

Abstract
Adrenomedullin (AM) is a 52 amino acid peptide that plays a regulatory role in the vasculature. Receptors for AM comprise the class B G protein-coupled receptor, the calcitonin-like receptor (CLR), in complex with one of three receptor activity-modifying proteins (RAMPs). The C-terminus of AM is involved in binding to the extracellular domain of the receptor, while the N-terminus is proposed to interact with the juxtamembranous portion of the receptor to activate signaling. There is currently limited information on the molecular determinants involved in AM signaling, thus we set out to define the importance of the AM N-terminus through five signaling pathways (cAMP production, ERK phosphorylation, CREB phosphorylation, Akt phosphorylation, and IP1 production). We characterized the three CLR:RAMP complexes through the five pathways, finding that each had a distinct repertoire of intracellular signaling pathways that it is able to regulate. We then performed an alanine scan of AM from residues 15–31 and found that most residues could be substituted with only small effects on signaling, and that most substitutions affected signaling through all receptors and pathways in a similar manner. We identify F18, T20, L26, and I30 as being critical for AM function, while also identifying an analogue (AM15–52 G19A) which has unique signaling properties relative to the unmodified AM. We interpret our findings in the context of new structural information, highlighting the complementary nature of structural biology and functional assays.
Keywords: calcitonin-like receptor, adrenomedullin, adrenomedullin 2/intermedin, receptor activity-modifying protein, signaling bias/functional selectivity, G protein-coupled receptor
Adrenomedullin (AM) is a 52 amino acid peptide hormone belonging to the structurally related calcitonin (CT) family of peptides (Figure 1).1 AM was originally isolated from human pheochromocytoma but is widely distributed across a range of tissues. AM has regulatory and protective effects in the cardiovascular system, while also being involved in a number of other physiological processes such as lymphatic regulation, tumor progression, and sepsis.2−7
Figure 1.

(A) Amino acid sequence alignment of AM and related peptides from humans. (B) Amino acid sequences of peptide fragments used during this study. (C) Amino acid sequences of alanine-substituted analogues used in this study, substitutions are highlighted in orange. All sequence alignments performed in Geneious 11, using in-built ClustalW alignment and amino acid comparison. Unless otherwise noted elsewhere, there is a disulfide bond between C16 and C21 of the AM peptides (or the corresponding conserved cysteines in other peptides), and all the peptides are amidated at the C-terminus.
AM has been suggested as a target for the treatment of various diseases; AM administration has been linked to positive outcomes in cases of myocardial infarction, pulmonary and systemic hypertension, and wound healing.2 Conversely, the administration of AM receptor antagonists has been linked to reduced tumor growth and invasion, indicating that antagonizing the activity of AM may be a route for developing antitumor agents.2 While AM and its receptors hold promise as therapeutic targets, this has yet to be realized. AM is rapidly metabolized in vivo and has poor bioavailability.8 Additionally, the on-target side effect of excessive vasodilation restricts its use in a clinical setting.9 Detailed examinations of AM structure–function relationships and signaling bias are important steps in the development of drugs that target the AM system.10
AM activates receptors that comprise the core class B G protein-coupled receptor (GPCR), the calcitonin-like receptor (CLR), in complex with one of three receptor activity-modifying proteins (RAMPs).1 Class B GPCRs comprise seven transmembrane (TM) domains, a large extracellular N-terminus, and an intracellular C-terminal tail. Similarly, RAMPs comprise a single TM pass domain, a large extracellular N-terminus, and a short intracellular tail. These CLR:RAMP interactions give rise to three distinct receptors: CLR:RAMP1 is known as the calcitonin gene-related peptide (CGRP) receptor, while CLR complexed with RAMP2 or RAMP3 creates the AM1 or AM2 receptors, respectively.1 RAMPs regulate all aspects of the CLR lifespan, exerting effects on ligand binding, G protein interactions, and receptor fate following agonist stimulation.1,11,12 RAMPs drive CLR pharmacology by allosterically modulating the receptor, while also providing direct ligand contact points.13−17
When activating class B GPCRs, the C-terminus of the peptide ligand interacts with the extracellular domain of the receptor, allowing the N-terminus of the peptide to adopt an α-helical conformation, burying itself into the TM domain. This stabilizes a conformation of the receptor that promotes receptor activity. This is known as the two-domain model of activation.18 Recent structures of several class B GPCRs in complex with their ligands support this broad mechanism; however, unlike other class B GPCRs, structures of CTR and CLR:RAMP complexes suggest that peptides of the CT family are not entirely α-helical when bound to these receptors.19−26 This indicates that these peptides may activate receptors through an alternative mechanism.
There have been extensive structure–function investigations exploring how the C-terminus of AM interacts with the extracellular domains of CLR:RAMP complexes.27,28 In contrast, we have much less information about how the N-terminus of AM is involved in receptor activation, which limits the development of novel agonists. Interestingly, sequence alignments of AM and related peptides indicate that AM has an unusual extended N-terminus containing residues 1–14, followed by the N-terminal region that we predict is important for receptor activation spanning residues ∼15–31 (Figure 1A). In an effort to characterize the peptide molecular signature that is necessary for receptor activation in CLR:RAMP complexes, we undertook an extensive analysis of AM, including an alanine scan to investigate the function of individual amino acids in activating a number of different signaling pathways. To increase the translational relevance of our work, we then profile our analogues in cells which endogenously express an AM-responsive receptor. We last move to interpret our results in light of the publication of AM receptor structures.
Results and Discussion
RAMPs Dictate the Signaling Pathways Regulated by CLR
To understand the mechanisms through which AM drives signaling, we first needed to define which pathways were regulated by CLR-based receptors in our cell models to establish a pharmacological framework for these receptors and pathways. Previous work has reported that the three CLR:RAMP complexes can couple to Gs, Gi, and Gq; however, there has been less characterization of pathway activation downstream of this G protein coupling.11 We therefore measured a number of signaling pathways selected based on their proposed importance for AM physiology (Table S1).29−31
This work was performed in Cos7 cells. For all signaling pathways, time-course experiments were first conducted using a saturating concentration of the cognate ligand to determine the optimal duration for subsequent concentration–response experiments (Figures S1 to S5). Taking into account the peak response relative to media controls, assay reproducibility, and the ability to compare the results derived from different pathways,32,33 the stimulation durations selected for concentration–response experiments were 15 min (cAMP production), 10 min (extracellular signal-regulated kinase 1/2 [ERK 1/2] phosphorylation, cAMP response element-binding protein [CREB] phosphorylation, Akt phosphorylation), and 120 min (inositol phosphate 1 [IP1]) production. For the concentration–response experiments we used the four peptides that are most commonly reported to activate CLR-based receptors: AM, AM2, αCGRP, and βCGRP (Figure 2).
Figure 2.

(A) activation of signaling pathways at the corresponding receptor by full-length AM, (B) Summary pEC50 values for the activation of signaling pathways at the corresponding receptor by AM, AM2, αCGRP, and βCGRP, (C) Δτ/KA values for pathway activation at the corresponding receptor, (D) ΔΔτ/KA values for pathway activation at the corresponding receptor. This figure shows results from transfected Cos7 cells. Results in panel A represent the mean ± s.e.m. of three or five independent experiments (flat-lines or curves, respectively). Results in panels B, C, D show the mean of at least five independent experiments for which a pathway could be measured. Results in panels C and D are presented as fold change relative to a reference ligand (αCGRP at the CGRP receptor, or AM at the AM1 and AM2 receptors). Results in panel D are normalized to a reference pathway (cAMP in all cases); in panel D, a value >1 indicates bias toward the named pathway over cAMP production. See Tables S2 to S6 and Figures S6 to S10 for all curves and values. Results in panels B and C analyzed by paired Student’s t tests (IP1 production at the CGRP receptor, Akt phosphorylation at the AM2 receptor), or repeated measures ANOVA with posthoc Tukey’s test (all other pathways). Results in panel D analyzed by one-way ANOVA with posthoc Dunnett’s test, comparing the ability of a peptide to activate a pathway relative to cAMP. A superscript letter above a pathway represents a significant (p < 0.05) difference between reference ligand and named ligand (panels B and C), or a significant difference between the named pathway and cAMP production (panel D). Superscript “w” indicates a significant difference for AM, “x” indicates a significant difference for AM2, “y” indicates a significant difference for αCGRP, and “z” indicates a significant difference for βCGRP.
There was a distinct pattern of pathway activation for each CLR:RAMP complex. The CGRP receptor was able to activate all five tested signaling pathways, although AM and AM2 were not able to stimulate IP1 production in the tested concentration range. The AM2 receptor was unable to stimulate IP1 production in response to endogenous ligands, and the AM1 receptor was unable to stimulate IP1 production or Akt phosphorylation in response to endogenous ligands (Figure 2; Figures S6 to S10, Tables S2 to S6). For all three receptors, CLR was expressed on the cell surface to similar levels, suggesting that there was no difference in complex expression (Figure S11). This suggests that differences in receptor activity were not due to differences in cell surface expression levels, and were more likely due to differences in the ability of different CLR:RAMP complexes to couple to signaling pathways. Although these signaling profiles seem absolute, it is unlikely that these receptors display “perfect bias” in which certain receptors are unable to activate certain signaling pathways. Instead, it is more likely pathways such as Akt phosphorylation and IP1 production are coupled with different strengths to the different CLR:RAMP complexes, and the functional response seen with these weakly coupled pathways is too small to be detected with current assays.30 These different signaling profiles could arise, at least in part, due to differences in the behavior of RAMP C-termini; molecular dynamics simulations report that RAMP3 is able to make transient interactions with the Gs αN helix, while RAMP2 cannot, which could manifest as differences in activation of signaling.34 Alternatively this could reflect a broader allosteric contribution of the RAMP on the CLR conformation.15,16,35 In our companion paper, coordinated, receptor-specific, motions of AM1 and AM2 receptors are observed in cryo-electron microscopy (cryo-EM) structures of these receptors that have been analyzed for conformation dynamics, supporting this latter hypothesis.34
The relative rank order of potency at the AM1 receptor for all pathways was AM ≥ AM2 > βCGRP ≥ αCGRP (Figure 2B). AM2 was a partial agonist of cAMP production at the AM1 receptor, this effect was less obvious for other pathways; αCGRP trended toward being a partial agonist for all tested pathways (Figures S6 to S8, Tables S2 to S4). The relative rank order of potency at the AM2 receptor for all pathways was AM = AM2 > βCGRP ≥ αCGRP (Figure 2B). AM2 was a full agonist for all pathways (Figures S6 to S8, Tables S2 to S4) except for Akt phosphorylation where it was a partial agonist with an Emax ∼ 50% of the AM Emax (Figure S9, Table S5). In most cases the relative rank order of potency at the CGRP receptor was βCGRP ≥ αCGRP > AM2 ≥ AM (Figure 2B, Figures S6, S8, S10, Tables S2, S4, S6); the exceptions to this were ERK phosphorylation, where all tested peptides were equipotent (Figure S7, Table S3), Akt phosphorylation, where αCGRP and βCGRP were equipotent (Figure S9, Table S5), and IP1 production, where AM and AM2 were unable to stimulate a measurable response (Figure 2, Figure S10, Table S6).
The apparent inability of the AM receptors to stimulate IP1 production (considered downstream of Gq activation) is in contrast with previous literature that suggests that all three CLR:RAMP complexes can couple to Gq in response to AM, AM2, and αCGRP.11 This previous study was performed in HEK293 cells. It is possible that there is a difference in relative abundance and/or distribution of Gq between HEK293 and Cos7 cells that could lead to this discrepancy in findings.36−38 Alternatively, this may reflect a difference in the assays used; the previous investigation measured Ca2+ influx as a proxy for Gq activation, while we measured IP1 production. Recent reports have highlighted that these two pathways, while both being used to measure Gq activation, can have discordant outcomes. This is thought to arise due to the Ca2+ measurement occurring before an equilibrium is reached, compared to the IP1 which is performed at equilibrium.33
Additionally, ERK phosphorylation appeared to differ from the currently understood paradigm of CGRP receptor activation defined by cAMP production and ligand binding, in that all tested peptides appeared to be equipotent through this receptor/pathway combination (Figure 2, Figure S7).39,40 This receptor profile caused AM to be biased toward ERK phosphorylation over cAMP production at this receptor; AM2 trended toward being biased toward ERK phosphorylation over cAMP production, and βCGRP trended toward being biased toward cAMP production over ERK phosphorylation (Figure 2D). To ensure that this was not an artifact of either the assay used to measure phosphorylation, or of the cell-line, both a second detection assay (CisBio homogeneous time-resolved fluorescence) and a second cell-line (HEK293S) were employed (Figures S12 and S13). In both cases, the pattern of ERK phosphorylation seen with the AlphaLISA kit in Cos7 cells was replicated in our other experimental paradigms, in that all peptides appeared to have similar potencies through this pathway:receptor combination. There was a slight difference in signaling profile obtained in HEK293S cells relative to the profile in Cos7 cells; however, the differences were small and the overall trend for a compression of potencies was retained. A similar compression of potencies is noted for ERK phosphorylation at CTR:RAMP complexes; amylin is more potent than CT when measuring cAMP production at CTR:RAMP complexes, but the two peptides are equipotent when measuring ERK phosphorylation at the same receptors.36 Similarly, αCGRP, amylin, pramlintide, and CT are equipotent when measuring ERK phosphorylation at the CTR:RAMP1 complex, while displaying a larger separation of potencies through other measured signaling pathways.41 Similar, but less pronounced effects are noted in investigations of the CTR:RAMP3 complex when compared to CTR on its own.41−43 ERK phosphorylation by CLR/CTR:RAMP complexes is known to be controlled by diverse signaling events, with Gq, Gs, β-arrestin recruitment, and receptor internalization all being at least partially involved in the measured response.36,44,45 Additionally, recent evidence suggests that GPCRs can rapidly transactivate the epidermal growth factor receptor to stimulate ERK phosphorylation; this could have an influence on our obtained signaling profile.46
The Extended N-Terminus of AM Is Dispensable for Receptor Activation
Compared to most of its family members, AM has an unusual extension N-terminal to the disulfide loop (residues 1–15, Figure 1A), CGRP and amylin have a single amino acid N-terminal to the disulfide loop, and CT has no additional residue. More similar in length to AM is AM2 but this is reported to have several molecular forms (AM2-53, AM2-47, and AM2-40) which have extensions to their N-termini of varying lengths.47 The importance of the extended AM N-terminus is unclear. Although the N-terminal extension is conserved across multiple species, an AM-like peptide in Ornithodoros ticks, which is thought to have arisen by horizontal gene transfer, lacks the N-terminal extension (Figure S14).48 There are previous reports that the extended N-terminus of AM is dispensable for peptide function; however, there is at least one instance where full length AM can exert an effect that an N-terminally truncated analogue cannot, namely, AM can exert dilation of precontracted aortic vessels, whereas an N-terminally truncated analogue (AM13–52) could not.49−55In vitro characterization of N-terminally truncated analogues had previously only been performed for cAMP production; thus, in order to more completely profile the role of the extension, we characterized AM15–52 at all three receptors through all five signaling pathways explored in Figure 2. AM15–52 was chosen because this fragment length is most similar to CGRP and amylin, having one amino acid prior to the disulfide loop structure (Figure 1A). AM and AM15–52 were functionally equivalent in Cos7 cells (Figure 3, Table S7), HEK293S cells (Figure S15, Table S8), and CHO-K1 cells (Figure S16). We also conducted competition binding assays to compare these peptides. Both peptides displaced 125I-AM13–52 with high affinity, although the AM data best fit a two-site model (Figure S17A). Circular dichroism (CD) spectroscopy showed no apparent differences in secondary structure (Figure S17B). A more limited characterization of AM16–52 was also conducted (Figure S18), which demonstrated similar potency to AM15–52 in cAMP production assays.
Figure 3.

Signaling of AM and AM15–52 at the three CLR:RAMP complexes in transfected Cos7 cells. All data are the mean ± s.e.m. of at least five or three independent experiments (curves or flat-lines, respectively) (see Table S7). Data are normalized to the maximum response observed for AM. The asterisk (∗) indicates that pathways that lacked an AM response are shown as fold-basal signaling.
A previous report suggested that the N-terminal disulfide loop structure alone was sufficient for receptor activation.56 We therefore synthesized and screened a series of N-terminal fragments containing this sequence (AM1–21, linear AM1–21[nonoxidized, lacking the disulfide bond between C16 and C21], and AM16–21); however, none of these fragments appeared to be able to stimulate cAMP production (Figure S19). To expand upon these results we sought to identify a minimal sequence required for receptor activation. We found that AM15–30 was a partial agonist and that AM15–34 was a full agonist, albeit with drastically reduced potency (Figure 1B, Figure S20). Adjacent to position 34, AM contains a succession of charged amino acids (DKDKD), which act as a linker between our active fragment AM15–34 and the extracellular domain binding C-terminus of the peptide.27 To determine the importance of the DKDKD region we then generated a peptide with these residues removed (AM15–52 Δ35–39), that directly joins the 15–34 and 40–52 fragments together, after first confirming that the C-terminal AM fragment AM40–52 could indeed bind to the receptors to act as a competitive antagonist (Figure S21). The AM15–52 Δ35–39 peptide had increased Emax compared to AM15–52 (∼170%) but reduced potency (∼100-fold) (Figure S20, Table S9).
Collectively, our work using AM fragments showed that the sequence required for full activation of the AM1 receptor is found between residues 15 and 34 of AM, with subsequent residues being more important for driving affinity than efficacy. This is relatively consistent with the proposed two-domain model of class B GPCR activation, in that our data shows the AM C-terminus is involved in receptor binding, and the N-terminus is involved in receptor activation.57
An Alanine Scan of the AM N-Terminus Highlights Residues Critical for Peptide Activity
Having profiled signaling across receptors, and determined the importance of different parts of the AM sequence, we proceeded to explore the contribution that each amino acid makes to the overall signaling profile of AM. This was achieved through alanine scanning in the AM15–52 sequence (Figure 1C), which was selected because of its functional similarity to full length AM (Figure 3) and its similarity in length to αCGRP and amylin (Figure 1A). We had a specific focus on the sequence from positions 15 to 31, guided by the recently published αCGRP-bound CGRP receptor structure.19 In this structure, residues 1–17 of αCGRP (corresponding to AM residues 15–31) were in close proximity to the juxtamembranous region of the receptor, and thus likely to drive differential signaling events;19,30 interactions of AM15–31 with the receptor TM core were confirmed in the structures of the AM1 and AM2 receptors described in our companion paper.34 We omitted substituting the cysteines in position 16 and 21 (as these are critically involved in forming the disulfide bond required for AM activity) and the alanine in position 28.
The majority of alanine-substituted AM15–52 analogues displayed conserved effects across all examined signaling pathways at all three CLR-based receptors (Figure 4, Figures S22 to S34, Tables S10 to S14). Most positions could tolerate alanine substitution with only a small effect on signaling (∼10-fold decrease); however, F18A, T20A, L26A, and I30A generally had large decreases in signaling at all tested pathways and receptors (30–600 fold; Figure 4, Figures S24, S25, S30, and S33). To confirm the results, we performed further characterization of analogues in a second cell-line (HEK293S) with consistent results (Figures S35 to S38, Table S5). We also extended our scan to residue 39 of AM to explore the importance of residues further along the peptide. We found that alanine substitution of residues past I30 did not decrease the Emax, but reduced peptide potency, indicating that these residues played a role in driving affinity for the receptor but not in driving peptide efficacy (Figure S39), a finding in line with the phenotype of the AM15–52 Δ35–39 peptide (Figure S20).
Figure 4.

(A) Relative effectiveness (Δτ/KA) values for pathway activation by alanine-substituted analogues of AM15–52; all values are presented as a fold change relative to unmodified AM15–52 and are derived from experiments performed in transfected Cos7 cells. (B) Bias factors (ΔΔτ/KA values) for pathway activation by alanine-substituted analogues of AM15–52, all values are presented relative to cAMP, with a positive value representing a bias toward the named pathway over cAMP. There was no IP1 production in response to unmodified AM15–52 at the CGRP or AM2 receptors, as such the values presented for AM15–52 G19A are estimates, and have not been used in statistical tests. Values in panel A were analyzed using paired Student’s t tests comparing the log(τ/KA) values of analogues to unmodified AM15–52. Values in panel B were analyzed using a one-way ANOVA with posthoc Dunnett’s test, comparing the activity of an analogue through the cAMP pathway to its activity at each other pathway. In both cases, the asterisk (∗) indicates a significant (p < 0.05) difference.
Our results showed that many of the residues most important for AM function were located within the disulfide loop structure. Thus, we examined the importance of this region through additional techniques. CD spectroscopy of analogues showed that AM15–52 F18A, G19A, and T20A had spectra associated with slight reductions in α-helical content (Figure S40); however, as these analogues have opposing effects on signaling (G19A increases signaling, while F18A and T20A reduce signaling), the α-helical content of the peptide does not seem to be a determinant of peptide function.
We also examined the pharmacology of these analogues through an additional cellular assay measuring β-arrestin recruitment. β-Arrestin recruitment is commonly measured in the context of biased signaling, as it is thought to be distinct from G protein signaling; β-arrestin recruitment is also linked to physiological outcomes distinct from physiological outcomes linked to G protein signaling.42,58,59 In this assay, AM15–52 G19A had improved recruitment at both the CGRP and AM1 receptors, while the other tested analogues had reduced activity at both tested receptors; the magnitude of this reduction was comparable to other signaling pathways, with R17A being best tolerated, F18A having a substantial decrease in potency and Emax at both receptors, and T20A being a weak partial agonist at the CGRP receptor, and unable to stimulate β-arrestin recruitment at the AM1 receptor (Figure S41, Table S16).
Position 19/5 is a Key Residue in AM/CGRP Pharmacology
Interestingly, AM15–52 G19A increased signaling through essentially all tested pathways and receptors (Figure 5, Figures S35, S38, S41). The increase was typically largest at the CGRP receptor. AM15–52G19A was also unique among AM based peptides for its ability to stimulate IP1 production, which was otherwise restricted to CGRP (Figure 2B, Figure S10). These findings are interesting because the residue in this position in αCGRP and βCGRP is natively alanine, therefore AM15–52 G19A could be thought of as a more “CGRP-like” AM. Adding to the “CGRP-like” pharmacology, AM15–52 G19A was a full agonist at stimulating β-arrestin recruitment at the CGRP receptor, as opposed to AM15–52 which was a partial agonist (Figure S41, Table S16), and trended toward being more potent than unmodified AM15–52 at the CTR:RAMP1 complex that can act as a second receptor for CGRP (Figure S42, Table S17). AM15–52 G19A was also interesting because it was the only analogue that appeared to be a biased agonist relative to the parent peptide, displaying a 13-fold preference for cAMP production over ERK phosphorylation at the CGRP receptor. This agonist profile arose because AM15–52 G19A was more potent than AM15–52 for cAMP production, but the two peptides were equipotent for ERK phosphorylation (Figure 5). This profile is likely to be driven by the bias intrinsic to the CGRP receptor, at which all endogenous peptides were equipotent for ERK phosphorylation, even when displaying differences in signaling profiles for other pathways (Figure 2).
Figure 5.
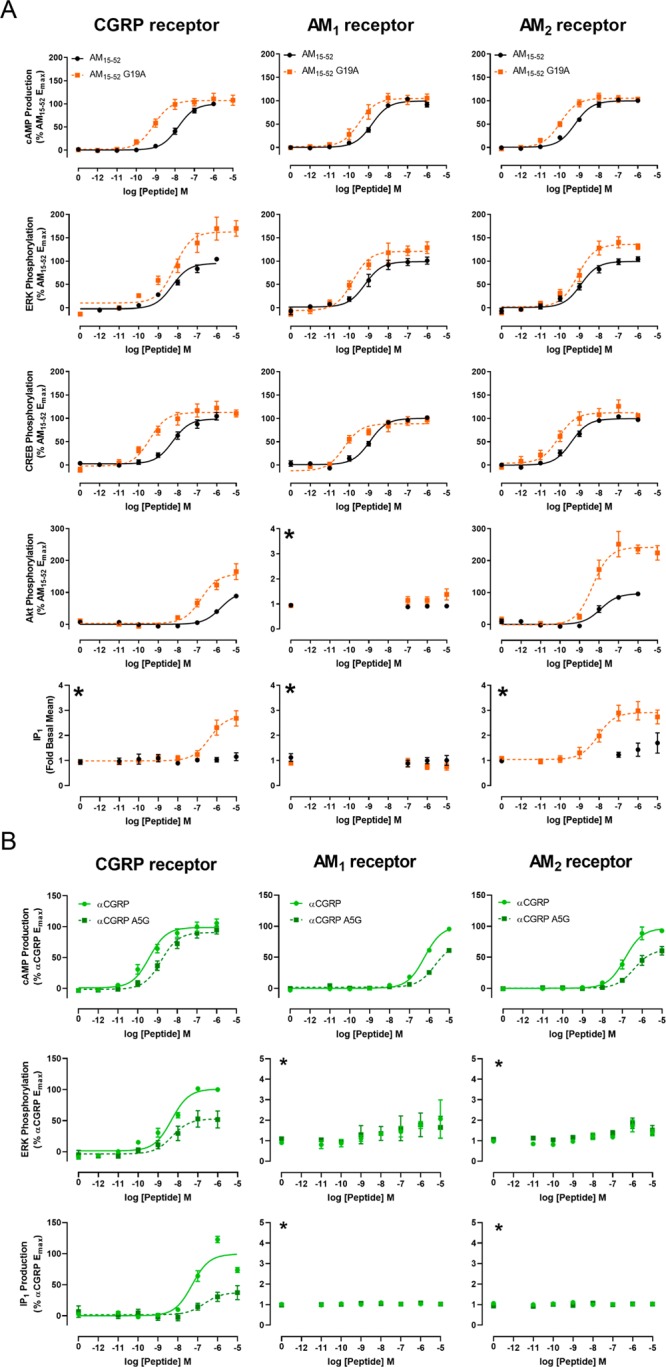
(A) Signaling of AM15–52 and AM15–52 G19A at the three CLR:RAMP complexes in transfected Cos7 cells. (B) Signaling of αCGRP A5G at the three CLR based receptors through select signaling pathways. All data are the mean ± s.e.m. of at least five or three independent experiments (curves or flat-lines, respectively) (see Tables S10 to S14 and S18). The asterisk (∗) denotes a pathway shown as fold-basal signaling as there was no measurable response to the control peptide.
To further explore the importance of this position for peptide function, we synthesized αCGRP A5G, which is the reciprocal amino acid exchange between AM and αCGRP. We tested this peptide at the three CLR based receptors through cAMP production, ERK phosphorylation, and IP1 production, as these were the pathways displaying AM15–52 G19A activity which differed from established AM signaling norms. αCGRP A5G was a weaker agonist than αCGRP through all tested pathways and receptors, indicating that making αCGRP more “AM-like” did not confer AM pharmacology at the AM1 and AM2 receptors. However, αCGRP A5G was more “AM-like” at the CGRP receptor, in that it was a weaker agonist than αCGRP through all tested pathways (Figure 5, Table S18). It is worth noting that the shifts in signaling ability were opposite to the effects noted with AM15–52 G19A. Specifically, while AM15–52 G19A had increased potency through cAMP production (but little change in Emax), and increased Emax for ERK phosphorylation (but little change in potency) at the CGRP receptor, αCGRP A5G had decreased potency for cAMP production (but no change in Emax), and a decreased Emax (with little change in potency) for ERK phosphorylation at the CGRP receptor. Similarly, while AM15–52 G19A gained the ability to stimulate IP1 production at the CGRP receptor, αCGRP A5G had a drastically reduced ability to stimulate IP1 production at this receptor.
Signaling Profiles of Peptide Analogues Are Similar in Cells That Endogenously Express AM-Responsive Receptors
To increase the translational relevance of this work, we investigated the alanine-substituted AM analogues in cells that endogenously express AM responsive receptors, reflecting the cellular context that AM may encounter in vivo. AM exerts its effects on the vasculature at least partially through activity on endothelial cells.6,60−63 HMEC-1 cells are an immortalized cell-line derived from human microvascular endothelial cells of dermal origin, and have been used to model various vascular processes including wound healing, angiogenesis, and vascular regulation.4,64−68 These are processes that AM regulates in vivo, indicating that HMEC-1 cells are an appropriate cell-line to model AM activity in the vasculature.69−72
HMEC-1 cells have variously been reported to express mRNA for CLR, CTR, and all three RAMPs; however, there is no consensus within the literature on which receptor components are expressed in these cells, or the relative expression levels.73−75 AM and αCGRP are both reported to exert functional effects in HMEC-1 cells; however, in these studies only a single peptide was tested, and often only at a single high concentration, meaning we have no information on which receptor is functionally expressed by this cell-line.4,74,76,77 Therefore, before profiling alanine-substituted analogues, we first characterized cAMP production using endogenous ligands. We first performed time-course experiments with AM to determine the optimal stimulation duration (7 min, Figure S43), before characterizing AM, AM2, αCGRP, and βCGRP in concentration–response experiments (Figure 6A, Table S19). Interestingly, the results from this pharmacological characterization did not exactly match the profiles obtained in transfected cells (Figure S6 and S44, Tables S2, S19, S20), or results from previous literature.1,11,78 The most noticeable difference between transfected cells and HMEC-1 cells was the extent to which AM2 and αCGRP were partial agonists; these were weaker agonists in HMEC-1 cells (Tables S2, S19, S20). This difference may be due to lower cell surface expression of receptors in HMEC-1 cells relative to cells transfected with receptor components, and aligns with previous reports that suggest that AM2 is a high affinity, low efficacy agonist of the AM1 receptor.79−81 Overall the profile of the HMEC-1 cells is most consistent with expression of an AM1 receptor, based on the cAMP signaling profile. This conclusion is based on the relative rank order of potency for cAMP production, being AM > AM2 > βCGRP ≥ αCGRP in both HMEC-1 cells (Figure 6A) and cells transfected with the AM1 receptor (Figure 2, Figure S6B, and Table S2 [Cos7], Figure S44B and Table S20 [HEK293S]). This profile is not consistent with an AM2 receptor, at which AM2 is consistently reported to be a full agonist that is equipotent to AM (Figure S6C and Table S2 [Cos7], Figure S44C and Table S20 [HEK293S]).1,11,82 Additionally, this profile is not consistent with expression of RAMP1, as αCGRP was a very weak agonist in HMEC-1 cells, while commonly reported to be a full agonist at CLR:RAMP1 and CTR:RAMP1 complexes.1 Preliminary experiments performed during initial characterization showed that expression of CTR in our HMEC-1 cells is unlikely, as neither amylin nor salmon CT elicited measurable cAMP production (data not shown).
Figure 6.
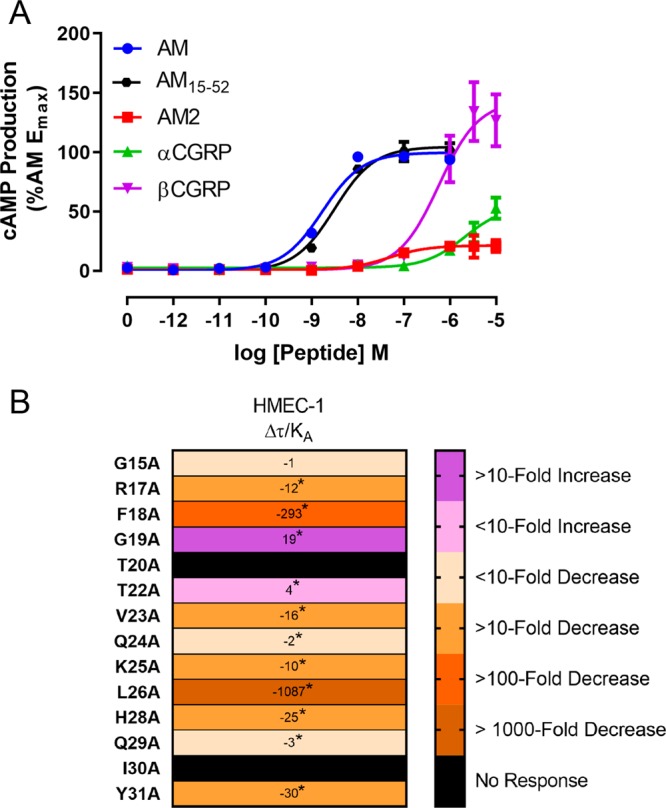
(A) cAMP production stimulated by endogenous peptides in HMEC-1 cells. Data points are the mean ± s.e.m. of seven independent experiments for all peptides except for AM2, which is n = 6, and AM15–52 which is n = 5. There was one other experiment where AM2 was unable to elicit a measurable response, and this experiment has been excluded from the current data set. For values derived from these curves, see Tables S19 and S21 and Figure S45. (B) relative effectiveness (Δτ/KA values) for alanine-substituted analogues of AM15–52 stimulating cAMP production in HMEC-1 cells. Values are presented as fold-change relative to AM15–52. Analogues were compared to AM15–52 using paired Student’s t-tests; this analysis was used because the experimental design created a paradigm in which the results from an individual analogue were linked to only the AM15–52 included on each plate, and not to the values of other peptides. The asterisk (∗) indicates a significant (p < 0.05) difference.
We also tested whether AM15–52 was equivalent to full-length AM in these HMEC-1 cells. AM15–52 was 2-fold weaker than full-length AM in these cells (Figure 6A); this difference was statistically significant; however, this reduction was very small (Table S21, Figure S45). Pharmacological characterization through other pathways was attempted (ERK phosphorylation, CREB phosphorylation, Akt phosphorylation, and IP1 production); however, we were unable to robustly measure the activation of any of these pathways (data not shown).
We then characterized the alanine-substituted AM15–52 analogues in the HMEC-1 cells (Figure 6B, Figure S46). Trends were generally conserved between transfected cells and the HMEC-1 cells, though effects of substitutions were often exaggerated in the HMEC-1 cells. For example, AM15–52 T20A was a partial agonist of cAMP production at the AM1 receptor in transfected Cos7 cells, but unable to stimulate cAMP production in HMEC-1 cells (Figures 4 and 6B). Likewise, the increase in potency for cAMP production associated with AM15–52 G19A was greater in HMEC-1 cells than in transfected cells (Figures 4 and 6B). This may be due to HMEC-1 cells expressing a lower density of receptors on the cell surface.79,83 Therefore, while there may be differences between transfected cell models and cells which endogenously express receptors, performing screening in transfected cell systems still holds utility as a tool.
Analysis of Our Results in Light of New Structures: Complementary Outcomes of Structural Biology and Functional Biology
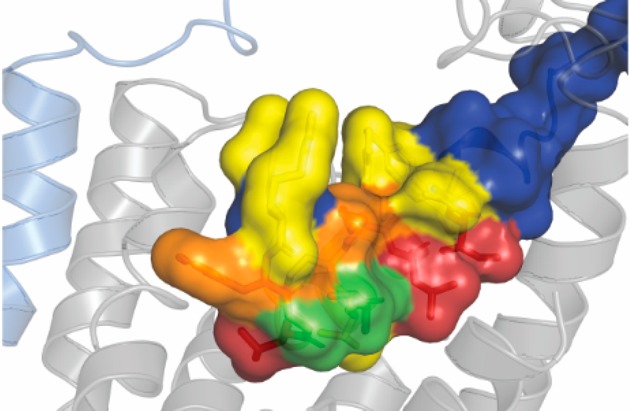
The current renaissance of cryo-EM is providing unprecedented insights into the structures of class B GPCRs.19,21−26,84 The cryo-EM structure of αCGRP bound to the CGRP receptor, combined with the new cryo-EM structures of AM bound to AM1 and AM2 receptor, and AM2 bound to the AM2 receptor allow insight into how ligands interact with this family of receptors.34 Combining these structures with structure–function studies allows a complementary approach to understand the dynamic molecular mechanisms of receptor engagement and activation. Functional data would suggest that the important residues in peptide function are conserved between αCGRP, AM, and AM2, even when these peptides are bound to different CLR based receptors; this functional data aligns well with new structural information that shows that these three peptides adopt a conserved N-terminal conformation on receptor interaction, though CGRP extends further toward CLR TMs 6/7 and makes more contact with this region of the receptor than AM (Figure 7). Additionally, it appears that the majority of residues which were important for stimulating signaling tend to project down into the binding pocket, generally in the direction of TM1, TM5, and to a lesser extent, TM6/ECL3/TM7 (Figure 7).
Figure 7.

Structural models showing the impact of individual amino acid substitutions across αCGRP, AM, and AM2. (A) αCGRP at the CGRP receptor,19 (B) AM at the AM1 receptor, (C) AM at the AM2 receptor, and (D) AM2 at the AM2 receptor.34 Results for AM are cAMP results from this paper at the AM1 receptor (Cos7 and HEK293S) and AM2 receptor (Cos7), results for AM2 and αCGRP are derived from previous publications (Tables S23 and S24). In this image CLR is gray, RAMP1 is pink, RAMP2 is light blue, RAMP3 is gold, and peptides are blue. Effects of substitutions are colored according to the legend.
These structures also allow insight into mechanisms of residue importance. L26 and I30 were critical for AM function, as substitution of either of these residues resulted in a large decrease in peptide activity. These residues are conserved as leucine or isoleucine in AM, AM2, αCGRP, βCGRP, amylin, and salmon CT (Figure 1A), and in human CT these residues are similarly bulky hydrophobic amino acids (tyrosine and phenylalanine). Structures of peptides bound to CLR/CTR based receptors show that the residues in this position sit at either side of TM1, essentially sandwiching CLR A138 (CTR A145) between two large hydrophobic residues on the peptide. Previous investigations into αCGRP signaling highlighted that alanine substitution of either L12 or L16 reduced the potency and affinity of the peptide, while mutational analysis of CLR suggests that this region of TM1 can be important for stimulating cAMP production.13,85 It is possible that the interaction of L26, I30, and CLR A138 plays an important role in maintaining a peptide conformation that allows for receptor activation, or in anchoring the peptide to the receptor.
AM15–52 F18A was interesting in that there was a large decrease in Emax with this peptide at the AM1 receptor, and a smaller decrease at the other CLR-based receptors. This finding is in line with other reports on the activity of this peptide, which have shown that substituting this residue with alanine reduces Emax at the AM1 receptor but not the AM2 receptor, and that interfering with this residue by olefin stapling F18 to T22 reduces the Emax at the AM1 receptor but not at the CGRP receptor.15,49 This residue sits in a similar environment in both the AM1 and AM2 receptors, projecting toward the extracellular face of CLR TM5/ECL2, suggesting that the relative importance of F18 is not because this residue makes differential contacts between receptors. Differences in relative importance between receptors are therefore most likely to arise from the extent of conformational ranges sampled by the AM1 receptor compared to the CGRP and AM2 receptors.34
Our results also showed that T20 plays a critical role in the activity of AM. T20 is conserved as a threonine both across species (Figure S14), and across other peptides in this family (Figure 1A).1,2 Previous investigations into related peptides have shown that this residue plays a critical role in activating receptors, as substitution of this residue consistently causes a large decrease in the ability of the peptide to stimulate a signaling response, while only having a small effect on affinity.41,82,86 Structures of peptides bound to their receptors show that this residue sits deep in the binding pocket, interacting with residues on TMs 4, 5, and 6.19,25 On the basis of MD simulations, the −OH group on αCGRP T6 forms persistent hydrogen bonds with CLR H295; similar hydrogen bonds are predicted between salmon CT T6 and CTR H302 (equivalent to CLR H295).19,25 The methyl group on this threonine also contributes to peptide function; αCGRP T6S, which retains the −OH group of threonine but lacks a methyl group, is a weaker agonist than unmodified αCGRP, but more potent than αCGRP T6A, which lacks both the methyl group and the hydrogen bond forming −OH group of threonine.86
New cryo-EM structures can also offer insight into why G19 plays such a critical role in peptide function. This residue is conserved as a small amino acid across the peptide family, being glycine in AM and AM2, serine in CT, and alanine in CGRP and amylin (Figure 1). Likewise, this position is well conserved as glycine across species of AM (Figure S14).2 On the basis of structural information, this residue binds deep in the receptor pocket projecting toward the juxtamembranous tips of CTR/CLR TM6 and TM7.19,25,34 Previous investigation into this position has shown that it can, to some extent, direct peptide specificity. Amylin A5S (a more “CT-like” amylin) was more potent than unmodified amylin at all CTR based receptors; however, the largest increase was noted at CTR alone (the receptor at which CT is most potent).41
The region of TM6/ECL3/TM7 has the largest differences in conformation between the currently reported CLR:RAMP complexes.19,25,34 Residues in CLR TM6/ECL3 can generally tolerate mutation to alanine with little-to-no effect on αCGRP signaling at the CGRP receptor, with the exception of E357 and I360, which both cause large decreases in αCGRP potency when mutated to alanine. In contrast, alanine mutation of residues in TM6/ECL3 negatively impacts the ability of AM to signal at the AM1 and AM2 receptors, though alanine mutation of I360 does not affect AM signaling at these receptors.15,87 At present, we do not know whether this is a ligand-specific effect, or a RAMP-specific effect. It is possible that ECL3 of the AM1 and AM2 receptors makes contacts with the lipid bilayer, while the CGRP receptor ECL3 does not, thus explaining the discrepancy between receptors. Alternatively, this effect could arise from differential interactions between the peptides and this region of CLR. An alanine in this position on the peptide is likely to make more contacts with the receptor than a glycine, and thus peptides incorporating an alanine may better tolerate receptor mutations as they can still contact the receptor. It is possible that this idea underlies the difference in pharmacology noted with AM15–52 G19A—the introduction of a methyl group (glycine to alanine) may allow for additional contacts to be made between the peptide and TM6/ECL3/TM7. Alternatively, it is possible that the effect noted with G19A results from an alteration to the flexibility of the disulfide loop. The native glycine found in AM is likely to provide conformational flexibility to the region.88 As such, replacing this glycine with an alanine could constrain the loop in a way that improves receptor activation by promoting a peptide conformation which makes more/stronger interactions with CLR, though given that most of the residues in the loop occupy similar positions when comparing between peptides, this may be unlikely.19,25,34 Regardless of the mechanism, this position seems to be a powerful determinant of peptide activity, and should be investigated further for developing novel agonists.
Our finding that substitutions affected signaling in a balanced way is at odds with literature on class B GPCR ligands. Previous investigations have shown that modifications (such as alanine substitutions) can have drastic and differential effects on signaling.89−94 A possible mechanistic explanation for this difference is that the peptides in the AM/CT peptide family make only limited contact with the receptor regions of TM6/ECL3/TM7, which have been highlighted as key areas for directing biased signaling in other class B GPCRs.34,95 Thus, this lack of interaction with the bias-directing portion of the receptor could account for the relatively balanced profile of our analogues.
Structures Provide Insight into the Role of Residues within the Disulfide Loop
Our findings highlighted interesting effects of residues within the disulfide loop structure. This is consistent with other studies on peptides from this family, and with the knowledge that truncated peptides which lack the disulfide loop structure act as competitive antagonists of these receptors.41,81,82,86,96,97 Within the loop, the residue immediately following the first cysteine (AM R17, AM2 V12, αCGRP D3) can tolerate modification with only small effects on signaling (Figure 4);86,98,99 this is explained by the residue in this position projecting into free space, thus having few structural constraints (as evidenced by the multiple conformations residues in this position can adopt between structures and the low densities reported in cryo-EM maps).19,25,34 Substitution of T20 (or the equivalent residue in other peptides) has a substantial effect in all peptides, this consistency is most likely due to it occupying a common position between peptides and receptors. Substitution of F18 and G19 had more differential effects, which given their constrained environments and adoption of similar positions between structures, may be due to differences in receptor dynamics.34
Likewise, structures provide an understanding as to the lack of importance of the extended N-terminus of AM. The N-termini of AM, AM2, and αCGRP all project up and out of the binding pocket, explaining why the N-terminal extensions associated with AM and AM2 are not required for signaling (Figure 3), and why lipidation of position 1 in αCGRP is tolerated with only minor effects on signaling.82,100 In contrast, the N-terminus of sCT projects back into the binding pocket in the region of ECL2, this effect is caused by CT having a larger loop (seven amino acids) than AM, AM2, and CGRP (six amino acids).25
Conclusion
We have characterized the three CLR:RAMP complexes, showing that each CLR:RAMP complex can regulate a different suite of intracellular proteins. This exploratory work lays the foundation for understanding how each signaling pathway contributes to the diverse roles associated with CLR-based receptors. Through our alanine scan we have emphasized the importance of the disulfide loop as a key determinant in peptide activity within this family, but also show that residues through to the midregion of AM are important for stimulating signaling. Our findings also highlight the importance of G19 in the pharmacology of AM, and the importance of this position across the entire peptide family; it is possible that this residue could be modified to alter selectivity. We have also highlighted that substitutions affected signaling in balanced ways, indicating that there may be less scope to design biased analogues for this family of receptors than for other class B GPCRs, although modifications that enhance engagement with TM6/ECL3/TM7 may provide an avenue to promote alternative signaling profiles. Although there were differences in the signaling profiles between transfected cells and those which endogenously express receptors of interest, we highlighted that the effects of substitutions were generally retained between the two cell-types, indicating that transfected cells still hold an important place in the screening process. We also highlighted the complementary nature of cryo-EM structures and structure–function investigations as two methods which, when combined, offer deep insights into the dynamic molecular mechanisms for receptor engagement, and subsequent activation.
Methods
Peptide Chemistry
Unmodified peptides were either bought commercially or synthesized in-house. AM and αCGRP were bought from American Peptide (Sunnyvale, CA, U.S.A.), Bachem (Bubendorf, Switzerland), or synthesized in-house, AM2-47 was bought from Bachem or synthesized in-house, and βCGRP was synthesized in-house. Synthesis of unmodified peptides has been described previously.13,41,82 All analogues and fragments were synthesized in-house.
Peptide synthesis was performed using an Fmoc solid phase peptide synthesis approach. A detailed description of the methodology is available in Supporting Information.
Cell Culture and Transfection
Multiple mammalian cell-lines were used in this study. For experiments involving transient expression of receptor constructs, Cos7 and HEK293S cells were used. The cell-lines used in this study have previously been characterized by our lab to show they lack endogenous expression of CLR, CTR, and RAMPs, thereby allowing us careful control of receptor expression.101 Both cell-lines were cultured as previously described.101,102 Briefly, cells were maintained in Dulbecco’s modified Eagle medium (DMEM; ThermoFisher, New Zealand) supplemented with 8% heat-inactivated fetal bovine serum (FBS) in a 37 °C/5% CO2 humidified incubator. Cells were seeded at a density of 20 000 cells per well (determined using a Countess Counter, ThermoFisher, New Zealand) into 96-well Spectraplates (Cos7 [all assays] and CHO-K1 [cAMP assays]; PerkinElmer, Waltham, MA, U.S.A), CellBind multiwell plates (HEK293S; Corning, NY, U.S.A), or white-walled clear bottomed 384-well microplates (CHO-K1 [β-arrestin assays]). Cos7 and HEK293S cells were transfected using polyethylenimine as previously described.101 All DNA constructs were encoded in pcDNA3.1. Receptor constructs used in this study were human. CTR(a), CLR, RAMP1, and RAMP2 were N-terminally tagged with HA (CTR and CLR), myc (RAMP1), and FLAG (RAMP2); these tags have been shown to not affect signaling.101−103 The RAMP3 construct used in this study was not tagged.
HMEC-1 Cell Culture
The HMEC-1 cell-line was also investigated during this study. HMEC-1 cells were grown in complete MCDB-131 comprising MCDB-131, no glutamine (Life Technologies, New Zealand), supplemented with 10% heat-inactivated FBS, 1 μg/mL hydrocortisone (Sigma-Alrich), 50 μg/mL endothelial cell growth supplement (Abacus Dx Limited, New Zealand), and 5% penicillin/streptomycin/glutamine (Gibco). For regular passaging, cells were grown in T-75 or T-175 flasks until 90% confluent. For T-75 flasks, upon reaching 90% confluency, the growth media was removed and cells were washed once with 5 mL of Dulbecco’s phosphate-buffered saline (DPBS). The DPBS was removed and replaced with 5 mL of TrypLE, cells were then incubated at 37 °C for 5 min. The flask was then agitated to suspend cells, and 5 mL of complete MCDB-131 was added to the flask. Cells were then transferred to a new flask containing fresh complete MCDB-131. Cells were then grown in a 37 °C/5% CO2 humidified incubator.
Cell seeding was performed essentially as described above for Cos7 and HEK293S cells. Cells were seeded at a density of 20 000 cells per well into 96-well Spectraplates. Cells were grown for 2 days before being used in experiments.
Experimental Design
For all signaling pathways, time-course experiments were first performed using a saturating concentration of peptide to determine the optimal duration for subsequent concentration–response experiments. There were two experimental designs used throughout this study. For characterization of endogenous peptides, all four peptides (AM, AM2, αCGRP, and βCGRP) were always included on each plate. This created a paradigm in which the results for a single peptide were directly related to the results of every other peptide. For characterization of analogues and fragments, peptides were randomly assigned to experimental plates; each experimental plate also contained a control peptide. This resulted in a paradigm where the response of each analogue/fragment was linked to the control peptide included on the same plate, but not to the results of other analogues/fragments. In all cases, duplicate, triplicate, or quadruplicate technical replicates were included for each independent experiment. Independent experiments involve plating cells from a distinct passage, separate transient transfections (where applicable), and separate peptide dilutions for stimulations.
Cellular Assays–cAMP Detection in Transfected Cos7 and HEK293S Cells
cAMP assays were performed using the LANCE cAMP detection kit (PerkinElmer) or the AlphaScreen cAMP assay kit (PerkinElmer) as described previously.13,104 Unless otherwise noted, cAMP detection in Cos7 cells was performed using the LANCE cAMP detection kit, and cAMP detection in HEK293S cells was performed using the AlphaScreen cAMP assay.
Cellular Assays–cAMP Detection in HMEC-1 Cells
cAMP assays were performed in accordance with previous literature with minor modifications.41 Two days after seeding, HMEC-1 cells were used in experiments. Briefly, on the day of the experiment growth media was aspirated from the cells and replaced with 50 μL of stimulation media (comprising MCDB-131 supplemented with 0.1% bovine serum albumin and 1 mM IBMX). Plates were then incubated at 37 °C for 30 min before being stimulated with peptides. Peptides were serially diluted in stimulation media. Cells were stimulated with peptide for 7 min. Wells were then thoroughly aspirated and replaced with 50 μL ice-cold ethanol. Plates were then placed at −20 °C for a minimum of 15 min and a maximum of 7 days.
Ethanol was evaporated from the wells by placing the plate in a fume hood. Cells were lysed by adding 25 μL of LANCE Ultra lysis buffer (provided with the kit) then shaken at room temperature for 10–15 min. A cAMP standard curve was created in kit lysis buffer by serially diluting a stock cAMP in kit lysis buffer. Cell lysate or standard curve was transferred to a 384-well OptiPlate (both 10 μL); standards were transferred in duplicate. Eu-cAMP (5 μL diluted 1:50 in LANCE Ultra lysis buffer) and Ulight reagent (5 μL diluted 1:150 in LANCE Ultra lysis buffer) were added to each well, and the plate was then sealed and centrifuged for 10 s at 400g. The plate was left to incubate for 1 h before being read on an EnVision plate reader with excitation at 340 nm and emissions detected at 620 and 665 nm.
Cellular Assays—IP1 Production
IP1 assays were performed as described previously with minor modifications.41 The stimulation duration was extended from 90 to 120 min, and other than that the protocol remained unchanged.
Cellular Assays—ERK Phosphorylation, CREB Phosphorylation, and Akt Phosphorylation
AlphaLISA SureFire Ultra kits were used to measure ERK phosphorylation on residues T202/Y204, CREB phosphorylation on S133, and Akt phosphorylation on S473. Assays were performed in accordance with previous literature.41 Stimulation durations for concentration–response experiments were 10 min in all instances, otherwise the protocol remained unchanged. For Akt phosphorylation, 50% FBS, and 200 nU insulin were used as positive controls.
Data Analysis—Concentration—Response Assays
Data were analyzed using GraphPad PRISM versions 6, 7, and 8. For each individual experiment, concentration response curves were fit using three-parameter nonlinear regression. A response was only deemed a curve when at least two data-points were above the response to media control, otherwise the response was deemed unquantifiable and referred to as a flat-line. If a response was deemed a curve, but did not appear to reach its maximal response within the tested concentration range, the curve-fit was constrained using the mean response at the highest concentration of peptide as the Emax for the peptide.
In the case of weak agonists or weakly coupled pathways, there were some instances in which a peptide could stimulate a measurable response during some experiments and not in others. In these cases the outcome from the majority of independent experiments has been reported. When reporting these results, the experiments from the minority are excluded from the reported n numbers, and a note is included in the legend to indicate this.
From curve fits we obtained the pEC50 and Emax. Individual pEC50 and Emax values were combined to generate mean data. pEC50 data were analyzed using either repeated measures one-way analysis of variance (ANOVA) with posthoc Tukey’s test (endogenous ligand characterization), or paired Student’s t-tests (characterization of analogues/fragments). These different approaches are justified in the above section “Experimental design”. To analyze the differences in Emax between endogenous ligands, the raw Emax values were log-transformed, then the resultant values were compared using a repeated measures one-way ANOVA with posthoc Tukey’s test.105 Raw Emax values for analogues and fragments were compared to the relevant control using a paired ratio Student’s t-test.
For ease of comparison, and to take into account day-to-day variability associated with transient transfections, data were normalized for presentation in the manuscript. This involved normalizing each experiment to the fitted maximum and minimum of the relevant control included on each plate. Normalized curves were then generated by combining the mean of data points from individual experiments.
Data analysis—Operational Model of Agonism
Transduction ratios and bias factors were quantified using the operational model of agonism as described previously.106 This analysis was only applied to results from Cos7 cells and HMEC-1 cells. Transduction ratios (log(τ/KA) values) were derived from individual experiments by fitting the operational model as described by van der Westhuizen et al., to normalized individual experiments.106 The maximal response window of the system was defined as the largest normalized Emax recorded across the entire data set. All curves were constrained by setting n to 1, and the Emax as the maximal response window of the system. All curves were then fit as “partial agonists” relative to this Emax. The derived log(τ/KA) values were then compared to a reference ligand to obtain Δ(τ/KA) values. When the ability of endogenous agonists to simulate a signaling pathway was compared, the reference ligand was αCGRP at the CGRP receptor and AM at the AM1 and AM2 receptors; data were analyzed using repeated measures one-way ANOVA with posthoc Tukey’s test, comparing each peptide to each other peptide. When the ability of analogues or fragments to stimulate signaling was compared, the reference ligand was unmodified AM15–52; data were analyzed using paired Student’s t-tests. In both cases, statistical significance was accepted at p < 0.05.
To obtain bias factors (ΔΔ(τ/KA) values), we normalized the Δ(τ/KA) values to a chosen reference pathway, in this case cAMP production. This allowed us to investigate whether peptides had a “biased” signaling profile, that is, a preference for activating one signaling pathway over another. ΔΔ(τ/KA) values were analyzed using a one-way ANOVA with posthoc Dunnett’s test, comparing the ability of each peptide to activate a signaling pathway relative to its ability to stimulate cAMP production. Statistical significance was accepted at p < 0.05.
Acknowledgments
This work was supported by the Health Research Council (New Zealand), Lottery Health, Maurice Wilkins Centre for Molecular Biodiscovery, Marsden Fund (Royal Society of New Zealand), and the New Zealand Heart Foundation. D.L.H. acknowledges receipt of a James Cook Research Fellowship from the Royal Society of New Zealand. C.S.W. acknowledges receipt of a Sir Charles Hercus Fellowship from the Health Research Council (New Zealand), M.L.G. acknowledges receipt of a University of Auckland Health Research Ph.D. Scholarship and E.R.H. acknowledges receipt of a Ph.D. scholarship from the Auckland Medical Research Foundation. P.M.S. is a Principal Research Fellow of the National Health and Medical Research Council of Australia (NHMRC). D.W. is a Career Development Fellow of the NHMRC. We thank David R. Poyner of Aston University for useful discussions and help with planning this project. We also thank Richard Kingston of The University of Auckland for help with CD spectroscopy.
Glossary
Abbreviations
- AM
adrenomedullin
- AM2
adrenomedullin 2/intermedin
- CD
circular dichroism
- CGRP
calcitonin gene-related peptide
- CLR
calcitonin receptor-like receptor
- CREB
cAMP response element-binding protein
- CT
calcitonin
- ECD
extracellular domain
- ECL
extracellular loops
- ERK
extracellular regulated kinase
- GPCR
G protein-coupled receptor
- IP
inositol phosphate
- RAMP
receptor activity-modifying protein
- TM
transmembrane
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.9b00083.
Additional biology methodology (time-course assays, CisBio HTRF assays, cell surface expression through ELISA, competition binding using radiolabeled peptide, CD spectroscopy, and β-arrestin recruitment assays); rationale for selection of signaling pathways; additional biology results (time-course assays, concentration–response assays of AM fragments and analogues, summary tables reporting values derived from concentration–response assays, amino acid alignments, competition binding assays using radiolabeled peptide, CD spectroscopy, and tables of previous modifications to αCGRP and AM2); additional chemistry methods (general peptide synthesis, purification, and analysis techniques); summary table of synthesized peptides; LCMS and HPLC and ESI-MS chromatograms for synthesized peptides (PDF)
Author Contributions
M.A.B., A.S., G.M.W., S.H.Y., and P.W.R.H. performed peptide synthesis. M.L.G., M.A., J.J.G., E.R.H., A.L., N.P., H.A.W., and D.L.H., performed biological experiments. M.L.G., P.M.S., D.W., C.S.W., P.W.R.H., and D.L.H. interpreted experiments and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hay D. L.; Garelja M. L.; Poyner D. R.; Walker C. S. (2018) Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br. J. Pharmacol. 175, 3–17. 10.1111/bph.14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönauer R.; Els-Heindl S.; Beck-Sickinger A. G. (2017) Adrenomedullin – new perspectives of a potent peptide hormone. J. Pept. Sci. 23, 472–485. 10.1002/psc.2953. [DOI] [PubMed] [Google Scholar]
- Trincot C. E.; Xu W.; Zhang H.; Kulikauskas M. R.; Caranasos T. G.; Jensen B. C.; Sabine A.; Petrova T. V.; Caron K. M. (2019) Adrenomedullin Induces Cardiac Lymphangiogenesis After Myocardial Infarction and Regulates Cardiac Edema Via Connexin 43. Circ. Res. 124, 101–113. 10.1161/CIRCRESAHA.118.313835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalfaoui-Bendriss G.; Dussault N.; Fernandez-Sauze S.; Berengeur-Daize C.; Sigaud R.; Delfino C.; Cayol M.; Metellus P.; Chinot O.; Mabrouk K.; Martin P. M.; Ouafik L. (2015) Adrenomedullin blockade induces regression of tumor neovessels through interference with vascular endothelial-cadherin signalling. Oncotarget 6, 7536–7553. 10.18632/oncotarget.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Hur M.; Struck J.; Bergmann A.; Di Somma S. (2019) Circulating Biologically Active Adrenomedullin Predicts Organ Failure and Mortality in Sepsis. Ann. Lab. Med. 39, 454–463. 10.3343/alm.2019.39.5.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iring A.; Jin Y. J.; Albarran-Juarez J.; Siragusa M.; Wang S.; Dancs P. T.; Nakayama A.; Tonack S.; Chen M.; Kunne C.; Sokol A. M.; Gunther S.; Martinez A.; Fleming I.; Wettschureck N.; Graumann J.; Weinstein L. S.; Offermanns S. (2019) Shear stress-induced endothelial adrenomedullin signaling regulates vascular tone and blood pressure. J. Clin. Invest. 129, 2775–2791. 10.1172/JCI123825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron K. M.; Smithies O. (2001) Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc. Natl. Acad. Sci. U. S. A. 98, 615–619. 10.1073/pnas.98.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönauer R.; Els-Heindl S.; Fischer J.-P.; Köbberling J.; Riedl B.; Beck-Sickinger A. G. (2016) Adrenomedullin 2.0: Adjusting Key Levers for Metabolic Stability. J. Med. Chem. 59, 5695–5705. 10.1021/acs.jmedchem.6b00126. [DOI] [PubMed] [Google Scholar]
- Kataoka Y.; Miyazaki S.; Yasuda S.; Nagaya N.; Noguchi T.; Yamada N.; Morii I.; Kawamura A.; Doi K.; Miyatake K.; Tomoike H.; Kangawa K. (2010) The first clinical pilot study of intravenous adrenomedullin administration in patients with acute myocardial infarction. J. Cardiovasc. Pharmacol. 56, 413–419. 10.1097/FJC.0b013e3181f15b45. [DOI] [PubMed] [Google Scholar]
- Bermudez M.; Nguyen T. N.; Omieczynski C.; Wolber G. (2019) Strategies for the discovery of biased GPCR ligands. Drug Discovery Today 24, 1031–1037. 10.1016/j.drudis.2019.02.010. [DOI] [PubMed] [Google Scholar]
- Weston C.; Winfield I.; Harris M.; Hodgson R.; Shah A.; Dowell S. J.; Mobarec J. C.; Woodlock D. A.; Reynolds C. A.; Poyner D. R.; Watkins H. A.; Ladds G. (2016) Receptor Activity-modifying Protein-directed G Protein Signaling Specificity for the Calcitonin Gene-related Peptide Family of Receptors. J. Biol. Chem. 291, 21925–21944. 10.1074/jbc.M116.751362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomberger J. M.; Parameswaran N.; Hall C. S.; Aiyar N.; Spielman W. S. (2005) Novel function for receptor activity-modifying proteins (RAMPs) in post-endocytic receptor trafficking. J. Biol. Chem. 280, 9297–9307. 10.1074/jbc.M413786200. [DOI] [PubMed] [Google Scholar]
- Garelja M. L.; Walker C. A.; Siow A.; Yang S. H.; Harris P. W. R.; Brimble M. A.; Watkins H. A.; Gingell J. J.; Hay D. L. (2018) Receptor Activity Modifying Proteins Have Limited Effects on the Class B G Protein-Coupled Receptor Calcitonin Receptor-Like Receptor Stalk. Biochemistry 57, 1410–1422. 10.1021/acs.biochem.7b01180. [DOI] [PubMed] [Google Scholar]
- Simms J.; Hay D. L.; Bailey R. J.; Konycheva G.; Bailey G.; Wheatley M.; Poyner D. R. (2009) Structure-function analysis of RAMP1 by alanine mutagenesis. Biochemistry 48, 198–205. 10.1021/bi801869n. [DOI] [PubMed] [Google Scholar]
- Watkins H. A.; Chakravarthy M.; Abhayawardana R. S.; Gingell J. J.; Garelja M.; Pardamwar M.; McElhinney J. M.; Lathbridge A.; Constantine A.; Harris P. W.; Yuen T. Y.; Brimble M. A.; Barwell J.; Poyner D. R.; Woolley M. J.; Conner A. C.; Pioszak A. A.; Reynolds C. A.; Hay D. L. (2016) Receptor Activity-modifying Proteins 2 and 3 Generate Adrenomedullin Receptor Subtypes with Distinct Molecular Properties. J. Biol. Chem. 291, 11657–11675. 10.1074/jbc.M115.688218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley M. J.; Reynolds C. A.; Simms J.; Walker C. S.; Mobarec J. C.; Garelja M. L.; Conner A. C.; Poyner D. R.; Hay D. L. (2017) Receptor activity-modifying protein dependent and independent activation mechanisms in the coupling of calcitonin gene-related peptide and adrenomedullin receptors to Gs. Biochem. Pharmacol. 142, 96–110. 10.1016/j.bcp.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins H. A.; Walker C. S.; Ly K. N.; Bailey R. J.; Barwell J.; Poyner D. R.; Hay D. L. (2014) Receptor activity-modifying protein-dependent effects of mutations in the calcitonin receptor-like receptor: implications for adrenomedullin and calcitonin gene-related peptide pharmacology. Br. J. Pharmacol. 171, 772–788. 10.1111/bph.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C.; Reedtz-Runge S.; Rudolph R.; Stubbs M. T. (2009) Passing the baton in class B GPCRs: peptide hormone activation via helix induction. Trends Biochem. Sci. 34, 303–310. 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Liang Y. L.; Khoshouei M.; Deganutti G.; Glukhova A.; Koole C.; Peat T. S.; Radjainia M.; Plitzko J. M.; Baumeister W.; Miller L. J.; Hay D. L.; Christopoulos A.; Reynolds C. A.; Wootten D.; Sexton P. M. (2018) Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497. 10.1038/s41586-018-0535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L. H.; Ma S.; Sutkeviciute I.; Shen D. D.; Zhou X. E.; de Waal P. W.; Li C. Y.; Kang Y.; Clark L. J.; Jean-Alphonse F. G.; White A. D.; Yang D.; Dai A.; Cai X.; Chen J.; Li C.; Jiang Y.; Watanabe T.; Gardella T. J.; Melcher K.; Wang M. W.; Vilardaga J. P.; Xu H. E.; Zhang Y. (2019) Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153. 10.1126/science.aav7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Qiao A.; Yang L.; Van Eps N.; Frederiksen K. S.; Yang D.; Dai A.; Cai X.; Yi C.; Cao C.; He L.; Yang H.; Lau J.; Ernst O. P.; Hanson M. A.; Stevens R. C.; Wang M. W.; Reedtz-Runge S.; Jiang H.; Zhao Q.; Wu B. (2018) Structure of the glucagon receptor in complex with a glucagon analogue. Nature 553, 106–110. 10.1038/nature25153. [DOI] [PubMed] [Google Scholar]
- Liang Y. L.; Khoshouei M.; Glukhova A.; Furness S. G. B.; Zhao P.; Clydesdale L.; Koole C.; Truong T. T.; Thal D. M.; Lei S.; Radjainia M.; Danev R.; Baumeister W.; Wang M. W.; Miller L. J.; Christopoulos A.; Sexton P. M.; Wootten D. (2018) Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125. 10.1038/nature25773. [DOI] [PubMed] [Google Scholar]
- Ehrenmann J.; Schoppe J.; Klenk C.; Rappas M.; Kummer L.; Dore A. S.; Pluckthun A. (2018) High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat. Struct. Mol. Biol. 25, 1086–1092. 10.1038/s41594-018-0151-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Sun B.; Feng D.; Hu H.; Chu M.; Qu Q.; Tarrasch J.; Li S.; Kobilka T. S.; Kobilka B. K.; Skiniotis G. (2017) Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253. 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y. L.; Khoshouei M.; Radjainia M.; Zhang Y.; Glukhova A.; Tarrasch J.; Thal D. M.; Furness S. G. B.; Christopoulos G.; Coudrat T.; Danev R.; Baumeister W.; Miller L. J.; Christopoulos A.; Kobilka B. K.; Wootten D.; Skiniotis G.; Sexton P. M. (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123. 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A.; Rappas M.; Brown A. J. H.; Kean J.; Errey J. C.; Robertson N. J.; Fiez-Vandal C.; Andrews S. P.; Congreve M.; Bortolato A.; Mason J. S.; Baig A. H.; Teobald I.; Dore A. S.; Weir M.; Cooke R. M.; Marshall F. H. (2017) Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 546, 254–258. 10.1038/nature22800. [DOI] [PubMed] [Google Scholar]
- Booe J. M.; Walker C. S.; Barwell J.; Kuteyi G.; Simms J.; Jamaluddin M. A.; Warner M. L.; Bill R. M.; Harris P. W.; Brimble M. A.; Poyner D. R.; Hay D. L.; Pioszak A. A. (2015) Structural Basis for Receptor Activity-Modifying Protein-Dependent Selective Peptide Recognition by a G Protein-Coupled Receptor. Mol. Cell 58, 1040–1052. 10.1016/j.molcel.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano S.; Kukimoto-Niino M.; Hino N.; Ohsawa N.; Okuda K.; Sakamoto K.; Shirouzu M.; Shindo T.; Yokoyama S. (2012) Structural basis for extracellular interactions between calcitonin receptor-like receptor and receptor activity-modifying protein 2 for adrenomedullin-specific binding. Protein Sci. 21, 199–210. 10.1002/pro.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness S. G. B.; Liang Y. L.; Nowell C. J.; Halls M. L.; Wookey P. J.; Dal Maso E.; Inoue A.; Christopoulos A.; Wootten D.; Sexton P. M. (2016) Ligand-Dependent Modulation of G Protein Conformation Alters Drug Efficacy. Cell 67, 739–749. 10.1016/j.cell.2016.09.021. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2019) Biased Receptor Signaling in Drug Discovery. Pharmacol. Rev. 71, 267–315. 10.1124/pr.118.016790. [DOI] [PubMed] [Google Scholar]
- Peterson Y. K.; Luttrell L. M. (2017) The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 69, 256–297. 10.1124/pr.116.013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Herenbrink C.; Sykes D. A.; Donthamsetti P.; Canals M.; Coudrat T.; Shonberg J.; Scammells P. J.; Capuano B.; Sexton P. M.; Charlton S. J.; Javitch J. A.; Christopoulos A.; Lane J. R. (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 7, 10842. 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bdioui S.; Verdi J.; Pierre N.; Trinquet E.; Roux T.; Kenakin T. (2018) Equilibrium Assays Are Required to Accurately Characterize the Activity Profiles of Drugs Modulating Gq-Protein-Coupled Receptors. Mol. Pharmacol. 94, 992–1006. 10.1124/mol.118.112573. [DOI] [PubMed] [Google Scholar]
- Liang Y. L., Belousoff M. J., Fletcher M. M., Zhang X., Khoshouei M., Deganutti G., Koole C., Furness S. G. B., Miller L. J., Hay D. L., Christopoulos A., Reynolds C. A., Danev R., Wootten D., and Sexton P. M. (2019) Structure and Dynamics of Adrenomedullin Receptors AM1 and AM2 Reveal Key Mechanisms in the Control of Receptor Phenotype by Receptor Activity-Modifying Proteins. ACS Pharmacol. Transl. Sci. 10.1021/acsptsci.9b00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. L.; Pioszak A. A. (2016) Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annu. Rev. Pharmacol. Toxicol. 56, 469–487. 10.1146/annurev-pharmtox-010715-103120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfis M.; Tilakaratne N.; Furness S. G.; Christopoulos G.; Werry T. D.; Christopoulos A.; Sexton P. M. (2008) Receptor activity-modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology 149, 5423–5431. 10.1210/en.2007-1735. [DOI] [PubMed] [Google Scholar]
- Drastichova Z.; Novotny J. (2012) Identification and subcellular localization of molecular complexes of Gq/11alpha protein in HEK293 cells. Acta Biochim. Biophys. Sin. 44, 641–649. 10.1093/abbs/gms050. [DOI] [PubMed] [Google Scholar]
- Costa-Neto C. M.; Parreiras E. S. L. T.; Bouvier M. (2016) A Pluridimensional View of Biased Agonism. Mol. Pharmacol. 90, 587–595. 10.1124/mol.116.105940. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Hay D. L.; Quirion R.; Poyner D. R. (2012) The pharmacology of adrenomedullin 2/intermedin. Br. J. Pharmacol. 166, 110–120. 10.1111/j.1476-5381.2011.01530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLatchie L. M.; Fraser N. J.; Main M. J.; Wise A.; Brown J.; Thompson N.; Solari R.; Lee M. G.; Foord S. M. (1998) RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393, 333–339. 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- Bower R. L.; Yule L.; Rees T. A.; Deganutti G.; Hendrikse E. R.; Harris P. W. R.; Kowalczyk R.; Ridgway Z.; Wong A. G.; Swierkula K.; Raleigh D. P.; Pioszak A. A.; Brimble M. A.; Reynolds C. A.; Walker C. S.; Hay D. L. (2018) Molecular Signature for Receptor Engagement in the Metabolic Peptide Hormone Amylin. ACS Pharmacol. Transl. Sci. 1, 32–49. 10.1021/acsptsci.8b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dal Maso E.; Glukhova A.; Zhu Y.; Garcia-Nafria J.; Tate C. G.; Atanasio S.; Reynolds C. A.; Ramírez-Aportela E.; Carazo J.-M.; Hick C. A.; Furness S. G. B.; Hay D. L.; Liang Y.-L.; Miller L. J.; Christopoulos A.; Wang M.-W.; Wootten D.; Sexton P. M. (2019) The Molecular Control of Calcitonin Receptor Signaling. ACS Pharmacol. Transl. Sci. 2, 31–51. 10.1021/acsptsci.8b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham V.; Zhu Y.; dal Maso E.; Reynolds C. A.; Deganutti G.; Atanasio S.; Hick C.; Yang D.; Christopoulos A.; Hay D. L.; Furness S. G.; Wang M.; Wootten D.; Sexton P. M. (2019) Deconvoluting the Molecular Control of Binding and Signaling at the Amylin 3 Receptor: RAMP3 Alters Signal Propagation through Extracellular Loops of the Calcitonin Receptor. ACS Pharmacol. Transl. Sci. 2, 183–197. 10.1021/acsptsci.9b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarwood R. E.; Imlach W. L.; Lieu T.; Veldhuis N. A.; Jensen D. D.; Klein Herenbrink C.; Aurelio L.; Cai Z.; Christie M. J.; Poole D. P.; Porter C. J. H.; McLean P.; Hicks G. A.; Geppetti P.; Halls M. L.; Canals M.; Bunnett N. W. (2017) Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc. Natl. Acad. Sci. U. S. A. 114, 12309–12314. 10.1073/pnas.1706656114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingell J. J.; Hendrikse E. R.; Hay D. L. (2019) New Insights into the Regulation of CGRP-Family Receptors. Trends Pharmacol. Sci. 40, 71–83. 10.1016/j.tips.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Forrester S. J.; Kawai T.; O’Brien S.; Thomas W.; Harris R. C.; Eguchi S. (2016) Epidermal Growth Factor Receptor Transactivation: Mechanisms, Pathophysiology, and Potential Therapies in the Cardiovascular System. Annu. Rev. Pharmacol. Toxicol. 56, 627–653. 10.1146/annurev-pharmtox-070115-095427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. Y.; Xu M. J.; Wang X. (2018) Adrenomedullin 2/intermedin: a putative drug candidate for treatment of cardiometabolic diseases. Br. J. Pharmacol. 175, 1230–1240. 10.1111/bph.13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga S.; Isawa H.; Yuda M. (2014) Horizontal gene transfer of a vertebrate vasodilatory hormone into ticks. Nat. Commun. 5, 3373. 10.1038/ncomms4373. [DOI] [PubMed] [Google Scholar]
- Fischer J. P.; Els-Heindl S.; Schonauer R.; Bierer D.; Kobberling J.; Riedl B.; Beck-Sickinger A. G. (2018) The Impact of Adrenomedullin Thr22 on Selectivity within the Calcitonin Receptor-like Receptor/Receptor Activity-Modifying Protein System. ChemMedChem 13, 1797–1805. 10.1002/cmdc.201800329. [DOI] [PubMed] [Google Scholar]
- Schönauer R.; Kaiser A.; Holze C.; Babilon S.; Köbberling J.; Riedl B.; Beck-Sickinger A. G. (2015) Fluorescently labeled adrenomedullin allows real-time monitoring of adrenomedullin receptor trafficking in living cells. J. Pept. Sci. 21, 905–912. 10.1002/psc.2833. [DOI] [PubMed] [Google Scholar]
- Champion H. C.; Nussdorfer G. G.; Kadowitz P. J. (1999) Structure-activity relationships of adrenomedullin in the circulation and adrenal gland. Regul. Pept. 85, 1–8. 10.1016/S0167-0115(99)00025-7. [DOI] [PubMed] [Google Scholar]
- Lin B.; Gao Y.; Chang J. K.; Heaton J.; Hyman A.; Lippton H. (1994) An adrenomedullin fragment retains the systemic vasodepressor activity of rat adrenomedullin. Eur. J. Pharmacol. 260, 1–4. 10.1016/0014-2999(94)90002-7. [DOI] [PubMed] [Google Scholar]
- Santiago J. A.; Garrison E. A.; Ventura V. L.; Coy D. H.; Bitar K.; Murphy W. A.; McNamara D. B.; Kadowitz P. J. (1994) Synthetic human adrenomedullin and adrenomedullin 15–52 have potent short-lived vasodilator activity in the hindlimb vascular bed of the cat. Life Sci. 55, 85–90. 10.1016/0024-3205(94)00652-0. [DOI] [PubMed] [Google Scholar]
- Yang B. C.; Lippton H.; Gumusel B.; Hyman A.; Mehta J. L. (1996) Adrenomedullin dilates rat pulmonary artery rings during hypoxia: role of nitric oxide and vasodilator prostaglandins. J. Cardiovasc. Pharmacol. 28, 458–462. 10.1097/00005344-199609000-00016. [DOI] [PubMed] [Google Scholar]
- Eguchi S.; Hirata Y.; Iwasaki H.; Sato K.; Watanabe T. X.; Inui T.; Nakajima K.; Sakakibara S.; Marumo F. (1994) Structure-activity relationship of adrenomedullin, a novel vasodilatory peptide, in cultured rat vascular smooth muscle cells. Endocrinology 135, 2454–2458. 10.1210/endo.135.6.7988431. [DOI] [PubMed] [Google Scholar]
- Watanabe T. X.; Itahara Y.; Inui T.; Yoshizawa-Kumagaye K.; Nakajima K.; Sakakibara S. (1996) Vasopressor activities of N-terminal fragments of adrenomedullin in anesthetized rat. Biochem. Biophys. Res. Commun. 219, 59–63. 10.1006/bbrc.1996.0181. [DOI] [PubMed] [Google Scholar]
- Hoare S. R. (2005) Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discovery Today 10, 417–427. 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- dal Maso E.; Zhu Y.; Pham V.; Reynolds C. A.; Deganutti G.; Hick C. A.; Yang D.; Christopoulos A.; Hay D. L.; Wang M. W.; Sexton P. M.; Furness S. G. B.; Wootten D. (2018) Extracellular loops 2 and 3 of the calcitonin receptor selectively modify agonist binding and efficacy. Biochem. Pharmacol. 150, 214–244. 10.1016/j.bcp.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisler J. W.; DeWire S. M.; Whalen E. J.; Violin J. D.; Drake M. T.; Ahn S.; Shenoy S. K.; Lefkowitz R. J. (2007) A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. U. S. A. 104, 16657–16662. 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geven C.; Pickkers P. (2018) The mechanism of action of the adrenomedullin-binding antibody adrecizumab. Crit. Care 22, 159. 10.1186/s13054-018-2074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinich N. O.; Hoopes S. L.; Kechele D. O.; Lenhart P. M.; Caron K. M. (2011) Adrenomedullin Function in Vascular Endothelial Cells: Insights from Genetic Mouse Models. Curr. Hypertens. Rev. 7, 228–239. 10.2174/157340211799304761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.; Moon S.-O.; Sung M. J.; Kim S. H.; Lee S. M.; S J.-N.; Park S. K. (2003) Angiogenic role of adrenomedullin through activation of Akt, mitogen-activated protein kinase, and focal adhesion kinase in endothelial cells. FASEB J. 17, 1937–1939. 10.1096/fj.02-1209fje. [DOI] [PubMed] [Google Scholar]
- Tanaka M.; Koyama T.; Sakurai T.; Kamiyoshi A.; Ichikawa-Shindo Y.; Kawate H.; Liu T.; Xian X.; Imai A.; Zhai L.; Hirabayashi K.; Owa S.; Yamauchi A.; Igarashi L.; Taniguchi S.; Shindo T. (2016) The endothelial adrenomedullin-RAMP2 system regulates vascular integrity and suppresses tumor metastasis. Cardiovasc. Res. 111, 398–409. 10.1093/cvr/cvw166. [DOI] [PubMed] [Google Scholar]
- Albiñana V.; Velasco L.; Zarrabeitia R.; Botella L. (2013) Immunosuppressor FK506 Increases Endoglin and Activin Receptor-Like Kinase 1 Expression and Modulates Transforming Growth Factor-beta1 Signaling in Endothelial Cells. Mol. Pharmacol. 79, 833–843. 10.1124/mol.110.067447. [DOI] [PubMed] [Google Scholar]
- Bertl E.; Bartsch H.; Gerhauser C. (2006) Inhibition of angiogenesis and endothelial cell functions are novel sulforaphane-mediated mechanisms in chemoprevention. Mol. Cancer Ther. 5, 575–585. 10.1158/1535-7163.MCT-05-0324. [DOI] [PubMed] [Google Scholar]
- Del Carratore R.; Carpi A.; Beffy P.; Lubrano V.; Giorgetti L.; Maserti B. E.; Carluccio M. A.; Simili M.; Iervasi G.; Balzan S. (2012) Itraconazole inhibits HMEC-1 angiogenesis. Biomed. Pharmacother. 66, 312–317. 10.1016/j.biopha.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Detaille D.; Guigas B.; Chauvin C.; Batandier C.; Fontaine E.; Wiernsperger N.; Leverve X. (2005) Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 54, 2179–2187. 10.2337/diabetes.54.7.2179. [DOI] [PubMed] [Google Scholar]
- Singh S.; Anupriya M. G.; Modak A.; Sreekumar E. (2018) Dengue virus or NS1 protein induces trans-endothelial cell permeability associated with VE-Cadherin and RhoA phosphorylation in HMEC-1 cells preventable by Angiopoietin-1. J. Gen. Virol. 99, 1658–1670. 10.1099/jgv.0.001163. [DOI] [PubMed] [Google Scholar]
- Harada K.; Yamahara K.; Ohnishi S.; Otani K.; Kanoh H.; Ishibashi-Ueda H.; Minamino N.; Kangawa K.; Nagaya N.; Ikeda T. (2011) Sustained-release adrenomedullin ointment accelerates wound healing of pressure ulcers. Regul. Pept. 168, 21–26. 10.1016/j.regpep.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Nagaya N.; Mori H.; Murakami S.; Kangawa K.; Kitamura S. (2005) Adrenomedullin: angiogenesis and gene therapy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R1432–R1437. 10.1152/ajpregu.00662.2004. [DOI] [PubMed] [Google Scholar]
- Ochoa-Callejero L.; Pozo-Rodrigalvarez A.; Martinez-Murillo R.; Martinez A. (2016) Lack of adrenomedullin in mouse endothelial cells results in defective angiogenesis, enhanced vascular permeability, less metastasis, and more brain damage. Sci. Rep. 6, 33495. 10.1038/srep33495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadivel A.; Abozaid S.; van Haaften T.; Sawicka M.; Eaton F.; Chen M.; Thebaud B. (2010) Adrenomedullin promotes lung angiogenesis, alveolar development, and repair. Am. J. Respir. Cell Mol. Biol. 43, 152–160. 10.1165/rcmb.2009-0004OC. [DOI] [PubMed] [Google Scholar]
- Nikitenko L. L.; Smith D. M.; Bicknell R.; Rees M. C. P. (2003) Transcriptional regulation of the CRLR gene in human microvascular endothelial cells. FASEB J. 17, 1499–1501. 10.1096/fj.02-0993fje. [DOI] [PubMed] [Google Scholar]
- Huang J.; Stohl L. L.; Zhou X.; Ding W.; Granstein R. D. (2011) Calcitonin gene-related peptide inhibits chemokine production by human dermal microvascular endothelial cells. Brain, Behav., Immun. 25, 787–799. 10.1016/j.bbi.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chigurupati S.; Kulkarni T.; Thomas S.; Shah G. (2005) Calcitonin stimulates multiple stages of angiogenesis by directly acting on endothelial cells. Cancer Res. 65, 8519–8529. 10.1158/0008-5472.CAN-05-0848. [DOI] [PubMed] [Google Scholar]
- Garcia Ponce A.; Citalan Madrid A. F.; Vargas Robles H.; Chanez Paredes S.; Nava P.; Betanzos A.; Zarbock A.; Rottner K.; Vestweber D.; Schnoor M. (2016) Loss of cortactin causes endothelial barrier dysfunction via disturbed adrenomedullin secretion and actomyosin contractility. Sci. Rep. 6, 29003. 10.1038/srep29003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz N.; Renshaw D.; Kapas S.; Hinson J. P. (2006) Adrenomedullin increases the expression of calcitonin-like receptor and receptor activity modifying protein 2 mRNA in human microvascular endothelial cells. J. Endocrinol. 190, 505–514. 10.1677/joe.1.06806. [DOI] [PubMed] [Google Scholar]
- Wunder F.; Rebmann A.; Geerts A.; Kalthof B. (2008) Pharmacological and kinetic characterization of adrenomedullin 1 and calcitonin gene-related peptide 1 receptor reporter cell lines. Mol. Pharmacol. 73, 1235–1243. 10.1124/mol.107.042283. [DOI] [PubMed] [Google Scholar]
- White C. W.; Johnstone E. K. M.; See H. B.; Pfleger K. D. G. (2019) NanoBRET ligand binding at a GPCR under endogenous promotion facilitated by CRISPR/Cas9 genome editing. Cell. Signalling 54, 27–34. 10.1016/j.cellsig.2018.11.018. [DOI] [PubMed] [Google Scholar]
- Gibson T. J.; Seiler M.; Veitia R. A. (2013) The transience of transient overexpression. Nat. Methods 10, 715–721. 10.1038/nmeth.2534. [DOI] [PubMed] [Google Scholar]
- Roehrkasse A. M.; Booe J. M.; Lee S. M.; Warner M. L.; Pioszak A. A. (2018) Structure-function analyses reveal a triple beta-turn receptor-bound conformation of adrenomedullin 2/intermedin and enable peptide antagonist design. J. Biol. Chem. 293, 15840–15854. 10.1074/jbc.RA118.005062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa H.; Hendrikse E. R.; Brimble M. A.; Garelja M. L.; Watkins H. A.; Harris P. W. R.; Hay D. L. (2019) Pharmacological Characterization and Investigation of N-Terminal Loop Amino Acids of Adrenomedullin 2 That Are Important for Receptor Activation. Biochemistry 58, 3468–3474. 10.1021/acs.biochem.9b00571. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2014) A Pharmacology Primer: Techniques for More Effective and Strategic Drug Discovery, Vol. 4, Elsevier, UK. [Google Scholar]
- Zhang H.; Qiao A.; Yang D.; Yang L.; Dai A.; de Graaf C.; Reedtz-Runge S.; Dharmarajan V.; Zhang H.; Han G. W.; Grant T. D.; Sierra R. G.; Weierstall U.; Nelson G.; Liu W.; Wu Y.; Ma L.; Cai X.; Lin G.; Wu X.; Geng Z.; Dong Y.; Song G.; Griffin P. R.; Lau J.; Cherezov V.; Yang H.; Hanson M. A.; Stevens R. C.; Zhao Q.; Jiang H.; Wang M. W.; Wu B. (2017) Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546, 259–264. 10.1038/nature22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms J.; Uddin R.; Sakmar T. P.; Gingell J. J.; Garelja M. L.; Hay D. L.; Brimble M. A.; Harris P. W.; Reynolds C. A.; Poyner D. R. (2018) Photoaffinity Cross-Linking and Unnatural Amino Acid Mutagenesis Reveal Insights into Calcitonin Gene-Related Peptide Binding to the Calcitonin Receptor-like Receptor/Receptor Activity-Modifying Protein 1 (CLR/RAMP1) Complex. Biochemistry 57, 4915–4922. 10.1021/acs.biochem.8b00502. [DOI] [PubMed] [Google Scholar]
- Hay D. L.; Harris P. W.; Kowalczyk R.; Brimble M. A.; Rathbone D. L.; Barwell J.; Conner A. C.; Poyner D. R. (2014) Structure-activity relationships of the N-terminus of calcitonin gene-related peptide: key roles of alanine-5 and threonine-6 in receptor activation. Br. J. Pharmacol. 171, 415–426. 10.1111/bph.12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwell J.; Conner A.; Poyner D. R. (2011) Extracellular loops 1 and 3 and their associated transmembrane regions of the calcitonin receptor-like receptor are needed for CGRP receptor function. Biochim. Biophys. Acta, Mol. Cell Res. 1813, 1906–1916. 10.1016/j.bbamcr.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts M. J., and Russel R. B. (2003) Amino acid properties and consequences of substitutions. In Bioinformatics for Geneticists (Barnes M.R., and Gray I. C., Eds.) 1st ed, Wiley. [Google Scholar]
- Hager M. V.; Clydesdale L.; Gellman S. H.; Sexton P. M.; Wootten D. (2017) Characterization of signal bias at the GLP-1 receptor induced by backbone modification of GLP-1. Biochem. Pharmacol. 136, 99–108. 10.1016/j.bcp.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager M. V.; Johnson L. M.; Wootten D.; Sexton P. M.; Gellman S. H. (2016) beta-Arrestin-Biased Agonists of the GLP-1 Receptor from beta-Amino Acid Residue Incorporation into GLP-1 Analogues. J. Am. Chem. Soc. 138, 14970–14979. 10.1021/jacs.6b08323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce M. L.; Mehrotra S.; Mustoe A. C.; French J. A.; Murray T. F. (2019) A comparison of the ability of Leu(8)- and Pro(8)-oxytocin to regulate intracellular Ca(2+) and Ca(2+)-activated K(+) channels at human and marmoset oxytocin receptors. Mol. Pharmacol. 95, 376–385. 10.1124/mol.118.114744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plisson F.; Hill T. A.; Mitchell J. M.; Hoang H. N.; de Araujo A. D.; Xu W.; Cotterell A.; Edmonds D. J.; Stanton R. V.; Derksen D. R.; Loria P. M.; Griffith D. A.; Price D. A.; Liras S.; Fairlie D. P. (2017) Helix constraints and amino acid substitution in GLP-1 increase cAMP and insulin secretion but not beta-arrestin 2 signaling. Eur. J. Med. Chem. 127, 703–714. 10.1016/j.ejmech.2016.10.044. [DOI] [PubMed] [Google Scholar]
- Deora G. S.; Qin C. X.; Vecchio E. A.; Debono A. J.; Priebbenow D. L.; Brady R. M.; Beveridge J.; Teguh S. C.; Deo M.; May L. T.; Krippner G.; Ritchie R. H.; Baell J. B. (2019) Substituted Pyridazin-3(2 H)-ones as Highly Potent and Biased Formyl Peptide Receptor Agonists. J. Med. Chem. 62, 5242–5248. 10.1021/acs.jmedchem.8b01912. [DOI] [PubMed] [Google Scholar]
- Holloway A. C.; Qian H.; Pipolo L.; Ziogas J.; Miura S.; Karnik S.; Southwell B. R.; Lew M. J.; Thomas W. G. (2002) Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol. Pharmacol. 61, 768–777. 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- Lei S.; Clydesdale L.; Dai A.; Cai X.; Feng Y.; Yang D.; Liang Y. L.; Koole C.; Zhao P.; Coudrat T.; Christopoulos A.; Wang M. W.; Wootten D.; Sexton P. M. (2018) Two distinct domains of the glucagon-like peptide-1 receptor control peptide-mediated biased agonism. J. Biol. Chem. 293, 9370–9387. 10.1074/jbc.RA118.003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heino P.; Oksala O.; Palkama A.; Valo T.; Vihavainen S.; Koskinen A.; Uusitalo H. (1998) Binding of CGRP analogs and their effect on adenylate cyclase activity in porcine iris-ciliary body. J. Ocul. Pharmacol. Ther. 14, 543–554. 10.1089/jop.1998.14.543. [DOI] [PubMed] [Google Scholar]
- Booe J. M.; Warner M. L.; Roehrkasse A. M.; Hay D. L.; Pioszak A. A. (2018) Probing the Mechanism of Receptor Activity-Modifying Protein Modulation of GPCR Ligand Selectivity through Rational Design of Potent Adrenomedullin and Calcitonin Gene-Related Peptide Antagonists. Mol. Pharmacol. 93, 355–367. 10.1124/mol.117.110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yule L.; Garelja M. L.; Hendrikse E. R.; Gingell J. J.; Poyner D.; Harris P. W. R.; Brimble M. A.; Hay D. L. (2019) A potent fluorescent CGRP analogue enables visualisation of receptor internalisation. Pept. Sci. e24126 10.1002/pep2.24126. [DOI] [Google Scholar]
- Stangl D.; Muff R.; Schmolck C.; Fischer J. A. (1993) Photoaffinity labeling of rat calcitonin gene-related peptide receptors and adenylate cyclase activation: identification of receptor subtypes. Endocrinology 132, 744–750. 10.1210/endo.132.2.8381072. [DOI] [PubMed] [Google Scholar]
- Sheykhzade M.; Abdolalizadeh B.; Koole C.; Pickering D. S.; Dreisig K.; Johansson S. E.; Abboud B. K.; Dreier R.; Berg J. O.; Jeppesen J. L.; Sexton P. M.; Edvinsson L.; Wootten D.; Sams A. (2018) Vascular and molecular pharmacology of the metabolically stable CGRP analogue, SAX. Eur. J. Pharmacol. 829, 85–92. 10.1016/j.ejphar.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Bailey R. J.; Hay D. L. (2006) Pharmacology of the human CGRP1 receptor in Cos 7 cells. Peptides 27, 1367–1375. 10.1016/j.peptides.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Qi T.; Dong M.; Watkins H. A.; Wootten D.; Miller L. J.; Hay D. L. (2013) Receptor activity-modifying protein-dependent impairment of calcitonin splice variant Δ(1–47)hCT(a) function. Br. J. Pharmacol. 168, 644–657. 10.1111/j.1476-5381.2012.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi T.; Christopoulos G.; Bailey R. J.; Christopoulos A.; Sexton P. M.; Hay D. L. (2008) Identification of N-terminal receptor activity-modifying protein residues important for calcitonin gene-related peptide, adrenomedullin, and amylin receptor function. Mol. Pharmacol. 74, 1059–1071. 10.1124/mol.108.047142. [DOI] [PubMed] [Google Scholar]
- Gingell J. J.; Qi T.; Bailey R. J.; Hay D. L. (2010) A key role for tryptophan 84 in receptor activity-modifying protein 1 in the amylin 1 receptor. Peptides 31, 1400–1404. 10.1016/j.peptides.2010.03.027. [DOI] [PubMed] [Google Scholar]
- GraphPad Software . (2019) GraphPad Statistics Guide: Ratio t-test, Graphpad. [Google Scholar]
- van der Westhuizen E. T.; Breton B.; Christopoulos A.; Bouvier M. (2014) Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 85, 492–509. 10.1124/mol.113.088880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.