

Summary
BRAF V600 mutation influences cellular signaling pathways for melanoma development. However, the role of oncogenic BRAF in adaptive stress response pathways is not fully understood. Here, we show that oncogenic BRAF plays an essential role in the induction of ATF4 following the activation of general control non-derepressible 2 (GCN2) kinase during nutrient stress and BRAF-targeted, therapeutic stress. Under GCN2 activation, BRAF ensures ATF4 induction by utilizing mTOR and eIF4B as downstream regulators. In contrast to the MEK-ERK pathway, this signaling pathway remains temporarily active even during treatment with BRAF inhibitors, thereby enabling the transient induction of ATF4. We also identify a chemical compound that prevents BRAF inhibitor-induced activation of the GCN2-ATF4 pathway and produces synergistic cell killing with BRAF inhibitors. Our findings establish a collaborative relationship between oncogenic BRAF and the GCN2-ATF4 signaling pathway, which may provide a novel therapeutic approach to target the adaptive stress response.
Subject Areas: Biological Sciences, Molecular Biology, Cancer
Graphical Abstract
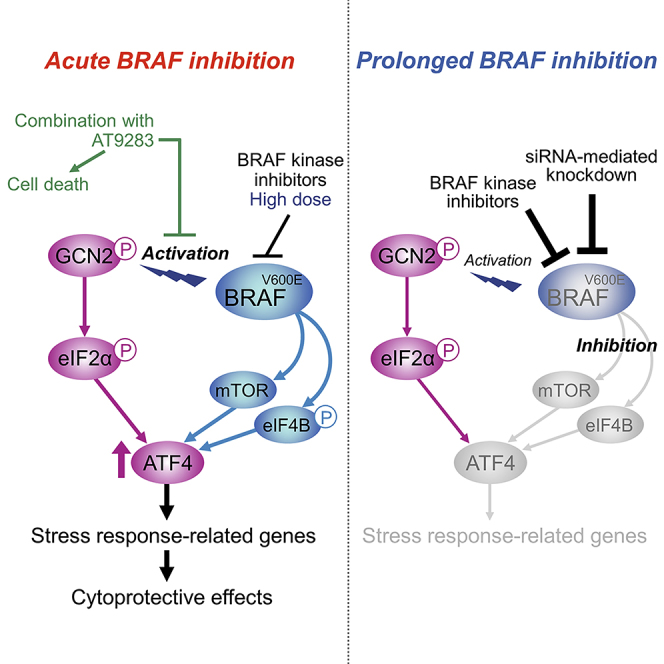
Highlights
-
•
Oncogenic BRAF signals mTOR and eIF4B to ensure ATF4 induction under GCN2 activation
-
•
The signaling pathway decays relatively slowly during BRAF kinase inhibition
-
•
The slow signaling decay enables adaptive response via the GCN2-ATF4 pathway
-
•
The GCN2-ATF4 activation mechanisms by BRAF inhibitors may provide druggable targets
Biological Sciences; Molecular Biology; Cancer
Introduction
Oncogenic mutations of BRAF are observed in approximately 50% of patients with melanoma (Schadendorf et al., 2018). The most common mutation is the substitution of valine at position 600 by glutamic acid (V600E), which results in constitutive activation of its kinase activity, leading to tumor initiation, progression, metastasis, and therapy resistance (Schadendorf et al., 2018). Mechanistically, the mutationally activated BRAF directly induces hyperactivation of the downstream MEK-ERK pathway (Davies et al., 2002) and also acts in cooperation with other genetic abnormalities, such as loss of function of PTEN, a negative regulator of the PI3K-Akt-mTOR pathway (The Cancer Genome Atlas Network, 2015). BRAF has also been reported to promote immune suppression (Bradley et al., 2016, Shabaneh et al., 2018). Thus, BRAF mutation can widely influence cellular signaling pathways and functions in the development of malignant melanoma.
BRAF inhibitors and the combination of BRAF and MEK inhibitors have shown impressive clinical efficacy in patients with BRAF-mutated melanoma (Chapman et al., 2011, Robert et al., 2015), but drug resistance generally remains inevitable. Resistant tumors frequently arise due to reactivation of the MEK-ERK pathway through multiple mechanisms, including amplification of BRAF, splice site mutations in BRAF, de novo mutations in NRAS or MEK, or upregulation of receptor tyrosine kinases (Das Thakur and Stuart, 2014, Van Allen et al., 2014). Drug resistance is also associated with non-mutational drug-tolerant states of cells, seen at the early phase of treatments (Raha et al., 2014, Sharma et al., 2010). Indeed, BRAF inhibitors, immediately after their administration to drug-sensitive cells, lead to reactivation of the MEK-ERK pathway (Hatzivassiliou et al., 2010) and metabolic reprogramming similar to that seen in drug-resistant cells (Parmenter et al., 2014). Thus, along with acquired vulnerability of drug-resistant cells (Hangauer et al., 2017, Wang et al., 2018), understanding the early response of drug-sensitive cells to BRAF inhibitors can provide clues to circumvent resistance and maximize the therapeutic efficacy.
To cope with diverse stresses, including environmental and therapeutic stresses, cancer cells often activate the integrated stress response (ISR) pathway, which is initiated by activation of eukaryotic initiation factor 2α (eIF2α) kinases (Pakos-Zebrucka et al., 2016). There are four different eIF2α kinases: general control non-derepressible 2 (GCN2), protein kinase-like endoplasmic reticulum (ER) kinase (PERK), protein kinase double-stranded RNA-dependent (PKR), and heme-regulated inhibitor (HRI), which sense distinct types of stress, typically, amino acid limitation, ER stress, viral infection, and heme deficiency, respectively (Pakos-Zebrucka et al., 2016). These eIF2α kinases catalyze the phosphorylation of eIF2α on serine 51, which attenuates general protein synthesis while promoting the translation of ATF4 mRNA with upstream open reading frames that enable preferential translation (Vattem and Wek, 2004). ATF4 is a key ISR transcription factor that induces the expression of genes involved in stress adaptation, such as amino acid and redox metabolism (Pakos-Zebrucka et al., 2016).
We previously demonstrated that the BRAF kinase inhibitors vemurafenib (10 μM) and dabrafenib (1 μM), at relatively high but clinically relevant concentrations (Falchook et al., 2014, Flaherty et al., 2010), can rapidly induce ATF4 in melanoma cells with BRAFV600E mutation (Nagasawa et al., 2017). As the silencing of ATF4 expression sensitizes cells to vemurafenib, this rapid induction of ATF4 can contribute to cell survival during treatments with BRAF inhibitors. Mechanistically, this ATF4 induction occurs via the GCN2 arm of the ISR, which is primarily activated in response to amino acid limitation. This observation, together with previous findings that metabolic reprogramming toward glutamine addiction occurred with the acquisition of resistance by chronic exposure to BRAF inhibitors (Baenke et al., 2016, Hernandez-Davies et al., 2015), suggests that modulating the metabolism of amino acids, especially glutamine, through ATF4 induction may be important for early adaptation to BRAF inhibition. However, the role of oncogenic BRAF in the adaptive mechanisms inducing ATF4 is not well understood.
We show herein that oncogenic BRAF exerts activity to drive the expression of ATF4 and can be associated with ATF4 target gene expression in patients with melanoma. In contrast to the MEK-ERK pathway, this signaling pathway that utilizes mTOR and eIF4B as downstream regulators for ATF4 expression remains temporarily active even during exposure to BRAF inhibitors, leading to ATF4 induction in cooperation with the GCN2 arm of the ISR pathway. We also identify a small compound that prevents the activation of the GCN2-ATF4 pathway and synergistically kills melanoma cells during BRAF inhibitor treatments. Our results demonstrate that BRAF-driven ATF4 expression mechanisms can provide a new strategy to circumvent resistance to BRAF-targeted therapy.
Results
BRAF Kinase Inhibitors Induce ATF4 Expression Transiently
Treatments with BRAF kinase inhibitors, 10 μM vemurafenib as well as 1 μM dabrafenib, for 4 h clearly induced GCN2 phosphorylation, eIF2α phosphorylation, and ATF4 expression in BRAF-mutated A375 and G-361 cells (Figures 1A and S1A), as we previously reported (Nagasawa et al., 2017). Activation of the GCN2-ATF4 pathway was also seen upon 4 h of treatment with PLX7904 (Figure 1B), a different type of BRAF kinase inhibitor that has overcome the paradoxical property of vemurafenib and dabrafenib for ERK signaling (Zhang et al., 2015). Thus, ATF4 induction through GCN2 activation appears to be a common feature of BRAF kinase inhibitors. However, the increases in ATF4 expression levels by BRAF kinase inhibitors were transient and they returned to the basal levels within 24 h, despite the phosphorylation of GCN2/eIF2α being kept at high levels (Figures 1A and S1A).
Figure 1.

BRAF Kinase Inhibitors Induce ATF4 Expression Transiently
(A and B) Immunoblot analysis of A375 and G-361 cells treated with vemurafenib (VEM, 10 μM) for the indicated times (A) or vemurafenib (10 μM) or PLX7904 (PLX, 1, 10 μM) for 4 h (B).
(C and D) Immunoblot analysis of A375 cells with BRAF knockdown (C) and treatment with vemurafenib (10 μM) or dabrafenib (DAB, 1 μM) for 4 h (D).
See also Figure S1.
BRAF Depletion or Prolonged BRAF Kinase Inhibition Prevents ATF4 Induction
Unlike kinase inhibitors, siRNA-mediated knockdown of BRAF did not activate the GCN2-ATF4 pathway in A375 cells at any of the time points examined, although ERK phosphorylation declined in accordance with the decrease in BRAF expression (Figure 1C). Instead, BRAF knockdown prevented the induction of ATF4 expression but not GCN2 phosphorylation during additional challenge of 4-h treatments with BRAF kinase inhibitors (Figures 1D and S1B: alternative siRNA). Similarly, BRAF knockdown in A375 and G-361 cells prevented ATF4 induction but not GCN2/eIF2α phosphorylation under conditions of stress due to L-histidinol, a chemical stressor that mimics histidine deprivation through inhibiting histidyl tRNA synthetase (De Sousa-Coelho et al., 2013) (Figures 2A and S2A: alternative siRNA). In the case of glutamine deprivation, both GCN2/eIF2α phosphorylation and ATF4 induction were suppressed by BRAF knockdown (Figure 2B). Such preventive effects of BRAF knockdown on the GCN2-ATF4 pathway, especially ATF4 induction, were not seen in SK-MEL-2 and MeWo cells expressing wild-type BRAF (Figures 2A and 2B). Thus, depletion of oncogenic BRAF, but not wild-type BRAF, prevents ATF4 induction in response to BRAF kinase inhibitors as well as nutrient deprivation stress.
Figure 2.

BRAF Depletion or Prolonged BRAF Kinase Inhibition Prevents ATF4 Induction
(A and B) Immunoblot analysis of the melanoma cell lines with BRAF knockdown and treatment with L-histidinol (HIS, 2 mM) for 4 h (A) or cultured in glutamine-free (GlnF) medium for 18 h (B).
(C) Immunoblot analysis of A375 and G-361 cells treated with vemurafenib (VEM, 10 μM) for 24 h and then treated with L-histidinol (2 mM) for 4 h.
(D) Immunoblot analysis of A375 and G-361 cells cultured in glutamine-free medium for 18 h in the presence or absence of vemurafenib (10 μM).
See also Figure S2.
We also found that ATF4 induction during nutrient stress can be inhibited by prolonged pretreatments of A375 and G-361 cells with BRAF kinase inhibitors. Indeed, pretreatment with vemurafenib or dabrafenib for 24 h prevented ATF4 induction, without ERK reactivation, under stress conditions of L-histidinol addition and glutamine withdrawal, although GCN2/eIF2α phosphorylation remained high (Figures 2C, 2D, and S2B). Thus, BRAF kinase inhibitors can exert biphasic effects on ATF4 expression: induction by short-term (4 h; see Figure 1) and inhibition by long-term (24 h) treatments, a unique property not seen for other stressors such as L-histidinol (Figure S2C).
Oncogenic BRAF Utilizes mTOR Signaling Pathway for ATF4 Induction
By treating A375 and G-361 cells with vemurafenib for 1 to 24 h, we found that the decay of ATF4 induction coincided with the attenuation of mTORC1 activity (Figure 3A). Unlike ERK dephosphorylation, the mTORC1 attenuation occurred relatively slowly. Actually, the phosphorylation levels of p70 ribosomal protein S6 kinase (p70 S6K) as well as its substrate ribosomal protein S6 (S6) (and, although less clearly, those of eIF4E binding protein 1 [4E-BP1]) steadily dropped from 8 h. Similar attenuation of mTORC1 activity was also provoked by BRAF knockdown in melanoma cells harboring oncogenic BRAF but not wild-type BRAF, as monitored by determining the phosphorylation status of S6 and 4E-BP1 (Figure 3B). Interestingly, compared with wild-type cells, BRAF-mutated cells showed resistance to L-histidinol-induced mTORC1 suppression, and this resistance was also diminished by BRAF knockdown (Figure 3B). Thus, the mTOR signaling pathway can be regulated by oncogenic BRAF under both normal and stress conditions.
Figure 3.

Oncogenic BRAF Utilizes mTOR Signaling Pathway for ATF4 Induction
(A) Immunoblot analysis of A375 and G-361 cells treated with vemurafenib (VEM, 10 μM) for the indicated times.
(B) Immunoblot analysis of the melanoma cell lines with BRAF knockdown and treatment with L-histidinol (HIS, 2 mM) for 4 h. The internal control bands are shown in Figure 2A.
(C) Immunoblot analysis of A375 cells treated with L-histidinol (2 mM) alone or in combination with pp242 (1 μM), rapamycin (0.1 μM), or everolimus (0.1 μM) for 4 h.
(D and E) Immunoblot analysis of A375 cells treated with vemurafenib (10 μM) or dabrafenib (DAB, 1 μM) alone or in combination with pp242 (1 μM) (D) or rapamycin (0.1 μM) (E) for 4 h.
See also Figure S3.
To assess the involvement of mTOR in regulating ATF4 expression, we used different types of mTOR inhibitor. The selective and ATP-competitive mTOR inhibitor pp242 potently suppresses both mTORC1 and mTORC2, whereas the rapalogs rapamycin and everolimus moderately inhibit mTORC1 (Benjamin et al., 2011). Consistent with the mTOR-inhibitory potential, pp242 broadly prevented ATF4 induction under short-term treatments with vemurafenib or dabrafenib in BRAF-mutated cells (Figures 3D and S3C), as well as under conditions of stress due to L-histidinol in both wild-type and mutant cells (Figures 3C and S3A). Compared with those of pp242, the preventive effects of rapalogs were relatively weak and uneven depending on the stress types (Figures 3C–3E, S3B, and S3D). Conversely, mTORC1 activity and ATF4 expression were partially rescued by knockdown of Tuberous Sclerosis Complex 2 (TSC2), a negative regulator of mTORC1 (Inoki et al., 2002), under L-histidinol (but not vemurafenib [4 h]) stress in BRAF-silenced cells as well as under long-term vemurafenib treatment (16 h) in BRAF-unsilenced cells (Figures S3E and S3F). Although the precise reason remains unknown, the lack of rescue of ATF4 expression by TSC knockdown in BRAF-silenced, vemurafenib (4 h)-treated cells might be related to insufficient reactivation of mTORC1 owing to profound inhibition of the BRAF-mTOR axis. Notably, the ATF4 repression by mTOR inhibitors accompanied a tendency (especially in the case of pp242) to dampen GCN2/eIF2α phosphorylation, suggesting cross talk between the mTOR and GCN2 pathways. Taken together, these results indicate that oncogenic BRAF utilizes mTOR as a downstream regulator of ATF4 expression.
Oncogenic BRAF Enhances eIF4B Phosphorylation for ATF4 Induction
During the above time course experiments with vemurafenib (see Figure 3A), delayed attenuation similar to that in the mTOR signaling pathway was observed in phosphorylation (Ser422) for the activation of eIF4B (Figure 4A), an accessory factor stimulating eIF4A RNA helicase (Rozovsky et al., 2008). BRAF knockdown also lowered the phosphorylation levels of eIF4B in BRAF-mutated cells specifically (Figures 4B and S4A: alternative siRNA). Conversely, overexpression of mutant BRAF, but not the wild-type, enhanced eIF4B phosphorylation levels (Figure 4C). However, eIF4B phosphorylation in A375 cells was not affected by mTOR inhibition or the inhibition of other BRAF downstream kinases MEK and Akt with each of the selective inhibitors (Figure S4B). Thus, eIF4B phosphorylation can be under the control of oncogenic BRAF but not wild-type BRAF, possibly in a manner independent of MEK, Akt, and mTOR.
Figure 4.

Oncogenic BRAF Enhances eIF4B Phosphorylation for ATF4 Induction
(A) Immunoblot analysis of A375 and G-361 cells treated with vemurafenib (VEM, 10 μM) for the indicated times.
(B) Immunoblot analysis of the melanoma cell lines with BRAF knockdown.
(C) Immunoblot analysis of HEK293T and A375 cells with overexpression of wild-type BRAF or mutant BRAF.
(D and E) Immunoblot analysis of A375 and G-361 cells with EIF4B knockdown and treatment with vemurafenib (10 μM) or dabrafenib (DAB, 1 μM) (D) or with L-histidinol (HIS, 2 mM) (E) for 4 h.
(F and G) Immunoblot analysis of A375 and G-361 cells with EIF4A1 knockdown and treated with vemurafenib (10 μM) (F) or with L-histidinol (2 mM), tunicamycin (TM, 1 μg/mL), or thapsigargin (TG, 0.3 μM) (G) for 4 h.
See also Figure S4.
Interestingly, eIF4B knockdown in A375 and G-361 cells attenuated ATF4 induction under conditions of short-term treatments with BRAF kinase inhibitors (Figures 4D and S4C: alternative siRNA) but not under nutrient stress conditions of L-histidinol addition and glutamine withdrawal (Figures 4E and S4D). This selectivity in ATF4 attenuation was more consistent with the decrease in phosphorylation levels of eIF4B rather than the levels of eIF4B or the GCN2/eIF2α phosphorylation state, raising the possibility that the loss of residual eIF4A-stimulating activity by dephosphorylation may be needed to attenuate ATF4 induction. Actually, hampering eIF4A with specific siRNAs or the selective inhibitor silvestrol impaired ATF4 induction very broadly, in both wild-type and mutant BRAF cells, under all conditions examined, including not only GCN2-activating (BRAF inhibitors and L-histidinol) but also PERK-activating conditions (tunicamycin and thapsigargin) (Figures 4F, 4G, and S4E–S4H). Thus, eIF4A likely functions as basic machinery for ATF4 translation under stress conditions. Taking these findings together, eIF4B, especially active, phosphorylated forms, appears to be involved in ATF4 induction, possibly through regulating the eIF4A-dependent translation in BRAF-mutated melanoma cells. Notably, this eIF4B-ATF4 axis may also contribute to cell survival upon BRAF inhibition because eIF4B knockdown can slightly but significantly enhance cellular sensitivity to vemurafenib (Figure S4I), as previously shown with ATF4 knockdown (Nagasawa et al., 2017).
Expression of ATF4 Target Genes Is Influenced by BRAF Inhibition
Gene expression analysis of A375 cells treated with vemurafenib for 6 or 16 h, or silenced with siRNA specific for BRAF, revealed that 3,214 probe sets, representing genes that are involved in various cellular processes, were altered more than 2-fold by either perturbation (Figure 5A and Tables S1 and S2). These gene expression changes were very similar and, indeed, the directions of expression changes were highly concordant between the BRAF-inhibiting perturbations (61.8%–82.4% concordance rates; Figure S5A). Notably, somewhat distinct changes in expression of particular gene sets were seen between vemurafenib treatment and BRAF knockdown (e.g., clusters 2–4 in Figure 5A), possibly, at least in some cases, reflecting the different modes of action of each perturbation (kinase inhibition versus protein depletion). As part of the effectiveness evaluation, BRAF knockdown was also found to suppress the transcriptional response in A375 cells deprived of glutamine (Figure S5B and Table S3), in line with the above findings that oncogenic BRAF regulates the ISR signaling pathway during nutrient stress.
Figure 5.

Expression of ATF4 Target Genes Is Influenced by BRAF Inhibition
(A) Signature of 3,214 probe sets (2,311 genes) that were either upregulated or downregulated by treatment with vemurafenib (VEM, 10 μM, 6 or 16 h) or BRAF knockdown in A375 cells. The log2 fold change of levels under the indicated conditions is shown for the condition of treatment with DMSO (for vemurafenib treatment) or transfection with nontargeting siRNA (siCont) (for BRAF knockdown). For probe lists and GO analysis results, see Tables S1 and S2.
(B) Signature of 155 probe sets (116 genes) that were downregulated by ATF4 knockdown during treatment with vemurafenib (10 μM) for 4 h in A375 cells. The log2 fold change of levels under the indicated conditions is shown for the condition of transfection with nontargeting siRNA and then treatment with DMSO. For probe lists, see Table S5.
(C) Enrichment plot of GSEA using the 107 genes defined from the BRAFi-ATF4 signature. We tested the enrichment of these genes in transcripts upregulated by BRAF V600 mutation in human melanomas. NES, normalized enrichment score. For gene lists, see Table S7. See also Figure S5.
Comparison of changes in gene expression upon vemurafenib treatment (4 h) with or without ATF4 knockdown suggested that ATF4 exerted only limited effects on the transcriptional response to vemurafenib in A375 cells (Figures S5C and S5D, and Table S4). We then extracted 155 probe sets (116 genes) as a BRAFi-ATF4 signature that was downregulated by ATF4 knockdown under vemurafenib treatment (Figure 5B and Table S5). Consistent with the time course changes in ATF4 levels (see Figure 3A), the BRAFi-ATF4 signature, especially 72 probe sets (56 genes) that overlapped with those altered by BRAF-inhibiting perturbations as above (Figure 5A), had a tendency to show lower expression levels at 16 h than at 6 h during vemurafenib treatment (Figure S5E and Table S6). Remarkably, the majority of 72 probe sets (56 genes) were downregulated by BRAF knockdown (Figure S5E). Furthermore, gene set enrichment analysis, using a public melanoma gene expression dataset, revealed that the BRAFi-ATF4 signature was highly enriched in melanoma harboring BRAF V600 mutations (p < 10−4) (Figure 5C and Table S7). Thus, oncogenic BRAF-mediated regulation of ATF4 target genes can be clinically relevant in melanoma.
Identification of a Chemical Compound that Suppresses GCN2-ATF4 Pathway Activation during BRAF Kinase Inhibition
By screening of a kinase inhibitor library with fluorescent immunostaining of ATF4, we identified a multi-kinase inhibitor, AT9283 (Figure 6A), that selectively prevented nuclear ATF4 accumulation induced by BRAF kinase inhibitors (Figure 6B). AT9283 dose-dependently inhibited GCN2/eIF2α phosphorylation induced by vemurafenib (Figure 6C) in A375 and G-361 cells and by dabrafenib (Figure S6A) in A375 cells, but did not inhibit GCN2/eIF2α phosphorylation induced by nutrient stresses (Figures 6D and S6B) and also PERK/eIF2α phosphorylation induced by tunicamycin or thapsigargin (Figure 6D). The prevention of ATF4 induction occurred with only marginal effects on the phosphorylation levels of eIF4B and S6 (Figures 6C and S6C) and was not recaptured by other kinase inhibitors targeting known AT9283-inhibitable kinases, such as Abl, JAK2/3, and Aurora (Howard et al., 2009) (Figure S6D). Gene expression analysis of A375 cells further supported GCN2-ATF4 pathway inhibition. Indeed, AT9283 counteracted vemurafenib (6 h)-induced activation of ATF4-target genes (p < 10−4 by Enrichr online tool with the Reactome pathway database for cluster 1 and 2 in Figure 6E) (Chen et al., 2013, Gjymishka et al., 2009, Kuleshov et al., 2016), especially ASNS, PSAT1, and PSPH that are well-known ATF4 targets involved in amino acid metabolism (Tables S8 and S9) (Adams, 2007). Consistently, AT9283 led to the downregulation of gene expression broadly in the BRAFi-ATF4 signature (86% and 62% of probe sets up- and downregulated by vemurafenib, respectively) (Figure S6E). Thus, AT9283 effectively prevents activation of the GCN2-ATF4 pathway in response to BRAF inhibitors.
Figure 6.

Identification of a Chemical Compound that Suppresses GCN2-ATF4 Pathway Activation during BRAF Kinase Inhibition
(A) Chemical structure of AT9283.
(B) A375 cells were treated with vemurafenib (VEM, 10 μM), L-histidinol (HIS, 2 mM), or tunicamycin (TM, 1 μg/mL) in the presence or absence of AT9283 (0.1, 0.2, 0.5, or 1 μM) for 6 h. The cells were fixed and stained with anti-ATF4 antibody (green). Bars represent the quantification of the intensity of ATF4 signals in the nucleus. Results are shown as the mean ± SD (n = 6).
(C and D) Immunoblot analysis of A375 and G-361 cells treated with AT9283 alone or in combination with vemurafenib (10 μM), L-histidinol (2 mM), tunicamycin (1 μg/mL), or thapsigargin (TG, 0.3 μM) for 4 h.
(E) Signature of 432 probe sets (309 genes) whose expression levels were upregulated or downregulated by combined treatment with vemurafenib (10 μM) and AT9283 (0.1 μM) for 6 h compared with the level upon treatment with vemurafenib alone for 6 h in A375 cells. The log2 fold change of levels under the indicated conditions is shown for the condition of treatment with DMSO. For probe lists and GO analysis results, see Tables S8 and S9.
See also Figure S6.
Combined treatment with vemurafenib and AT9283 for 16 h led to profound growth inhibition in A375 and G-361 cells when the cell growth ability was assessed by reseeding and culturing cells in fresh medium for 96 h or until colony formation (Figures 7A and 7B). During the drug treatments, cell attachment was largely unaffected, although a decrease in cell number was seen (Figure S7C). In fact, in the BRAF-mutated cell lines, AT9283 also enhanced apoptosis induction in combination with vemurafenib or dabrafenib for 16 h, as determined by increased caspase 3/7 activity and PARP cleavage (Figures 7C, 7D, S7A, and S7B). The effects of AT9283, however, were not phenocopied by other kinase inhibitors targeting Abl, JAK2/3, Aurora, or their combinations (Figure S7D). In addition, such enhancement of apoptosis induction by AT9283 was not seen in BRAF wild-type cells co-treated with vemurafenib (Figure S7E) or in BRAF mutant cells co-treated with L-histidinol (Figure S7F). These results indicate that AT9283 can induce vulnerability to BRAF kinase inhibitors in BRAF-mutated melanoma cells (Figure 7E).
Figure 7.

Enhanced Growth Inhibition by Combined Treatments with BRAF Inhibitors and AT9283
(A and B) A375 and G-361 cells were treated with AT9283 alone or in combination with vemurafenib (VEM) for 16 h. The cells were reseeded in 96-well plates (A) or 6-well plates (B) and cultured in drug-free medium for 96 h (A), or 7 (A375) or 14 (G-361) days (B). Results are shown as the mean ± SD (n = 3).
(C) A375 and G-361 cells were treated with AT9283 alone or in combination with vemurafenib for 16 h. The caspase-3/7 activities were measured by the Caspase-Glo 3/7 assay. Results are shown as the mean ± SD (n = 3).
(D) Immunoblot analysis of A375 and G-361 cells treated with AT9283 alone or in combination with vemurafenib for 16 h.
(E) Mechanisms of AT9283 involved in prevention of the GCN2-ATF4 pathway activation by BRAF kinase inhibitors.
See also Figure S7.
Discussion
Using melanoma cell lines with BRAFV600E mutation, we have shown that oncogenic BRAF signaling controls the expression of ATF4, the major ISR transcription factor. Indeed, siRNA-mediated knockdown of BRAF prevents ATF4 induction during nutrient stress. ATF4 induction is also inhibited by relatively long-term treatments (16–24 h) with the BRAF kinase inhibitors vemurafenib and dabrafenib. Thus, oncogenic BRAF positively regulates ATF4 expression. We further identified mTOR and eIF4B as downstream regulators in BRAF-mediated ATF4 regulation. Curiously, ATF4 was rapidly induced by short-term treatments (4–8 h) with BRAF kinase inhibitors, depending on the stress kinase GCN2 (Nagasawa et al., 2017), before onset of the downregulation of mTOR and eIF4B. This paradoxical response was effectively inhibited by a small compound, resulting in sensitization to BRAF kinase inhibitors. Taken together, our findings indicate that oncogenic BRAF-mediated regulation of ATF4 expression plays a central role in stress response and cell survival.
In addition to BRAF, several oncogenes or oncogenic signals have been shown to regulate ATF4 expression (Gwinn et al., 2018, Heydt et al., 2018, Zhao et al., 2016). Although ATF4 can be cytoprotective and proapoptotic depending on the cell conditions (Pakos-Zebrucka et al., 2016), loss of ATF4 has been repeatedly shown to suppress tumor growth in xenograft models (Dey et al., 2015, Ye et al., 2010). In line with this, increased expression of ATF4 has been observed in certain tumors in clinical settings (Chen et al., 2017, Dey et al., 2015). Using a TCGA dataset, we also found that the BRAFi-ATF4 signature, an ATF4-regulated gene set defined under short-term BRAF kinase inhibition, is enriched in BRAF-mutated tumors derived from patients with skin cutaneous melanoma (Figure 5C). Thus, it is conceivable that the BRAF-ATF4 axis contributes to tumor formation in malignant melanoma, possibly through modulating the transcriptional response to nutrient status in tumors. Indeed, oncogenic BRAF modulates various types of cellular metabolism (Haq et al., 2013, Kang et al., 2015), and we demonstrated that knockdown of BRAF not only prevents ATF4 induction but also abrogates transcriptional alterations in response to glutamine deprivation (Figure S5B). Similar oncogenic regulation of glutamine response has been seen in KRAS mutant lung cancer, and in those cells, KRAS regulates ATF4 expression to support amino acid homeostasis (Gwinn et al., 2018). These observations collectively suggest that oncogene-mediated regulation of ATF4 can be a common mechanism to manage metabolic homeostasis, particularly amino acid metabolism, in tumors.
It is conceivable that mTOR and eIF4B, as downstream regulators of oncogenic BRAF, cooperate to regulate ATF4 expression. Actually, both proteins, in common, modulate the eIF4F translation initiation complex, consisting of the eIF4E cap-binding protein, the eIF4G scaffolding protein, and the eIF4A RNA helicase (Silvera et al., 2010). Inhibition of mTOR kinase can lead to disruption of the eIF4F complex by activating 4E-BP1 that blocks eIF4E-eIF4G interaction (Richter and Sonenberg, 2005), whereas inactivation of eIF4B can lead to loss of function to stimulate eIF4A activity (Rozovsky et al., 2008). In agreement with this, 4E-BP1 can negatively regulate ATF4 expression under the control of mTORC1 (Park et al., 2017), and, as we showed herein, inactivation of eIF4A can prevent ATF4 induction. Thus, the eIF4F complex is likely one of the focal points where oncogenic BRAF effectively regulates ATF4 expression. In this regard, persistent formation of active eIF4F complex has been identified as a mechanism for both innate and acquired resistance to BRAF-targeted therapy (Boussemart et al., 2014). Together with this, our present findings raise the possibility that control of ATF4 translation initiation through the eIF4F complex plays an important role in resistance to BRAF-targeted therapy.
Contrary to relatively long-term treatments (16–24 h), short-term treatments (4–8 h) with BRAF kinase inhibitors paradoxically lead to GCN2 activation and subsequent induction of ATF4. Importantly, ATF4 knockdown enhances cellular sensitivity to the anti-proliferative effect of vemurafenib (Nagasawa et al., 2017), suggesting that the GCN2-mediated ATF4 induction functions as part of the cellular stress response to BRAF inhibition. Consistent with this notion, we herein found that the BRAFi-ATF4 signature contained many genes expressed in a BRAF-dependent manner (Figure S5E). In this context, it would be conceivable that abrupt inhibition of BRAF kinase activity, per se, becomes a stressor to trigger the GCN2-ATF4 stress response pathway. Actually, in the presence of BRAF kinase inhibitors, paradoxical induction of ATF4 occurs after the rapid inhibition of ERK phosphorylation and continues until the onset of mTOR and eIF4B inhibition, which can lead to prevention of the translation initiation of ATF4 as above. Thus, differential regulation of the ERK and mTOR/eIF4B signaling pathways during BRAF kinase inhibition may provide a time window that enables cells to activate the GCN2-mediated stress response and induce ATF4 transiently. However, it is likely that activation of the GCN2-ATF4 pathway is uncoupled from the MEK-ERK pathway inhibition, as the MEK inhibitor trametinib does not activate the GCN2-ATF4 pathway in BRAF-mutated cells (Nagasawa et al., 2017).
Finally, we found that rapid activation of the GCN2-ATF4 pathway by BRAF kinase inhibitors can be effectively prevented by AT9283, a broad-spectrum kinase inhibitor that inhibits kinases such as JAK2, JAK3, Aurora A, Aurora B, and c-ABL (Howard et al., 2009). Our efforts to drive clinical applications of this finding, including to identify the target kinase(s) of AT9283 for this unexpected property and to determine the appropriate conditions for its administration to tumor-bearing mice, have been unsuccessful. Nonetheless, our results already show that AT9283 possesses unique features as a lead compound against BRAF-mutated cells. The prevention of GCN2-ATF4 pathway activation by this compound has been seen selectively in BRAF-mutated cells treated with BRAF kinase inhibitors, but not with amino acid stress, revealing that different signaling mechanisms can operate between each type of stressor. In agreement with this, combinations of AT9283 with BRAF kinase inhibitors, but not with amino acid stress, lead to the synergistic induction of apoptosis in BRAF-mutated cells. Meanwhile, the potent efficacy of AT9383, as compared with that of ATF4 knockdown (Nagasawa et al., 2017), suggests that the synergistic apoptosis induction may be attributed to ATF4-dependent and -independent mechanisms.
In fact, although somewhat less effective, AT9283 can also enhance apoptosis induction in combination with the MEK inhibitor trametinib (Figure S7G). This observation, together with the fact that trametinib does not activate the GCN2-ATF4 pathway (Nagasawa et al., 2017), suggests that mechanisms other than preventing ATF4 induction are also involved in the enhanced cell death by combined treatments with BRAF inhibitors and AT9283. Although further studies are needed to understand the whole mechanisms of AT9283-induced apoptosis during BRAF inhibition, these features shed light on the existence of druggable mechanisms to improve melanoma therapy using BRAF inhibitor together with or without MEK inhibitor. A deeper understanding of the mechanisms for GCN2-ATF4 pathway activation upon the acute inhibition of BRAF kinase activity, as well as for oncogenic BRAF-mediated ATF4 regulation, would provide a novel therapeutic strategy to combat melanoma harboring mutated BRAF.
Limitations of the Study
We demonstrated that oncogenic BRAF promotes cellular stress adaptation by collaborating with the ISR pathway and that disruption of this collaborative, adaptive mechanism results in sensitization of melanoma cells to BRAF inhibitors. However, the detailed mechanisms of BRAF-driven stress adaptation as well as BRAF inhibitor-induced ISR activation remain to be fully elucidated. The chemical compound, which we identified to inhibit activation of the ISR and to sensitize melanoma cells to BRAF inhibitors, may have pleiotropic, as-yet-unidentified mechanisms of action. Although we showed that the expression of ISR target genes is enriched in BRAF-mutated melanomas, future studies will be needed to determine the clinical relevance of our findings and their potential implications in BRAF-targeted therapy.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported in part by JSPS KAKENHI Grant Numbers 16J10435, 16H04717, and 19H03526, and by the Nippon Foundation. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors thank Dr. Dustin Maly for the gift of the pFLAG-B-Raf plasmid (Addgene plasmid #40775; http://n2t.net/addgene:40775; RRID: Addgene_40775). The authors also thank Edanz (www.edanzediting.co.jp) for editing the English text of a draft of this manuscript.
Author Contributions
I.N. and A.T. conceived the study and analyzed the data. I.N. and Y.T. performed immunoblot analysis. S.T. and K.K. performed microarray experiments. M.K. performed gene expression profiling. I.N. performed other experiments. I.N. and A.T. wrote the manuscript.
Declaration of Interests
The authors declare no conflicts of interest.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101028.
Data and Code Availability
The accession number for the microarray data reported in this paper is National Center for Biotechnology Information Gene Expression Omnibus: GSE136615.
Supplemental Information
References
- Adams C.M. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J. Biol. Chem. 2007;282:16744–16753. doi: 10.1074/jbc.M610510200. [DOI] [PubMed] [Google Scholar]
- Baenke F., Chaneton B., Smith M., Van Den Broek N., Hogan K., Tang H., Viros A., Martin M., Galbraith L., Girotti M.R. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol. Oncol. 2016;10:73–84. doi: 10.1016/j.molonc.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D., Colombi M., Moroni C., Hall M.N. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011;10:868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- Boussemart L., Malka-Mahieu H., Girault I., Allard D., Hemmingsson O., Tomasic G., Thomas M., Basmadjian C., Ribeiro N., Thuaud F. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–109. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- Bradley S.D., Melendez B., Talukder A., Lizee G. Trouble at the core: BRAF(V600E) drives multiple modes of T-cell suppression in melanoma. Oncoimmunology. 2016;5:e1078966. doi: 10.1080/2162402X.2015.1078966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P.B., Hauschild A., Robert C., Haanen J.B., Ascierto P., Larkin J., Dummer R., Garbe C., Testori A., Maio M. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Fan Z., Rauh M., Buchfelder M., Eyupoglu I.Y., Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36:5593–5608. doi: 10.1038/onc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M., Stuart D.D. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin. Cancer Res. 2014;20:1074–1080. doi: 10.1158/1078-0432.CCR-13-0103. [DOI] [PubMed] [Google Scholar]
- Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M.J., Bottomley W. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- De Sousa-Coelho A.L., Relat J., Hondares E., Perez-Marti A., Ribas F., Villarroya F., Marrero P.F., Haro D. FGF21 mediates the lipid metabolism response to amino acid starvation. J. Lipid Res. 2013;54:1786–1797. doi: 10.1194/jlr.M033415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S., Sayers C.M., Verginadis, Lehman S.L., Cheng Y., Cerniglia G.J., Tuttle S.W., Feldman M.D., Zhang P.J., Fuchs S.Y. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Invest. 2015;125:2592–2608. doi: 10.1172/JCI78031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook G.S., Long G.V., Kurzrock R., Kim K.B., Arkenau H.T., Brown M.P., Hamid O., Infante J.R., Millward M., Pavlick A. Dose selection, pharmacokinetics, and pharmacodynamics of BRAF inhibitor dabrafenib (GSK2118436) Clin. Cancer Res. 2014;20:4449–4458. doi: 10.1158/1078-0432.CCR-14-0887. [DOI] [PubMed] [Google Scholar]
- Flaherty K.T., Puzanov I., Kim K.B., Ribas A., McArthur G.A., Sosman J.A., O’Dwyer P.J., Lee R.J., Grippo J.F., Nolop K. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjymishka A., Su N., Kilberg M.S. Transcriptional induction of the human asparagine synthetase gene during the unfolded protein response does not require the ATF6 and IRE1/XBP1 arms of the pathway. Biochem. J. 2009;417:695–703. doi: 10.1042/BJ20081706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn D.M., Lee A.G., Briones-Martin-Del-Campo M., Conn C.S., Simpson D.R., Scott A.I., Le A., Cowan T.M., Ruggero D., Sweet-Cordero E.A. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell. 2018;33:91–107.e106. doi: 10.1016/j.ccell.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangauer M.J., Viswanathan V.S., Ryan M.J., Bole D., Eaton J.K., Matov A., Galeas J., Dhruv H.D., Berens M.E., Schreiber S.L. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–250. doi: 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R., Shoag J., Andreu-Perez P., Yokoyama S., Edelman H., Rowe G.C., Frederick D.T., Hurley A.D., Nellore A., Kung A.L. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G., Song K., Yen I., Brandhuber B.J., Anderson D.J., Alvarado R., Ludlam M.J., Stokoe D., Gloor S.L., Vigers G. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Hernandez-Davies J.E., Tran T.Q., Reid M.A., Rosales K.R., Lowman X.H., Pan M., Moriceau G., Yang Y., Wu J., Lo R.S. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J. Transl. Med. 2015;13:210. doi: 10.1186/s12967-015-0581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydt Q., Larrue C., Saland E., Bertoli S., Sarry J.E., Besson A., Manenti S., Joffre C., Mansat-De Mas V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene. 2018;37:787–797. doi: 10.1038/onc.2017.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard S., Berdini V., Boulstridge J.A., Carr M.G., Cross D.M., Curry J., Devine L.A., Early T.R., Fazal L., Gill A.L. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J. Med. Chem. 2009;52:379–388. doi: 10.1021/jm800984v. [DOI] [PubMed] [Google Scholar]
- Inoki K., Li Y., Zhu T., Wu J., Guan K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Kang H.B., Fan J., Lin R., Elf S., Ji Q., Zhao L., Jin L., Seo J.H., Shan C., Arbiser J.L. Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF-MEK1 signaling. Mol. Cell. 2015;59:345–358. doi: 10.1016/j.molcel.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa I., Kunimasa K., Tsukahara S., Tomida A. BRAF-mutated cells activate GCN2-mediated integrated stress response as a cytoprotective mechanism in response to vemurafenib. Biochem. Biophys. Res. Commun. 2017;482:1491–1497. doi: 10.1016/j.bbrc.2016.12.062. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A.M. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y., Reyna-Neyra A., Philippe L., Thoreen C.C. mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep. 2017;19:1083–1090. doi: 10.1016/j.celrep.2017.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmenter T.J., Kleinschmidt M., Kinross K.M., Bond S.T., Li J., Kaadige M.R., Rao A., Sheppard K.E., Hugo W., Pupo G.M. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014;4:423–433. doi: 10.1158/2159-8290.CD-13-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D., Wilson T.R., Peng J., Peterson D., Yue P., Evangelista M., Wilson C., Merchant M., Settleman J. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014;74:3579–3590. doi: 10.1158/0008-5472.CAN-13-3456. [DOI] [PubMed] [Google Scholar]
- Richter J.D., Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- Robert C., Karaszewska B., Schachter J., Rutkowski P., Mackiewicz A., Stroiakovski D., Lichinitser M., Dummer R., Grange F., Mortier L. Improved overall survival in melanoma with combined dabrafenib and trametinib. New Engl. J. Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Rozovsky N., Butterworth A.C., Moore M.J. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA. 2008;14:2136–2148. doi: 10.1261/rna.1049608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D., van Akkooi A.C.J., Berking C., Griewank K.G., Gutzmer R., Hauschild A., Stang A., Roesch A., Ugurel S. Melanoma. Lancet. 2018;392:971–984. doi: 10.1016/S0140-6736(18)31559-9. [DOI] [PubMed] [Google Scholar]
- Shabaneh T.B., Molodtsov A.K., Steinberg S.M., Zhang P., Torres G.M., Mohamed G.A., Boni A., Curiel T.J., Angeles C.V., Turk M.J. Oncogenic BRAF(V600E) governs regulatory T-cell recruitment during melanoma tumorigenesis. Cancer Res. 2018;78:5038–5049. doi: 10.1158/0008-5472.CAN-18-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.V., Lee D.Y., Li B., Quinlan M.P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M.A. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvera D., Formenti S.C., Schneider R.J. Translational control in cancer. Nat. Rev. Cancer. 2010;10:254–266. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen E.M., Wagle N., Sucker A., Treacy D.J., Johannessen C.M., Goetz E.M., Place C.S., Taylor-Weiner A., Whittaker S., Kryukov G.V. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem K.M., Wek R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U S A. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Leite de Oliveira R., Huijberts S., Bosdriesz E., Pencheva N., Brunen D., Bosma A., Song J.Y., Zevenhoven J., Los-de Vries G.T. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell. 2018;173:1413–1425.e4. doi: 10.1016/j.cell.2018.04.012. [DOI] [PubMed] [Google Scholar]
- Ye J., Kumanova M., Hart L.S., Sloane K., Zhang H., De Panis D.N., Bobrovnikova-Marjon E., Diehl J.A., Ron D., Koumenis C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010;29:2082–2096. doi: 10.1038/emboj.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Spevak W., Zhang Y., Burton E.A., Ma Y., Habets G., Zhang J., Lin J., Ewing T., Matusow B. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- Zhao E., Ding J., Xia Y., Liu M., Ye B., Choi J.H., Yan C., Dong Z., Huang S., Zha Y. KDM4C and ATF4 cooperate in transcriptional control of amino acid metabolism. Cell Rep. 2016;14:506–519. doi: 10.1016/j.celrep.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the microarray data reported in this paper is National Center for Biotechnology Information Gene Expression Omnibus: GSE136615.