

Abstract
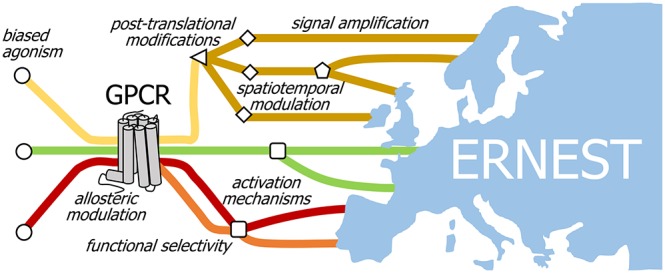
G protein-coupled receptors (GPCRs) are intensively studied due to their therapeutic potential as drug targets. Members of this large family of transmembrane receptor proteins mediate signal transduction in diverse cell types and play key roles in human physiology and health. In 2013 the research consortium GLISTEN (COST Action CM1207) was founded with the goal of harnessing the substantial growth in knowledge of GPCR structure and dynamics to push forward the development of molecular modulators of GPCR function. The success of GLISTEN, coupled with new findings and paradigm shifts in the field, led in 2019 to the creation of a related consortium called ERNEST (COST Action CA18133). ERNEST broadens focus to entire signaling cascades, based on emerging ideas of how complexity and specificity in signal transduction are not determined by receptor–ligand interactions alone. A holistic approach that unites the diverse data and perspectives of the research community into a single multidimensional map holds great promise for improved drug design and therapeutic targeting.
Keywords: G protein-coupled receptor, GPCR, signal transduction, biased agonism, functional selectivity, structure based drug design
The importance of research on G protein-coupled receptor (GPCR) signal transduction is known and appreciated by most in the biomedical field. Over one-third of all prescribed drugs target a GPCR,1 which is not surprising considering the substantial number of GPCRs expressed in the human body and their prevalence in mediating signal transduction in nearly every cell. Since the first crystal structure of a GPCR2 was published in the year 2000, there has been intense activity elucidating how these receptors work on the molecular level. The field has benefited immensely from a blossoming of structural data on different GPCRs in various states of activation, bound to different types of ligands and/or to intracellular binding partners.3−5 Currently, the “resolution revolution”6 in cryo-electron microscopy is rapidly producing unprecedented insights into the structures of GPCRs in native-like and functionally relevant states and complexes.7,8
Simultaneous to these advancements, strategies for structure-based GPCR drug design are moving in an exciting new direction, based on the discovery that different ligands can bind to the same receptor yet stimulate the association of different effector proteins to different extents. These concepts of biased agonism and the broadly more general functional selectivity (discussed more below) open significant possibilities for creating more effective drugs that have desired therapeutic effects while avoiding deleterious side-effects. Indeed, examples of such biased GPCR ligands for improved treatment of various diseases have already been identified and tested in clinical trials.9 The potential of signaling pathway-specific drugs to improve human health worldwide cannot be understated. Despite this fact, as a field we still know very little about the mechanisms governing ligand bias and functional selectivity. To explain these phenomena, multiple approaches that encompass a wider perspective including information on where and when signaling events take place—while taking into account the unique environment inside different cell types—is required. Moreover, recent studies cast doubt on key established dogmas underlying ligand bias and functional selectivity. As a field, we stand at a critical turning point and must re-evaluate our assumptions and seek innovative and interdisciplinary approaches to understand the mechanisms governing GPCR signal transduction. Our efforts at designing better drugs and therapeutics will be severely hampered until we do so.
In this perspective piece, we introduce the recently launched COST (1) Action CA18133 “European Research Network on Signal Transduction” (ERNEST), the primary goal of which is to address these challenges faced by the research community. We describe the origins of ERNEST and how these roots led to a more holistic approach to understand GPCR signal transduction. We outline the specific objectives of ERNEST and how these will be met by the large and diverse community of researchers that compose the Network. We discuss the current state of knowledge in the field, the major unresolved questions, and our reasoning why our planned approach holds the best promise for untangling these issues. Finally, we provide an outlook of the anticipated long-lasting impacts of ERNEST on science and society.
GLISTEN and the Dawn of ERNEST
Launched in 2013, the COST Action CM1207 GLISTEN (GPCR-Ligand Interactions, Structures, and Transmembrane Signaling: a European Research Network) was dedicated to deepening the understanding of GPCRs, especially with regard to the receptor activation mechanism, ligand binding, and effects of the membrane and other interaction partners on GPCR signaling. A central motivation of GLISTEN was to utilize this information to identify and design chemical modulators of GPCR signaling. In line with this focus, many pharmacologists, structure-based drug designers, and computational and medicinal chemists, joined the network early on. As the network grew, membership became more diverse as those with other perspectives in structural and cell biology, (patho)physiology, and translational medicine became aware of GLISTEN. Recruitment and diversification were made possible by decentralization of semiannual meeting organization. Local organizers were free to designate the thematic focus of their respective meetings according to their own research interests and perspectives. Hence, a wide variety of topics in the GPCR field was covered in the course of eight GLISTEN meetings. The trajectory went from a focus on computational methods at the inaugural meeting in Warsaw (2013),10 followed by meetings with emphasis on biophysics, structural biology, and drug discovery in Barcelona (2014),11 Budapest (2014),12 and Allschwil (2015),13 to meetings focused on medicinal chemistry and pharmacology in Amsterdam (2015),14 Erlangen (2016),15 and Prague (2016)16 to more (patho)physiological aspects at the last meeting in Porto, Portugal (2017).17 GLISTEN meetings were put together by enthusiastic local organizers representing different academic institutes and companies in Europe (see Acknowledgments). Notably, several industry partners were closely involved in scientific program development and hosting companies to stimulate and integrate academic and biotech/pharmaceutical drug discovery cooperation in GLISTEN. GPCR research training was further advanced by a series of workshops and training schools, including GPCR Computational Chemistry Workshops in Warsaw (2013) (Computer-Aided Drug Design, Molecular Dynamics, Protein Homology Modeling, Structural Chemogenomics and Chemoinformatics), GPCRdb Structural Bioinformatics tutorials at every GLISTEN meeting from 2013 to 2017, a training school on GPCR Fragment-Based Drug Discovery (FBDD), and Molecular Pharmacology in Budapest (2014), a GLISTEN-Lorentz Workshop in Leiden (2014) on in vitro to in vivo GPCR biology, a workshop on GPCR Research Valorization in Amsterdam (2015), and contributions of GLISTEN researchers at the Biophysics Training School in Croatia in 2016.18
The eventual composition of the network (more than 200 research groups from 31 countries) fulfilled the main purpose of GLISTEN to connect researchers and thereby enable cross-disciplinary cooperation. In addition, information exchange was promoted by cross-border exchanges of investigators between laboratories and training schools that connected experts and novices. The strategies employed by GLISTEN in its activities were highly successful at fostering collaborative research, in particular between experimental and theoretical groups, and ultimately provided valuable examples for similar consortia. Many leading experts of the GPCR field in Europe and worldwide, as well as the key pharmaceutical companies, were regular participants at GLISTEN events. One significant indicator of the importance of GLISTEN to the research community was the popularity of the semiannual meetings. Meetings were often oversubscribed within hours of the opening of online registration, most notably for the meeting in Erlangen that featured Nobel laureate Brian Kobilka as keynote speaker.
GLISTEN formally came to an end in 2017, yet its many successes are still being realized. First, more than 65 joint publications stemming from collaborations established within the network have been released. Notable GLISTEN publications are cited here.19−24 In addition, a multitude of joint grants for research funding was acquired by network members, many of which will continue to produce significant scientific findings in the coming years (e.g., Oncornet 2.025 and PSYBIAS26,27). Technology and know-how were spread by GLISTEN-sponsored exchanges of investigators between research groups in different countries and training schools, and the impact of this dissemination will continue to benefit diverse groups and individuals for a long time to come. Relatedly, GLISTEN positively influenced the careers of many early career investigators, which will serve to strengthen the future scientific output of the field. Last but certainly not least, GLISTEN pushed the development and use of GPCR-focused web-based databases and tools for the benefit of the international research community. GPCRdb, which was originally headed by Gert Vriend at the EMBL in Heidelberg, Germany,28 relocated to the group of David Gloriam at the University of Copenhagen, Denmark in 2013. Several investigator-exchanges and the contributions of GLISTEN members helped this database to grow to contain GPCR reference data, visualization/analysis suites, and tools to design new experiments.1,21,29−31−33 GLISTEN also spurred the creation of GPCRmd, an online repository and visualization platform for molecular dynamics (MD) simulation data and their analysis. This database arose from the need to organize and standardize this information including experimental setups/protocols and to provide intuitive analysis tools. By these means, GPCRmd promotes transparency, consistency, and reproducibility in the field of GPCR dynamics and also facilitates data exchange between GPCR scientists from different disciplines.24
Well before the official end of COST funding, the inclusiveness and influence of GLISTEN drove an intense community-wide desire to evolve a new COST Action. At the same time, developments in the field made clear that in order to meet current challenges, a wider perspective must be taken (explained in detail below). Hence, ERNEST came into existence, continuing the excellent tradition of GLISTEN and leading the field in an exciting new direction.
Unresolved Questions in Signal Transduction
To survive and reproduce, all living cells must be able to sense their environments and respond in an appropriate way. Nature accomplishes this task by signal transduction, the process of passing external stimuli into and through the cell in order to induce cellular responses. Every signaling pathway essentially consists of a series of macromolecular interactions, and the signal is carried through the chain of interactions by biochemical events (e.g., changes in protein structure, association or dissociation of small molecules, or chemical modification of proteins). Most signaling pathways consist of a transmembrane receptor protein that binds an extracellular ligand (e.g., small molecule, peptide, or ion). This event triggers the binding of intracellular effector proteins that then produce second messengers, usually small molecules or protein modifications, which activate the next level of effector proteins. Each molecular interaction along a pathway can be considered a node at which the signal can be modulated and amplified, and different signaling cascades can consist of any number of nodes.
The spectrum of stimuli encountered by cells is both sizable and diverse, as is the number and type of cellular responses elicited by these stimuli. Yet, remarkably, signal transduction systems use a relatively limited repertoire of intracellular signaling components. This small number of effector proteins nevertheless enables a cellular signaling apparatus that is flexible and versatile. Versatility is achieved by modulation at the molecular, spatial, and temporal levels of the macromolecular interactions at each node in the pathway. In effect, a limited number of nodes, each with several alternative downstream pathways, can give rise to a vast number of distinct signaling pathways. Although versatile and complex, the biological role of signal transduction demands specificity and precision in signaling. This is, in part, achieved through a large number of receptors (about 1000 GPCRs34), each of which binds only one or a few endogenous ligands.
On the atomic level, the receptor is a dynamic structure that exists in different conformations or states, and ligand binding enhances the presence of some receptor states over others. States derived from experimental structural determination techniques are usually described as either active or inactive, depending on whether they can couple to effectors or not. Moreover, ligands can selectively stabilize receptor states that preferentially interact with certain primary effectors over others, a concept called biased agonism. Put another way, different biased ligands can stimulate distinct signaling pathways with different efficacies, a concept also referred to as functional selectivity. Biased agonists differentially influence the conformational dynamics of GPCRs,35−38 yet it is not fully understood how these signals control interactions with effector proteins and their downstream cellular functions.
The influence of molecular, temporal, and spatial factors is widely appreciated in the field and actively discussed.39−43 However, the underlying mechanisms by which these factors and the cellular environment modulate signal transduction remain unexplained. A few recent studies have approached this gap in knowledge by using experimental or computational systems.44,45 However, in order to progress as a field toward molecular modulators that have predictable and reproducible effects in living cells (and eventually patients), the field requires a detailed map of signal transduction that takes into account all known factors that influence pathway selectivity. To build such a holistic multidimensional map, diverse types of data must be integrated in a way to allow mechanistic insight and formulation of general principles that govern signal transduction modulation in different cell types. This ambitious endeavor, which is out of reach for individual research groups and smaller research consortia, now forms a key objective of ERNEST. The network is in a unique position to harness the diverse data and expertise of hundreds of researchers in different disciplines.
There is an urgent need for a holistic mapping of signal transduction. Signal transduction plays a ubiquitous and critical role in normal physiology, and aberrations in pathways controlled by GPCRs lead to disease. Currently many research groups are unravelling signaling pathways that contribute to different disease states, and there is great excitement for the promise of biased ligands to treat these diseases more effectively and with fewer side effects. For example, ligands that promote sustained G protein signaling from β-adrenergic and angiotensin receptors lead to deleterious effects on the heart, while those that stimulate arrestin activity bring many cardioprotective effects.46 In the case of the dopamine D2 receptor, drug candidates for the treatment of schizophrenia are being developed that selectively antagonize arrestin activity (leading to antipsychotic effects) while still promoting G protein signaling and thus avoiding motoric side effects.47−49 For the opioid receptors, many academic research groups and companies have sought biased agonists, based on the belief that analgesic effects are supported by G protein-signaling, while unpleasant side-effects (e.g., respiratory depression and constipation) arise from arrestin-mediated effects.50,51 A few such G protein-biased agonists have shown promise as potential drug candidates.50,52 However, recent publications cast doubt on whether some of these drug candidates are actually biased,53,54 and moreover, on whether arrestin activity at opioid receptors is responsible for side-effects.55,56 The developing controversies in biased agonism at opioid receptors illustrate our significant gaps in knowledge in GPCR signal transduction,57,53 often exacerbated by inconsistent or incorrectly applied quantitative pharmacology,58,59 that undermine efforts to translate candidate drugs into the clinic.60
Objectives of ERNEST
The main purpose of ERNEST is to establish and support a diverse network of signal transduction investigators through regular meetings, training schools, cross-border research group exchanges, and other dissemination activities. ERNEST promotes communication, knowledge exchange, and cooperation between scientists from different training levels, disciplines, institutions, and countries to address unresolved questions in signal transduction. Specific points to be addressed are described below.
Clarification of Biased Agonism and Functional Selectivity
The ability of certain ligands to elicit distinct cellular responses is broadly referred to as functional selectivity, the first determinate of which is the receptor–ligand interaction. Biased agonism, where certain GPCR conformations are preferentially stabilized that recruit certain effector proteins, can certainly influence the functional selectivity of a ligand. However, many other factors have significant influence over the signaling outcome, including allosteric modulation, membrane environment, crosstalk, intracellular location, and cell type, among others.58 Biased agonism at GPCRs, and its specific influence on functional selectivity, is widely discussed in the field.59,61−63 However, there is much confusion about the precise definition of biased agonism and how it relates to functional selectivity. The field requires community-wide accepted methods to assess and report ligand bias in GPCR signaling, and to distinguish signaling bias (i.e., functional selectivity) from bias in terms of preferential receptor coupling to intracellular proteins. For example, most studies report bias by comparing the relative abilities of different ligands to stimulate activation of different G proteins and/or arrestin recruitment. Since these events are measured experimentally in different ways, often using indirect readouts at different time points and in different cell types, clear conclusions and comparisons to other studies are neither possible nor sound. Another fundamental issue is the choice of reference ligands for each GPCR, and whether endogenous ligands should preferentially be used as reference, since they themselves might be biased. Moreover, the widely accepted paradigm of arrestin as a GPCR signal transducer in its own right is being re-evaluated in recent publications,64−66 which complicates the interpretation of “arrestin-biased” ligands. ERNEST, in coordination with other authoritative organisations and groups, is addressing these issues by gathering leading experts to constructively dissect the root issues from their diverse perspectives and then publish collective strategies for resolving discrepancies by suggesting best practices. In addition, ERNEST will push the development of better methods, technologies, and database resources for evaluating biased agonism and, eventually, functional selectivity.
Multidimensional Signaling Map
As described above, future development of signaling pathway-specific molecular modulators is hindered by our incomplete understanding of the complexities of GPCR-mediated signaling. ERNEST is addressing this challenge by coordinating and supporting development of a holistic multidimensional map of signal transduction that will be comparable to previous and ongoing efforts to map interactomes and signaling pathways with respect to scale and level of detail,67−75 yet will be distinctive in its focus on elucidating the mechanisms of biased agonism in GPCR signal transduction. ERNEST has clear advantages for accomplishing this ambitious task, namely a substantial membership of diverse researchers and the ability to bring the key players together to work in interdisciplinary cooperation. Development of the signaling map is at present ongoing and will proceed in three phases (Figure 1). First, the data and expertise of all Network members will be curated, catalogued, and (when possible) standardized with the help of experts in data integration and database development. A “blueprint” of all signaling pathways studied within the Network will be generated, thereby allowing the identification of the pathways containing a sufficient amount of validated data (as well as highlighting gaps in information that need to be addressed) for the development of a comprehensive map. ERNEST efforts will be guided by similar web-based resources for mapping biomolecule interactions and signaling pathways.76,77 Next, a focused “Mapping Group” within ERNEST will facilitate communication between data contributors and the systems biologists who will eventually create the signaling map. Communication between these participants will be bidirectional. Systems biologists will receive and evaluate substantial data from Network members and then give feedback regarding the usefulness of the data and shortcomings that need to be resolved. Again, the size and interdisciplinarity of ERNEST affords the participating system biologists a rare opportunity to optimize the necessary parameters (e.g., binding affinities, rates and kinetics of interactions, cellular localization, cell type) for their computational modeling approaches. Since signaling responses differ between different organisms and also within different cell types (primary and cultured), all gathered data will be labeled with great detail according to their origin and environment. With enough data at hand, signaling map(s) could be visualized for different cell types and growth conditions. Comparison of signaling pathways in different cell types may provide further information on how signaling is modulated by cell-specific characteristics, as well as indicate gaps in understanding. Armed with this large and well-curated data set, the systems biologists will develop signaling map(s). Multiple approaches will be encouraged and supported by ERNEST by facilitating joint grant applications. The envisioned signaling maps will serve three main purposes: (1) Provide an overview of available GPCR knowledge and missing links; (2) Visualize and understand how molecular, spatiotemporal, and cell environment factors control signal transduction; (3) Allow prediction of which cellular responses are elicited by particular GPCR ligands (depending on cell type). The predictive capability of the signaling maps—of utmost importance for the improved development of pathway-specific drugs—will be verified and further optimized by collaborations between map developers, drug designers, and experimentalists within ERNEST.
Figure 1.
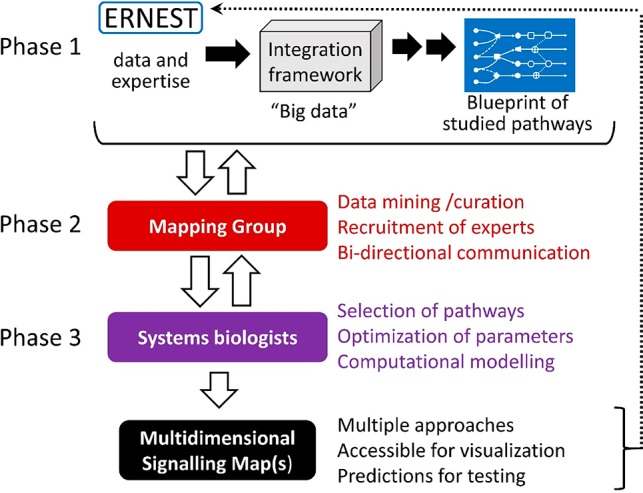
Schematic overview of how ERNEST will develop a holistic multidimensional map of GPCR-mediated signal transduction. See text for full description.
Pathway-Specific Chemical Modulators of Signal Transduction
Biased ligands might provide improved efficacy and/or side effect and safety profiles.78 A holistic signaling map will obviously greatly advance the development strategies of such ligands for therapeutically significant GPCR targets. We anticipate that participating computational drug designers will benefit from this map already in the early stages of development, and ERNEST invites such researchers to join the Network in order to stimulate progress. Besides drug discovery efforts, the field requires chemical probes to study signal transduction, that is, specific inhibitors to determine which pathways receptors signal through, or how different signaling networks are interconnected. Such tools will benefit the entire GPCR field, and their application will further assist development and refinement of the signaling map.
An advantage of the large ERNEST network will be to extend the characterization of developed molecular modulators into animal models, both to study pharmacology and test the treatment of disease states, and eventually to translate these findings to patients in clinical trials. Such work will be carried out by linking the drug developers to physiologists, clinicians and those in industry.
Advanced Methods, Technologies, and Database Resources
Over the last 10 years, research on signal transduction has been propelled forward by new methods and technologies spanning many disciplines. These include advancements in structural biology (e.g., nanobodies for stabilization of proteins and protein complexes, free-electron lasers for poorly diffracting crystals, atomic resolution improvements in cryo-electron microscopy), computational biology (longer molecular dynamics simulations of complex systems, advanced sampling methods, novel methodologies), cell biology (visualization of cellular structures and proteins, including super-resolution microscopy, biosensors, single-molecule fluorescence), chemical biology (ad-hoc chemical probes, for example, new radioligands and radiotracers, covalent ligands, affinity-based probes, fluorescent probes, allosteric and bitopic ligands) and systems biology (“omics” and bioinformatics methods, such as deep-mutational scanning, data mining, coevolution, and machine learning approaches). In addition, the field has benefited immensely from public web-based resources, such as GPCRdb and GPCRmd. In order to advance all aspects of signal transduction research, to promote international cooperation and to facilitate its scientific objectives, ERNEST will further develop and disseminate new methods and technologies. These goals will be accomplished by (1) cataloguing known expertise within the Network in an open platform for participants to find collaborators; (2) identifying new methods and technologies that are needed by the community through constructive discussions at Network meetings; (3) pushing the development of new methods and technologies by recruiting the right experts and supporting them in grant applications; (4) establishing “best practice” principles for new and existing approaches by recruiting experts and encouraging publication on this topic. ERNEST will foster further development of GPCRdb and GPCRmd by enabling communication between database users and developers regarding which features and tools are needed by the research community.
Cooperation between Academia and Industry
The potential of ERNEST to improve the design of signaling pathway-specific drugs naturally attracts many researchers from both academia and industry. On the basis of experience from GLISTEN, the close inclusion and involvement of participants from pharmaceutical and biotech companies will be valuable for ERNEST and its goals. Pathway-specific chemical modulators would be of interest due to the challenging target validation and target engagement efforts in early phase drug discovery programs. Advanced methods and technologies developed in ERNEST might attract significant attention in pharmaceutical and biotech settings. Cooperation between industry and academic researchers will be fostered at dedicated sessions on the topic at ERNEST meetings. Besides promoting collaboration between researchers in academia and industry, ERNEST will support cross-sectoral training of early career investigators by sponsoring exchange programs between academic groups and research-oriented companies.
Working Groups of ERNEST
Network participants belong to one or more Working Groups (WG), whose task is to coordinate members and their expertise in order to achieve the objectives of the Network. The focus areas of ERNEST are designed to be inclusive of all researchers in the signal transduction field, from the molecular to the cellular to the physiological perspectives, as well as those focused on pushing forward new methods, technologies, and resources for the advancement of research (Figure 2).
Figure 2.
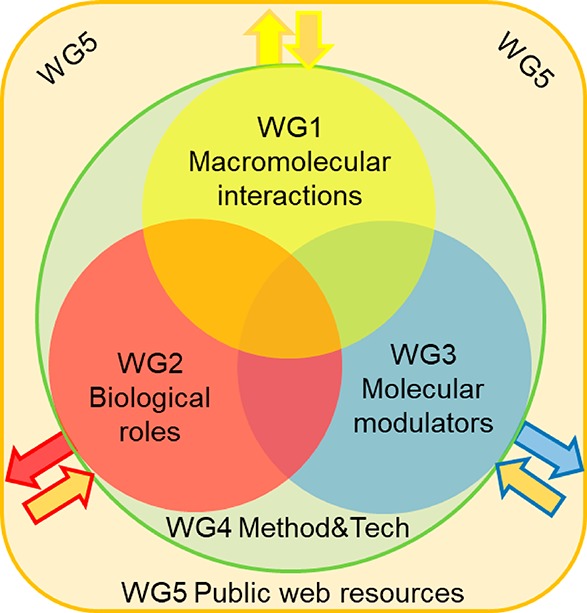
Thematic synergism between the Working Groups (WG) of ERNEST. WGs 1, 2, and 3 form the core of scientific knowledge of the Action, and the overlaps represent shared focus and potential for interdisciplinary cooperation. WG4 will support the three core WGs with new methods and technologies and also establish best practice standards for their application. Output from the three core workgroups (arrows out) will be incorporated by WG5 into database resources for public dissemination, and WG5 will generate database tools that will feedback into the three core WGs (arrows in). Figure reprinted in part from COST Action CA18133 ERNEST Memorandum of Understanding with permission from the COST Association.80
WG1: Macromolecular Interactions in Signaling Pathways
Signal transduction is mediated and dependent on macromolecular interactions. The protein players in signal transduction cascades are molecular machines, the conformations and movements of which are modulated by interactions with small molecules and other biological macromolecules, resulting in different signaling states. The main objective of WG1 is elucidation of structural dynamics and molecular interactions at the atomic level that give rise to signal transduction, with emphasis on how the modulation of interactions generates specificity in transmembrane receptor-mediated signal transduction.
WG2: Biological Roles of Signal Transduction
The biological importance of macromolecular interactions can only be understood within the context of a living cell. The main objective of WG2 is to connect the molecular interactions and their subcellular localization to the cellular response and (patho)physiological states. Signaling pathways must be defined and characterized for different cell types and systems, involving experts in different physiological systems (e.g., neurobiology, cardiovascular system, cancer, and immunity) within the Network. This activity has the benefit that new biological model systems can be developed and new therapeutic targets can be identified. Signaling pathways must also be defined in order to understand how disease results from imbalances in signal transduction.
WG3: Molecular Modulators of Signal Transduction
WG3 will harness chemical space for modulation of protein interactions and signaling. The core objective is the design and optimization of molecules that interact with components of the signal transduction cascade. The design of functionally selective therapeutics will be possible through information about relevant receptor conformations or residues provided by WG1 and WG2. WG3 will also work closely with WG4 (advanced technologies and methods) and industry partners to develop chemical probes to investigate cell signaling mechanisms and to stabilize GPCR complexes with binding partners for high-resolution structural analysis and hit/lead compounds for drug discovery programs.
WG4: Advanced Methodologies and Technologies
The main objective of WG4 is to promote advanced methodologies and technologies within ERNEST, and to coordinate sharing through collaboration. Included are novel approaches that can help achieve the aims of the Network, as well as repurposing of existing methods and technologies from other fields of biomedical research. This WG will also establish best practice principles for the application of commonly used methodologies across groups within and beyond the Network. These advancements in experimental approach and technology will support the other WGs and overall objectives of ERNEST.
WG5: Public Web Resources
The key objective of WG5 is to structure, integrate, and make accessible different types of data emanating from ERNEST and its members and the global GPCR community. Online databases have greatly benefited the signal transduction research community in the past decade. WG5 will contribute key public online resources in this area for reference data and analysis tools. Experts from all WGs will design and use this resource and thus increase the dissemination of their scientific results.
Future Outlook and Anticipated Impacts to the Field and beyond
ERNEST was created by a diverse group of researchers to directly address and collectively resolve the major challenges facing the field of GPCR-mediated signal transduction. The support of the COST Association in the endeavor will prove highly worthwhile, as several positive impacts of this Action are anticipated. The large membership of the Network, including most world-leading GPCR experts in Europe, united in close cooperation with a holistic concept of how to best understand signal transduction and harness this knowledge, is likely to achieve many successes. In the short term, ERNEST will stimulate collaborations between academic groups of quite disparate fields, which will be brought together because of the unprecedented holistic approach to understanding signal transduction. In particular, collaborations between academia and industry will be pushed by the new targeting possibilities offered by the signaling map. In the long term, ERNEST will advance this holistic view of signal transduction and establish new paradigms for modulation of signaling pathways. The detailed multidimensional map and database tools that will be developed during the lifetime of the Network will be valuable resources for the research community for many years to come. In addition, new chemical modulators developed through ERNEST activities will be invaluable for future research and as starting points for drug development. These therapeutic agents will have a significant and positive socio-economic impact by improving human health worldwide.
ERNEST will have further long-term impact through the cross-disciplinary training of early career investigators, as these individuals will drive signal transduction research into the future. ERNEST-sponsored cross-border exchanges will open up career perspectives and opportunities for many researchers. These exchanges will have further lasting benefits by spreading technologies and knowhow throughout Europe and beyond. Finally, ERNEST will promote diversity in the leadership ranks of European research by promoting target groups, especially women and scientists from less research-intensive countries. In summary, the future looks promising for signal transduction research, and time will tell the importance of being ERNEST.79
Acknowledgments
The authors are grateful for the continued support of the European Cooperation in Science and Technology (COST) through Actions CM1207 GLISTEN and CA18133 ERNEST. On behalf of ERNEST, M.E.S. thanks the Max Delbrück Center for Molecular Medicine Berlin for support in managing the Action. M.E.S. is supported by the Deutsche Forschungsgemeinschaft (DFG) (SO1037/1-3) and the Berlin Institute of Health (Delbrück Fellowship BIH_PRO_314). J.S. acknowledges support from the Instituto de Salud Carlos III FEDER (PI18/00094) and the ERA-NET NEURON & Ministry of Economy, Industry and Competitiveness (AC18/00030). J.C. receives funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 715052). D.E.G. is supported by the Lundbeck Foundation (R313-2019-526) and Novo Nordisk Foundation (NNF17OC0031226). G.M.K. is funded by the National Brain Research Program (2017-1.2.1-NKP-2017-00002). M.K. acknowledges support from the Israel Science Foundation (Grants 1454/13 and 3512/19) and the DS Research Center at the University of Haifa. S.M. is supported by the Alfred Benzon Foundation (ABF-0-0-312) and Polish National Science Center (HARMONIA 2015/18/M/NZ2/00423). M.M.R. acknowledges support from the European Research Council: VIREX Grant agreement 682549, Call ERC-2105-CoG, the Independent Research Fund Denmark, the NovoNordisk Foundation (NNF17OC0029222:) and the Lundbeck Foundation (R268-2017-409). E.S. thanks the Xunta de Galicia (Centro singular de Investigación de Galicia acreditación 2019-2022, ED431G 2019/03 and GI-1597 2017-2019 ED431B2017/70) and the European Union (European Regional Development Fund - ERDF) for financial support. J.K.S.T. acknowledges support from the DFG (HI1502/1-2) and the Novo Nordisk Foundation (Challenge Grant PRISM). N.V. is funded by grants from the Slovenian Research Agency (P3-310, J3-7605, BI-DE/18-19-015). P.K. is supported by the DFG (KO4095/4-1 and Heisenberg professorship KO4095/5-1). All coauthors thank the stellar organizers of the eight GLISTEN meetings for their vital contributions and their associated institutes and companies for support, including the University of Warsaw (Poland), Pompeu Fabra University and Autonomous University of Barcelona (Spain), Research Centre for Natural Sciences of the Hungarian Academy of Sciences (Budapest, Hungary), Actelion Pharmaceuticals (Allschwil, Switerland), Vrije Universiteit (Amsterdam, The Netherlands), Friedrich Alexander University Erlangen and Philipps-University Marburg (Germany), University of Chemistry and Technology Prague (Czech Republic), the University of Porto (Portugal), and Sosei Heptares (Cambridge, UK). Parts of this paper are derived from the Memorandum of Understanding for the implementation of the COST Action “European Research Network on Signal Transduction” (ERNEST) CA18133.80
Author Contributions
The manuscript was written by M.E.S. with contributions from all authors. Except for the first, second and last, authors are listed alphabetically with respect to last name. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Footnotes
The European Cooperation in Science and Technology (COST), founded in 1971, funds research networks (COST Actions) created by researchers themselves to address specific and significant challenges in their respective fields. COST supports networking activities, e.g. conferences, training schools, international exchange of investigators between research groups, dissemination, and communication activities. COST Actions are highly interdisciplinary, open to new participants throughout their lifetime of 4 years, and active in promoting early career investigators and investigators from less research-intensive countries. Hence, COST plays a key role in stimulating international cooperation that pushes scientific progress and benefits a diverse community of researchers. For more information, see: https://www.cost.eu.
References
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schioth H. B.; Gloriam D. E. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 16 (12), 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K.; Kumasaka T.; Hori T.; Behnke C. A.; Motoshima H.; Fox B. A.; Le Trong I.; Teller D. C.; Okada T.; Stenkamp R. E.; Yamamoto M.; Miyano M. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289 (5480), 739–45. 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Wacker D.; Stevens R. C.; Roth B. L. (2017) How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170 (3), 414–427. 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. E.; Melcher K.; Xu H. E. (2017) Understanding the GPCR biased signalling through G protein and arrestin complex structures. Curr. Opin. Struct. Biol. 45, 150–159. 10.1016/j.sbi.2017.05.004. [DOI] [PubMed] [Google Scholar]
- Hilger D.; Masureel M.; Kobilka B. K. (2018) Structure and dynamics of GPCR signalling complexes. Nat. Struct. Mol. Biol. 25 (1), 4–12. 10.1038/s41594-017-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. (2015) The revolution will not be crystallized: a new method sweeps through structural biology. Nature 525 (7568), 172–4. 10.1038/525172a. [DOI] [PubMed] [Google Scholar]
- Safdari H. A.; Pandey S.; Shukla A. K.; Dutta S. (2018) Illuminating GPCR Signalling by Cryo-EM. Trends Cell Biol. 28 (8), 591–594. 10.1016/j.tcb.2018.06.002. [DOI] [PubMed] [Google Scholar]
- Garcia-Nafria J.; Tate C. G. (2020) Cryo-Electron Microscopy: Moving Beyond X-Ray Crystal Structures for Drug Receptors and Drug Development. Annu. Rev. Pharmacol. Toxicol. 60, 51–71. 10.1146/annurev-pharmtox-010919-023545. [DOI] [PubMed] [Google Scholar]
- Schrage R.; Kostenis E. (2017) Functional selectivity and dualsteric/bitopic GPCR targeting. Curr. Opin. Pharmacol. 32, 85–90. 10.1016/j.coph.2016.12.001. [DOI] [PubMed] [Google Scholar]
- www.biomodellab.eu/1glisten (accessed 2020/03/02).
- http://eventia.upf.edu/Barcelona-GPCR-Conference-2014 (accessed 2020/03/02).
- http://glisten.ttk.hu/ (accessed 2020/03/02).
- https://sites.google.com/site/glisten2015/home (accessed 2020/03/02).
- http://www.medchemsymposium.org/Home.html (accessed 2020/03/02).
- http://grk1910.de/glisten-2016.html (accessed 2020/03/02).
- www.glisten2016.cz (accessed 2020/03/02).
- https://glistensymposium.wordpress.com (accessed 2020/03/02).
- http://school.ifs.hr/2016 (accessed 2020/03/02).
- Rodriguez D.; Brea J.; Loza M. I.; Carlsson J. (2014) Structure-based discovery of selective serotonin 5-HT(1B) receptor ligands. Structure 22 (8), 1140–1151. 10.1016/j.str.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Piscitelli C. L.; Kean J.; de Graaf C.; Deupi X. (2015) A Molecular Pharmacologist’s Guide to G Protein-Coupled Receptor Crystallography. Mol. Pharmacol. 88 (3), 536–51. 10.1124/mol.115.099663. [DOI] [PubMed] [Google Scholar]
- Munk C.; Isberg V.; Mordalski S.; Harpsoe K.; Rataj K.; Hauser A. S.; Kolb P.; Bojarski A. J.; Vriend G.; Gloriam D. E. (2016) GPCRdb: the G protein-coupled receptor database - an introduction. Br. J. Pharmacol. 173 (14), 2195–207. 10.1111/bph.13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally C. C.; Bauer B.; Selent J.; Sommer M. E. (2017) C-edge loops of arrestin function as a membrane anchor. Nat. Commun. 8, 14258. 10.1038/ncomms14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillard F.; Stotani S.; Karawajczyk A.; Hristeva S.; Pardon E.; Steyaert J.; Tzalis D.; Kolb P. (2019) Interrogating dense ligand chemical space with a forward-synthetic library. Proc. Natl. Acad. Sci. U. S. A. 116 (23), 11496–11501. 10.1073/pnas.1818718116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Espigares I.; Torrens-Fontanals M.; Tiemann J. K. S.; Aranda-García D.; Ramírez-Anguita J. M.; Stepniewski T. M.; Worp N.; Varela-Rial A.; Morales-Pastor A.; Lacruz B. M.; Pándy-Szekeres G.; Mayol E.; Giorgino T.; Carlsson J.; Deupi X.; Filipek S.; Filizola M.; Gómez-Tamayo J. C.; Gonzalez A.; Gutierrez-de-Teran H.; Jimenez M.; Jespers W.; Kapla J.; Khelashvili G.; Kolb P.; Latek D.; Marti-Solano M.; Matricon P.; Matsoukas M.-T.; Miszta P.; Olivella M.; Perez-Benito L.; Provasi D.; Ríos S.; Rodríguez-Torrecillas I.; Sallander J.; Sztyler A.; Vaidehi N.; Vasile S.; Weinstein H.; Zachariae U.; Hildebrand P. W.; Fabritiis G. D.; Sanz F.; Gloriam D. E.; Cordomi A.; Guixà-González R.; Selent J. (2019) GPCRmd uncovers the dynamics of the 3D-GPCRome. bioRxiv 839597. [Google Scholar]
- http://www.oncornet.eu/ (accessed 2020/03/02).
- https://www.era-learn.eu/network-information/networks/neuron-cofund/call-for-proposals-for-transnational-research-projects-on-mental-disorders/a-novel-paradigm-for-effective-and-safer-treatment-of-schizophrenia-biased-ant-agonists-with-a-characterized-polypharmacological-profile (accessed 2020/03/02).
- https://www.neuron-eranet.eu/_media/PSYBIAS_summary.pdf (accessed 2020/03/02).
- Horn F.; Weare J.; Beukers M. W.; Horsch S.; Bairoch A.; Chen W.; Edvardsen O.; Campagne F.; Vriend G. (1998) GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 26 (1), 275–9. 10.1093/nar/26.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg V.; Mordalski S.; Munk C.; Rataj K.; Harpsoe K.; Hauser A. S.; Vroling B.; Bojarski A. J.; Vriend G.; Gloriam D. E. (2016) GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res. 44 (D1), D356–64. 10.1093/nar/gkv1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flock T.; Hauser A. S.; Lund N.; Gloriam D. E.; Balaji S.; Babu M. M. (2017) Selectivity determinants of GPCR-G-protein binding. Nature 545 (7654), 317–322. 10.1038/nature22070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. S.; Chavali S.; Masuho I.; Jahn L. J.; Martemyanov K. A.; Gloriam D. E.; Babu M. M. (2018) Pharmacogenomics of GPCR Drug Targets. Cell 172 (1–2), 41–54 e19. 10.1016/j.cell.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandy-Szekeres G.; Munk C.; Tsonkov T. M.; Mordalski S.; Harpsoe K.; Hauser A. S.; Bojarski A. J.; Gloriam D. E. (2018) GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 46 (D1), D440–D446. 10.1093/nar/gkx1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk C.; Mutt E.; Isberg V.; Nikolajsen L. F.; Bibbe J. M.; Flock T.; Hanson M. A.; Stevens R. C.; Deupi X.; Gloriam D. E. (2019) An online resource for GPCR structure determination and analysis. Nat. Methods 16 (2), 151–162. 10.1038/s41592-018-0302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S.; Kadowaki S.; Haga T.; Takaesu H.; Mitaku S. (2002) Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. 520 (1–3), 97–101. 10.1016/S0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- Rahmeh R.; Damian M.; Cottet M.; Orcel H.; Mendre C.; Durroux T.; Sharma K. S.; Durand G.; Pucci B.; Trinquet E.; Zwier J. M.; Deupi X.; Bron P.; Baneres J. L.; Mouillac B.; Granier S. (2012) Structural insights into biased G protein-coupled receptor signalling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 109 (17), 6733–8. 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X. P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F. Y.; Liu W.; Xu H. E.; Cherezov V.; Roth B. L.; Stevens R. C. (2013) Structural features for functional selectivity at serotonin receptors. Science 340 (6132), 615–9. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler L. M.; Elgeti M.; Hilger D.; Latorraca N. R.; Lerch M. T.; Staus D. P.; Dror R. O.; Kobilka B. K.; Hubbell W. L.; Lefkowitz R. J. (2019) Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 176 (3), 468–478 e11. 10.1016/j.cell.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane R.; Liu J. J.; White K. L.; Katritch V.; Stevens R. C.; Wuthrich K.; Millar D. P. (2020) Biased Signalling of the G-Protein-Coupled Receptor beta2AR Is Governed by Conformational Exchange Kinetics. Structure 28, 371. 10.1016/j.str.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen A.; Larsen O.; Thiele S.; Rosenkilde M. M. (2014) Biased and g protein-independent signalling of chemokine receptors. Front. Immunol. 5, 277. 10.3389/fimmu.2014.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S.; Kholodenko B. N.; Smit M. J.; Bruggeman F. J. (2015) G Protein-Coupled Receptor Signalling Networks from a Systems Perspective. Mol. Pharmacol. 88 (3), 604–16. 10.1124/mol.115.100057. [DOI] [PubMed] [Google Scholar]
- Grundmann M.; Kostenis E. (2017) Temporal Bias: Time-Encoded Dynamic GPCR Signalling. Trends Pharmacol. Sci. 38 (12), 1110–1124. 10.1016/j.tips.2017.09.004. [DOI] [PubMed] [Google Scholar]
- Irannejad R.; Pessino V.; Mika D.; Huang B.; Wedegaertner P. B.; Conti M.; von Zastrow M. (2017) Functional selectivity of GPCR-directed drug action through location bias. Nat. Chem. Biol. 13 (7), 799–806. 10.1038/nchembio.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K.; von Zastrow M. (2018) Subcellular Organization of GPCR Signalling. Trends Pharmacol. Sci. 39 (2), 200–208. 10.1016/j.tips.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw W. M.; Yamauchi H.; Mead J.; Gowers G. F.; Bell D. J.; Oling D.; Larsson N.; Wigglesworth M.; Ladds G.; Ellis T. (2019) Engineering a Model Cell for Rational Tuning of GPCR Signalling. Cell 177 (3), 782–796 e27. 10.1016/j.cell.2019.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolakou A. E.; Baltoumas F. A.; Stravopodis D. J.; Iconomidou V. A. (2020) Extended Human G-Protein Coupled Receptor Network: Cell-Type-Specific Analysis of G-Protein Coupled Receptor Signalling Pathways. J. Proteome Res. 19 (1), 511–524. 10.1021/acs.jproteome.9b00754. [DOI] [PubMed] [Google Scholar]
- Wang J.; Gareri C.; Rockman H. A. (2018) G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 123 (6), 716–735. 10.1161/CIRCRESAHA.118.311403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J. A.; Yost J. M.; Setola V.; Chen X.; Sassano M. F.; Chen M.; Peterson S.; Yadav P. N.; Huang X. P.; Feng B.; Jensen N. H.; Che X.; Bai X.; Frye S. V.; Wetsel W. C.; Caron M. G.; Javitch J. A.; Roth B. L.; Jin J. (2011) Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U. S. A. 108 (45), 18488–93. 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller D.; Kling R. C.; Skultety M.; Leuner K.; Hubner H.; Gmeiner P. (2014) Functionally selective dopamine D(2), D(3) receptor partial agonists. J. Med. Chem. 57 (11), 4861–75. 10.1021/jm5004039. [DOI] [PubMed] [Google Scholar]
- Weiwer M.; Xu Q.; Gale J. P.; Lewis M.; Campbell A. J.; Schroeder F. A.; Van de Bittner G. C.; Walk M.; Amaya A.; Su P.; L D. O.; Sacher J. R.; Skepner A.; Fei D.; Dennehy K.; Nguyen S.; Faloon P. W.; Perez J.; Cottrell J. R.; Liu F.; Palmer M.; Pan J. Q.; Hooker J. M.; Zhang Y. L.; Scolnick E.; Wagner F. F.; Holson E. B. (2018) Functionally Biased D2R Antagonists: Targeting the beta-Arrestin Pathway to Improve Antipsychotic Treatment. ACS Chem. Biol. 13 (4), 1038–1047. 10.1021/acschembio.8b00168. [DOI] [PubMed] [Google Scholar]
- DeWire S. M.; Yamashita D. S.; Rominger D. H.; Liu G.; Cowan C. L.; Graczyk T. M.; Chen X. T.; Pitis P. M.; Gotchev D.; Yuan C.; Koblish M.; Lark M. W.; Violin J. D. (2013) A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 344 (3), 708–17. 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- Schmid C. L.; Kennedy N. M.; Ross N. C.; Lovell K. M.; Yue Z.; Morgenweck J.; Cameron M. D.; Bannister T. D.; Bohn L. M. (2017) Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 171 (5), 1165–1175 e13. 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James I. E.; Skobieranda F.; Soergel D. G.; Ramos K. A.; Ruff D.; Fossler M. J. (2020) A First-in-Human Clinical Study With TRV734, an Orally Bioavailable G-Protein-Biased Ligand at the mu-Opioid Receptor. Clin. Pharmacol. Drug Dev. 9 (2), 256–266. 10.1002/cpdd.721. [DOI] [PubMed] [Google Scholar]
- Conibear A. E.; Kelly E. (2019) A Biased View of mu-Opioid Receptors?. Mol. Pharmacol. 96 (5), 542–549. 10.1124/mol.119.115956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen M. F.; Wrobel T. M.; Marcher-Rorsted E.; Pedersen D. S.; Moller T. C.; Gabriele F.; Pedersen H.; Matosiuk D.; Foster S. R.; Bouvier M.; Brauner-Osborne H. (2020) Biased agonism of clinically approved mu-opioid receptor agonists and TRV130 is not controlled by binding and signalling kinetics. Neuropharmacology 166, 107718. 10.1016/j.neuropharm.2019.107718. [DOI] [PubMed] [Google Scholar]
- Kliewer A.; Schmiedel F.; Sianati S.; Bailey A.; Bateman J. T.; Levitt E. S.; Williams J. T.; Christie M. J.; Schulz S. (2019) Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 10 (1), 367. 10.1038/s41467-018-08162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A.; Gillis A.; Hill R.; Schmidel F.; Bailey C.; Kelly E.; Henderson G.; Christie M. J.; Schulz S. (2020) Morphine-induced respiratory depression is independent of beta-arrestin2 signalling. Br. J. Pharmacol. 10.1111/bph.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M. C.; Charlton S. J. (2018) Biased Agonism in Drug Discovery-Is It Too Soon to Choose a Path?. Mol. Pharmacol. 93 (4), 259–265. 10.1124/mol.117.110890. [DOI] [PubMed] [Google Scholar]
- Wootten D.; Christopoulos A.; Marti-Solano M.; Babu M. M.; Sexton P. M. (2018) Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19 (10), 638–653. 10.1038/s41580-018-0049-3. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2019) Biased Receptor Signalling in Drug Discovery. Pharmacol. Rev. 71 (2), 267–315. 10.1124/pr.118.016790. [DOI] [PubMed] [Google Scholar]
- Pang P. S.; Butler J.; Collins S. P.; Cotter G.; Davison B. A.; Ezekowitz J. A.; Filippatos G.; Levy P. D.; Metra M.; Ponikowski P.; Teerlink J. R.; Voors A. A.; Bharucha D.; Goin K.; Soergel D. G.; Felker G. M. (2017) Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: a randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur. Heart J. 38 (30), 2364–2373. 10.1093/eurheartj/ehx196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin J. D.; Crombie A. L.; Soergel D. G.; Lark M. W. (2014) Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci. 35 (7), 308–16. 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Costa-Neto C. M.; Parreiras E. S. L. T.; Bouvier M. (2016) A Pluridimensional View of Biased Agonism. Mol. Pharmacol. 90 (5), 587–595. 10.1124/mol.116.105940. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Lefkowitz R. J.; Rajagopal S. (2018) Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discovery 17 (4), 243–260. 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M.; Eichel K.; Avino S.; Zhao X.; Steffen D. J.; Feng X.; Kawakami K.; Aoki J.; Messer K.; Sunahara R.; Inoue A.; von Zastrow M.; Gutkind J. S. (2017) Genetic evidence that beta-arrestins are dispensable for the initiation of beta2-adrenergic receptor signalling to ERK. Sci. Signaling 10 (484), eaal3395. 10.1126/scisignal.aal3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann M.; Merten N.; Malfacini D.; Inoue A.; Preis P.; Simon K.; Ruttiger N.; Ziegler N.; Benkel T.; Schmitt N. K.; Ishida S.; Muller I.; Reher R.; Kawakami K.; Inoue A.; Rick U.; Kuhl T.; Imhof D.; Aoki J.; Konig G. M.; Hoffmann C.; Gomeza J.; Wess J.; Kostenis E. (2018) Lack of beta-arrestin signalling in the absence of active G proteins. Nat. Commun. 9 (1), 341. 10.1038/s41467-017-02661-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell L. M.; Wang J.; Plouffe B.; Smith J. S.; Yamani L.; Kaur S.; Jean-Charles P. Y.; Gauthier C.; Lee M. H.; Pani B.; Kim J.; Ahn S.; Rajagopal S.; Reiter E.; Bouvier M.; Shenoy S. K.; Laporte S. A.; Rockman H. A.; Lefkowitz R. J. (2018) Manifold roles of beta-arrestins in GPCR signalling elucidated with siRNA and CRISPR/Cas9. Sci. Signaling 11 (549), eaat7650. 10.1126/scisignal.aat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott A. (1999) Alliance of US labs plans to build map of cell signalling pathways. Nature 402 (6759), 219–20. 10.1038/46111. [DOI] [PubMed] [Google Scholar]
- Rual J. F.; Venkatesan K.; Hao T.; Hirozane-Kishikawa T.; Dricot A.; Li N.; Berriz G. F.; Gibbons F. D.; Dreze M.; Ayivi-Guedehoussou N.; Klitgord N.; Simon C.; Boxem M.; Milstein S.; Rosenberg J.; Goldberg D. S.; Zhang L. V.; Wong S. L.; Franklin G.; Li S.; Albala J. S.; Lim J.; Fraughton C.; Llamosas E.; Cevik S.; Bex C.; Lamesch P.; Sikorski R. S.; Vandenhaute J.; Zoghbi H. Y.; Smolyar A.; Bosak S.; Sequerra R.; Doucette-Stamm L.; Cusick M. E.; Hill D. E.; Roth F. P.; Vidal M. (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437 (7062), 1173–8. 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Hein M. Y.; Hubner N. C.; Poser I.; Cox J.; Nagaraj N.; Toyoda Y.; Gak I. A.; Weisswange I.; Mansfeld J.; Buchholz F.; Hyman A. A.; Mann M. (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163 (3), 712–23. 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Huttlin E. L.; Ting L.; Bruckner R. J.; Gebreab F.; Gygi M. P.; Szpyt J.; Tam S.; Zarraga G.; Colby G.; Baltier K.; Dong R.; Guarani V.; Vaites L. P.; Ordureau A.; Rad R.; Erickson B. K.; Wuhr M.; Chick J.; Zhai B.; Kolippakkam D.; Mintseris J.; Obar R. A.; Harris T.; Artavanis-Tsakonas S.; Sowa M. E.; De Camilli P.; Paulo J. A.; Harper J. W.; Gygi S. P. (2015) The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162 (2), 425–440. 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Fagerberg L.; Hallstrom B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson A.; Kampf C.; Sjostedt E.; Asplund A.; Olsson I.; Edlund K.; Lundberg E.; Navani S.; Szigyarto C. A.; Odeberg J.; Djureinovic D.; Takanen J. O.; Hober S.; Alm T.; Edqvist P. H.; Berling H.; Tegel H.; Mulder J.; Rockberg J.; Nilsson P.; Schwenk J. M.; Hamsten M.; von Feilitzen K.; Forsberg M.; Persson L.; Johansson F.; Zwahlen M.; von Heijne G.; Nielsen J.; Ponten F. (2015) Proteomics. Tissue-based map of the human proteome. Science 347 (6220), 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Huttlin E. L.; Bruckner R. J.; Paulo J. A.; Cannon J. R.; Ting L.; Baltier K.; Colby G.; Gebreab F.; Gygi M. P.; Parzen H.; Szpyt J.; Tam S.; Zarraga G.; Pontano-Vaites L.; Swarup S.; White A. E.; Schweppe D. K.; Rad R.; Erickson B. K.; Obar R. A.; Guruharsha K. G.; Li K.; Artavanis-Tsakonas S.; Gygi S. P.; Harper J. W. (2017) Architecture of the human interactome defines protein communities and disease networks. Nature 545 (7655), 505–509. 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O.; Stubbington M. J. T.; Regev A.; Teichmann S. A. (2017) The Human Cell Atlas: from vision to reality. Nature 550 (7677), 451–453. 10.1038/550451a. [DOI] [PubMed] [Google Scholar]
- Fabregat A.; Jupe S.; Matthews L.; Sidiropoulos K.; Gillespie M.; Garapati P.; Haw R.; Jassal B.; Korninger F.; May B.; Milacic M.; Roca C. D.; Rothfels K.; Sevilla C.; Shamovsky V.; Shorser S.; Varusai T.; Viteri G.; Weiser J.; Wu G.; Stein L.; Hermjakob H.; D’Eustachio P. (2018) The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46 (D1), D649–D655. 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla J.; McClary K. M.; White K. L.; Alber F.; Sali A.; Stevens R. C. (2018) Opportunities and Challenges in Building a Spatiotemporal Multi-scale Model of the Human Pancreatic beta Cell. Cell 173 (1), 11–19. 10.1016/j.cell.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://reactome.org/ (accessed 2020/03/02).
- https://string-db.org/ (accessed 2020/03/02).
- Tan L.; Yan W.; McCorvy J. D.; Cheng J. (2018) Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. J. Med. Chem. 61 (22), 9841–9878. 10.1021/acs.jmedchem.8b00435. [DOI] [PubMed] [Google Scholar]
- Wilde O.The Importance of Being Earnest: a Trivial Comedy for Serious People; Leonard Smithers and Co.: London, 1898. [Google Scholar]
- https://www.cost.eu/actions/CA18133/ (accessed 2020/03/02).