of human malaria species is more expensive and produces less sporozoites per mosquito, decreasing screening efficiency. This limitation to throughput is compounded by a decreased in vitro infection rate of human-infecting malaria parasites. Furthermore, the prophylactic drug combination Malarone (specifically the atovaquone component) shows significant potency against our in vitro model, while the liver stage activity of other clinical candidates17 was identified by similar methods.
An open question is: which proteins are targeted by these compounds? Biological parsimony suggests that targets are conserved across stages (e.g., cytochrome bc1 function is needed in the blood and liver stages), but the determination of how the complete set acts could be challenging, especially for compounds that have no activity against the asexual blood stage. Options for such compounds include proteomic methodologies, such as the cellular thermal shift assay (CETSA),67 or in vitro evolution and whole genome analysis in other species, such as the related apicomplexan parasite Toxoplasma gondii.68 Metabolomic profiling69 in gametocytes or tests against specific targets that are needed for P. berghei development in the liver stage70 could also be informative.
In conjunction with limited access to clinics and an abundance of counterfeit pharmaceuticals,71 the lack of patient adherence to a full course of therapy is a continual problem that promotes selection pressure in favor of drug resistance. While the search for a Single Exposure Radical Cure and Prophylaxis (SERCaP)72 therapy continues, the fact remains that all asexual blood stage treatments to date have varying degrees of resistant malaria parasites in the field.73 Thus, there is intrinsic value in the pursuit of prophylactic and transmission blocking therapies as a means of global malaria eradication, starting points for which could be contained in this work.
Methods
Compound Library
Global Health Chemical Diversity Library
This collection of 68,614 compounds was compiled by University of Dundee, Scotland, and generously provided for screening against the Bill and Melinda Gates Foundation priority pathogens. Compounds were sourced from commercial vendors following the filtration of undesirable chemotypes. These include structural alert filters from Eli Lilly, University of Dundee, and Pan-Assay Interference Compounds (PAINS).74,75 The library was curated to interact with a diverse range of biological targets and is available for 25 screens over 3 years.
Parasite and Cell Culturing
Asexual Blood Stage Parasites
P. falciparum intraerythrocytic parasites were continuously grown in complete media but assayed in screening media, the latter being devoid of human serum. Complete media is composed of RPMI Medium 1640 (Thermo Fischer cat# 11835-030) supplemented with 0.05 mg/mL gentamicin, 0.015 mg/mL hypoxanthine, 3.4 mM NaOH, 38.5 mM HEPES, 0.19% sodium bicarbonate, 0.21% glucose, 0.21% albumax, and 4.3% human serum (O+). The lack of human serum in screening media is offset by an increased albumax (0.44%) and HEPES (39.8 mM) concentration, while remaining similar otherwise.
Gametocyte Production
Mature gametocytes were generated using previously described methods.51 Briefly, asexual P. falciparum parasites (NF54) were grown to 7–10% parasitemia after triple synchronization with 5% (w/v) d-sorbitol in T225 (100 mL volume) culture flasks. Gametocyte induction was performed by adding 50% spent media to cultures for 24 h, followed by daily media changes supplemented with 50 mM NAG. Culture progression was monitored daily by a blood smear, and NAG was excluded from the media after 10 days.
EEF Sporozoites
P. berghei ANKA-GFP-Luc-SMcon (reference clone 15cy1) and P. berghei GFP were freshly obtained by salivary gland dissection of A. stephensi mosquitoes (New York University Insectary).41 Salivary glands were suspended in cold DMEM (Invitrogen, Carlsbad, USA) and kept on ice until the time of infection. A 15 mL glass Dounce tissue grinder (Wheaton Industries) was used to free the sporozoites from the salivary glands, which were sequentially vacuum filtered through 20 μm (Millipore SCNY00020) and 11 μm (Millipore NY1104700) nylon net filters. Parasite concentration was determined by manual counting using a Neubauer hemocytometer.
Hepatocyte Line
The immortalized HepG2-A16-CD81EGFP line42 was used for all liver stage infection screening. Huh7.5.1 hepatocytes used specifically for high content imaging assays were propagated using identical methods to HepG2 cells. Culture maintenance is performed with phenol red containing DMEM (Invitrogen cat# 11965-092), 10% FBS (Corning cat# 35-011-CV), and 1× Pen Strep Glutamine (100 units/mL penicillin, 100 μg/mL streptomycin, and 0.292 mg/mL l-glutamine) (Invitrogen cat# 10378-016). Conversely, screening conditions require phenol naive DMEM (Invitrogen cat# 31053-028), 5% FBS (Corning), and 5× Pen Strep Glutamine (Invitrogen). Culture maintenance and screening conditions both occur at 37 °C and 5% CO2.
Asexual Blood Stage Screening
A transgenic line of Plasmodium falciparum (Dd2) constitutively expressing firefly luciferase was used for primary and reconfirmation screening. Cultures were maintained in complete media at 5% hematocrit (Human Whole Blood O+) and incubated at 37 °C in a low oxygen environment (3% O2, 5% CO2, and 92% N2). Culture health and parasitemia were tracked by Giemsa staining and bright field microscopy. Prior to dispensing parasites for primary screening, 20 nL of test compounds and 10 nL of control compounds (artemisinin and DMSO) were seeded per well using an Echo 550 acoustic dispenser (Labcyte) for final assay concentrations of 5 and 10 μM, respectively. When parasite cultures reached 3% to 8% parasitemia, as determined by a blood smear, infected blood was centrifuged at 800g for 5 min. A predetermined volume was taken from the resulting pellet and transferred to generate a mixture of 0.1% parasitemia and 2.5% hematocrit in screening media. From this blood mixture, 8 μL was dispensed per well in 1536-well, white, solid-bottom plates (789173-F, Greiner) using a MultiFlo FX Multi-Mode dispenser (BioTek). Plates were covered with a porous metal lid and incubated at 37 °C inside large plastic Ziploc bags filled with a blood gas mixture (3% O2, 5% CO2, and 92% N2) for 96 h. To prevent evaporation, a clean tray of water was included within each bag. Following incubation, BrightGlo was prepared by manufacturer specifications and dispensed at 2 μL per well using the MultiFlo dispenser. Plates were left for 20 min before being read with the ViewLux uHTS microplate imager (PerkinElmer) with a 90 s exposure time at the High Sensitivity Luminescence setting. Raw data were processed using GeneData (v12.0.5), whereby activity was normalized to negative minus positive controls. The correction of Runwise Pattern (Multiplicative) was applied, and compounds were considered hits when activity was reduced by ≥70%. The procedure for dose response testing was similar, except compounds were diluted 1:3 for both 8-point (5 μM top assay concentration) and 11-point (12.5 μM peak assay concentration) titrations. Also, plates tested in 11-point dose response format were incubated at both 48 and 96 h for delayed-death studies.
Stage V Gametocyte Screening
Prior to parasite addition, 10 nL (final concentration of 2 μM) of each compound was dispensed using an Echo 550 acoustic dispenser (Labcyte) in 1536-well, black, clear-bottom plates (ref# 789866-691-2B, Greiner Bio-One). High-content imaging experiments were performed on stage V gametocytes as previously described.51 Briefly, mature gametocytes were diluted to 0.5–0.75% gametocytemia and 1.25% hematocrit in serum free screening media. Cultures were dispensed at 10 μL per well using a MultiFlo dispenser (BioTek) and incubated at 37 °C for 72 h (3% O2, 5% CO2, and 92% N2). A solution of 2.5 μM MitoTracker Red CMXRos and 0.13% saponin (w/v) was made in screening media and added to each well for 1–2 h at 37 °C to allow for complete red blood cell lysis. The Operetta (PerkinElmer) high content imaging platform was used to read all 1536-well plates with the analysis performed by the accompanying Harmony software.
| 1 |
Compounds with >50% inhibition were resourced from younger stocks and plated in 10-point dose response format. All screening and analysis steps were then repeated as previously described.
EEF Liver Stage Screening (Pbluc)
The P. berghei sporozoite (PbLuc) infection and maturation assay was conducted as previously described with some modifications.19 Using the Gen 4 Plus Acoustic Transfer System (Biosero, San Diego, USA), 50 nL of compounds diluted in DMSO (final concentration of 10 μM) was dispensed into 1536-well, white, opaque-bottom plates (ref# 789173-F, Greiner Bio-One). Similarly, atovaquone (final concentration of 5 μM/well) and DMSO (final concentration of 0.5%/well) were added as positive and negative controls, respectively. HepG2-A16-CD81EGFP cells (3 × 103 cells per 5 μL volume dispensed to each well) were added within 12 h of the compound being dispensed. Within 20–24 h, day 23 PbLuc sporozoites (103 purified sporozoites per 5 μL volume dispensed to each well) were dissected, purified, and dispensed using a bottle valve liquid handler (GNF). Plates were centrifuged (Eppendorf 5810R) for 3 min (RT) at 330g and incubated for 48 h at 37 °C and 5% CO2. A combination of high incubator humidity and custom ventilated metal lids (Wako Automation, San Diego, USA) with tight sealing rubber gaskets was used to mitigate the edge effect. All other methods were as previously described.
| 2 |
P. berghei Stage High Content Imaging
For the fluorescent liver stage validation, roughly 2500 P. berghei-GFP (PbGFP) sporozoites76 were dissected from mosquitoes provided by the NYU Insectary Core Facility and seeded onto a lawn of 5000 Huh7.5.1 hepatocytes in 384-well plates. These assay plates were pretreated with DDD01061024 in 8-point dose response (10–4.57 × 10–3 μM), with atovaquone (0.5–3.91 × 10–3 μM) and DMSO (0.5%) as positive and negative controls, respectively. Unlike our standard HepG2 cell line, these Huh7.5.1 cells lack GFP expression, making it preferable for these experiments. After a 48 h incubation in the presence of our test compounds, the presence of GFP positive infected cells were imaged and counted using the Operetta High-content Imaging System (PerkinElmer).
Immunofluorescence and Confocal Microscopy for Plasmodium vivax
For immunofluorescence and confocal microscopy assays, 384-well glass-bottom plates (Greiner Bio-one, #781091) or 8-well Nunc Lab-Tek Chamber slides (Thermo Scientific) were coated with Poly-l-Lysine 0.01% (v/v) (SIGMA) and subsequently seeded with HC04 cells (5000 cells per well for 384-well plates and 50,000 cells per well for 8-well Lab-tek slides), and the different antimalarial compounds (in serial dilutions) were also added 24 h before infection. P. vivax sporozoites were freshly isolated from saliva glands of infected An. darlingi mosquitoes from a laboratory-established colony in the Peruvian Amazon region. P. vivax sporozoites were diluted in antibiotics and antifungals supplemented with DMEM as above. Plates or slides were infected using a 1:2 infection ratio (sporozoite to cell) and incubated for 4 h at 37 °C in 5% CO2. After 4 h, media were replaced and plates were incubated for 7 days; the cell culture media with the respective antimalarial drugs were replaced every 24 h.
After 7 days, the cells were fixed with 4% paraformaldehyde-PBS (Affymetrix) for 20 min at room temperature (RT), permeabilized with 0.1% Triton X-100 (Sigma) for 10 min at RT, blocked with 1% BSA (Sigma) for 30 min, and stained overnight at 4 °C using anti-PbUIS4 Goat polyclonal antibody (dilution of 1:500 from a 2 mg/mL stock, LS-C204260, LifeSpan BioSciences, Inc.). Then, Bovine-anti-Goat IgG fragment specific secondary antibody (Jackson Immunoresearch Laboratories, Inc. #805-297-008) was added at a 1:1000 dilution; the cells were incubated for 2 h at RT in a humid chamber covered from light. The hepatocyte plasma membrane was detected using 1× CellMask deep red (Thermo Fisher Scientific). After immunofluorescence staining, plates were mounted with Vectashield with DAPI (Vector Laboratories, dilution 1:100) or chambers were removed from the P. vivax-infected Lab-Tek systems; slides were mounted with Vectashield with DAPI, and #1.5 glass coverslips were affixed using nail polish. Images were acquired using a Zeiss LSM880 with Airyscan Confocal Microscope (63× oil immersion lens); laser power was set to 5% for 405, 488, 561, and 640 nm. The images were captured and processed using the confocal Zen software (Blue and Black edition, Zeiss).
HepG2 Cytotoxicity Screening
At the same time, hepatocytes were dispensed for PbLuc infection, and HepG2-A16-CD81EGFP cells (3 × 103 cells per 5 μL volume dispensed to each well) were dispensed into compound treated, uninfected, 1536-well plates. An additional 5 μL of parasite-free screening media was added to match the infected plate final volume (Vf = 10 μL). Cytotoxicity plates were stamped with identical concentrations of compounds earmarked for infection, with the inclusion of puromycin (final concentration of 10 μM) (Cayman Chemical, Ann Arbor, USA) as a positive control. All other methods were as previously described.19
| 3 |
Firefly Luciferase Inhibition
To quantify the rate of false positives reported during the PbLuc liver stage reconfirmation screen, hits selected for tertiary round screening (405 compounds from fresh DMSO stocks) were also tested for luminescent interference between recombinant firefly luciferase and luciferin. Initially, 40 nL of selected compounds was dispensed using a Gen 4 Plus Acoustic Transfer System (Biosero, San Diego, USA) at 1:3 in 12-point dose response format (25 to 141.13 × 10–6 μM; final DMSO concentration of 0.5% per well) in 1536-well, white, opaque-bottom plates (ref# 789173-F, Greiner Bio-One). A 24-point single concentration series of Luciferase Inhibitor-II (9.8 μM per well) was dispensed as a positive control, while 96 wells of DMSO at 0.5% per well acted as the negative control. Separately, a solution of 20 pM recombinant luciferase (Promega cat# E170A) was prepared on ice by successive dilutions (first 1:999 then into final solution at 20 pM) in phenol naive DMEM (Invitrogen cat# 31053-028), 5% FBS (Corning), and 5× Pen Strep Glutamine (Invitrogen). This solution was dispensed at 8 μL per well into the previously spotted plates using a MultiFlo dispenser (BioTek) and incubated at room temperature for 1 h prior to the addition of 1 μL of BrightGlo (Promega). Immediately after the addition of BrightGlo, plates were read using the EnVision Microplate reader (PerkinElmer).
Robustness of HTS Methods
The assay reproducibility was measured by the Z′-factor as previously described with modifications.77
| 4 |
where σp = standard deviation (SD) pos control, σn = SD neg control, μp = mean pos control, and μn = mean neg control.
Due to the low overall infection rate and uncontrollable nature of the hepatocyte invasion by the sporozoites, luminescence values one standard deviation from either tail of distribution were removed prior to calculating Z′-factors. Intraerythrocytic assays were calculated normally, as their end points are less influenced by weak infection rates.
Inhibitory Curve Calculations
Pbluc and stage V GAM single point as well as all dose response inhibition values were calculated using custom protocol definitions on the Collaborative Drug Discovery Vault database (www.collaborativedrug.com). Single point assays measure inhibition by normalizing each test well against the positive control in a given life cycle stage (for example, artemisinin in the asexual blood stage) and the negative control (DMSO in all screening assays). IC50’s were calculated by first normalizing the data to positive and negative controls and then by fitting a Hill equation using the Levenberg–Marquardt algorithm to the dose response data. Constraints were applied to the fitted curve such that minimum values fall between −5 and 5, while max values are between 80 and 120 (slope value ≥ 0).
Cheminformatic Analysis
Fingerprints (ECFP4) for all 68,614 library compounds were generated using a custom pipeline in Knime (v3.5.2). Hierarchical clustering was then performed using these fingerprints whereby structures with a Dice similarity coefficient of 0.7 were preserved. Edges were assigned between similar scaffolds and a central parent node. Scaffolds that had no parent node at the given similarity cutoff were considered structural singletons. Clusters were visualized using Cytoscape (v3.6.0) and the Y-files organic layout. The enrichment for potent clusters was calculated using the hypergeometric distribution equation:
| 5 |
where x = sample success, n = size of sample, M = population success, and N = total population.
Enrichment scores were generated by comparing the number of submicromolar inhibitors from a given stage to the number of equally potent inhibitors in a given cluster.
Homology Modeling
A homology model of P. falciparum cytochrome bc1 was constructed with SWISS-MODEL using a bovine crystal structure template (1BE3; 3 Å).78 Docking studies were performed by designating a search space in MGL Tools (v1.5.6) and calculating free energy binding positions with Autodock Vina (v1.1.2). Substrate protein interactions were visualized using PyMOL (v2.0.7).
Whole Genome Sequencing and Analysis
To extract genomic DNA (gDNA) from DDD01061024-resistant Dd2-B2 clones, infected RBCs were washed with 0.05% saponin and gDNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen) according to the standard protocols. Sequencing libraries were prepared with the Nextera XT kit (cat# FC-131-1024, Illumina) via the standard dual index protocol and sequenced on the Illumina HiSeq 2500 in RapidRun mode to generate paired-end reads of 100 bp length (Table S7). Reads were aligned to the P. falciparum 3D7 reference genome (PlasmoDB v13.0) using a previously described pipeline.44 A total of 4 clones (3 resistant, 1 nonresistant parent) were sequenced to an average coverage of 82× (Table S7), with an average of 91.7% of reads mapping to the 3D7 reference genome. Following alignment, SNVs and INDELs were called using GATK HaplotypeCaller and filtered according to GATK’s best practice recommendations.79 Variants were annotated using SnpEff80 and further filtered by comparing those from resistant clones to the parent clone, such that only a mutation present in the resistant clone but not the sensitive parent clone would be retained (Tables S8 and S9).
Acknowledgments
This work was supported by a grant from the Bill & Melinda Gates Foundation (OPP1107194) as well as NIH (5R01AI090141 and R01AI103058). M.R.L. was supported in part by a Ruth L. Kirschstein Institutional National Research Award from the National Institute for General Medical Sciences, T32 GM008666. J.C. was supported from a Fogarty-International Center GloCal fellowship. We thank our colleagues from the Bill & Melinda Gates Foundation for sponsoring the development of the GHCDL, in addition to providing meaningful advice throughout the screening efforts. We would also like to thank Calibr’s Compound Management Group and the HTS team for supplying compound stocks for reconfirmation and for expertly conducting the asexual blood stage screens. These efforts were possible thanks to the University of Dundee Drug Discovery Unit carefully designing and curating the Global Health Chemical Diversity Library. DNA library preparation and sequencing were performed at the UCSD Institute for Genomic Medicine sequencing core. These data have been deposited in ChEMBL Neglected Tropical Disease (ChEMBL-NTD) archive under set 23 “Probing the Open Global Health Chemical Diversity Library for multistage-active starting points for next-generation antimalarials”.
Glossary
Abbreviations
- GHCDL
Global Health Chemical Diversity Library
- EEF
exoerythrocytic form
- ABS
asexual blood stage
- Pbluc
P. berghei luciferase assay
- Pfluc
P. falciparum ABS luciferase assay
- GAM
gametocytes
- WHO
World Health Organization
- SNV
single nucleotide variant
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.9b00482.
Table S1: cheminformatic and primary screening (XLSX)
Table S3: antimalarials assessment (XLSX)
Table S4: ABS reconfirmation (XLSX)
Table S5: liver reconfirmation (XLSX)
Table S6: gametocyte reconfirmation (XLSX)
Table S2: GHCDL library characteristics; Table S7: genome sequencing; Table S8: whole genome variant-calling; Table S9: genotyping; Figure S1: primary screen; Figure S2: luciferase inhibitors; Figure S3: Bc1 inhibitor; Figure S4: singletons (PDF)
Author Present Address
∇ D.M.P.: Apros Therapeutics Inc., 10210 Campus Point Drive, Suite 150, San Diego, CA 92121, USA.
Author Present Address
○ M.L.M.: Thermo Fisher Scientific, 5823 Newton Drive, Carlsbad, CA 92008, USA.
Author Contributions
M.A. wrote the manuscript, designed the experiments, performed Pbluc HTS assays, and analyzed data. D.M.P., M.W., and A.Y.D. performed and analyzed data from the sexual stage HTS assay. Y.A.-K. and K.E. performed exoerythrocytic stage screens. M.V. performed target identification selections in the asexual blood stage. M.R.L. analyzed sequencing data. K.G. and M.L.M. performed erythrocytic stage screens and analyzed data. S.O. procured compounds and performed data management. J.C. performed P. vivax liver stage assays, and K.P.G.-M. created figures and analyzed data. D.A.F. and C.W.M. provided advice and wrote the manuscript. E.A.W. wrote the manuscript and analyzed data.
The authors declare no competing financial interest.
Notes
The dataset can be downloaded from https://chembl.gitbook.io/chembl-ntd/downloads/deposited-set-23-ucsd-ghcdl-dataset-28th-january-2020.
Supplementary Material
References
- World Health Organization . (2018) World malaria report 2018, World Health Organization, Geneva, http://www.who.int/iris/handle/10665/275867. [Google Scholar]
- Shanks G. D.; Mohrle J. J. (2017) Treating malaria: new drugs for a new era. Lancet Infect. Dis. 17 (12), 1223–1224. 10.1016/S1473-3099(17)30475-9. [DOI] [PubMed] [Google Scholar]
- Killeen G. F.; Tatarsky A.; Diabate A.; Chaccour C. J.; Marshall J. M.; Okumu F. O.; Brunner S.; Newby G.; Williams Y. A.; Malone D.; Tusting L. S.; Gosling R. D. (2017) Developing an expanded vector control toolbox for malaria elimination. BMJ. Glob Health 2 (2), e000211. 10.1136/bmjgh-2016-000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills A.; Lubell Y.; Hanson K. (2008) Malaria eradication: the economic, financial and institutional challenge. Malar. J. 7 (Suppl 1), S11. 10.1186/1475-2875-7-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard D.; Dondorp A. (2017) Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harbor Perspect. Med. 7 (7), a025619. 10.1101/cshperspect.a025619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg R.; Wirtz R. A.; Schneider I.; Burge R. (1990) An estimation of the number of malaria sporozoites ejected by a feeding mosquito. Trans. R. Soc. Trop. Med. Hyg. 84 (2), 209–12. 10.1016/0035-9203(90)90258-G. [DOI] [PubMed] [Google Scholar]
- Medica D. L.; Sinnis P. (2005) Quantitative dynamics of Plasmodium yoelii sporozoite transmission by infected anopheline mosquitoes. Infect. Immun. 73 (7), 4363–9. 10.1128/IAI.73.7.4363-4369.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. J. (2008) Malarial proteases and host cell egress: an ’emerging’ cascade. Cell. Microbiol. 10 (10), 1925–34. 10.1111/j.1462-5822.2008.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amino R.; Thiberge S.; Martin B.; Celli S.; Shorte S.; Frischknecht F.; Menard R. (2006) Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat. Med. 12 (2), 220–4. 10.1038/nm1350. [DOI] [PubMed] [Google Scholar]
- Eichner M.; Diebner H. H.; Molineaux L.; Collins W. E.; Jeffery G. M.; Dietz K. (2001) Genesis, sequestration and survival of Plasmodium falciparum gametocytes: parameter estimates from fitting a model to malariatherapy data. Trans. R. Soc. Trop. Med. Hyg. 95 (5), 497–501. 10.1016/S0035-9203(01)90016-1. [DOI] [PubMed] [Google Scholar]
- Talman A. M.; Domarle O.; McKenzie F. E.; Ariey F.; Robert V. (2004) Gametocytogenesis: the puberty of Plasmodium falciparum. Malar. J. 3, 24. 10.1186/1475-2875-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba K. S.; Jacobs-Lorena M. (2012) Production of Plasmodium falciparum gametocytes in vitro. Methods Mol. Biol. 923, 17–25. 10.1007/978-1-62703-026-7_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueangweerayut R.; Bancone G.; Harrell E. J.; Beelen A. P.; Kongpatanakul S.; Mohrle J. J.; Rousell V.; Mohamed K.; Qureshi A.; Narayan S.; Yubon N.; Miller A.; Nosten F. H.; Luzzatto L.; Duparc S.; Kleim J. P.; Green J. A. (2017) Hemolytic Potential of Tafenoquine in Female Volunteers Heterozygous for Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency (G6PD Mahidol Variant) versus G6PD-Normal Volunteers. Am. J. Trop. Med. Hyg. 97 (3), 702–711. 10.4269/ajtmh.16-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diagana T. T. (2015) Supporting malaria elimination with 21st century antimalarial agent drug discovery. Drug Discovery Today 20 (10), 1265–70. 10.1016/j.drudis.2015.06.009. [DOI] [PubMed] [Google Scholar]
- Okombo J.; Chibale K. (2018) Correction: Recent updates in the discovery and development of novel antimalarial drug candidates. MedChemComm 9, 590. 10.1039/C8MD90009D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S.; Sarma P.; Sehgal R.; Medhi B. (2017) Development in Assay Methods for in Vitro Antimalarial Drug Efficacy Testing: A Systematic Review. Front. Pharmacol. 8, 754. 10.3389/fphar.2017.00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister S.; Plouffe D. M.; Kuhen K. L.; Bonamy G. M.; Wu T.; Barnes S. W.; Bopp S. E.; Borboa R.; Bright A. T.; Che J.; Cohen S.; Dharia N. V.; Gagaring K.; Gettayacamin M.; Gordon P.; Groessl T.; Kato N.; Lee M. C.; McNamara C. W.; Fidock D. A.; Nagle A.; Nam T. G.; Richmond W.; Roland J.; Rottmann M.; Zhou B.; Froissard P.; Glynne R. J.; Mazier D.; Sattabongkot J.; Schultz P. G.; Tuntland T.; Walker J. R.; Zhou Y.; Chatterjee A.; Diagana T. T.; Winzeler E. A. (2011) Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 334 (6061), 1372–7. 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almela M. J.; Lozano S.; Lelievre J.; Colmenarejo G.; Coteron J. M.; Rodrigues J.; Gonzalez C.; Herreros E. (2015) A New Set of Chemical Starting Points with Plasmodium falciparum Transmission-Blocking Potential for Antimalarial Drug Discovery. PLoS One 10 (8), e0135139. 10.1371/journal.pone.0135139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann J.; Corey V.; Scherer C. A.; Kato N.; Comer E.; Maetani M.; Antonova-Koch Y.; Reimer C.; Gagaring K.; Ibanez M.; Plouffe D.; Zeeman A. M.; Kocken C. H.; McNamara C. W.; Schreiber S. L.; Campo B.; Winzeler E. A.; Meister S. (2016) High-Throughput Luciferase-Based Assay for the Discovery of Therapeutics That Prevent Malaria. ACS Infect. Dis. 2 (4), 281–293. 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhen K. L.; Chatterjee A. K.; Rottmann M.; Gagaring K.; Borboa R.; Buenviaje J.; Chen Z.; Francek C.; Wu T.; Nagle A.; Barnes S. W.; Plouffe D.; Lee M. C.; Fidock D. A.; Graumans W.; van de Vegte-Bolmer M.; van Gemert G. J.; Wirjanata G.; Sebayang B.; Marfurt J.; Russell B.; Suwanarusk R.; Price R. N.; Nosten F.; Tungtaeng A.; Gettayacamin M.; Sattabongkot J.; Taylor J.; Walker J. R.; Tully D.; Patra K. P.; Flannery E. L.; Vinetz J. M.; Renia L.; Sauerwein R. W.; Winzeler E. A.; Glynne R. J.; Diagana T. T. (2014) KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob. Agents Chemother. 58 (9), 5060–7. 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M.; McNamara C.; Yeung B. K.; Lee M. C.; Zou B.; Russell B.; Seitz P.; Plouffe D. M.; Dharia N. V.; Tan J.; Cohen S. B.; Spencer K. R.; Gonzalez-Paez G. E.; Lakshminarayana S. B.; Goh A.; Suwanarusk R.; Jegla T.; Schmitt E. K.; Beck H. P.; Brun R.; Nosten F.; Renia L.; Dartois V.; Keller T. H.; Fidock D. A.; Winzeler E. A.; Diagana T. T. (2010) Spiroindolones, a potent compound class for the treatment of malaria. Science 329 (5996), 1175–80. 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coteron J. M.; Marco M.; Esquivias J.; Deng X.; White K. L.; White J.; Koltun M.; El Mazouni F.; Kokkonda S.; Katneni K.; Bhamidipati R.; Shackleford D. M.; Angulo-Barturen I.; Ferrer S. B.; Jimenez-Diaz M. B.; Gamo F. J.; Goldsmith E. J.; Charman W. N.; Bathurst I.; Floyd D.; Matthews D.; Burrows J. N.; Rathod P. K.; Charman S. A.; Phillips M. A. (2011) Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 54 (15), 5540–61. 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet T.; Le Manach C.; Cabrera D. G.; Younis Y.; Henrich P. P.; Abraham T. S.; Lee M. C. S.; Basak R.; Ghidelli-Disse S.; Lafuente-Monasterio M. J.; Bantscheff M.; Ruecker A.; Blagborough A. M.; Zakutansky S. E.; Zeeman A. M.; White K. L.; Shackleford D. M.; Mannila J.; Morizzi J.; Scheurer C.; Angulo-Barturen I.; Martinez M. S.; Ferrer S.; Sanz L. M.; Gamo F. J.; Reader J.; Botha M.; Dechering K. J.; Sauerwein R. W.; Tungtaeng A.; Vanachayangkul P.; Lim C. S.; Burrows J.; Witty M. J.; Marsh K. C.; Bodenreider C.; Rochford R.; Solapure S. M.; Jimenez-Diaz M. B.; Wittlin S.; Charman S. A.; Donini C.; Campo B.; Birkholtz L. M.; Hanson K. K.; Drewes G.; Kocken C. H. M.; Delves M. J.; Leroy D.; Fidock D. A.; Waterson D.; Street L. J.; Chibale K. (2017) Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci. Transl. Med. 9 (387), eaad9735. 10.1126/scitranslmed.aad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. (2008) In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U. S. A. 105 (26), 9059–64. 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jimenez-Diaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; El Mazouni F.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; Derisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. (2010) Chemical genetics of Plasmodium falciparum. Nature 465 (7296), 311–315. 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilkstein M.; Sriwilaijaroen N.; Kelly J. X.; Wilairat P.; Riscoe M. (2004) Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48 (5), 1803–6. 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. (2010) Thousands of chemical starting points for antimalarial lead identification. Nature 465 (7296), 305–10. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- University of Dundee (2018) Gates Library screen for HepG2 cell viability. ChEMBL, https://www.ebi.ac.uk/chembl/document_report_card/CHEMBL3507680/.
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46 (1–3), 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Hou T.; Wang J.; Zhang W.; Xu X. (2007) ADME evaluation in drug discovery. 7. Prediction of oral absorption by correlation and classification. J. Chem. Inf. Model. 47 (1), 208–18. 10.1021/ci600343x. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. (2009) Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52 (21), 6752–6. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Love M. S.; Beasley F. C.; Jumani R. S.; Wright T. M.; Chatterjee A. K.; Huston C. D.; Schultz P. G.; McNamara C. W. (2017) A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Neglected Trop. Dis. 11 (2), e0005373. 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delves M. J.; Miguel-Blanco C.; Matthews H.; Molina I.; Ruecker A.; Yahiya S.; Straschil U.; Abraham M.; Leon M. L.; Fischer O. J.; Rueda-Zubiaurre A.; Brandt J. R.; Cortes A.; Barnard A.; Fuchter M. J.; Calderon F.; Winzeler E. A.; Sinden R. E.; Herreros E.; Gamo F. J.; Baum J. (2018) A high throughput screen for next-generation leads targeting malaria parasite transmission. Nat. Commun. 9 (1), 3805. 10.1038/s41467-018-05777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonova-Koch Y.; Meister S.; Abraham M.; Luth M. R.; Ottilie S.; Lukens A. K.; Sakata-Kato T.; Vanaerschot M.; Owen E.; Jado Rodriguez J. C.; Maher S. P.; Calla J.; Plouffe D.; Zhong Y.; Chen K.; Chaumeau V.; Conway A. J.; McNamara C. W.; Ibanez M.; Gagaring K.; Serrano F. N.; Eribez K.; Taggard C. M.; Cheung A. L.; Lincoln C.; Ambachew B.; Rouillier M.; Siegel D.; Nosten F.; Kyle D. E.; Gamo F.-J.; Zhou Y.; Llinás M.; Fidock D. A.; Wirth D. F.; Burrows J.; Campo B.; Winzeler E. A. (2018) Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science 362 (6419), eaat9446. 10.1126/science.aat9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delves M.; Plouffe D.; Scheurer C.; Meister S.; Wittlin S.; Winzeler E. A.; Sinden R. E.; Leroy D. (2012) The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 9 (2), e1001169. 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D. M.; Wree M.; Du A. Y.; Meister S.; Li F.; Patra K.; Lubar A.; Okitsu S. L.; Flannery E. L.; Kato N.; et al. (2016) High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe 19 (1), 114–126. 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekland E. H.; Schneider J.; Fidock D. A. (2011) Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 25 (10), 3583–93. 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E. L.; Rosenthal P. J. (2007) Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 51 (10), 3485–90. 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt D.; Wiesner J.; Stoesser N.; Ramharter M.; Uhlemann A. C.; Issifou S.; Jomaa H.; Krishna S.; Kremsner P. G.; Borrmann S. (2007) Delayed parasite elimination in human infections treated with clindamycin parallels ’delayed death’ of Plasmodium falciparum in vitro. Int. J. Parasitol. 37 (7), 777–85. 10.1016/j.ijpara.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Ramya T. N.; Mishra S.; Karmodiya K.; Surolia N.; Surolia A. (2007) Inhibitors of nonhousekeeping functions of the apicoplast defy delayed death in Plasmodium falciparum. Antimicrob. Agents Chemother. 51 (1), 307–16. 10.1128/AAC.00808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse C. J.; Franke-Fayard B.; Mair G. R.; Ramesar J.; Thiel C.; Engelmann S.; Matuschewski K.; van Gemert G. J.; Sauerwein R. W.; Waters A. P. (2006) High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol. Biochem. Parasitol. 145 (1), 60–70. 10.1016/j.molbiopara.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Yalaoui S.; Zougbede S.; Charrin S.; Silvie O.; Arduise C.; Farhati K.; Boucheix C.; Mazier D.; Rubinstein E.; Froissard P. (2008) Hepatocyte permissiveness to Plasmodium infection is conveyed by a short and structurally conserved region of the CD81 large extracellular domain. PLoS Pathog. 4 (2), e1000010. 10.1371/journal.ppat.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke-Fayard B.; Janse C. J.; Cunha-Rodrigues M.; Ramesar J.; Buscher P.; Que I.; Lowik C.; Voshol P. J.; den Boer M. A.; van Duinen S. G.; Febbraio M.; Mota M. M.; Waters A. P. (2005) Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc. Natl. Acad. Sci. U. S. A. 102 (32), 11468–73. 10.1073/pnas.0503386102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell A. N.; Istvan E. S.; Lukens A. K.; Gomez-Lorenzo M. G.; Vanaerschot M.; Sakata-Kato T.; Flannery E. L.; Magistrado P.; Owen E.; Abraham M.; LaMonte G.; Painter H. J.; Williams R. M.; Franco V.; Linares M.; Arriaga I.; Bopp S.; Corey V. C.; Gnadig N. F.; Coburn-Flynn O.; Reimer C.; Gupta P.; Murithi J. M.; Moura P. A.; Fuchs O.; Sasaki E.; Kim S. W.; Teng C. H.; Wang L. T.; Akidil A.; Adjalley S.; Willis P. A.; Siegel D.; Tanaseichuk O.; Zhong Y.; Zhou Y.; Llinas M.; Ottilie S.; Gamo F. J.; Lee M. C. S.; Goldberg D. E.; Fidock D. A.; Wirth D. F.; Winzeler E. A. (2018) Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science 359 (6372), 191–199. 10.1126/science.aan4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Rathod P. K. (2010) Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect. Disord.: Drug Targets 10 (3), 226–239. 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne N.; Auld D. S.; Inglese J. (2010) Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 14 (3), 315–24. 10.1016/j.cbpa.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld D. S.; Thorne N.; Maguire W. F.; Inglese J. (2009) Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. U. S. A. 106 (9), 3585–90. 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siregar J. E.; Kurisu G.; Kobayashi T.; Matsuzaki M.; Sakamoto K.; Mi-ichi F.; Watanabe Y.; Hirai M.; Matsuoka H.; Syafruddin D.; Marzuki S.; Kita K. (2015) Direct evidence for the atovaquone action on the Plasmodium cytochrome bc1 complex. Parasitol. Int. 64 (3), 295–300. 10.1016/j.parint.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Stickles A. M.; Ting L. M.; Morrisey J. M.; Li Y.; Mather M. W.; Meermeier E.; Pershing A. M.; Forquer I. P.; Miley G. P.; Pou S.; Winter R. W.; Hinrichs D. J.; Kelly J. X.; Kim K.; Vaidya A. B.; Riscoe M. K.; Nilsen A. (2015) Inhibition of cytochrome bc1 as a strategy for single-dose, multi-stage antimalarial therapy. Am. J. Trop. Med. Hyg. 92 (6), 1195–201. 10.4269/ajtmh.14-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane K. D.; Mu J.; Lu J.; Windle S. T.; Liu A.; Sun P. D.; Wellems T. E. (2018) Selection of Plasmodium falciparum cytochrome B mutants by putative PfNDH2 inhibitors. Proc. Natl. Acad. Sci. U. S. A. 115 (24), 6285–6290. 10.1073/pnas.1804492115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D. M.; Wree M.; Du A. Y.; Meister S.; Li F.; Patra K.; Lubar A.; Okitsu S. L.; Flannery E. L.; Kato N.; Tanaseichuk O.; Comer E.; Zhou B.; Kuhen K.; Zhou Y.; Leroy D.; Schreiber S. L.; Scherer C. A.; Vinetz J.; Winzeler E. A. (2016) High-Throughput Assay and Discovery of Small Molecules that Interrupt Malaria Transmission. Cell Host Microbe 19 (1), 114–26. 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro S.; Camarda G.; Corbett Y.; Siciliano G.; Parapini S.; Cevenini L.; Michelini E.; Roda A.; Leroy D.; Taramelli D.; Alano P. (2016) A chemical susceptibility profile of the Plasmodium falciparum transmission stages by complementary cell-based gametocyte assays. J. Antimicrob. Chemother. 71 (5), 1148–58. 10.1093/jac/dkv493. [DOI] [PubMed] [Google Scholar]
- Pendergrass W.; Wolf N.; Poot M. (2004) Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry 61A (2), 162–169. 10.1002/cyto.a.20033. [DOI] [PubMed] [Google Scholar]
- Yang H. Y.; Tae J.; Seo Y. W.; Kim Y. J.; Im H. Y.; Choi G. D.; Cho H.; Park W. K.; Kwon O. S.; Cho Y. S.; Ko M.; Jang H.; Lee J.; Choi K.; Kim C. H.; Pae A. N.; et al. (2013) Novel pyrimidoazepine analogs as serotonin 5-HT(2A) and 5-HT(2C) receptor ligands for the treatment of obesity. Eur. J. Med. Chem. 63, 558–569. 10.1016/j.ejmech.2013.02.020. [DOI] [PubMed] [Google Scholar]
- Hain A. U.; Bartee D.; Sanders N. G.; Miller A. S.; Sullivan D. J.; Levitskaya J.; Meyers C. F.; Bosch J. (2014) Identification of an Atg8-Atg3 protein-protein interaction inhibitor from the medicines for Malaria Venture Malaria Box active in blood and liver stage Plasmodium falciparum parasites. J. Med. Chem. 57 (11), 4521–31. 10.1021/jm401675a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeza Zaidi S.; Agarwal S. M.; Chavalitshewinkoon-Petmitr P.; Suksangpleng T.; Ahmad K.; Avecilla F.; Azam A. Y. (2016) Thienopyrimidine sulphonamide hybrids: design, synthesis, antiprotozoal activity and molecular docking studies. RSC Adv. 93, 90371. 10.1039/C6RA15181G. [DOI] [Google Scholar]
- Kennedy K.; Cobbold S. A.; Hanssen E.; Birnbaum J.; Spillman N. J.; McHugh E.; Brown H.; Tilley L.; Spielmann T.; McConville M. J.; Ralph S. A. (2019) Delayed death in the malaria parasite Plasmodium falciparum is caused by disruption of prenylation-dependent intracellular trafficking. PLoS Biol. 17 (7), e3000376. 10.1371/journal.pbio.3000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen J.; Silvie O.; Putrianti E. D.; Hafalla J. C.; Matuschewski K.; Borrmann S. (2010) Natural immunization against malaria: causal prophylaxis with antibiotics. Sci. Transl. Med. 2 (40), 40ra49. 10.1126/scitranslmed.3001058. [DOI] [PubMed] [Google Scholar]
- Wu T.; Nagle A.; Kuhen K.; Gagaring K.; Borboa R.; Francek C.; Chen Z.; Plouffe D.; Goh A.; Lakshminarayana S. B.; Wu J.; Ang H. Q.; Zeng P.; Kang M. L.; Tan W.; Tan M.; Ye N.; Lin X.; Caldwell C.; Ek J.; Skolnik S.; Liu F.; Wang J.; Chang J.; Li C.; Hollenbeck T.; Tuntland T.; Isbell J.; Fischli C.; Brun R.; Rottmann M.; Dartois V.; Keller T.; Diagana T.; Winzeler E.; Glynne R.; Tully D. C.; Chatterjee A. K. (2011) Imidazolopiperazines: hit to lead optimization of new antimalarial agents. J. Med. Chem. 54 (14), 5116–30. 10.1021/jm2003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N.; Comer E.; Sakata-Kato T.; Sharma A.; Sharma M.; Maetani M.; Bastien J.; Brancucci N. M.; Bittker J. A.; Corey V.; Clarke D.; Derbyshire E. R.; Dornan G. L.; Duffy S.; Eckley S.; Itoe M. A.; Koolen K. M.; Lewis T. A.; Lui P. S.; Lukens A. K.; Lund E.; March S.; Meibalan E.; Meier B. C.; McPhail J. A.; Mitasev B.; Moss E. L.; Sayes M.; Van Gessel Y.; Wawer M. J.; Yoshinaga T.; Zeeman A. M.; Avery V. M.; Bhatia S. N.; Burke J. E.; Catteruccia F.; Clardy J. C.; Clemons P. A.; Dechering K. J.; Duvall J. R.; Foley M. A.; Gusovsky F.; Kocken C. H.; Marti M.; Morningstar M. L.; Munoz B.; Neafsey D. E.; Winzeler E. A.; Wirth D. F.; Scherer C. A.; Schreiber S. L.; et al. (2016) Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 538 (7625), 344–349. 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; Duparc S.; Gutteridge W. E.; Hooft van Huijsduijnen R.; Kaszubska W.; Macintyre F.; Mazzuri S.; Mohrle J. J.; Wells T. N. C. (2017) New developments in anti-malarial target candidate and product profiles. Malar. J. 16 (1), 26. 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair G. R.; Braks J. A.; Garver L. S.; Wiegant J. C.; Hall N.; Dirks R. W.; Khan S. M.; Dimopoulos G.; Janse C. J.; Waters A. P. (2006) Regulation of sexual development of Plasmodium by translational repression. Science 313 (5787), 667–9. 10.1126/science.1125129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes S.; Bunnik E. M.; Saraf A.; Conner C. M.; Escalante A.; Sardiu M. E.; Ponts N.; Prudhomme J.; Florens L.; Le Roch K. G. (2014) The multifunctional autophagy pathway in the human malaria parasite, Plasmodium falciparum. Autophagy 10 (1), 80–92. 10.4161/auto.26743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delves M. J.; Ruecker A.; Straschil U.; Lelièvre J.; Marques S.; López-Barragán M. J.; Herreros E.; Sinden R. E. (2013) Male and female Plasmodium falciparum mature gametocytes show different responses to antimalarial drugs. Antimicrob. Agents Chemother. 57 (7), 3268–74. 10.1128/AAC.00325-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte G. M., Marapana D. S., Gnadig N., Ottilie S., Luth M. R., Worgall T. S., Rocamora F., Goldgof G. M., Mohunlal R., Kumar T. R. S., Thompson J. K., Vigil E., Yang J., Hutson D., Johnson T., Huang J., Williams R. M., Zou B. Y., Cheung A. L., Kumar P., Egan T. J., Lee M. C. S., Siegel D., Cowman A. F., Fidock D. A., and Winzeler E. A. (2019) Pan-active imidazolopiperazine antimalarials target the Plasmodium falciparum intracellular secretory pathway. bioRxiv, 735894, https://doi.org/10.1101/735894. [DOI] [PMC free article] [PubMed]
- Antonova-Koch Y.; Meister S.; Abraham M.; Luth M. R.; Ottilie S.; Lukens A. K.; Sakata-Kato T.; Vanaerschot M.; Owen E.; Jado J. C.; Maher S. P.; Calla J.; Plouffe D.; Zhong Y.; Chen K.; Chaumeau V.; Conway A. J.; McNamara C. W.; Ibanez M.; Gagaring K.; Serrano F. N.; Eribez K.; Taggard C. M.; Cheung A. L.; Lincoln C.; Ambachew B.; Rouillier M.; Siegel D.; Nosten F.; Kyle D. E.; Gamo F. J.; Zhou Y.; Llinas M.; Fidock D. A.; Wirth D. F.; Burrows J.; Campo B.; Winzeler E. A. (2018) Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science 362 (6419), eaat9446. 10.1126/science.aat9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen A. J.; Martinez Molina D.; Lundback T. (2015) CETSA: a target engagement assay with potential to transform drug discovery. Future Med. Chem. 7 (8), 975–978. 10.4155/fmc.15.50. [DOI] [PubMed] [Google Scholar]
- Rosenberg A.; Luth M. R.; Winzeler E. A.; Behnke M.; Sibley L. D. (2019) Evolution of resistance in vitro reveals mechanisms of artemisinin activity in Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A. 116, 26881. 10.1073/pnas.1914732116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman E. L.; Painter H. J.; Samra J.; Carrasquilla M.; Llinas M. (2016) Metabolomic Profiling of the Malaria Box Reveals Antimalarial Target Pathways. Antimicrob. Agents Chemother. 60, 6635. 10.1128/AAC.01224-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanway R. R.; Bushell E.; Chiappino-Pepe A.; Roques M.; Sanderson T.; Franke-Fayard B.; Caldelari R.; Golomingi M.; Nyonda M.; Pandey V.; Schwach F.; Chevalley S.; Ramesar J.; Metcalf T.; Herd C.; Burda P. C.; Rayner J. C.; Soldati-Favre D.; Janse C. J.; Hatzimanikatis V.; Billker O.; Heussler V. T. (2019) Genome-Scale Identification of Essential Metabolic Processes for Targeting the Plasmodium Liver Stage. Cell 179 (5), 1112–1128.e26. 10.1016/j.cell.2019.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroise-Thomas P. (2012) The tragedy caused by fake antimalarial drugs. Mediterr J. Hematol Infect Dis 4 (1), e2012027. 10.4084/mjhid.2012.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2011) A research agenda for malaria eradication: drugs. PLoS Med. 8 (1), e1000402. 10.1371/journal.pmed.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure N. S.; Day T. (2014) A theoretical examination of the relative importance of evolution management and drug development for managing resistance. Proc. R. Soc. London, Ser. B 281 (1797), 20141861. 10.1098/rspb.2014.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53 (7), 2719–40. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Bruns R. F.; Watson I. A. (2012) Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 55 (22), 9763–72. 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- McNamara C. W.; Lee M. C.; Lim C. S.; Lim S. H.; Roland J.; Simon O.; Yeung B. K.; Chatterjee A. K.; McCormack S. L.; Manary M. J.; Zeeman A. M.; Dechering K. J.; Kumar T. S.; Henrich P. P.; Gagaring K.; Ibanez M.; Kato N.; Kuhen K. L.; Fischli C.; Nagle A.; Rottmann M.; Plouffe D. M.; Bursulaya B.; Meister S.; Rameh L.; Trappe J.; Haasen D.; Timmerman M.; Sauerwein R. W.; Suwanarusk R.; Russell B.; Renia L.; Nosten F.; Tully D. C.; Kocken C. H.; Glynne R. J.; Bodenreider C.; Fidock D. A.; Diagana T. T.; Winzeler E. A. (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504 (7479), 248–253. 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 4 (2), 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Iwata S.; Lee J. W.; Okada K.; Lee J. K.; Iwata M.; Rasmussen B.; Link T. A.; Ramaswamy S.; Jap B. K. (1998) Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281 (5373), 64–71. 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- McKenna A.; Hanna M.; Banks E.; Sivachenko A.; Cibulskis K.; Kernytsky A.; Garimella K.; Altshuler D.; Gabriel S.; Daly M.; DePristo M. A. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P.; Platts A.; Wang L. L.; Coon M.; Nguyen T.; Wang L.; Land S. J.; Lu X.; Ruden D. M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.