

Summary
Chemical modifications and adducts at DNA double-strand break (DSB) ends must be cleaned before re-joining by non-homologous end-joining (NHEJ). MRE11 nuclease is essential for efficient removal of Topoisomerase II (TOP2)-DNA adducts from TOP2 poison-induced DSBs. However, mechanisms in MRE11 recruitment to DSB sites in G1 phase remain poorly understood. Here, we report that TOP2-DNA adducts are expeditiously removed through UBC13-mediated polyubiquitination, which promotes DSB resection in G2 phase. We found that this ubiquitin signaling is required for efficient recruitment of MRE11 onto DSB sites in G1 by facilitating localization of RAP80 and BRCA1 to DSB sites and complex formation between BRCA1 and MRE11 at DSB sites. UBC13 and MRE11 are dispensable for restriction-enzyme-induced “clean” DSBs repair but responsible for over 50% and 70% of NHEJ-dependent repair of γ-ray-induced “dirty” DSBs, respectively. In conclusion, ubiquitin signaling promotes nucleolytic removal of DSB blocking adducts by MRE11 before NHEJ.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
Highlights
-
•
We establish a bioassay to identify the proteins that remove blocking adducts
-
•
UBC13 facilitates the removal of blocking adducts by recruiting MRE11 to DSB sites
-
•
UBC13 and RAP80 are required for BRCA1-MRE11 interaction in G1 phase
-
•
The UBC13-BRCA1-MRE11 axis functions independently of TDP2
Biological Sciences; Molecular Biology; Cell Biology
Introduction
A DNA double-strand break (DSB) is the most genotoxic type of DNA lesion. If misrepaired or left unrepaired, DSBs can lead to carcinogenesis and cell death. DSBs are repaired by two major repair pathways: homology-directed repair (HDR) and non-homologous end joining (NHEJ) (O'Driscoll and Jeggo, 2006). HDR is active only in the S/G2 phases, whereas NHEJ functions throughout the cell cycle (Moynahan and Jasin, 2010) (Chang et al., 2017) (Nickoloff et al., 2017) (Shrivastav et al., 2008). Note that NHEJ plays the dominant role in the G0/G1 phases and repairs approximately 80% of ionizing-radiation (IR)-induced DSBs even in the S/G2 phases (Beucher et al., 2009) (Shibata et al., 2014). NHEJ is initiated by the binding of a KU70/KU80 heterodimer and a DNA-PK-dependent protein kinase catalytic subunit (DNA-PKcs) to the DSB ends and finishes with ligation by a complex involving DNA LIGASE 4 (LIG4) and XRCC4 (Chang et al., 2017) (Dynan and Yoo, 1998). Ligation by LIG4 requires 3′-OH and 5′-phosphate DSB ends, termed “clean” DSBs (Robins and Lindahl, 1996) (Chappell et al., 2002). Thus, direct ligation by NHEJ requires the prior removal of blocking adducts and the restoration of ligatable (clean) DSB ends.
DSBs induced by endogenous and exogenous sources are generally accompanied by chemical adducts at DSB ends and are termed “dirty” DSBs (Asaithamby et al., 2011) (Woodbine et al., 2011). A typical exogenous source, IR, generates such dirty DSBs bearing multiple DNA lesions, including abasic sites and damaged bases at DSB ends (Davis and Chen, 2014) (Schipler and Iliakis, 2013) (Averbeck et al., 2014) (Jeggo et al., 2011) (Mladenov et al., 2018). Dirty DSBs are also produced by an anti-cancer Topoisomerase II (TOP2) poison, etoposide, which stabilizes the covalent association of TOP2 adducts at the 5′ ends of DSBs, referred to as a pathological TOP2 cleavage complex (TOP2cc) and also TOP2-DNA adducts (Pommier et al., 2016). Since chemical modifications as well as 5′ TOP2 adducts obstruct direct ligation of ends by LIG4, these blocking adducts need to be removed prior to ligation by NHEJ. Removal of such blocking modifications from DSBs is the rate-limiting step in DSB repair. This is evidenced by data showing that the vast majority of restriction-enzyme (RE) (AsiSI)-induced ligatable clean DSBs are repaired within an hour (Caron et al., 2015), whereas it takes several hours for DSB-repair pathways to ligate the majority of dirty DSBs induced by IR and etoposide (Hoa et al., 2016, Woodbine et al., 2014). However, the molecular mechanisms underlying the removal of the blocking adducts remains poorly understood, in part owing to the complexity of blocking adducts present at IR-induced DSBs.
MRE11 forms a complex with RAD50 and NBS1 (Xrs2 in Saccharomyces cerevisiae), and the resulting MRN(X) complex is involved in the initial DSB resection step of HDR, which generates 3′ single-stranded tails at DSBs (Shibata et al., 2014) (Roques et al., 2009) (Mimitou and Symington, 2008, Zhu et al., 2008) (Garcia et al., 2011) (reviewed in Oh and Symington, 2018, Paull, 2018). Yeast genetic studies suggest an important role for MRE11 endonuclease in the repair of DSBs with dirty ends as evidenced by the following study. Although Saccharomyces cerevisiae mutants expressing nuclease-deficient MRE11 perform HDR of RE-induced clean DSBs with nearly normal kinetics, the mutant is extremely sensitive to IR (Oh and Symington, 2018) (Moreau et al., 1999) (Westmoreland and Resnick, 2013), suggesting that MRE11 plays an important role in DSB repair other than DSB resection, potentially by removing blocking adducts. This is supported by data showing that the nuclease activity of Schizosaccharomyces pombe MRE11 removes both 3′ TOP1 and 5′ TOP2 adducts from DSBs in vivo (Hartsuiker et al., 2009). Moreover, the nuclease activity of purified MRN complex is capable of removing both 3′ and 5′ blocking adducts from DSBs (Cannavo and Cejka, 2014) (Deshpande et al., 2016) (Deshpande et al., 2018) (reviewed in (Paull, 2018)). Although the role of yeast MRE11 in creating ligatable ends has been established by comparing the repair kinetics of IR-induced dirty DSBs with those of RE-induced clean DSBs (Lisby et al., 2004, Westmoreland and Resnick, 2013), similar studies have not yet been performed in mammalian cells, because human cells deficient in MRE11 nuclease activity display severe genome instability and are inviable (Hoa et al., 2016).
The removal of blocking TOP2 adducts provides an excellent way to examine the molecular mechanism by which dirty DSBs are processed and clean DSBs are restored for the following reasons. Pulse exposure to etoposide specifically generates DSBs bearing a well-characterized blocking adduct, a pathological TOP2cc, and its number can be accurately measured (Hoa et al., 2016). TOP2 normally resolves DNA catenanes by transiently forming a DSB, a TOP2cc in duplex DNA, which allows the intact DNA duplex to pass through the DSB, followed by religation of the DSB by TOP2 (Cowell and Austin, 2012) (Nitiss, 2009). The repair of etoposide-induced DSBs is performed by NHEJ as well as by HDR (Hoa et al., 2016) (Aparicio et al., 2016). Repair by NHEJ requires prior removal of 5′ TOP2 adducts by tyrosyl-DNA-phosphodiesterase 2 (TDP2) and MRE11 (Hoa et al., 2016) (Ledesma et al., 2009) (Lee et al., 2018). MRE11 contributes to the removal of 5′ TOP2 adducts to a considerably greater extent than does TDP2, as MRE11 nuclease-deficient cells, but not TDP2−/− cells, display an endogenous accumulation of pathological TOP2ccs, which leads to cell death (Hoa et al., 2016). In HDR, MRE11 nuclease activity is tightly controlled by the ubiquitination and phosphorylation pathways during DSB resection in the S/G2 phases (Jachimowicz et al., 2019) (Dong et al., 1999) (Costanzo et al., 2001) (Falck et al., 2012) (Kijas et al., 2015). It remains unclear exactly how MRE11 nuclease is activated at pathological TOP2ccs during the G1 phase.
The E3 ligase RNF168 catalyzes H2A K15 monoubiquitination and K63-linked polyubiquitination near DSB sites (Kolas et al., 2007) (Huen et al., 2008) (Stewart et al., 2009) (Doil et al., 2009) (Mattiroli et al., 2012) (Uckelmann and Sixma, 2017). UBC13 plays a crucial role in K63-linked polyubiquitination (Stewart et al., 2009) (Mattiroli et al., 2012) (Zhao et al., 2007). RAP80 is a reader of K63-linked ubiquitin chains and facilitates the recruitment of the BRCA1-A complex onto DSB sites (Sato et al., 2009) (Wang et al., 2007) (Kim et al., 2007) (Sobhian et al., 2007) (Hu et al., 2011). The induction of DSBs leads to the formation of a complex between BRCA1 and MRE11 at DSB sites in a UBC13-dependent manner and promotes DSB resection (Zhao et al., 2007) (Greenberg et al., 2006) (Chen et al., 2008). RNF168 and BRCA1 play a redundant role in promoting HDR (Zong et al., 2019). It remains unclear whether UBC13-dependent K63 ubiquitin signaling also contributes to DSB repair during the G1 phase (reviewed in Nakada, 2016, Uckelmann and Sixma, 2017). Although NHEJ is required for both V(D)J recombination of antigen receptor genes and development of B and T lymphocytes (Alt et al., 2013), V(D)J recombination is not impaired by the loss of MRE11, RAP80, RNF168, or UBC13, indicating that all four are dispensable for NHEJ of clean DSBs carrying 3′-OH and 5′-phosphate moieties (Yamamoto et al., 2006) (Bohgaki et al., 2011) (Yin et al., 2012) (Dinkelmann et al., 2009).
To date, no bioassay has accurately assessed the capability of NHEJ except analysis of V(D)J recombination. Previous studies have evaluated the efficiency of NHEJ by measuring the repair kinetics of IR-induced DSBs as well as IR sensitivity in the G1 phase. However, these phenotypic assays do not distinguish NHEJ from the preceding step, which involves the processing of dirty DSBs to restore clean ends. Another widely used phenotypic analysis of NHEJ measures the repair of I-Sce1 RE-induced DSBs in reporter genes (reviewed in Jasin and Haber, 2016). However, this assay does not assess the frequency of the accurate DSB-repair events that restore the I-Sce1 site, even though the vast majority of the NHEJ events are accurate (Bétermier et al., 2014). Here we describe a new assay for assessing the capability of NHEJ. To this end, we expressed a regulatable AsiSI RE coupled to the estrogen receptor (ER-AsiSI) in cells (Caron et al., 2015) (Aymard et al., 2014); pulse-exposed the cells to an estrogen antagonist, 4-hydroxytamoxifen (4-OHT), to transiently activate AsiSI; and measured the number of unrepaired DSBs during the G1 phase. We verified over a 100-fold delay in DSB repair in the absence of LIG4. We, therefore, conclude that our repair kinetics analysis of ER-AsiSI-induced DSBs may measure virtually all NHEJ events in wild-type cells.
We show that UBC13 promotes the recruitment of MRE11 nuclease to remove 5′ TOP2 adducts from pathological TOP2ccs for subsequent NHEJ. We tested the involvement of UBC13 and MRE11 nuclease activity in the repair of IR-induced DSBs in the G1 phase. Although both these factors are essential for the efficient repair of dirty DSBs generated by etoposide and IR, they are both dispensable for the repair of AsiSI-induced clean DSBs. These results indicate that the UBC13 promotes MRE11-dependent removal of blocking adducts from IR-induced dirty DSBs in addition of that of TOP2 adducts prior to their ligation by NHEJ. We propose that K63-linked ubiquitin signaling involving MRE11 is indicated as the key step to determine the repair kinetics of dirty DSBs.
Results
UBC13 Contributes to DSB Repair during the G1 Phase
We exposed an asynchronous population of MCF-7 human breast cancer cells to etoposide for 30 min, then counted the number of conjugated-ubiquitin FK2 foci, which represent various types of ubiquitin chains (Shi et al., 2008). Table S1 shows the list of mutant cells analyzed in this study. We detected FK2 foci colocalizing with 53BP1 foci in virtually all cells (Figure S1A). We also detected etoposide-induced FK2 foci in serum-starved G1 cells (Figures S1A and S1B). We depleted UBC13 with shRNA (shUBC13) in the serum-starved cells (Figure S1C) and found an ~80% decrease in the number of etoposide-induced FK2 foci (Figures 1A, S1A, and S1D). Since UBC13 promotes K63-linked polyubiquitination at DSB sites (Stewart et al., 2009), this result indicates that a majority of the FK2 foci contain K63-linked polyubiquitination. We found that UBC13 depletion impaired H2AX ubiquitination in G1 phase, suggesting that H2AX is one of the targets of UBC13 (Figure S1E). Thus, UBC13 generates K63-linked polyubiquitin chains of the substrates, including H2A/H2AX, at DSB sites in the G1 as well as in the S/G2 phases.
Figure 1.

UBC13 Contributes to DSB Repair by Recruiting BRCA1 and RAP80 onto DSB Sites during the G1 Phase
(A) Average number of etoposide-induced FK2 foci in MCF-7 cells synchronized during the G1 phase by serum starvation (24 h). We examined wild-type MCF-7 cells treated with shRNA targeting UBC13 (shUBC13) and non-targeting shRNA (shControl). Synchronized cells were treated with etoposide (10 μM) for 30 min, washed, and incubated with etoposide-free media. Error bars show the standard deviation (SD) from three independent experiments. At least 50 G1-phase (cyclin A-negative) cells per experiment were counted. Single asterisk indicates p = 1.1 × 10−3, calculated by Student's t test. Representative images and box plots of FK2 foci are shown in Figures S1A and S1D, respectively.
(B) DSB-repair kinetics of G1-phase MCF-7 cells after pulse exposure (0–0.5 h) to etoposide (10 μM). Average number of γH2AX foci was counted at 0.5 and 8.5 h after addition of etoposide. Data are as shown in (A). Single, double, and triple asterisks indicate p = 1.2 × 10−3, p = 5.6 × 10−4, and p = 3.2 × 10−5, respectively, calculated by Student's t test. The box plots of γH2AX foci are shown in Figure S1F.
(C and D) Average number of etoposide-induced BRCA1 (C) and RAP80 (D) foci per cell. The experimental procedure and data are as shown in (A). Asterisks indicate p = 9.3 × 10−4 in (C) and p = 8.0 × 10−3 in (D). Representative images and box plots of BRCA1 and RAP80 foci are shown in Figures S1I and S1J, respectively.
To confirm the role of UBC13 in DSB repair in the G1 phase, we exposed serum-starved MCF-7 cells to etoposide for 30 min, then monitored the resolution kinetics of the γH2AX foci (Figures 1B and S1F). Pulse exposure (0.5 h) to etoposide caused similar increases in the number of γH2AX foci in all cell types tested. At 8 h after removal of etoposide, the number of γH2AX foci had diminished almost to background levels in the control cells but persisted in the DNA-PKcs−/− mutant cells (Figures 1B and S1F–S1H). The delay in DSB repair in the shUBC13 cells was also more prominent than in the control cells. These results suggest that UBC13 contributes to DSB repair through a pathway other than HDR. We next assessed the possible functional interaction between UBC13 and canonical NHEJ by depleting UBC13 in DNA-PKcs−/− cells (Figure S1C) and examining the etoposide-induced γH2AX foci. The depletion of UBC13 did not affect the repair kinetics of DNA-PKcs−/− cells (Figures 1B and S1F). Since UBC13 is dispensable for efficient NHEJ (Yamamoto et al., 2006), it is surprising that UBC13 shows a phenotype similar to DNA-PKcs deficiency for the repair of TOP2cc lesions caused by etoposide. Moreover, this epistatic relationship between UBC13 and NHEJ suggests that UBC13 contributes to NHEJ-mediated repair of these lesions.
UBC13-Dependent Focus Formation of BRCA1 and RAP80 in G1 Cells
UBC13-dependent ubiquitination promotes the recruitment of various factors, including BRCA1 and RAP80, to DSB sites in the S/G2 phases (reviewed in Nakada, 2016, Uckelmann and Sixma, 2017). We asked whether the formation of BRCA1 and RAP80 foci could also occur in the G1 phase. Pulse exposure (0.5 h) to etoposide caused 2.9- and 2.4-fold increases in the number of BRCA1 and RAP80 foci, respectively, in serum-starved wild-type MCF-7 cells (Figures 1C, 1D, S1I, and S1J). UBC13 depletion reduced the numbers of BRCA1 foci and RAP80 foci by 5 and 1.5 folds, respectively, in G1 cells. Additionally, the truncated form of RAP80 lacking ubiquitin-interacting motifs (RAP80-UIMΔ), which specifically recognize K63-linked polyubiquitin chains of H2A/H2AX (Figures S1K and S1L) (Mattiroli et al., 2012, Sato et al., 2009), completely abolished the ability to form foci after etoposide treatment in G1 phase. These results indicate that, similar to what has been observed during the S/G2 phases (Wang and Elledge, 2007), UBC13-dependent K63-linked ubiquitination plays a pivotal role in the recruitment of BRCA1 and RAP80 onto DSB sites during the G1 phase.
RAP80 and BRCA1 Contribute to DSB Repair during G1 Phase
To test the role of RAP80 in DSB repair in the G1 phase, we generated RAP80−/− cells by disrupting exon 5 of RAP80 in TK6 human B cells and in MCF-7 cells (Figures S2A and S2B). NHEJ is preferred over HDR to repair etoposide-induced DSBs (Maede et al., 2014). The RAP80−/− cells showed a higher sensitivity to etoposide than did the wild-type cells (Figures S2C and S2D). To analyze the role of RAP80 in DSB repair in the G1 phase, we enriched G1-phase MCF-7 cells by serum starvation, exposed the cells to etoposide for 30 min, then monitored the resolution kinetics of the γH2AX foci (Figures 2A and S2E). Remarkably, the NHEJ-deficient DNA-PKcs−/− and the RAP80−/− MCF-7 cells exhibited a very similar phenotype: a strong defect in the repair of etoposide-induced DSBs (Figures 2A and S2E). Thus, RAP80 plays an important role in NHEJ-dependent repair of etoposide-induced DSBs in the G1 phase. Since RAP80 is dispensable for NHEJ of clean DSBs (Yin et al., 2012), our results suggest that RAP80 is involved in the repair of DSBs with TOP2 adducts, similar to BRCA1 (Sasanuma et al., 2018).
Figure 2.

RAP80 and BRCA1 Contribute to DSB Repair during the G1 Phase
(A) Analysis of DSB-repair kinetics (as presented in Figure 1B). Single and double asterisks indicate p = 1.3 × 10−4 and p = 3.8 × 10−4, respectively. Representative images and box plots of γH2AX foci are shown in Figure S2E.
(B) Average number of etoposide-induced BRCA1 foci in wild-type and RAP80−/− MCF-7 cells before and after pulse exposure (0.5 h) to etoposide. Standard deviation (SD) was calculated from three independent experiments. Asterisk indicates p = 4.3 × 10−5. Representative images and box plots of BRCA1 foci are shown in Figure S2F.
(C) DSB-repair kinetics of MCF-7 cells in the G1 phase following pulse exposure (0.5 h) to etoposide (10 μM). Experimental procedure and data presentation are as shown in Figure 1B. Single and double asterisks indicate p = 6.5 × 10−3 and p = 3.2 × 10−5, respectively. Representative images and box plots of γH2AX foci are shown in Figure S2H.
We next investigated the impact of RAP80 disruption on BRCA1-focus formation at etoposide-induced DSB sites in serum-starved G1 MCF-7 cells. The loss of RAP80 caused a 20-fold decrease in the number of etoposide-induced BRCA1 foci (Figures 2B and S2F), indicating that RAP80 is required for the recruitment of BRCA1 onto DSB sites in the G1 phase. We previously showed that BRCA1 plays an important role in the removal of 5′ TOP2 adducts from pathological TOP2ccs in the G1 phase (Sasanuma et al., 2018). In agreement with this, shRNA-mediated depletion of BRCA1 (Figure S2G) delayed the repair kinetics of etoposide-induced DSBs in wild-type cells to a level very similar to that found in DNA-PKcs−/− cells (Figures 2C and S2H). The depletion did not further delay repair kinetics in DNA-PKcs−/− cells (Figures 2C and S2H). Thus, both UBC13 and RAP80 play a crucial role in the NHEJ-mediated repair of etoposide-induced DSBs, most likely by recruiting BRCA1 onto DSB sites and promoting the removal of 5′ TOP2 adducts from DSBs.
UBC13, RAP80, and BRCA1 Promote the Removal of Etoposide-Induced TOP2 Adducts Independent of TDP2
We measured the number of TOP2ccs in the G1 phase by analyzing serum-starved MCF-7 cells. We lysed cells and separated TOP2cc from free TOP2 in cellular lysates by subjecting them to sedimentation by means of cesium chloride (CsCl) density-gradient ultra-centrifugation, as described previously (Hoa et al., 2016). TOP2ccs were detected as single or double dots in the middle fractions of the TOP2-DNA complex, shown at the bottom of the blot in Figure S3A. As demonstrated previously, we detected a greater number of etoposide-induced TOP2ccs in both nuclease-deficient MRE11-/H129N and BRCA1-depleted TK6 cells, compared with wild-type cells (Hoa et al., 2016) (Sasanuma et al., 2018) (Figures 3A, S3B, and S3E). To identify the role played by UBC13 in removing etoposide-induced TOP2ccs, we depleted UBC13 in serum-starved MCF-7 cells and subjected these cells to the same assay. The MCF-7 cells also showed a greater number of TOP2ccs (Figures 3B and S3F) compared with shControl cells. UBC13-depleted cells reconstituted with wild-type UBC13 (shUBC13-WT) did not accumulate etoposide-induced TOP2ccs, whereas expression of catalytically inactive UBC13 C87A mutant protein (shUBC13-C87A) resulted in accumulation of TOP2ccs compared with that of shUBC13 cells (Figures 3B, S3D, and S3F). These results indicate that E2 ubiquitin-conjugating activity of UBC13 is required for the efficient removal of 5′ TOP2 adducts. Likewise, RAP80−/− cells and RAP80−/− cells expressing RAP80-UIMΔ caused an increase in the number of TOP2ccs in serum-starved MCF-7 cells (Figures 3B and S3G). We therefore conclude that ubiquitin signaling pathway involving BRCA1, RAP80, and UBC13 is required for efficient removal of 5′ TOP2 adducts from etoposide-induced TOP2ccs.
Figure 3.

UBC13, RAP80, and BRCA1 Promote the Removal of Etoposide-Induced TOP2 Adducts Independent of TDP2
(A) Quantification of TOP2-DNA-cleavage-complexes (TOP2ccs) in TK6 lymphoid cells carrying the indicated genotypes relative to the amount of TOP2ccs in wild-type cells. Schematic of in vivo TOP2cc measurement by immunodetection with α-TOP2 antibody is shown in Figure S3A. Cells were treated with etoposide (10 μM) (“+”) or DMSO (“-”) for 2 h. BRCA1AID/AID cells were pretreated with auxin for 2 h, then incubated with etoposide (10 μM) plus auxin for an additional 2 h. MRE11+/H129N cells were treated with 4-hydroxytamoxifen (4-OHT) for 3 days to inactivate the wild-type MRE11 allele, then treated with etoposide (10μM) for 2 h. Error bars represent standard deviation (SD) of three independent experiments. Asterisk indicates p = 2.8 × 10−2, calculated by Student's t test. Representative images of dot plots are shown in Figure S3E.
(B) Quantification of TOP2ccs in MCF-7 cells with the indicated genotypes relative to the amount of TOP2ccs in wild-type MCF-7 cells. Cells were incubated with serum-free medium for 24 h then treated with etoposide for 2 h. Each experiment was performed independently at least three times. Error bars represent SD. Single, double, triple, and quadruple asterisks indicate p = 4.7 × 10−4, p = 6.4 × 10−3, p = 5.3 × 10−3, and p = 1.9 × 10−2 respectively, calculated by Student's t -test. Representative images of dot plots are shown in Figures S3F and S3G.
Since TDP2 is implicated in removal of TOP2ccs (Ledesma et al., 2009) we tested if UBC13 and RAP80 facilitate the removal of 5′ TOP2 adducts in a manner dependent on TDP2. To this end, we depleted UBC13 and BRCA1 in TDP2−/− MCF-7 cells and generated RAP80−/−/TDP2−/− MCF-7 cells (Figures S1C, S2B, and S2G), then measured the number of TOP2ccs in the G1 phase. Consistent with earlier reports (Hoa et al., 2016) (Ledesma et al., 2009), depletion of TDP2 caused an increase in TOP2ccs upon etoposide treatment (Figures 3B and S3F). UBC13-depletion, RAP80−/− mutation, and BRCA1-depletion further enhanced the accumulation of etoposide-induced TOP2ccs in TDP2−/− cells (Figures 3B, S3F, and S3G). Thus, like BRCA1, UBC13 and RAP80 promote the removal of 5′ TOP2 adducts in a TDP2-independent manner in the G1 phase.
To examine the genetic interaction between MRE11 and UBC13, we depleted UBC13 in MRE11-/H129N cells (shUBC13/MRE11-/H129N) and analyzed etoposide-induced TOP2ccs. The accumulation of TOP2ccs in shUBC13/MRE11-/H129N cells is very similar to those of MRE11-/H129N cells (Figures 3A and S3E). This epistatic relationship suggests that UBC13-dependent ubiquitination pathway promotes the removal of TOP2ccs through MRE11 nuclease activity.
Ubiquitin Signaling Involving UBC13, RAP80, and BRCA1 Is Required for Efficient Recruitment of MRE11 Nuclease onto DSB Sites in G1 Cells
We investigated the role played by UBC13-dependent ubiquitin signaling in the recruitment of MRE11 to DSB sites. To this end, we examined MRE11 foci following exposure of G1-phase MCF-7 cells to etoposide. These treatments caused increases in the number of MRE11 foci colocalizing with 53BP1, a marker of DSB sites (Figure S4). MRE11-focus formation was impaired in BRCA1-depleted cells after etoposide treatment, as shown previously (Sasanuma et al., 2018) (Figure 4A). The depletion of UBC13 reduced the percentage of MRE11-positive cells by ~60% upon treatment with etoposide (Figures 4A and S4). Similarly, RAP80−/− cells showed an ~70% reduction in the percentage of MRE11-positive cells upon treatment with etoposide. These results indicate that in ubiquitin signaling involving UBC13, RAP80, and BRCA1, all three facilitate the recruitment of MRE11 onto DSB sites for the efficient removal of the adducts of dirty DSBs.
Figure 4.

UBC13-Mediated Ubiquitin Signaling Involving UBC13, RAP80, and BRCA1 is Required for Efficient Recruitment of MRE11 Nuclease onto DSB Sites in G1 Cells
(A) Quantification of MRE11-positive MCF-7 cells with at least 10 foci per nucleus for the indicated genotypes. Serum-starved MCF-7 cells were treated with etoposide (10 μM) for 30 min. Error bars were plotted for standard deviation (SD) from three independent experiments. Single, double, and triple asterisks indicate p = 2.3 × 10−4, p = 5.3 × 10−5, and p = 1.3 × 10−4, respectively, calculated by Student's t test. Representative images of etoposide-induced MRE11/53BP1 foci are shown in Figure S4.
(B) Etoposide-induced complex formation of BRCA1 and MRE11 in G1-phase MCF-7 cells. Whole-cell extracts (WCEs) were prepared from the indicated cells, untreated (“-”) or treated (“+”) with etoposide (10 μM) for 0.5 h. “Input” indicates 5% of WCEs used for immunoprecipitation. BRCA1 was immunoprecipitated from the WCEs. Intensities of the immunoprecipitated MRE11 bands for the indicated genotypes were normalized to those of the input. The graph indicates relative band intensities of the MRE11 bands in comparison with the untreated wild-type (Lane 1). Single and double asterisks indicate p = 2.1 × 10−2 and p = 1.5 × 10−3, respectively, calculated by Student's t test.
UBC13 Is Essential for DNA-Damage-Induced Stable Complex Formation between BRCA1 and MRE11 in the G1 Phase
Upon DNA damage in the S/G2 phases, BRCA1 physically interacted with the MRN complex (Greenberg et al., 2006, Polanowska et al., 2006) in a UBC13-dependent manner (Zhao et al., 2007), which promotes DSB resection by MRE11. This finding prompted us to analyze DNA-damage-induced complex formation between BRCA1 and MRE11, specifically in G1-phase cells. We exposed serum-starved MCF-7 cells to etoposide for 30 min, immunoprecipitated BRCA1, and tested for the presence of co-immunoprecipitated MRE11 by western blotting. No interactions between BRCA1 and MRE11 were seen in the absence of etoposide, whereas etoposide exposure induced interactions (lanes 2 and 4 of Figure 4B). Remarkably, the depletion of UBC13 abolished the DNA-damage-induced interaction (lane 6 of Figure 4B). RAP80 deletion also decreased the amount of MRE11 associating with BRCA1 (lane 10 of Figure 4B). These results indicate that, upon DNA damage in the G1 phase, UBC13 and RAP80 facilitate a stable interaction between BRCA1 and MRE11. These data support the notion that UBC13-mediated ubiquitin signaling activates the nuclease activity of MRE11 via interaction with BRCA1 at DSB sites. Considering the vital role played by UBC13, RAP80, and BRCA1 in NHEJ-dependent repair of etoposide-induced DSBs (Figures 1B, 2A, and 2C), these data suggest that UBC13-mediated ubiquitin signaling promotes the removal of 5′ TOP2 adducts by activating MRE11.
The Loss of LIG4 Reduces Repair of RE-Induced Clean DSBs over 100-Fold in the G1 Phase
The I-Sce1 reporter assays currently used to measure NHEJ events do not correctly measure the capability of NHEJ, because the I-Sce1 RE can re-cleave the accurately repaired junction, introducing a bias in favor of inaccurate repair that deletes the I-Sce1 site (Bétermier et al., 2014). We thus sought to establish a method to measure the frequency of all NHEJ events correctly. To this end, we expressed ER-AsiSI RE in TK6 cells and introduced clean DSBs via pulse exposure (4 h) of cells to 4-OHT (Iacovoni et al., 2010). The 4-OHT treatment caused an increase by over eight times in the number of γH2AX foci in all genotypes (0 h in Figures 5A and S5A). The number of γH2AX foci did not decrease from 0 to 1 h after 4-OHT removal owing to residual cleavage activity of ER-AsiSI RE. From 1 to 2 h, the number of γH2AX foci had dropped almost to background levels in the wild-type cells (Figures 5A and S5A), which agrees with the previous finding that AsiSI-induced DSBs are repaired within an hour (Caron et al., 2015). In contrast, even at 4 h after 4-OHT removal, essentially all γH2AX foci persisted in the LIG4−/− cells, indicating that the AsiSI-induced DSBs are repaired through the NHEJ pathway. The data indicated over 100-fold delay in DSB repair in the absence of LIG4 (Figure 5A), which is in sharp contrast with only up to several folds decrease in the number of (inaccurate) NHEJ events measured by reporter genes carrying the I-Sce1 site in NHEJ-deficient cells in comparison with wild-type cells (Biehs et al., 2017, Delacote et al., 2002, Gupta et al., 2018, Schipler and Iliakis, 2013, Zhang et al., 2011).
Figure 5.

MRE11 Nuclease and UBC13-Mediated Ubiquitin Signaling Are Required for Efficient Repair of IR-Induced Dirty DSBs in the G1 Phase
(A) Repair rate of DSBs induced by AsiSI restriction enzyme in G1-phase TK6 cells. Cells expressing AsiSI fused with estrogen receptor (ER) were treated with 4-OHT for 4 h for DSB induction. We analyzed γH2AX foci in cyclin A-negative cells after 4-OHT was removed (time 0 h). We subtracted the average number of foci in the 4-OHT-untreated cells from the average number of foci in the 4-OHT-treated cells. Values correspond to the percentage of unrepaired DSBs relative to the value at time 0 h, set to 100%. Error bars were plotted for standard deviation (SD) from three independent experiments. More than 50 G1 cells (cyclin A negative) were analyzed for each experiment. Single, double, and triple asterisks indicate p = 1.9 × 10−5, p = 2.9 × 10−5, and p = 4.3 × 10−5, respectively, calculated by Student's t test. Box plots of γH2AX foci at the indicated time points (0, 1, 2, and 4 h) are shown in Figure S5A.
(B–E) Repair rate of DSBs induced by IR (1 Gy) in G1-phase TK6 and HCT116 cells. Average number of γH2AX foci in cyclin A-negative cells was counted at the indicated time points. We subtracted the average number of foci in IR-untreated cells from the average number of foci in IR-treated cells. Values correspond to the percentage of unrepaired DSBs relative to the value at time 0.3 h, set to 100%. Error bars were plotted for standard deviation (SD) from three independent experiments. In (B), p values were 2.0 × 10−4 (shUBC13 versus wild-type), 2.1 × 10−3 (shBRCA1 versus wild-type), 1.9 × 10−5 (MRE11-/H129N versus wild-type), 9.5 × 10−5 (LIG4−/−/MRE11-/H129N versus wild-type), 4.1 × 10−4 (EXD2−/− versus wild-type), 3.6 × 10−2 (ARTEMIS−/− versus wild-type), 6.3 × 10−6 (EXO1−/− versus wild-type), and 1.7× 10−4 (shUBC13/MRE11-/H129N versus wild-type), calculated by Student's t test. Box plots of γH2AX foci at the indicated time points (0, 0.3, and 6 h) are shown in Figure S5D. In (C), asterisks indicate p = 1.3 × 10−4 (single) and p = 7.9 × 10−5 (double), calculated by Student's t test. Box plots of γH2AX foci at the indicated time points (0, 0.3, and 6 h) are shown in Figure S6D. In (D), asterisks indicate p = 5.1× 10−4 (single) and p = 3.8 × 10−2 (double), calculated by Student's t test. Box plots of γH2AX foci at the indicated time points (0, 0.3, and 6 h) are shown in Figure S5F. In (E), asterisks indicate p = 1.2× 10−2 (single) and p = 3.2 × 10−3 (double), calculated by Student's t test. Box plots of γH2AX foci at the indicated time points (0, 0.3, and 6 h) are shown in Figure S5H.
MRE11 Nuclease and UBC13-Dependent Ubiquitin Signaling Are Dispensable for NHEJ but Required for Efficient Repair of IR-Induced DSBs
Similar to wild-type cells, at 2 h after 4-OHT removal, the number of γH2AX foci per cell had declined to near background levels in UBC13-depleted (Figure S5B), BRCA1-depleted (Figure S5C), and MRE11 nuclease-deficient cells (Figures 5A and S5A). Thus, UBC13, BRCA1, and the nuclease activity of MRE11 are all dispensable for NHEJ. In contrast to AsiSI-mediated DSBs, IR-induced foci require a longer time (6 h) to drop back to background levels in wild-type cells (Figure 5B). The repair was NHEJ dependent as seen by the persistence of foci in LIG4−/− cells (Figure 5B). Interestingly, UBC13-depleted and MRE11-/H129N cells also showed the persistence of foci, approximately 54% and 69% of that in LIG4−/− mutant cells, respectively, at 6 h after IR exposure (Figures 5B and S5D). These values are probably an under-estimation of the actual contributions of UBC13 and MRE11 to the NHEJ of IR-induced DSBs since there were some residual UBC13 and MRE11 proteins left in their depleted cells. LIG4−/−/MRE11-/H129N mutant cells (Figure S5E) did not show an increase in γH2AX foci compared with LIG4−/− mutant cells (Figures 5B and S5D). Thus, MRE11 nuclease activity collaborates with NHEJ-dependent repair, most likely by removing blocking adducts prior to NHEJ of clean DSBs. We conclude that MRE11 and UBC13 play a dominant role in the removal of blocking adducts from DSB ends preceding canonical NHEJ.
Although a previous study using shRNA to knockdown MRE11 reported nearly normal repair of IR-induced DSBs in MRE11-depleted cells (Biehs et al., 2017), we found a very severe defect in IR-induced DSB repair in MRE11 nuclease-deficient TK6 cells (Figures 5B and S5D). We thus tested the effect of MRE11 depletion on the repair of IR-induced DSBs in another cell line: HCT116 cells. To achieve a sufficient depletion of MRE11, we inserted auxin-induced-degron (AID) tag sequences into the endogenous MRE11 allelic genes, generating MRE11AID/AID HCT116 cells (Figures S6A and S6B). When we depleted MRE11 using shRNA alone in HCT116 cells (Figure S6C), the depletion had very little effect on the repair of IR-induced DSBs during the G1 phase (Figures 5C and S6D), as shown previously (Biehs et al., 2017). In marked contrast, cells simultaneously treated with auxin and shRNA showed delayed repair kinetics very similar to that of HCT116 cells treated with an inhibitor of DNA-PKcs to inhibit NHEJ (Figures 5C and S6D). The present results indicate that shRNA-mediated depletion of MRE11 might not be sufficient to identify the critical role of MRE11 in DSB repair. This observation of HCT116 cells is reminiscent of previous data indicating that a mere ~1% of the endogenous MRE11 protein is sufficient to effectively prevent the prominent phenotype, an accumulation of spontaneously arising mitotic chromosome breaks in TK6 cells (Hoa et al., 2015). We conclude that MRE11 is required for the removal of blocking adducts from IR-induced DSBs for subsequent NHEJ.
RNF8, RNF168, and BRCA1 Contribute to Cellular Tolerance to IR
Although BRCA1-depleted cells and DNA-PKcs−/− cells showed the same prominent delays in the repair of etoposide-induced DSBs in the G1 phase (Figure 2C), BRCA1's contribution to the repair of IR-induced DSBs was less prominent than that of LIG4 (Figures 5B and S5D). We hypothesized that another enzyme(s) substituted for lack of BRCA1 and chose to analyze BRCA1AID/AID/RNF168−/− TK6 cells because BRCA1 and RNF168 are compensatory in HDR (Zong et al., 2019). Like BRCA1-depleted cells (BRCA1AID/AID cells treated with auxin), BRCA1AID/AID/RNF168−/− cells without auxin treatment showed the delayed repair of IR-induced DSBs in the G1 phase (Figures 5D and S5F). The addition of auxin to deplete BRCA1 in the BRCA1AID/AID/RNF168−/− cells caused a greater delay in the repair of IR-induced DSBs than the BRCA1-depleted BRCA1AID/AID cells (Figures 5D and S5F). We also examined the contribution of RNF8, another ubiquitin E3 ligase involved in DSB repair, to the repair of IR-induced DSBs in G1-phase cells. We observed the delayed DSB repair in RNF8−/− cells (Figures 5E and S5H). The depletion of BRCA1 in RNF8−/− cells (shBRCA1/RNF8−/−) caused a further delay in the repair of IR-induced DSBs, indicating a compensatory function of BRCA1 and RNF8 in the repair of IR-induced DSBs. Taken together, the data support the idea that removal of blocking adducts from IR-induced DSBs involves more complex ubiquitin signaling than that from etoposide-induced DSBs. RNF8 and RNF168 may mask the important role played by BRCA1 in the repair of IR-induced DSBs in the G1 phase.
A Collaboration of ARTEMIS, EXD2, and EXO1 with MRE11 in the Repair of IR-Induced DSBs
We explored the role played by enzymes that are implicated in the processing of IR-induced DSBs in TK6 cells. We analyzed DNA polymerases β and θ, both of which have 5’ -deoxyribose phosphate (dRP) lyase activity (Prasad et al., 2009), TDP1, TDP2, PARP1, and XRCC1, the last two playing a key role in microhomology-mediated end joining (MMEJ) (Figures S7A–S7D) (Saha et al., 2018) (Sfeir and Symington, 2015). TK6 cells deficient in these proteins were all tolerant to IR (Figure S7E). We also analyzed nucleases that process DSB ends during the G1 phase, including ARTEMIS, EXD2, and EXO1 (Figures S7F–S7H) (Biehs et al., 2017). TK6 cells null-deficient in any of these three nucleases were all sensitive to IR (Figure S7E), but the phenotypes were milder than those of LIG4-deficient and MRE11 nuclease-deficient cells (Figures 5B and S5D). ARTEMIS has an overhang endonucleolytic processing activity (Ma et al., 2002, Pannunzio et al., 2018) and might remove blocking adducts attached to overhang sequences at DSBs. Neither EXD2 nor EXO1 may be able to remove blocking adducts from IR-induced DSBs. It is more likely that these two nucleases generate ligatable blunt-end and cohesive-end breaks (Pannunzio et al., 2018). In summary, these data indicate MRE11 nuclease plays a dominant role in the removal of blocking adducts to generate clean ends and ARTEMIS, EXD2, and EXO1 may subsequently process the clean DSBs for direct ligation by NHEJ.
Discussion
We herein demonstrate that UBC13-mediated ubiquitin signaling plays a pivotal role in the removal of 5′ TOP2 adducts preceding NHEJ (Figures 1, 2, 3, and 4). This signaling is carried out most likely by RAP80 and BRCA1 and activates the MRE11 nuclease to remove 5′ TOP2 adducts. We also show that UBC13-mediated ubiquitin signaling and MRE11 are dispensable for rejoining of the AsiSI RE-induced DSBs (Figures 5A and S5A) but required for most NHEJ-dependent repair events in IR-irradiated G1 cells (Figures 5B, 5C, S5D, and S6D). This finding indicates the crucial role played by both UBC13-mediated ubiquitin signaling and MRE11 in removing IR-induced blocking adducts in addition to TOP2 adducts from DSB ends. NHEJ repairs approximately 80% of the IR-induced DSBs even in the S/G2 phases (Beucher et al., 2009) (Shibata et al., 2014) and plays a more important role in repairing etoposide-induced DSBs than does HDR (Maede et al., 2014). Moreover, the repair time of dirty DSBs induced by etoposide (Figures 1B, 2A, and 2C) and IR (Figures 5B–5E) was several times longer than that of RE-induced clean DSBs (Figure 5A). Considering these data, the current study sheds light on the removal of blocking adducts from dirty DSBs as the key rate-limiting step in the repair of DSBs generated by radiotherapy and a chemotherapy via etoposide during all phases of the cell cycle.
We have shown that MRE11 plays a dominant role in the removal of blocking adducts prior to NHEJ of clean DSBs in cells. Our results are supported by several biochemical studies that showed that non-covalent DNA-bound KU70/80 proteins stimulate the endonuclease activity of MRE11, leading to the removal of adducts from both 3′ and 5′ termini at DSBs (Deshpande et al., 2016) (Deshpande et al., 2018) (Anand et al., 2019) (reviewed in Paull, 2018). Thus, MRE11-dependent elimination of various chemical adducts attached to DSB ends may be a common mechanism for generating ligatable clean ends prior to direct ligation by canonical NHEJ. We also examined the contributions of other nucleases to the processing of dirty DSBs in the G1 phase. ARTEMIS, EXD2, EXO1, and MRE11 contribute to approximately 36%, 39%, 45%, and 69%, respectively, of the NHEJ events during 6-h repair time post-IR in the G1 phase (Figure 5B). Although exonucleases EXD2 or EXO1 may not be capable of removing various blocking adducts from DSB ends, the loss of ARTEMIS increases sensitivity to camptothecin, a TOP1 poison, but not etoposide (Maede et al., 2014), suggesting that ARTEMIS removes various 3′ blocking adducts including TOP1 adducts from DSBs. ARTEMIS, EXD2, and EXO1 might generate blunt-end and cohesive-end breaks, which can be ligated by LIG4 after the removal of blocking adducts from DSB ends by MRE11. This notion is supported by the previous findings that DSB resection is executed by these nucleases in G1 phase (Biehs et al., 2017). Identifying the roles played by ARTEMIS, EXD2, and EXO1 in the repair of IR-induced DSBs is an important area of study for the future. In contrast with the four nucleases, DNA polymerases β and θ, both of which have 5′-dRP lyase activity, TDP1, and TDP2 contribute only a little to the repair of IR-induced DSBs (Figure S7E). PARP1 and XRCC1, which play a key role in microhomology-mediated end joining (MMEJ) (Sfeir and Symington, 2015) are dispensable for the repair of IR-induced DSBs (Figure S7E), as previously reported (Simsek et al., 2011). In conclusion, ARTEMIS, EXD2, EXO1, and MRE11 all contribute to the repair of IR-induced DSBs in preparation for subsequent NHEJ. Given the dominant role played by MRE11, ARTEMIS, EXD2, and EXO1 may collaborate with MRE11 in the repair of IR-induced DSBs.
To protect genomic DNA from excess degradation, nuclease activity is strictly regulated (Symington, 2016). The mechanisms underlying MRE11 nuclease activity in the S/G2 phases have been thoroughly studied, but it is unclear how MRE11 nuclease is regulated in the removal of chemical adducts from DSB ends during the G1 phase. MRE11 nuclease activity is regulated by phosphorylation during DSB resection in the S/G2 phases (Costanzo et al., 2001) (Falck et al., 2012) (Kijas et al., 2015). DSB resection is also promoted by UBC13-dependent ubiquitination of histones at DSB sites (reviewed in Uckelmann and Sixma, 2017), although the usage of the lysine residues on the ubiquitin is unclear. In the present study, we show that UBC13-mediated ubiquitination signaling is activated not only during the S/G2 phases but also during the G1 phase (Figure 1). Activation of ubiquitin signaling in the G1 phase is required for the physical interaction between BRCA1 and MRE11 (Figure 4B), similar to S/G2 phases (Zhao et al., 2007). The resulting BRCA1-MRE11 complex that forms at the DSB site is essential for the removal of etoposide-induced 5′ TOP2 adducts. In addition to ubiquitin signaling-dependent BRCA1-MRE11 complex formation, CtIP, a regulatory factor of DSB end resection, also plays a critical role in the removal of blocking adducts. Biochemical studies demonstrated that phosphorylated CtIP stimulates MRE11 nuclease activity for the removal of blocking adducts (Anand et al., 2019, Anand et al., 2016, Quennet et al., 2011). CDK-dependent phosphorylation in CtIP is required for the removal of 5′ TOP2 adducts by promoting the interaction between CtIP and BRCA1 (Aparicio et al., 2016, Nakamura et al., 2010). The phosphorylated CtIP is recognized by NBS1, which regulates the nuclease activity of MRE11-RAD50 complex (Anand et al., 2019, Deshpande et al., 2016). It is possible that phosphorylated CtIP regulates MRE11 nuclease activity by facilitating complex formation among BRCA1-MRN-CtIP at etoposide-induced DSB sites in G1 phase. BRCA1 also promotes the removal of IR-induced chemical adducts by MRE11 prior to direct ligation of these processed DSBs by NHEJ. UBC13-mediated ubiquitination seems to activate complex signaling pathways involving multiple proteins, including BRCA1, RNF8, and RNF168 (Figures 5D and 5E), and contribute to the repair of IR-induced DSBs in the G1 phase. In sum, UBC13-mediated ubiquitin signaling activates MRE11-dependent DSB end-processing. Future biochemical studies are needed to clarify how BRCA1 stimulates the endonuclease activity of MRE11.
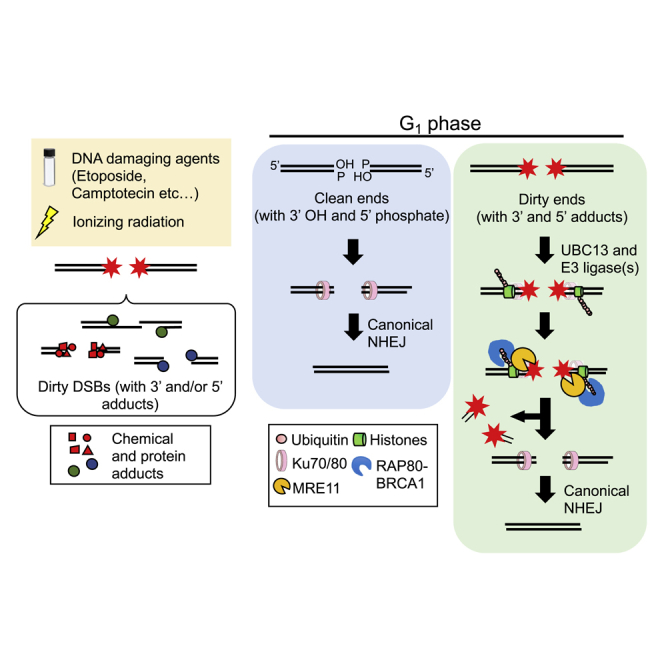
We propose a model in which dirty DSBs, induced by etoposide and IR, are repaired during the G1 phase. DSBs are rapidly recognized by the KU70/80 complex (step 2, Figures 6A and 6B). It remains unclear whether the KU70/80 complex interacts with DSB ends bearing intact TOP2 or interacts after TOP2 adducts are degraded by proteasome (Isik et al., 2003, Lee et al., 2018). TDP2 removes TOP2 adducts after its degradation by the proteasome (Lee et al., 2018). UBC13 catalyzes ubiquitination of histones at DSB sites in collaboration with ubiquitin E3 ligase(s), such as RNF8 and RNF168 (step 3, Figure 6B). The clean DSB ends induced by RE are quickly rejoined by NHEJ (step 3, Figure 6A). This ubiquitination (step 3, Figure 6B) facilitates the recruitment of multiple proteins such as BRCA1 and RAP80 onto the dirty DSB ends (step 4, Figure 6B). The recruited BRCA1 forms a stable complex with MRE11, with the resulting complex perhaps stimulating the endonuclease activity of MRE11 (step 5, Figure 6B). It remains unclear how RNF168 promotes the repair of IR-induced DSBs. Non-covalent DNA-bound KU70/80 proteins also stimulate MRE11 endonuclease activity, as previously suggested (Deshpande et al., 2018) (Reginato et al., 2017). This stimulated MRE11 endonuclease engages in sequential endonucleolytic processing of both 5′ and 3′ termini and may completely remove both blocking adducts and KU70/80 proteins from the DSBs (steps 6 and 7, Figure 6B) (Deshpande et al., 2018) (reviewed in Paull, 2018). The resulting clean DSB ends are bound by KU70/80 proteins (step 9, Figure 6B), leading to rejoining of the DSBs by canonical NHEJ (step 10, Figure 6B). The mechanism by which a variety of chemicals adducts at IR-induced DSBs is likely to be much more complicated than that by which 5′ TOP2 adducts are removed, with multiple mechanisms in play, depending on the structure of the dirty DSB ends. Future study will shed light on the multiple mechanisms underlying the removal of a variety of chemical adducts prior to NHEJ.
Figure 6.

Proposed Model for Ten-Step Elimination of the Adducts Attached to DSB Ends
(A) Restriction enzyme generates “clean” DSBs with 3′- hydroxyl groups and 5′-phosphate ends (step 1). The DSB ends are rapidly recognized by the KU70/80 complex (step 2) and rejoined by canonical NHEJ (step 3).
(B) Ionizing radiation generates “dirty” DSBs associated with 3′ and 5′ adducts (step 1). The DSB ends are rapidly recognized by the KU70/80 complex (step 2). UBC13 promotes K63 ubiquitination at DSB sites (step 3), where this ubiquitination is recognized by the BRCA1-RAP80 complex (step 4). UBC13 and RAP80 are required for stable complex formation between BRCA1 and MRE11 at DSB sites (step 5). Endonucleolytic cleavage by MRE11 releases 5′ and -3′ adducts from the DSB ends (steps 6–8). The resulting clean DSB ends are again recognized by the KU70/80 complex (step 9), then ligated by canonical NHEJ (step 10).
Limitations of the Study
In this study, we identified the involvement of the UBC13-mediated ubiquitination pathway in the removal of blocking adducts generated not only by etoposide but also by ionizing radiation. Mechanistically, UBC13-mediated ubiquitination signaling strongly promotes the interaction between BRCA1 and MRE11, thereby stimulating nuclease activity of MRE11 for the removal of blocking adducts from DSB sites. The current study has not clearly demonstrated the molecular targets ubiquitinated by UBC13 at the DSB sites. This topic should be investigated in future work.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Junji Itou for making boxplots in R language and technical assistance. We thank Drs. Tanya T. Paull, Rajashree A. Deshpande, Penny A. Jeggo, and the members of the Radiation Genetics lab for their helpful comments on the manuscript. For technical assistance with the cell sorter and confocal microscope, we are grateful to the staff of the Medical Research Support Center (supported by Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS), AMED Grant JP19am0101092). This study was conducted through the Joint Research Program of the Radiation Biology Center, Kyoto University. This work was supported by JSPS KAKENHI Grant Number 23221005 and JP16H06306 (S.T.) and 16H02953, 18H04900, and JP19H04267 (H.S.). This study was supported by JSPS Core-to-Core Program. This work was also supported by grants from the Takeda Research and Mitsubishi Foundation (to H.S.)
Author Contributions
This study was conceived by H.S. as well as S.T. Experiments and data analysis were performed by R.A., H.T.T., L.K.S., M.T., K.H., S.Y., A.S., M.T.K., S.N., and H.S. The paper was written by S.T. and H.S.
Declaration of Interests
The authors declare no competing interests.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101027.
Contributor Information
Shunichi Takeda, Email: stakeda@rg.med.kyoto-u.ac.jp.
Hiroyuki Sasanuma, Email: hiroysasa@rg.med.kyoto-u.ac.jp.
Supplemental Information
References
- Alt F.W., Zhang Y., Meng F.-L., Guo C., Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R., Jasrotia A., Bundschuh D., Howard S.M., Ranjha L., Stucki M., Cejka P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019;38:e101005. doi: 10.15252/embj.2018101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R., Ranjha L., Cannavo E., Cejka P. Phosphorylated CtIP functions as a Co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell. 2016;64:940–950. doi: 10.1016/j.molcel.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Aparicio T., Baer R., Gottesman M., Gautier J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J. Cell Biol. 2016;212:399–408. doi: 10.1083/jcb.201504005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaithamby A., Hu B., Chen D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. U S A. 2011;108:8293–8298. doi: 10.1073/pnas.1016045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averbeck N.B., Ringel O., Herrlitz M., Jakob B., Durante M., Taucher-Scholz G. DNA end resection is needed for the repair of complex lesions in G1-phase human cells. Cell Cycle. 2014;13:2509–2516. doi: 10.4161/15384101.2015.941743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P., Legube G. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétermier M., Bertrand P., Lopez B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014;10:e1004086. doi: 10.1371/journal.pgen.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beucher A., Birraux J., Tchouandong L., Barton O., Shibata A., Conrad S., Goodarzi A.A., Krempler A., Jeggo P.A., Löbrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biehs R., Steinlage M., Barton O., Juhász S., Künzel J., Spies J., Shibata A., Jeggo P.A., Löbrich M. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell. 2017;65:671–684.e5. doi: 10.1016/j.molcel.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohgaki T., Bohgaki M., Cardoso R., Panier S., Zeegers D., Li L., Stewart G.S., Sanchez O., Hande M.P., Durocher D. Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the RIDDLE syndrome. PLoS Genet. 2011;7:e1001381. doi: 10.1371/journal.pgen.1001381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E., Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- Caron P., Choudjaye J., Clouaire T., Bugler B., Daburon V., Aguirrebengoa M., Mangeat T., Iacovoni J.S., Álvarez-Quilón A., Cortés-Ledesma F., Legube G. Non-redundant functions of ATM and DNA-PKcs in response to DNA double-strand breaks. Cell Rep. 2015;13:1598–1609. doi: 10.1016/j.celrep.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.H.Y., Pannunzio N.R., Adachi N., Lieber M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017;18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell C., Hanakahi L.A., Karimi-Busheri F., Weinfeld M., West S.C. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Nievera C.J., Lee A.Y.-L., Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- Costanzo V., Robertson K., Bibikova M., Kim E., Grieco D., Gottesman M., Carroll D., Gautier J. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol. Cell. 2001;8:137–147. doi: 10.1016/s1097-2765(01)00294-5. [DOI] [PubMed] [Google Scholar]
- Cowell I.G., Austin C.A. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health. 2012;9:2075–2091. doi: 10.3390/ijerph9062075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A.J., Chen D.J. Complex DSBs: a need for resection. Cell Cycle. 2014;13:3796–3797. doi: 10.4161/15384101.2014.986630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacote F., Han M., Stamato T.D., Jasin M., Lopez B.S. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002;30:3454–3463. doi: 10.1093/nar/gkf452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande R.A., Lee J.-H., Arora S., Paull T.T. Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol. Cell. 2016;64:593–606. doi: 10.1016/j.molcel.2016.10.010. [DOI] [PubMed] [Google Scholar]
- Deshpande R.A., Myler L.R., Soniat M.M., Makharashvili N., Lee L., Lees-Miller S.P., Finkelstein I.J., Paull T.T. DNA-PKcs promotes DNA end processing. bioRxiv. 2018;64:395731. doi: 10.1126/sciadv.aay0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkelmann M., Spehalski E., Stoneham T., Buis J., Wu Y., Sekiguchi J.M., Ferguson D.O. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D.H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- Dong Z., Zhong Q., Chen P.L. The Nijmegen breakage syndrome protein is essential for Mre11 phosphorylation upon DNA damage. J. Biol. Chem. 1999;274:19513–19516. doi: 10.1074/jbc.274.28.19513. [DOI] [PubMed] [Google Scholar]
- Dynan W.S., Yoo S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998;26:1551–1559. doi: 10.1093/nar/26.7.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J., Forment J.V., Coates J., Mistrik M., Lukas J., Bartek J., Jackson S.P. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012;13:561–568. doi: 10.1038/embor.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V., Phelps S.E.L., Gray S., Neale M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011;479:241–244. doi: 10.1038/nature10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg R.A., Sobhian B., Pathania S., Cantor S.B., Nakatani Y., Livingston D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R., Somyajit K., Narita T., Maskey E., Stanlie A., Kremer M., Typas D., Lammers M., Mailand N., Nussenzweig A. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018;173:972–988.e23. doi: 10.1016/j.cell.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsuiker E., Neale M.J., Carr A.M. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell. 2009;33:117–123. doi: 10.1016/j.molcel.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoa N.N., Akagawa R., Yamasaki T., Hirota K., Sasa K., Natsume T., Kobayashi J., Sakuma T., Yamamoto T., Komatsu K. Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells. 2015;20:1059–1076. doi: 10.1111/gtc.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoa N.N., Shimizu T., Zhou Z.W., Wang Z.-Q., Deshpande R.A., Paull T.T., Akter S., Tsuda M., Furuta R., Tsutsui K. Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Mol. Cell. 2016;64:580–592. doi: 10.1016/j.molcel.2016.10.011. [DOI] [PubMed] [Google Scholar]
- Hu Y., Scully R., Sobhian B., Xie A., Shestakova E., Livingston D.M. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25:685–700. doi: 10.1101/gad.2011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen M.S.Y., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J. The E3 ubiquitin ligase RNF8 transduces the DNA damage signal via an ubiquitin-dependent signaling pathway. Cell. 2008;13190:901–914. [Google Scholar]
- Iacovoni J.S., Caron P., Lassadi I., Nicolas E., Massip L., Trouche D., Legube G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isik S., Sano K., Tsutsui K., Seki M., Enomoto T., Saitoh H., Tsutsui K. The SUMO pathway is required for selective degradation of DNA topoisomerase IIβ induced by a catalytic inhibitor ICRF-193(1) FEBS Lett. 2003;546:374–378. doi: 10.1016/s0014-5793(03)00637-9. [DOI] [PubMed] [Google Scholar]
- Jachimowicz R.D., Beleggia F., Isensee J., Velpula B.B., Goergens J., Bustos M.A., Doll M.A., Shenoy A., Checa-Rodriguez C., Wiederstein J.L. UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell. 2019;176:505–519.e22. doi: 10.1016/j.cell.2018.11.024. [DOI] [PubMed] [Google Scholar]
- Jasin M., Haber J.E. The democratization of gene editing: insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst.) 2016;44:6–16. doi: 10.1016/j.dnarep.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeggo P.A., Geuting V., Löbrich M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother. Oncol. 2011;101:7–12. doi: 10.1016/j.radonc.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Kijas A.W., Lim Y.C., Bolderson E., Cerosaletti K., Gatei M., Jakob B., Tobias F., Taucher-Scholz G., Gueven N., Oakley G. ATM-dependent phosphorylation of MRE11 controls extent of resection during homology directed repair by signalling through Exonuclease 1. Nucleic Acids Res. 2015;43:8352–8367. doi: 10.1093/nar/gkv754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Chen J., Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Kolas N.K., Chapman J.R., Nakada S., Ylanko J., Chahwan R., Sweeney F.D., Panier S., Mendez M., Wildenhain J., Thomson T.M. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma F.C., El Khamisy S.F., Zuma M.C., Osborn K., Caldecott K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature. 2009;461:674–678. doi: 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- Lee K., Swan R., Sondka Z., Padget K., Cowell I., Austin C. Effect of TDP2 on the level of TOP2-DNA complexes and SUMOylated TOP2-DNA complexes. Int. J. Mol. Sci. 2018;19:2056. doi: 10.3390/ijms19072056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M., Barlow J.H., Burgess R.C., Rothstein R. Choreography of the DNA damage response. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Ma Y., Pannicke U., Schwarz K., Lieber M.R. Hairpin opening and overhang processing by an artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- Maede Y., Shimizu H., Fukushima T., Kogame T., Nakamura T., Miki T., Takeda S., Pommier Y., Murai J. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 2014;13:214–220. doi: 10.1158/1535-7163.MCT-13-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F., Vissers J.H.A., van Dijk W.J., Ikpa P., Citterio E., Vermeulen W., Marteijn J.A., Sixma T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150:1182–1195. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Mimitou E.P., Symington L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–U3. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladenov E., Saha J., Iliakis G. Springer; 2018. Processing-Challenges Generated by Clusters of DNA Double-Strand Breaks Underpin Increased Effectiveness of High-LET Radiation and Chromothripsis; pp. 149–168. [DOI] [PubMed] [Google Scholar]
- Moreau S., Ferguson J.R., Symington L.S. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan M.E., Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada S. Opposing roles of RNF8/RNF168 and deubiquitinating enzymes in ubiquitination-dependent DNA double-strand break response signaling and DNA-repair pathway choice. J. Radiat. Res. 2016;57:i33–i40. doi: 10.1093/jrr/rrw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K., Kogame T., Oshiumi H., Shinohara A., Sumitomo Y., Agama K., Pommier Y., Tsutsui K.M., Tsutsui K., Hartsuiker E. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010;6:e1000828. doi: 10.1371/journal.pgen.1000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff J.A., Jones D., Lee S.-H., Williamson E.A., Hromas R. Drugging the cancers addicted to DNA repair. J. Natl. Cancer Inst. 2017;109:1–13. doi: 10.1093/jnci/djx059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer. 2009;9:327–337. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M., Jeggo P.A. The role of double-strand break repair — insights from human genetics. Nat. Rev. Genet. 2006;7:45. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- Oh J., Symington L.S. Role of the Mre11 complex in preserving genome integrity. Genes (Basel) 2018;9:589. doi: 10.3390/genes9120589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannunzio N.R., Watanabe G., Lieber M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018;293:10512–10523. doi: 10.1074/jbc.TM117.000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull T.T. 20 Years of Mre11 Biology: no end in sight. Mol. Cell. 2018;71:419–427. doi: 10.1016/j.molcel.2018.06.033. [DOI] [PubMed] [Google Scholar]
- Polanowska J., Martin J.S., Garcia-Muse T., Petalcorin M.I.R., Boulton S.J. A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J. 2006;25:2178–2188. doi: 10.1038/sj.emboj.7601102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y., Sun Y., Huang S.Y.N., Nitiss J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016;17:703–721. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R., Longley M.J., Sharief F.S., Hou E.W., Copeland W.C., Wilson S.H. Human DNA polymerase possesses 5’-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res. 2009;37:1868–1877. doi: 10.1093/nar/gkp035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quennet V., Beucher A., Barton O., Takeda S., Löbrich M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 2011;39:2144–2152. doi: 10.1093/nar/gkq1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato G., Cannavo E., Cejka P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017;31:2325–2330. doi: 10.1101/gad.308254.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins P., Lindahl T. DNA ligase IV from HeLa cell nuclei. J. Biol. Chem. 1996;271:24257–24261. doi: 10.1074/jbc.271.39.24257. [DOI] [PubMed] [Google Scholar]
- Roques C., Coulombe Y., Delannoy M., Vignard J., Grossi S., Brodeur I., Rodrigue A., Gautier J., Stasiak A.Z., Stasiak A. MRE11–RAD50–NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO J. 2009;28:2400–2413. doi: 10.1038/emboj.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha L.K., Kim S., Kang H., Akter S., Choi K., Sakuma T., Yamamoto T., Sasanuma H., Hirota K., Nakamura J. Differential micronucleus frequency in isogenic human cells deficient in DNA repair pathways is a valuable indicator for evaluating genotoxic agents and their genotoxic mechanisms. Environ. Mol. Mutagen. 2018;59:529–538. doi: 10.1002/em.22201. [DOI] [PubMed] [Google Scholar]
- Sasanuma H., Tsuda M., Morimoto S., Saha L.K., Rahman M.M., Kiyooka Y., Fujiike H., Cherniack A.D., Itou J., Callen Moreu E. BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II-DNA complexes. Proc. Natl. Acad. Sci. U S A. 2018;115:E10642–E10651. doi: 10.1073/pnas.1803177115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Yoshikawa A., Mimura H., Yamashita M., Yamagata A., Fukai S. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by tandem UIMs of RAP80. EMBO J. 2009;28:2461–2468. doi: 10.1038/emboj.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipler A., Iliakis G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013;41:7589–7605. doi: 10.1093/nar/gkt556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A., Symington L.S. Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 2015;40:701–714. doi: 10.1016/j.tibs.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W., Ma Z., Willers H., Akhtar K., Scott S.P., Zhang J., Powell S., Zhang J. Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J. Biol. Chem. 2008;283:31608–31616. doi: 10.1074/jbc.M801082200. [DOI] [PubMed] [Google Scholar]
- Shibata A., Moiani D., Arvai A.S., Perry J., Harding S.M., Genois M.M., Maity R., van Rossum-Fikkert S., Kertokalio A., Romoli F. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M., De Haro L.P., Nickoloff J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- Simsek D., Furda A., Gao Y., Artus J., Brunet E., Hadjantonakis A.-K., Van Houten B., Shuman S., McKinnon P.J., Jasin M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. 2011;471:245–248. doi: 10.1038/nature09794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B., Shao G., Lilli D.R., Culhane A.C., Moreau L.A., Xia B., Livingston D.M., Greenberg R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G.S., Panier S., Townsend K., Al-Hakim A.K., Kolas N.K., Miller E.S., Nakada S., Ylanko J., Olivarius S., Mendez M. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- Symington L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016;51:195–212. doi: 10.3109/10409238.2016.1172552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uckelmann M., Sixma T.K. Histone ubiquitination in the DNA damage response. DNA Repair (Amst.) 2017;56:92–101. doi: 10.1016/j.dnarep.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Wang B., Elledge S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. U S A. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Matsuoka S., Ballif B.A., Zhang D., Smogorzewska A., Gygi S.P., Elledge S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland J.W., Resnick M.A. Coincident resection at both ends of random, γ-induced double-strand breaks requires MRX (MRN), Sae2 (Ctp1), and Mre11-nuclease. PLoS Genet. 2013;9:e1003420. doi: 10.1371/journal.pgen.1003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L., Brunton H., Goodarzi A.A., Shibata A., Jeggo P.A. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011;39:6986–6997. doi: 10.1093/nar/gkr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L., Gennery A.R., Jeggo P.A. Reprint of “The clinical impact of deficiency in DNA non-homologous end-joining”. DNA Repair (Amst.) 2014;17:9–20. doi: 10.1016/j.dnarep.2014.04.002. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Okamoto T., Takeda K., Sato S., Sanjo H., Uematsu S., Saitoh T., Yamamoto N., Sakurai H., Ishii K.J. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 2006;7:962–970. doi: 10.1038/ni1367. [DOI] [PubMed] [Google Scholar]
- Yin Z., Menendez D., Resnick M.A., French J.E., Janardhan K.S., Jetten A.M. RAP80 is critical in maintaining genomic stability and suppressing tumor development. Cancer Res. 2012;72:5080–5090. doi: 10.1158/0008-5472.CAN-12-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Yajima H., Huynh H., Zheng J., Callen E., Chen H.-T., Wong N., Bunting S., Lin Y.-F., Li M. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J. Cell Biol. 2011;193:295–305. doi: 10.1083/jcb.201009074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.Y., Sonoda E., Barber L.J., Oka H., Murakawa Y., Yamada K., Ikura T., Wang X., Kobayashi M., Yamamoto K. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol. Cell. 2007;25:663–675. doi: 10.1016/j.molcel.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Zhu Z., Chung W.-H., Shim E.Y., Lee S.E., Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong D., Adam S., Wang Y., Sasanuma H., Callén E., Murga M., Day A., Kruhlak M.J., Wong N., Munro M. BRCA1 haploinsufficiency is masked by RNF168-mediated chromatin ubiquitylation. Mol. Cell. 2019;73:1267–1281.e7. doi: 10.1016/j.molcel.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.