

Summary
MCL-1 is a well-characterized inhibitor of cell death that has also been shown to be a regulator of mitochondrial dynamics in human pluripotent stem cells. We used cardiomyocytes derived from human-induced pluripotent stem cells (hiPSC-CMs) to uncover whether MCL-1 is crucial for cardiac function and survival. Inhibition of MCL-1 by BH3 mimetics resulted in the disruption of mitochondrial morphology and dynamics as well as disorganization of the actin cytoskeleton. Interfering with MCL-1 function affects the homeostatic proximity of DRP-1 and MCL-1 at the outer mitochondrial membrane, resulting in decreased functionality of hiPSC-CMs. Cardiomyocytes display abnormal cardiac performance even after caspase inhibition, supporting a nonapoptotic activity of MCL-1 in hiPSC-CMs. BH3 mimetics targeting MCL-1 are promising anti-tumor therapeutics. Progression toward using BCL-2 family inhibitors, especially targeting MCL-1, depends on understanding its canonical function not only in preventing apoptosis but also in the maintenance of mitochondrial dynamics and function.
Subject Areas: Molecular Biology, Cell Biology
Graphical Abstract
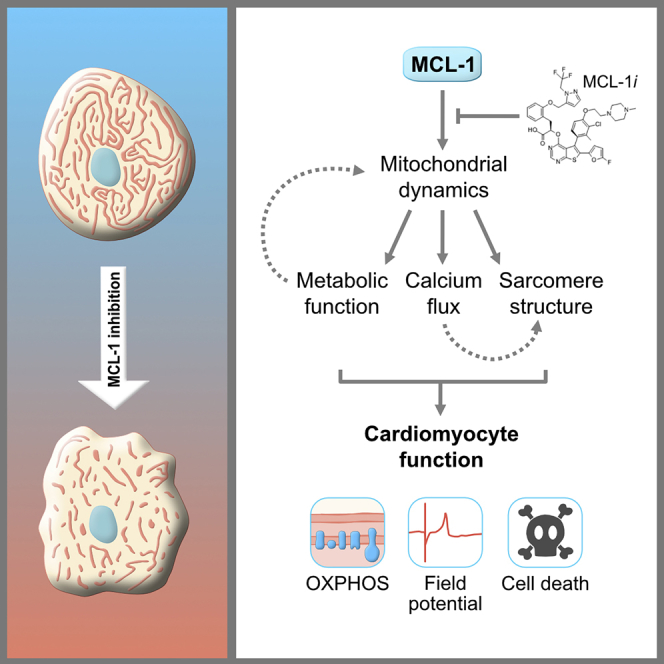
Highlights
-
•
BH3 mimetics targeting MCL-1 disrupt the mitochondrial network of human iPSC-CMs
-
•
The BH3-mimetic-mediated effects on mitochondrial dynamics are DRP-1-dependent
-
•
Targeting MCL-1 affects the survival and function of human cardiomyocytes
-
•
Human iPSC-derived cardiomyocytes can be used to reveal toxicity of MCL-1 inhibitors
Molecular Biology; Cell Biology
Introduction
Myeloid cell leukemia-1 (MCL-1) was originally identified as an early induced gene in human myeloid leukemia cell differentiation (Kozopas et al., 1993, Reynolds et al., 1996, Yang et al., 1996). MCL-1 is structurally similar to other anti-apoptotic BCL-2 (B cell lymphoma-2) family proteins (i.e. BCL-2, BCL-XL [B cell lymphoma extra-large]) (Chipuk et al., 2010). However, its larger, unstructured N-terminal domain and shorter half-life likely indicated that MCL-1 was not completely functionally redundant with other anti-apoptotic proteins (Perciavalle and Opferman, 2013). Supporting this idea, MCL-1 has been shown to be essential for embryonic development and for the survival of various cell types, including cardiomyocytes, neurons, and hematopoietic stem cells (Rinkenberger et al., 2000, Opferman et al., 2005, Arbour et al., 2008, Weber et al., 2010, Wang et al., 2013, Thomas et al., 2013).
MCL-1 is one of the most amplified genes in human cancers and is frequently associated with resistance to chemotherapy (Beroukhim et al., 2010, Perciavalle and Opferman, 2013). Earlier work demonstrated that MCL-1 genetic deletion is peri-implantation lethal in embryogenesis, not due to defects in apoptosis, but rather due to a combination of an embryonic developmental delay and an implantation defect (Rinkenberger et al., 2000). However, the nonapoptotic mechanism by which MCL-1 functions in normal and cancerous cells is still unclear. We previously reported that MCL-1 regulates mitochondrial dynamics in human pluripotent stem cells (hPSCs, which refer to both human embryonic stem cells [hESCs] and induced pluripotent stem cells [hiPSCs]) (Rasmussen et al., 2018). We found that MCL-1 maintains mitochondrial network homeostasis in hPSCs through interactions with dynamin-related protein 1 (DRP-1) and optic atrophy type 1 (OPA1), two GTPases responsible for maintaining mitochondrial morphology and dynamics. In this study, we investigated whether this nonapoptotic role of MCL-1 remains as stem cells differentiate, using cardiomyocytes derived from human-induced pluripotent stem cells (hiPSC-CMs).
Mitochondrial fusion promotes elongation of the mitochondrial network, which is key for mitochondrial DNA (mtDNA) homogenization and efficient assembly of the electron transport chain (ETC) (Westermann, 2010, Friedman and Nunnari, 2014). Loss of mitochondrial fusion has been implicated as a mechanism for the onset of dilated cardiomyopathy and reported to also contribute to hypertrophic cardiomyopathy and other heart diseases (Dorn, 2013, Dorn et al., 2015, Ong et al., 2017). Mitochondrial homeostasis is essential during cardiomyocyte differentiation and embryonic cardiac development (Kasahara et al., 2013, Kasahara and Scorrano, 2014, Cho et al., 2014). However, there is limited information about the mechanisms used by cardiomyocytes to minimize the risks for apoptosis, especially in cells derived from highly sensitive stem cells (Imahashi et al., 2004, Murriel et al., 2004, Gama and Deshmukh, 2012, Dumitru et al., 2012, Walensky, 2012).
Ultrastructural changes in mitochondria have long been observed in response to alterations in oxidative metabolism (Hackenbrock, 1966, Khacho et al., 2016). It has become increasingly clear that individual mitochondrial shape changes can also have dramatic effects on cellular metabolism (Chan, 2007, Hsu et al., 2016, Itoh et al., 2013, Burté et al., 2015). Several studies in the heart suggest that alterations in mitochondrial dynamics cause abnormal mitochondrial quality control, resulting in the buildup of defective mitochondria and reactive oxygen species (ROS) (Galloway and Yoon, 2015, Song et al., 2017). Interestingly, it has been shown that modulating the production of ROS can favor or prevent differentiation into cardiomyocytes (Buggisch et al., 2007, Murray et al., 2014). Thus, specific metabolic profiles controlled by mitochondrial dynamics are likely critical for hiPSC-CMs, because they can influence cell cycle, biomass, metabolite levels, and redox state (Zhang et al., 2012).
It is not completely understood how dynamic changes in metabolism affect cardiomyocyte function. Deletion of MCL-1 in murine heart muscle resulted in lethal cardiomyopathy, reduction of mitochondrial DNA (mtDNA), and mitochondrial dysfunction (Thomas et al., 2013, Wang et al., 2013). Inhibiting apoptosis via concurrent BAK/BAX knockout allowed for the survival of the mice; conversely, the mitochondrial ultrastructure abnormalities and respiratory deficiencies were not rescued. These results indicate that MCL-1 also has a crucial function in maintaining cell viability and metabolic profile in cardiomyocytes. Despite these efforts, the nonapoptotic mechanism by which MCL-1 specifically functions in cardiomyocytes is still unknown. Furthermore, the role for MCL-1 in the regulation of mitochondrial dynamics in cardiac cells has not yet been defined. Here we report that MCL-1 inhibition via BH3 mimetics caused severe contractility defects and impaired long-term survival of hPSC-CMs, due to MCL-1's essential function regulating mitochondrial morphology and dynamics.
Results
MCL-1 Inhibition Causes Severe Defects in hiPSC-CM Mitochondrial Network
Recently published small molecule inhibitors of MCL-1 have been anticipated as potent anti-tumor agents against MCL-1-dependent cancers with limited cardiotoxicity in mouse models (Cohen et al., 2012, Kotschy et al., 2016, Letai, 2016). Thus, we chose to use hiPSC-CMs to examine the effects of MCL-1 inhibition on mitochondrial morphology using the small molecule inhibitor S63845 (Kotschy et al., 2016), combined with structured illumination microscopy (SIM) to observe mitochondria at high resolution. Cardiomyocytes were imaged after 4 days of treatment with vehicle (DMSO) or MCL-1 inhibitor (S63845) and the caspase inhibitor Q-VD-OPh (QVD) to prevent downstream effects of apoptosis on mitochondrial morphology (Figures 1A–1C). Mitochondrial networks in S63845-treated cells were severely disrupted, with individual mitochondria becoming shorter in length and more globular on average, as opposed to elongated networks in control cells. Quantification of SIM images shows a significant reduction in average mitochondrial length (Figure 1D) and a significant increase in mitochondria sphericity (Figure 1E) compared with control cells. In addition to S63845, we also tested two other small molecule MCL-1 inhibitors, AMG-176 (AMG) and AZD5991 (AZD) (Caenepeel et al., 2018, Tron et al., 2018). Although we observed mitochondrial defects in both inhibitor conditions (Figures 1F–1H), mitochondrial morphology in AMG-treated cells was not significantly different compared with control cells (Figures 1I and 1J). Quantification of SIM images shows a significant reduction in average mitochondrial length in cells treated with AZD (Figure 1I).
Figure 1.

MCL-1 Inhibition Causes Mitochondrial Fragmentation
(A) Schematic of cell treatment paradigm used throughout this study. Structured Illumination Microscopy (SIM) was used for acquisition of all super-resolution images. hiPSC-CMs were treated with vehicle DMSO (B) or 2 μM S63845 (C) and Q-VD-Oph (QVD). Quantification of average mitochondrial length (D) and mitochondrial sphericity (E) are shown, in which a spherical object would have a value of 1.0. hiPSC-CMs were treated with vehicle DMSO (F), 2 μM AMG-176 (G), or 2 μM AZD5991 (H) and QVD. Insets show magnification of individual mitochondria morphology. Scale: 5 μm for all mitochondria images. Representative images are shown for all panels. Quantification of mitochondrial length (I) and mitochondrial sphericity (J) are shown, with AZD5991 treatment significantly decreasing average mitochondrial length. Graphs represent mean ± SEM from at least three independent experiments (n > 20 cells per condition). See also Figure S1.
Corresponding with the fragmented mitochondrial phenotypes seen in S63845-treated cells, we also observed impaired mitochondrial respiration as measured by the Seahorse XFe96 analyzer. MCL-1 inhibition significantly lowered the maximum oxygen consumption rate (OCR) after addition of FCCP, an uncoupler of oxidative phosphorylation (OXPHOS) (Figure S1A). ATP production was significantly reduced in S63845-treated cells as calculated from the OCR trace (Figure S1B). QVD was added to account for any effects on metabolism due to downstream apoptosis, but cells displayed similar OCR and ATP production as with S63845 alone (Figures S1C and S1D).
Recent reports have determined that MCL-1 functions not only as an apoptosis regulator but also as a modulator of mitochondrial morphology and dynamics (Perciavalle et al., 2012, Morciano et al., 2016, Rasmussen et al., 2018). Thus, we hypothesized that inhibiting MCL-1 with BH3 mimetics would affect the functionality of human cardiomyocytes, due to the disruption of crucial MCL-1 interactions with the mitochondrial dynamics machinery, which will ultimately lead to cell death.
MCL-1 Inhibition Affects Contractility of hiPSC-CMs and Myofibril Assembly in a Caspase-Independent Manner
Previous studies focused on human cardiomyocytes have suggested an effect of MCL-1 inhibition on mitochondrial morphology and mild effects on overall cardiac function (Guo et al., 2018). However, MCL-1 inhibition by S63845 was shown to have minimal effects on murine ejection fraction (Kotschy et al., 2016). These results are intriguing, considering previous studies reporting that MCL-1 deletion from murine cardiomyocytes has severe effects on mitochondrial morphology and cardiac function, which were not rescued by co-deletion of BAK and BAX (Wang et al., 2013). We treated hiPSC-CMs with S63845, while inhibiting caspase activity using QVD, and measured spontaneous cardiac beating using the AxionBiosystems analyzer (Clements and Thomas, 2014) (Figure 2A). We also used the BCL-2 inhibitor, Venetoclax (ABT-199) (Souers et al., 2013), to probe whether these cells are also sensitive to BCL-2 inhibition. We observed that only MCL-1 inhibition caused severe defects in cardiomyocyte functionality within 48 h of treatment (Figures 2B–2F). In particular, spike amplitude mean (Figure 2B) and spike slope mean (Figure 2C) were significantly decreased at 20 and 48 h, whereas beat period irregularity was significantly increased at 20 h post-treatment (Figure 2D). BCL-2 inhibition did not cause changes in cardiac beating ability compared with DMSO control in any of the measured parameters (Figures 2B–2F), suggesting that the function of hiPSC-CMs is highly dependent on MCL-1, but not BCL-2. S63845-treated cells stopped beating completely at 48 h, accounting for the decrease in beat period mean and beat period irregularity at this time point (Figures 2D and 2E). The field potential duration (FPD) was not detectable after 20 h of MCL-1 inhibition (Figure 2F). We also observed similar defects in cardiomyocyte functionality in cells treated with AZD, but not AMG (Figures S2A–S2E).
Figure 2.

MCL-1 Inhibition Causes Functional Defects and Disruption of Myofibrils
(A–F) (A) Schematic of AxionBiosystems MEA paradigm for recording cardiac performance in live cells. hiPSC-CMs were plated on a CytoView MEA plate (AxionBiosystems) and treated with either vehicle (DMSO), 0.5 μM S63845, or 0.5 μM ABT-199 and QVD. Live-cell activity was recorded at baseline (0 h), 2 h, 20 h, and 48 h post-initial treatment for 5 min each. Spike amplitude mean (B) and spike slope mean (C) were both decreased by 20 h and completely reduced by 48 h in the S63845 condition. Beat period irregularity (D) was increased at 20 h in S63845-treated cells before cells stopped beating at 48 h, whereas DMSO and ABT-199 had low levels of beat period irregularity at 48 h. Beat period mean (E) remained at levels comparable to DMSO control before cells stopped beating at 48 h, whereas field potential duration (FPD) mean (F) was not detectable after 20 h. p values show significance as follows: ∗ = DMSO + QVD versus S63845 + QVD, # = ABT-199 + QVD versus S63845 + QVD. One symbol indicates p < 0.05, two symbols indicate p < 0.01, three symbols indicate p < 0.001, and four symbols indicate p < 0.0001. p values were determined by two-way ANOVA. Graphs represent mean ± SEM.
(G) Representative montage from GCaMP calcium indicator time-lapse imaging in hiPSC-CMs treated with DMSO + QVD or S63845 + QVD (top). Representative kymographs of calcium pulses from individual cells treated with DMSO + QVD or S63845 + QVD (bottom).
(H) Quantification of calcium influx from hiPSC-CMs treated with QVD and either vehicle DMSO, S63845, AMG-176, or AZD5991, in which a value of 1.0 indicates no change in fluorescence intensity. n = 60 cells from three independent experiments. Symbols indicate mean and error bars indicate ±SD.
(I) Proportion of beating versus not beating hiPSC-CMs in each condition as observed by light microscopy. Significance between beating versus not beating cells was determined by Chi-square test. Error bars indicate ±SD and percentages were pooled from three experiments.
(J) Vehicle-treated hiPSC-CMs have organized myofibril structure as shown by maximum intensity projections. hiPSC-CMs treated with 2 μMS63845 have myofibrils that are unorganized and poorly defined Z-lines. Scale: 5 μm. Representative images are shown for all panels.
(K) Quantification of myofibril phenotypes represented in panel I (n > 20 cells per condition from three separate experiments). Error bars indicate ±SD.
See also Figure S2.
In a previous report, MCL-1 inhibition using RNAi also resulted in mitochondria morphology defects including severe cristae disruption and remarkable vacuolation in the mitochondrial matrix (Guo et al., 2018). In this study, MCL-1 knockdown by siRNA (Figures S2F and S2G) also caused increased beat period irregularity (Figure S2H), increased max delay mean (Figure S2I), and decreased FPD mean (Figure S2J). These results suggest that MCL-1 inhibition in human cardiomyocytes causes bradycardia- and arrhythmia-like phenotypes.
Because MCL-1 inhibition disrupted hiPSC-CM spike amplitude mean, we decided to probe whether calcium influx was also impaired in these cells. We visualized calcium dynamics using the GCaMP5G calcium reporter in hiPSC-CMs (Figure 2G). In DMSO-treated cells, we measured approximately a 1.9-fold increase in signal intensity (Figure 2H). As a positive control, we treated cells with cadmium chloride (CdCl2), a calcium channel blocker, which completely disrupted calcium intake, giving a ratio of 1.0. Consistent with our results with the MEA recordings, calcium signal intensity was significantly reduced in hiPSC-CMs treated with S63845 or AZD. AMG treatment caused a significant, but less severe, reduction in calcium intake. We also measured beating by light microscopy and found that MCL-1 inhibition reduced the proportion of beating cells in a dose-dependent manner (Figure 2I).
Intriguingly, we also observed significant changes in the structure of the actin network and subsequent myofibril organization in cells treated with any of the MCL-1 inhibitors (Figures 2J and S2K). hiPSC-CMs treated with MCL-1 inhibitors displayed poor Z-line organization, lower density of F-actin, and increased presence of stress fibers. Blinded quantification of F-actin organization revealed that MCL-1-inhibitor-treated cells had significantly less organized myofibril structure (Figures 2J and S2K).
MCL-1 Co-localizes with Mitochondrial Dynamics Proteins in hiPSC-CMs, and S63845 Disrupts MCL-1:DRP-1 Co-localization
Because MCL-1 inhibition disrupted mitochondrial network integrity in hiPSC-CMs and MCL-1 depletion affects mitochondrial dynamics proteins DRP-1 and OPA1 (Rasmussen et al., 2018), we next examined the effects of MCL-1 inhibition on the expression levels of these proteins in hiPSC-CMs. S63845-treated cells had a significant increase in the expression levels of DRP-1 (Figures 3A, 3B, and S3A–S3C) but not in phospho-DRP-1 (pDRP-1 S616) (Figure 3C). MCL-1 expression levels were significantly increased (Figures 3D and 3E). Previous studies also reported this induction of MCL-1 protein expression upon MCL-1 inhibition (Kotschy et al., 2016). There were no significant changes in OPA1 (Figures 3D and 3E) or TOM20 (Figures 3D and S3D). We then assessed whether MCL-1 interacts with DRP-1 and OPA1 using in situ proximity ligation assay (PLA). Our data show that MCL-1 is in close proximity to both DRP-1 and OPA1 (Figures 3G–3J). PLA puncta were quantified and normalized to the number of puncta in the control sample (no primary antibody) (Figure S3E). The co-localization of MCL-1 with DRP-1 (Figures 3G and 3H), but not OPA1 (Figures 3I and 3J), was disrupted upon inhibition of MCL-1 with S63845, suggesting that MCL-1 interacts with DRP-1 through its BH3-binding groove.
Figure 3.

MCL-1 is in Close Proximity to Mitochondrial Dynamics Proteins
(A–C) (A) Western blot showing total and phospho-DRP-1 expression in hiPSC-CMs treated with DMSO + QVD or S63845 + QVD. Quantification of DRP-1 (B) and pDRP-1 S616 (C) band density relative to α-Tubulin (n = 3 independent experiments).
(D–F) (D) Western blot showing OPA1, MCL-1, and TOM20 levels in hiPSC-CMs treated with S63845 + QVD. Quantification of MCL-1 (E) and OPA1 (F) band density relative to α-Tubulin (n = 3 independent experiments).
(G–J) Representative ROIs of PLA showing MCL-1:DRP-1 (G) or MCL-1:OPA1 (I) puncta in vehicle- or S63845-treated hiPSC-CMs. Scale: 5 μm. Quantification of PLA puncta from MCL-1:DRP-1 (H) or MCL-1:OPA1 (J) interactions (n = 10–15 ROIs per condition from three independent experiments).
All graphs represent mean ± SD. See also Figure S3.
To further assess the disruption of the mitochondrial network caused by MCL-1 inhibition, we employed an assay using a photo-convertible plasmid (mito-tdEos) to assess connectivity and fusion/motility of mitochondria. After photo-converting an area of the mitochondrial network, we assessed the spread of red signal, which we used as a proxy for mitochondrial fusion. We observed that in cells treated with MCL-1 inhibitor, both the initial converted area and the spread of the converted red signal after 20 min were significantly decreased, indicating impaired mitochondrial fusion (Figures 4A–4D). This phenomenon was DRP-1 dependent, because cells deficient in DRP-1 maintained an elongated network even when treated with S63845 (Figures 4E, 4F, S4A, and S4B).
Figure 4.

MCL-1 Inhibition Results in Mitochondrial Fragmentation in a DRP-1-Dependent Manner
(A–D) (A) Vehicle- and (B) S63845-treated hiPSC-CMs were transfected with mito-tdEos and a small area was photoconverted (see methods). Cells were imaged for 20 min post-conversion to assess mitochondrial network connectivity. Scale: 5 μm. Quantification of the initial converted area normalized to total unconverted area (C) and fold change in converted area after 20 min (D) shows decreased initial connectivity and mitochondrial fusion after treatment with 2 μM S63845 and QVD (MCL-1i).
(E) Quantification of initial converted area normalized to total unconverted area in hiPSC-CMs transfected with control siRNA or siRNA targeting DRP-1 ±MCL-1i (2 μM S63845) and QVD.
(F) Quantification of fold change in converted area after 20 min in same treatments from Figure 4E. Boxplots represent median of three independent experiments and Tukey whiskers.
See also Figure S4.
MCL-1 Inhibition Results in hiPSC-CM Death
To examine whether hiPSC-CMs treated with MCL-1 inhibitor were undergoing apoptosis, we treated the cells with increasing doses of S63845 and examined the activation of caspase-3 and caspase-7. Cells responded to S63845 in a dose-dependent manner after 48 h, with 1–2 μM inducing the most caspase activity (Figure 5A). We observed a similar dose response with both AMG and AZD (Figure 5B). It is important to note that S63845 and AZD5991 have a similar chemical structure, and that these two inhibitors had more severe effects on cardiomyocytes overall (Hird and Tron, 2019). To confirm that cells were undergoing a caspase-dependent cell death, we performed long-term live cell imaging in the presence of MCL-1 inhibitors with and without caspase inhibition (Figures S4A–S4D). We observed similar levels of death regardless of caspase inhibition. These results indicate that hiPSC-CMs are also committing to a caspase-independent cell death in response to MCL-1 inhibition. To assess the type of death caused by MCL-1 inhibition, we treated the cells with IM-54, a known inhibitor of necrosis. IM-54 treatment rescued the toxicity caused by MCL-1 inhibition (Figure 5C).
Figure 5.

MCL-1 Inhibition Causes Cell Death in hiPSC-CMs that is Blocked by a Necrosis Inhibitor
(A and B) hiPSC-CMs were treated with increasing doses of S63845 (A) or AMG-176, and AZD5991 (B) for 48 h before caspase activity was measured by CaspaseGlo 3/7 assay (Promega). Quantification shows results from at least three independent experiments performed in duplicate and were normalized to DMSO control. Graphs represent mean ± SEM.
(C) CellTiter-Blue assay (Promega) was used to assess cell viability in hiPSC-CMs. p values were calculated by one-way ANOVA. Data was quantified from three independent experiments performed in triplicate. Boxplots show median with Tukey whiskers.
See also Figure S5.
Previous reports have established that iPSC-derived cardiomyocytes mimic immature progenitor cells. To test the possibility that the effects of the MCL-1 inhibitors were exacerbated by the immature state of hiPSC-CMs, we used a previously published hormone-based method for cardiomyocyte maturation (Figures 6A and 6B) (Gentillon et al., 2019, Parikh et al., 2017). We tested for caspase-3/7 activation after 24 h of treatment with increasing doses of S63845 and detected similar effects of MCL-1 inhibition in hormone-matured hiPSC-CMs and vehicle-treated hiPSC-CMs (Figures 6C and 6D). Importantly, treatment of these hormone-matured hiPSC-CMs also results in decreased functionality in response to a low dose of MCL-1 inhibitor (100 nM S63845) (Figures 6E–6G).
Figure 6.

MCL-1 Inhibition Induces Caspase Activity and Reduces Functionality of Matured hiPSC-CMs
(A) Schematic of maturation protocol for hiPSC-CMs using hormone method.
(B) hiPSC-CMs treated with dexamethasone (Dex) and triiodothyronine (T3) display more mature myofibril phenotype compared with vehicle-treated control cells. Scale: 10 μm.
(C and D) (C) Vehicle- or (D) Dex + T3-treated hiPSC-CMs were exposed to S63845 at increasing doses for 24 h. Caspase activity was measured as in Figures 5A and 5B using the CaspaseGlo 3/7 assay. Quantification was performed from three independent maturation experiments in duplicate. Graphs represent mean ± SEM.
(E–G) CardioExcyte96 recordings show disruption of cardiac activity in Dex + T3-matured hiPSC-CMs when treated with 100 nMS63845. Data were quantified for Time to Peak (E), Time to Fall (TFall) (F), and Width50 (G). Quantification was performed from three independent maturation experiments for at least 5 wells per condition. p values were calculated by two-way ANOVA and graphs show mean ± SEM.
Long-term MCL-1 Inhibition, but Not BCL-2 Inhibition, Causes Defects in Cardiomyocyte Functionality
MCL-1 inhibition has significant effects on hiPSC-CM contractility and functionality when used at higher doses. To test if MCL-1 inhibition still affects cardiac functionality at lower doses, we treated hiPSC-CMs for two weeks (with treatments every two days) with 100 nM S63845. We also treated cells with the BCL-2 inhibitor ABT-199 (100 nM) and a combination of S63845 + ABT-199 (100 nM each). MCL-1 inhibition significantly disrupted hiPSC-CM spike amplitude mean and spike slope mean (Figures 7A and 7B). Although there were minimal differences between treatments in the beat period mean or FPD mean (Figures S6A and S6B), spike slope mean (Figure 7B), conduction velocity mean (Figure 7C), max delay mean (Figure 7D), and propagation consistency (Figure 7E) were significantly lowered in both the S63845 condition and when combined with ABT-199. Cells treated with ABT-199 appeared healthy and were functionally similar to control cells throughout the experiment (Figures 7A–7E, S6A, and S6B). Cells displayed mitochondrial network and actin disruption in the S63845-treated condition, and even more severe phenotypes were observed in cells treated with both inhibitors when compared with control cells (Figures 7F–7I and S6C–S6F). BCL-2 inhibition had little effect on mitochondrial network organization and virtually no effect on myofibril organization (Figures 7H and S6E). Our results further support the idea that MCL-1 is essential for maintaining mitochondrial homeostasis of human cardiomyocytes (Figure S7).
Figure 7.

Long-term MCL-1 Inhibition Causes Defects in Functionality and Mitochondrial Morphology of hiPSC-CMs
(A–E) Chronic inhibition of MCL-1, but not BCL-2, results in cardiac activity defects. hiPSC-CMs were treated every 2 days with DMSO, 100 nM S63845 (orange), 100 nM ABT-199 (green), or both inhibitors (magenta) for 14 days. MEA recordings were taken 2 h following each treatment for 5 min, and results were normalized to baseline recording for each respective well, followed by normalization to DMSO (gray dotted line). Results of recordings for spike amplitude mean (A), spike slope mean (B), conduction velocity mean (C), max delay mean (D), and propagation consistency (E) are shown. p values show significance as follows: ∗ = DMSO versus S63845, † = DMSO versus Combination, # = S63845 versus ABT-199, ‡ = S63845 versus Combination, • = ABT-199 versus Combination. One symbol indicates p < 0.05, two symbols indicate p < 0.01, three symbols indicate p < 0.001, and four symbols indicate p < 0.0001. p values were determined by two-way ANOVA. Graphs represent mean ± SEM.
(F–I) Mitochondria and F-actin were imaged at the end of the treatment paradigm in Figures 6A–6E. Representative images are shown of cells treated with DMSO (F), 100 nM S63845 (G), 100 nM ABT-199 (H), and 100 nM S63845 + 100 nM ABT-199 (Combination) (I). Scale: 10 μm. See also Figure S6.
Discussion
Recent studies have implicated MCL-1 in the maintenance of mitochondrial homeostasis in various cell types (Perciavalle and Opferman, 2013, Rasmussen et al., 2018, Senichkin et al., 2019). In this report, we show that MCL-1 inhibition affects human cardiomyocyte functionality potentially due to MCL-1's nonapoptotic role in modulating mitochondrial dynamics. Inhibition of MCL-1 using BH3 mimetics is a promising strategy to treat tumors (Hird and Tron, 2019), because resistance to chemotherapy is often associated with MCL-1 upregulation (Kotschy et al., 2016). To optimize the use of MCL-1 inhibitors, a deeper understanding of the biology of MCL-1 is crucial. Our studies show that MCL-1 inhibition affects human cardiomyocyte functional parameters such as spike amplitude, beat propagation, and conduction velocity, which overlap with disruption of the mitochondrial and actin networks, ultimately leading to cell death.
Cardiomyocytes exposed to MCL-1 inhibitors appear to exhibit bradycardia and arrhythmia phenotypes. Interestingly, these phenotypes were not seen in cells treated with Venetoclax, indicating that hiPSC-CMs are less dependent on BCL-2 and highlighting a potential role for MCL-1 beyond its canonical function in apoptosis. This is further supported by the finding that hiPSC-CMs treated with 100 nM S63845 alone were alive, but not beating, after two weeks of treatment.
We hypothesize that this alternate function of MCL-1 in maintaining mitochondrial homeostasis is due to its interactions with DRP-1 and OPA1, which are essential regulators of mitochondrial morphology and dynamics (Labbé et al., 2014, Nishimura et al., 2018). Treatment of iPSC-CMs with MCL-1 inhibitor caused disruption of the mitochondrial network and significantly decreased MCL-1 proximity to DRP-1 at the mitochondria. Because the interaction with OPA1 was not disturbed, it is possible that MCL-1 interacts with OPA1 either through a different domain or with a different isoform of OPA1 in hiPSC-CMs than in hPSCs (Rasmussen et al., 2018). Another possibility is that, upon differentiation, the small molecule can no longer penetrate the inner mitochondrial membrane. We also confirmed that the mitochondrial network disruption is dependent on DRP-1, because DRP-1 knockdown prevented the fragmentation caused by MCL-1 inhibition. The recruitment of DRP-1 to the mitochondria has been proposed to be a critical inducer of mitophagy (Lee et al., 2011, Kageyama et al., 2014, Burman et al., 2017). Thus, an interesting possibility is that inhibition of MCL-1 is decreasing clearing of damaged mitochondria in cardiomyocytes. It will be important to test if key proteins involved in mitophagy are affected in the presence of MCL-1 inhibitors.
The photo-conversion experiments in this study did not test for fragmentation directly; thus it is possible that the mitochondrial phenotypes are caused by a lack of fusion or mitochondrial motility. Further studies into the mechanism of MCL-1's interaction with DRP-1 and OPA1 could shed light on this possibility. In contrast to iPSCs (Rasmussen et al., 2018), S63845 did not affect the proximity of MCL-1 with OPA1 at the mitochondria. MCL-1 at the mitochondrial matrix has been proposed to regulate β-oxidation of long fatty acids through interactions with VLCAD, and deletion of MCL-1 from the matrix caused hyperactivity of β-oxidation (Escudero et al., 2018). It is tempting to speculate that inhibiting MCL-1 at the matrix of human cardiomyocytes would result in significant damage to the heart.
We investigated the effects of MCL-1 inhibition on calcium flux using the GCaMP5G calcium reporter. Our data show a significant decrease in calcium flux with all inhibitors tested. These results could help explain the loss of functionality caused by MCL-1 inhibition. In addition to this loss of cardiomyocyte functionality, MCL-1 inhibition also caused significant disruption of actin networks within hiPSC-CMs. There are many possible mechanisms that could drive this phenotype. One hypothesis is that disruption of actin networks could be a result of decreased calcium flux and resultant loss of cardiomyocyte beating. Indeed, previous studies have shown that the maintenance of proper myofibril organization requires functional calcium channels and cellular contractility (Sharp et al., 1997, Simpson et al., 1993). Future studies should aim to test other potential hypotheses, such as altered metabolism and ROS production. Furthermore, cardiomyocytes treated with BH3 mimetics show a mild dose-dependent activation of caspase-3 that was inhibited by QVD. However, when measuring overall cell viability, the most significant rescue of viability was achieved by the necrosis inhibitor, IM-54. How is inhibition of MCL-1 triggering caspase-independent cell death that is blocked by this necrosis inhibitor? Mitochondrial disruption induced by BH3 mimetics may cause increased oxidative stress that results in the loss of function and viability of cardiomyocytes. This is in agreement with a previous report (Thomas et al., 2013) that demonstrated the induction of necrosis in Mcl-1-deficient murine hearts. The data in this study showed that Mcl-1 deletion did not result in the massive loss of myocytes due to apoptosis (Thomas et al., 2013). It would be of interest to examine the molecular mechanisms behind this phenotype.
MCL-1 inhibition also caused cardiomyocyte death in hormone-matured cells (Parikh et al., 2017). Although apoptosic sensitivity has been shown to decrease throughout development (Wright and Deshmukh, 2006, Sarosiek et al., 2017), these matured cells were more sensitive to S63845 treatment than vehicle-treated cells. It would be of interest to determine whether MCL-1 function in mitochondrial dynamics affects the maturation of iPSC-CMs or heart development in vivo (Kasahara et al., 2013, Feaster et al., 2015, Parikh et al., 2017). We speculate that other determinants of mitochondrial homeostasis, including mitochondrial biogenesis and mitophagy, may be affected by MCL-1 deficiency as cardiomyocytes mature. Although previous studies reported limited effects of S63845 on mouse heart function, a recent study using a humanized mouse model demonstrated that S63845 binds human MCL-1 with higher affinity than mouse MCL-1 (Kotschy et al., 2016, Brennan et al., 2018). Although this study did not report significant effects to the heart of humanized mice treated with S63845, the potential interactions of human MCL-1 with mitochondrial dynamics proteins and VLCAD may be species specific and not completely recapitulated in this mouse model. Collectively these reports highlight the importance of further research on the effect of MCL-1 on human-specific mitochondrial dynamics and metabolism. These results together with previous work from other groups (Thomas et al., 2013, Wang et al., 2013) suggest that hiPSC-CMs may be an appropriate platform to assess the safety and potential off-target effects of MCL-1 inhibitors on adult human hearts.
Studies from our laboratory suggest that inhibition of MCL-1 induces the differentiation of iPSCs (Rasmussen et al., 2018), which is likely associated with changes in metabolism to support cell-type-specific processes (Folmes et al., 2016). Because mitochondrial morphology is tightly coupled to metabolic adaptations, future studies will aim to investigate whether MCL-1 inhibition may cause a metabolic switch from fatty acid β-oxidation to glycolysis. Cardiac contractions depend on energy from these metabolic pathways, and thus cardiac mitochondria are forced to work constantly and likely require strict quality control mechanisms to maintain a functioning state (Dorn et al., 2015). This quality control process could depend in part on MCL-1. In support of this idea, our studies indicate that MCL-1 activity is essential for hiPSC-CM viability and contractility, which could be linked to MCL-1's nonapoptotic function at the mitochondrial matrix. The eventual apoptotic response detected at later time points could be the result of mitochondrial ROS signaling to trigger translocation and activation of BAX (Chaudhari et al., 2007). The disruption of actin and myofibril morphology could also be explained by heightened ROS induction and downstream ROS-mediated damage. Another important aspect that needs to be evaluated is the prevalence of phenotypically normal genetic variants that could predispose otherwise healthy individuals to stress-related cardiomyopathy (Garcia-Pavia et al., 2019). Our results emphasize the need for a more complete molecular understanding of MCL-1's mechanism of action in human cardiomyocytes, as it may reveal new approaches to prevent potential cardiac toxicities associated with chemotherapeutic inhibition of MCL-1.
Limitations of the Study
The protocol for myocyte maturation used in this study involves treatment with both tri-iodo-L-thyronine (T3) and dexamethasone, which leads to the generation of extensive T-tubule network and other functional traits of mature myocytes (Parikh et al., 2017). Although effective in generating “matured” T-tubule structures, we did not fully evaluate other traits of cardiomyocyte maturation. Thus, our study could be complemented by other approaches to achieve myocyte maturation (e.g. altering the metabolic state, co-culturing with mesenchymal stem cells, or using three-dimensional approaches) (Machiraju and Greenway, 2019, Karbassi et al., 2020). Collective data, however, suggest that MCL-1 activity is required for normal cardiac myocyte mitochondrial activity. The results reported here further support the validity for testing small molecule MCL-1 inhibitors in human iPSC-derived model systems that could reveal potential toxicity prior to admission in phase 1 clinical trials.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Dr. Kevin Ess and John Snow for providing access and critical technical support with the AxionBiosystems MEA analyzer. We would also like to thank Dr. Ian Macara for providing access to the GloMax luminometer and Dr. Bryan Millis for providing expertise with high-resolution microscopy. This work was supported by 1R35 GM128915-01NIGMS (to VG), 4R00CA178190NCI (to VG), 1R21CA227483-01A1NCI (to VG), 19PRE34380515AHA (to MLR), 18PRE33960551AHA (to NT), 19POST34380182AHA (to LW), and R35 GM125028-01NIGMS (to DTB). The Vanderbilt Cell Imaging Shared Resource is supported by NIH grants 1S10OD012324-01 and 1S10OD021630-01. The authors declare no competing financial interests.
Author Contributions
V. Gama, M. Rasmussen, and N. Taneja conceived the study, designed experiments, interpreted data, and wrote the manuscript. M. Rasmussen and N. Taneja designed and carried out all the cell biology experiments, with input from D. Burnette. A. Neininger performed mitochondrial morphology analysis. L. Wang performed CM differentiation and maturation, and technical support was provided by A. Neininger, G. Robertson, S. Riffle, and L. Shi. V. Gama designed and supervised the project. The manuscript was prepared by M. Rasmussen and V. Gama and revised by N. Taneja and D. Burnette. D. Burnette and B. Knollmann provided vital reagents and critical expertise.
Declaration of Interests
The authors declare no competing interests.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101015.
Supplemental Information
References
- Arbour N., Vanderluit J.L., Grand J.N.L., Jahani-Asl A., Ruzhynsky V.A., Cheung E.C.C., Kelly M.A., MacKenzie A.E., Park D.S., Opferman J.T., Slack R.S. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J. Neurosci. 2008;28:6068–6078. doi: 10.1523/JNEUROSCI.4940-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan M.S., Chang C., Tai L., Lessene G., Strasser A., Dewson G., Kelly G.L., Herold M.J. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood. 2018;132:1573–1583. doi: 10.1182/blood-2018-06-859405. [DOI] [PubMed] [Google Scholar]
- Buggisch M., Ateghang B., Ruhe C., Strobel C., Lange S., Wartenberg M., Sauer H. Stimulation of ES-cell-derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. J. Cell Sci. 2007;120:885–894. doi: 10.1242/jcs.03386. [DOI] [PubMed] [Google Scholar]
- Burman J.L., Pickles S., Wang C., Sekine S., Vargas J.N.S., Zhang Z., Youle A.M., Nezich C.L., Wu X., Hammer J.A., Youle R.J. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017;216:3231–3247. doi: 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burté F., Carelli V., Chinnery P.F., Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Caenepeel S., Brown S.P., Belmontes B., Moody G., Keegan K.S., Chui D., Whittington D.A., Huang X., Poppe L., Cheng A.C. AMG 176, a selective MCL1 inhibitor, is effective in hematological cancer models alone and in combination with established therapies. Cancer Discov. 2018 doi: 10.1158/2159-8290.CD-18-0387. CD-18-0387. [DOI] [PubMed] [Google Scholar]
- Chan D.C. Mitochondrial dynamics in disease. N. Engl. J. Med. 2007;356:1707–1709. doi: 10.1056/NEJMp078040. [DOI] [PubMed] [Google Scholar]
- Chaudhari A.A., Seol J.-W., Kim S.-J., Lee Y.-J., Kang H., Kim I., Kim N.-S., Park S.-Y. Reactive oxygen species regulate Bax translocation and mitochondrial transmembrane potential, a possible mechanism for enhanced TRAIL-induced apoptosis by CCCP. Oncol. Rep. 2007;18:71–76. [PubMed] [Google Scholar]
- Chipuk J.E., Moldoveanu T., Llambi F., Parsons M.J., Green D.R. The BCL-2 family reunion. Mol. Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Park J., Heo H.J., Park S., Song S., Kim I., Han Y., Yamashita J.K., Youm J.B., Han J., Koh G.Y. Dual modulation of the mitochondrial permeability transition pore and redox signaling synergistically promotes cardiomyocyte differentiation from pluripotent stem cells. J. Am. Heart Assoc. 2014;3 doi: 10.1161/JAHA.113.000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements M., Thomas N. High-throughput multi-parameter profiling of electrophysiological drug effects in human embryonic stem cell derived cardiomyocytes using multi-electrode arrays. Toxicol. Sci. 2014;140:445–461. doi: 10.1093/toxsci/kfu084. [DOI] [PubMed] [Google Scholar]
- Cohen N.A., Stewart M.L., Gavathiotis E., Tepper J.L., Bruekner S.R., Koss B., Opferman J.T., Walensky L.D. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem. Biol. 2012;19:1175–1186. doi: 10.1016/j.chembiol.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn G.W. Mitochondrial dynamics in heart disease. Biochim.Biophys.Acta. 2013;1833:233–241. doi: 10.1016/j.bbamcr.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn G.W., Vega R.B., Kelly D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015;29:1981–1991. doi: 10.1101/gad.269894.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitru R., Gama V., Fagan B.M., Bower J.J., Swahari V., Pevny L.H., Deshmukh M. Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol. Cell. 2012;46:573–583. doi: 10.1016/j.molcel.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero S., Zaganjor E., Lee S., Mill C.P., Morgan A.M., Crawford E.B., Chen J., Wales T.E., Mourtada R., Luccarelli J. Dynamic regulation of long-chain fatty acid oxidation by a noncanonical interaction between the MCL-1 BH3 helix and VLCAD. Mol. Cell. 2018;65:729–743.e7. doi: 10.1016/j.molcel.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feaster T.K., Cadar A.G., Wang L., Williams C.H., Chun Y.W., Hempel J.E., Bloodworth N., Merryman W.D., Lim C.C., Wu J.C. Matrigel Mattress: a method for the generation of single contracting human-induced pluripotent stem cell–derived cardiomyocytes. Circ. Res. 2015;117:995–1000. doi: 10.1161/CIRCRESAHA.115.307580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes C.D., Ma H., Mitalipov S., Terzic A. Mitochondria in pluripotent stem cells: stemness regulators and disease targets. Curr.Opin.Genet. Dev. 2016;38:1–7. doi: 10.1016/j.gde.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J.R., Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–343. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway C.A., Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid.Redox Signal. 2015;22:1545–1562. doi: 10.1089/ars.2015.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama V., Deshmukh M. Human embryonic stem cells: living on the edge. Cell Cycle Georget.Tex. 2012;11:3905–3906. doi: 10.4161/cc.22233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pavia P., Kim Y., Restrepo-Cordoba M.A., Lunde I.G., Wakimoto H., Smith A.M., Toepfer C.N., Getz K., Gorham J., Patel P. Genetic variants associated with cancer therapy–induced cardiomyopathy. Circulation. 2019;140:31–41. doi: 10.1161/CIRCULATIONAHA.118.037934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentillon C., Li D., Duan M., Yu W.-M., Preininger M.K., Jha R., Rampoldi A., Saraf A., Gibson G.C., Qu C.-K. Targeting HIF-1α in combination with PPARα activation and postnatal factors promotes the metabolic maturation of human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2019;132:120–135. doi: 10.1016/j.yjmcc.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Eldridge S., Furniss M., Mussio J., Davis M. Role of Mcl-1 in regulation of cell death in human induced pluripotent stem cell-derived cardiomyocytes in vitro. Toxicol. Appl. Pharmacol. 2018;360:88–98. doi: 10.1016/j.taap.2018.09.041. [DOI] [PubMed] [Google Scholar]
- Hackenbrock C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. J. Cell Biol. 1966;30:269–297. doi: 10.1083/jcb.30.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird A.W., Tron A.E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol.Ther. 2019;198:59–67. doi: 10.1016/j.pharmthera.2019.02.007. [DOI] [PubMed] [Google Scholar]
- Hsu Y.-H.R., Yogasundaram H., Parajuli N., Valtuille L., Sergi C., Oudit G.Y. MELAS syndrome and cardiomyopathy: linking mitochondrial function to heart failure pathogenesis. Heart Fail. Rev. 2016;21:103–116. doi: 10.1007/s10741-015-9524-5. [DOI] [PubMed] [Google Scholar]
- Imahashi K., Schneider M.D., Steenbergen C., Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ. Res. 2004;95:734–741. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- Itoh K., Nakamura K., Iijima M., Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y., Hoshijima M., Seo K., Bedja D., Sysa-Shah P., Andrabi S.A., Chen W., Höke A., Dawson V.L., Dawson T.M. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbassi E., Fenix A., Marchiano S., Muraoka N., Nakamura K., Yang X., Murry C.E. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020:1–19. doi: 10.1038/s41569-019-0331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara A., Cipolat S., Chen Y., Dorn G.W., Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and notch signaling. Science. 2013;342:734–737. doi: 10.1126/science.1241359. [DOI] [PubMed] [Google Scholar]
- Kasahara A., Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Khacho M., Clark A., Svoboda D.S., Azzi J., MacLaurin J.G., Meghaizel C., Sesaki H., Lagace D.C., Germain M., Harper M.-E. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcription program. Cell Stem Cell. 2016;19:1–16. doi: 10.1016/j.stem.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Kotschy A., Szlavik Z., Murray J., Davidson J., Maragno A.L., Le Toumelin-Braizat G., Chanrion M., Kelly G.L., Gong J.-N., Moujalled D.M. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- Kozopas K.M., Yang T., Buchan H.L., Zhou P., Craig R.W. MCLI, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA. 1993;5:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé K., Murley A., Nunnari J. Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 2014;30:357–391. doi: 10.1146/annurev-cellbio-101011-155756. [DOI] [PubMed] [Google Scholar]
- Lee Y., Lee H.-Y., Hanna R.A., Gustafsson Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1924–H1931. doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A. S63845, an MCL-1 selective BH3 mimetic: another arrow in our quiver. Cancer Cell. 2016;30:834–835. doi: 10.1016/j.ccell.2016.11.016. [DOI] [PubMed] [Google Scholar]
- Machiraju P., Greenway S.C. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J. Stem Cells. 2019;11:33–43. doi: 10.4252/wjsc.v11.i1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morciano G., Giorgi C., Balestra D., Marchi S., Perrone D., Pinotti M., Pinton P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol. Biol. Cell. 2016;27:20–34. doi: 10.1091/mbc.E15-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray T.V.A., Ahmad A., Brewer A.C. Reactive oxygen at the heart of metabolism. Trends Cardiovasc. Med. 2014;24:113–120. doi: 10.1016/j.tcm.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Murriel C.L., Churchill E., Inagaki K., Szweda L.I., Mochly-Rosen D. Protein Kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage. A mechanism involving Bad and the mitochondria. J. Biol. Chem. 2004;279:47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- Nishimura A., Shimauchi T., Tanaka T., Shimoda K., Toyama T., Kitajima N., Ishikawa T., Shindo N., Numaga-Tomita T., Yasuda S. Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission–associated myocardial senescence. Sci. Signal. 2018;11:eaat5185. doi: 10.1126/scisignal.aat5185. [DOI] [PubMed] [Google Scholar]
- Ong S.-B., Kalkhoran S.B., Hernández-Reséndiz S., Samangouei P., Ong S.-G., Hausenloy D.J. Mitochondrial-shaping proteins in cardiac health and disease – the long and the short of it! Cardiovasc. Drugs Ther. 2017;31:87–107. doi: 10.1007/s10557-016-6710-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman J.T., Iwasaki H., Ong C.C., Suh H., Mizuno S., Akashi K., Korsmeyer S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- Parikh S.S., Blackwell D.J., Gomez-Hurtado N., Frisk M., Wang L., Kim K., Dahl C.P., Fiane A., Tønnessen T., Kryshtal D.O. Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell–derived cardiomyocytes. Circ. Res. 2017;121:1323–1330. doi: 10.1161/CIRCRESAHA.117.311920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perciavalle R.M., Opferman J.T. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013;23:22–29. doi: 10.1016/j.tcb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perciavalle R.M., Stewart D.P., Koss B., Lynch J., Milasta S., Bathina M., Temirov J., Cleland M.M., Pelletier S., Schuetz J.D. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M.L., Kline L.A., Park K.P., Ortolano N.A., Romero-Morales A.I., Anthony C.C., Beckermann K.E., Gama V. A non-apoptotic function of MCL-1 in promoting pluripotency and modulating mitochondrial dynamics in stem cells. Stem Cell Rep. 2018;10:684–692. doi: 10.1016/j.stemcr.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds J.E., Li J., Craig R.W., Eastman A. BCL-2 and MCL-1 expression in Chinese hamster ovary cells inhibits intracellular acidification and apoptosis induced by staurosporine. Exp. Cell Res. 1996;225:430–436. doi: 10.1006/excr.1996.0194. [DOI] [PubMed] [Google Scholar]
- Rinkenberger J.L., Horning S., Klocke B., Roth K., Korsmeyer S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;6:23–27. [PMC free article] [PubMed] [Google Scholar]
- Sarosiek K.A., Fraser C., Muthalagu N., Bhola P.D., Chang W., McBrayer S.K., Cantlon A., Fisch S., Golomb-Mello G., Ryan J.M. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell. 2017;31:142–156. doi: 10.1016/j.ccell.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senichkin V.V., Streletskaia A.Y., Zhivotovsky B., Kopeina G.S. Molecular comprehension of Mcl-1: from gene structure to cancer therapy. Trends Cell Biol. 2019;29:549–562. doi: 10.1016/j.tcb.2019.03.004. [DOI] [PubMed] [Google Scholar]
- Sharp W.W., Simpson D.G., Borg T.K., Samarel A.M., Terracio L. Mechanical forces regulate focal adhesion and costamere assembly in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 1997;273:H546–H556. doi: 10.1152/ajpheart.1997.273.2.H546. [DOI] [PubMed] [Google Scholar]
- Simpson D.G., Decker M.L., Clark W.A., Decker R.S. Contractile activity and cell-cell contact regulate myofibrillar organization in cultured cardiac myocytes. J. Cell Biol. 1993;123:323–336. doi: 10.1083/jcb.123.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M., Franco A., Fleischer J.A., Zhang L., Dorn G.W. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 2017;26:872–883.e5. doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., Dayton B.D., Ding H., Enschede S.H., Fairbrother W.J. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Thomas R.L., Roberts D.J., Kubli D.A., Lee Y., Quinsay M.N., Owens J.B., Fischer K.M., Sussman M.A., Miyamoto S., Gustafsson Å.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27:1365–1377. doi: 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tron A.E., Belmonte M.A., Adam A., Aquila B.M., Boise L.H., Chiarparin E., Cidado J., Embrey K.J., Gangl E., Gibbons F.D. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018;9:1–14. doi: 10.1038/s41467-018-07551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky L.D. Stemming danger with golgified BAX. Mol. Cell. 2012;46:554–556. doi: 10.1016/j.molcel.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Bathina M., Lynch J., Koss B., Calabrese C., Frase S., Schuetz J.D., Rehg J.E., Opferman J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27:1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A., Boger R., Vick B., Urbanik T., Haybaeck J., Zoller S., Teufel A., Krammer P., Opferman J., Galle P. Hepatocyte-specific deletion of the anti-apoptotic protein Mcl-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatol.Baltim.Md. 2010;51:1226–1236. doi: 10.1002/hep.23479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Wright K.M., Deshmukh M. Restricting apoptosis for postmitotic cell survival and its relevance to cancer. Cell Cycle. 2006;5:1616–1620. doi: 10.4161/cc.5.15.3129. [DOI] [PubMed] [Google Scholar]
- Yang T., Buchan H.L., Townsend K.J., Craig R.W. MCL-1, a member of the BCL-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J. Cell. Physiol. 1996;166:523–536. doi: 10.1002/(SICI)1097-4652(199603)166:3<523::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Zhang J., Nuebel E., Daley G.Q., Koehler C.M., Teitell M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589–595. doi: 10.1016/j.stem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.