

Abstract
While cells offer numerous inspiring examples in which membrane morphology and function are controlled by interactions with viruses or proteins, we still lack design principles for controlling membrane morphology in synthetic systems. With experiments and simulations, we show that spherical nanoparticles binding to lipid-bilayer membrane vesicles results in a remarkably rich set of collective morphologies that are controllable via the particle binding energy. We separately study cationic and anionic particles, where the adhesion is tuned by addition of oppositely charged lipids to the vesicles. When the binding energy is weak relative to a characteristic membrane-bending energy, vesicles adhere to one another and form a soft solid gel, a novel and useful platform for controlled release. With larger binding energy, a transition from partial to complete wrapping of the nanoparticles causes a remarkable vesicle destruction process culminating in rupture, nanoparticle-membrane tubules, and an apparent inversion of the vesicles. These findings help unify the diverse phenomena observed previously. They also open the door to a new class of vesicle-based, closed-cell gels that are more than 99% water and can encapsulate and release on demand, and show how to drive intentional membrane remodeling for shape-responsive systems.
Keywords: membrane morphology, lipid bilayer membrane, membrane physics, nanoparticle-membrane interactions, membrane nanotube
Graphical Abstract
Tuning the strength of binding between nanoparticles and lipid membranes controls the membrane deformation process, leading to a new form of closed-cell gel or to a remarkable vesicle-inversion and destruction process.
The lipid-bilayer membrane offers an enormous range of applications because it is thin, flexible, impermeable to most solutes, and fluid-like in its plane.1,2 Its flexibility allows the membrane to curve around binding particles or proteins or viruses, leading to the potential for major shape reorganization. Live cells harness these interactions to tune morphology and function, such as in the bicontinuous structure of the endoplasmic reticulum, protrusions leading to cell mobility,3 or enwrapped objects in phagocytosis or endocytosis.4,5 Despite the enormous progress in the application of synthetic lipid bilayers for encapsulation and delivery, there is considerably greater (and still undeveloped) potential if we can learn how to trigger changes in membrane geometry and topology in synthetic systems. This knowledge would lead to new responsive, bioinspired materials that could modulate morphology and function in complex ways, on demand.
When a single nanoparticle, virus, or protein binds to a membrane, the adhesion can force the membrane to deform. Treating the membrane as a continuum elastic body, deformation is driven by free-energy reduction from binding and opposed by the free-energy increase from bending or stretching the membrane.6–8 Defining the binding free energy per unit area of contact as w, the radius of a spherical particle or virus as a and the membrane bending modulus8 as κ, earlier theory work predicted a crossover from mild deformation to full wrapping of the bound object by the membrane when wa2/κ =2.6,9 On the other hand, if the membrane were subjected to high mechanical tension, τ, this could play the dominant role instead of κ if the dimensionless ratio τa2/κ ≫ 1.10,11 For lipid bilayer membranes having non-covalent interactions (e.g., electrostatic double-layer), particles that are a few nm in size lead to τa2/κ values that are typically small, while wa2/κ can vary from ≪ 1 to ≫ 1. Therefore, for objects of this interesting size scale (which are relevant for biology and nanoparticle applications), deformations should be tunable via w and should range from weak adhesion to partial or full wrapping under common conditions. For individual particles, calculations and simulations6,9,12–14 and experiments support the idea that wa2/κ is the key control parameter.7,15–17
When many particles or viruses are present, cooperativity leads to richer phenomenology. Experiments showed that cooperative interactions lead to in-plane attraction between particles16 and particle clustering,18,19 tubulation or pearling of the membrane,20–23 and internalization of particles within the vesicles.7,15 Similarly, simulations and calculations found hexagonal or chain-like particle aggregates,24,25 budding or tubulation of the membrane,13,23,26–31 or internalization.12,32 Despite the wide range of reported phenomenology and theory, it is still not known how to predict and control these particle-membrane behaviors – especially when many particles are present.
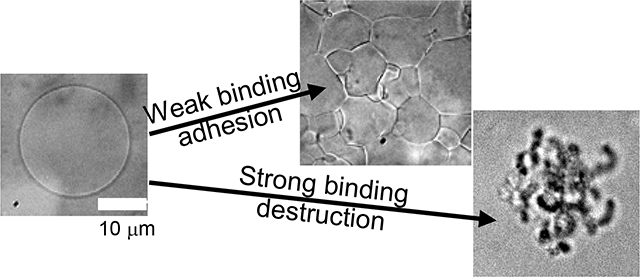
Here we report the results of three different well-defined systems of lipid membrane and nanoparticles that allowed us to tune the interaction strength, w, between the two components. We used giant lipid-bilayer vesicles (10–100 μm) with varied amounts of charged lipid to induce adhesion to oppositely charged nanoparticles. The majority of our studies focused on 6.7-nm-diameter cationic Au-TTMA nanoparticles (Fig. 1A).33 We chose these particles because they have a dense ligand coating, are stable against aggregation, and have a permanent positive charge on the quaternary ammonium group at the ligand terminus. We made the vesicles with a mixture of zwitterionic DOPC and anionic DOPS, so that the molar ratio of DOPS could be tuned to set the binding energy per unit area, w. When the DOPS fraction and w were small, the nanoparticles caused the vesicles to adhere to one another and form a soft but solid gel (Fig. 1B). By contrast, when w exceeded a threshold value, the vesicles were destroyed in a remarkably complex but highly repeatable process that included vesicle shrinkage, invagination, pore formation, runaway tubule formation, and possibly vesicle inversion (Fig. 1C). We also carried out experiments with negatively charged silica nanoparticles mixed with vesicles doped with positively charged DOTAP lipid and found similar results. With this silica system, we investigated two slightly different particle sizes and found that the threshold lipid composition was noticeably lower for the larger particles. Our computer simulations also showed a transition from partial to complete wrapping of nanoparticles and subsequent membrane rupture when the dimensionless ratio wa2/κ exceeded a threshold value of approximately 0.5. The sequence of morphologies leading to destruction was consistent in each case.
Fig. 1.

(A) Schematic overview of giant unilamellar vesicles (GUVs) with controllable anionic charge density exposed to cationic gold nanoparticles (Au-TTMA). The microscope image shows GUVs composed of 96 mol% DOPC and 4 mol% anionic DOPS without nanoparticles. (B) Microscope image of GUVs + nanoparticles that have adhered to one another forming a solid gel. (C) Microscope images of a single GUV undergoing rapid tubule formation and destruction upon Au-TTMA nanoparticle binding. We also report similar behaviors for anionic nanoparticles with cationic vesicles.
The ability to tune the morphology and shape-changing dynamics of vesicles provides a useful experimental model of cell lysis and opens the door to new applications. These findings could be used to create cargo-carrying vesicles with the ability to rupture when bound particles are stimulated34 or, potentially, when w is tuned by an external trigger. These results also show how to engineer soft solid gels that can encapsulate cargo. They may also provide a unified picture for the wide variety of phenomena reported in cells and vesicles, which likely correspond to different regions of a phase space defined chiefly by w, κ, a, and particle concentration.
Results
In this section, we describe the phenomenology of the gel and destruction regimes. Throughout, we focus primarily on the cationic Au-TTMA-nanoparticle results, and then later show a comparison to the anionic silica nanoparticle results. We then describe molecular dynamics simulations that show a similar crossover from weak binding to destruction. Finally, in the Discussion section, we describe the underlying mechanisms and compare to prior work.
Overview of the phenomenology
We studied the response of vesicles in situ after nanoparticles were added to the surrounding suspension. By adjusting the charged-lipid content of the vesicles, we tuned their average surface charge and thereby the adhesion energy per area, w, between the lipid bilayer and the oppositely charged nanoparticles. We took care to match the osmotic strength of the added nanoparticle suspension to that of the vesicle suspension, so that osmotic shock did not play a role in these processes. Figure 1 summarizes the two distinct behaviors that we observed with cationic Au-TTMA particles: adhesion and vesicle-gel formation at low DOPS fraction, and vesicle tubulation and destruction at high DOPS fraction. Remarkably, these two regimes of behavior were separated by a well-defined threshold charged-lipid fraction. Figure 2 shows ‘state diagrams’ for these systems, in the form of images of the steady-state structure as a function of dopant lipid mol%. For Au-TTMA (a = 3.4 nm) and DOPS without added salt, the threshold value was 4 mol% (Fig. 2A). With 4 mol% DOPS, a minority of vesicles survived in the steady state, whereas (for example) with 8 mol% DOPS, a negligible number of GUVs survived in the steady state. We attribute the surviving vesicles to statistical variations in lipid composition of individual GUVs, so that a few individual vesicles may have been below the threshold 4 mol%. The behavior of the GUVs was found to be unchanged if the exterior sugar solution was 180 mOsm/L (as described in the experimental section) or lowered to 170 mOsm/L before exposure to the nanoparticles. Figure 2B shows the Au-TTMA particles with 20 mM NaCl in solution (while still balancing osmolarity inside and outside). Here we found a higher threshold, 5 mol% DOPS. Other observed phenomena such as increased contrast and dark mobile particle aggregates also appeared at threshold, similar to the case without added NaCl. We attribute the threshold shift to screening of the electrostatic attraction, which meant that more DOPS was needed to achieve the same adhesion energy. All observed interactions were consistent with an effective decrease in the electrostatic interaction realized as a shifting of all charge-dependent behaviors uniformly. Below and in SI we describe the electrostatic interaction in terms of charged double-layer theory. This treatment of the role of added salt, though simplistic, captures the main effect. A similar threshold behavior was found for anionic silica particles of two different sizes, with added cationic DOTAP lipid (Fig. 2C,D). In these cases, the thresholds were 8 mol% and 7 mol% for particles with a = 11.3 and 12.6 nm.
Fig. 2.

Bright-field optical micrographs show state diagrams of GUVs with varying lipid composition in the presence of three different types of nanoparticles. The fraction of charged dopant lipid increases from 0 to 9 mol% from left to right. The heavy black lines indicate the boundary between samples that showed adhesion and gel formation vs. those that underwent tubulation and destruction. (A) Cationic Au-TTMA particles with anionic DOPS lipid. (B) Same as (A), but with 20 mM NaCl added (still osmotically balanced), which shifts the threshold. (C) Anionic silica (Ludox AS-30, a = 11.3 nm radius by DLS) with cationic DOTAP lipid. (D) Anionic silica (Ludox SM, a = 12.6 nm by DLS) with DOTAP lipid. Scale bars are 20 μm and each applies to images in the same row.
Weak binding: Vesicle adhesion and gel formation
When the DOPS content in the membrane was < 4 mol%, the Au-TTMA nanoparticles bound to the vesicles’ surfaces without any discernible deformation. The particles were able to spread laterally on the membrane with no observable aggregation. Evidence of particle binding is provided by dark-field optical microscopy, which showed enhanced scattering of light by the bound nanoparticles (Fig. S2). When the concentration of vesicles was high enough so that vesicles touched one another, the membranes adhered to one another owing to the nanoparticles’ forming an adhesive bridge between them. The adhesive contact area grew over a typical time on the order a few minutes before reaching a steady state (Fig. S3).
Even in the absence of DOPS, we still observed nanoparticle binding, consistent with earlier findings that DOPC vesicles have a slightly negative electrostatic (zeta) potential of –9 mV (electrophoretic mobility with 0.1 mM NaCl 35) and that they adhere to cationic particles.23,36
Adhesion led to a network of fluid vesicles, which we call a “vesicle-gel.” In appearance, the approximately polyhedral vesicles (Fig. 1B) resembled bubbles in a dry soap foam, except that here the interior and continuous phases were both aqueous. Figure S4 shows a dark-field image of a steady-state gel, composed of DOPC vesicles, in which strong light scattering is clearly visible at the adhesion sites between vesicles. Nanoparticles accumulated at the vesicle-vesicle junctions because of their ability to bind to both membranes. No systematic variation of morphology was found in images of samples where the DOPS fraction varied between 0 and 3 mol%.
We made larger-volume samples using a food-grade lipid and added cationic polymer (instead of nanoparticles) to induce adhesion. We found that a 50-mL-volume sample of this gel was able to support 270-μm-diameter copper beads against gravity for several hours, which indicates a low-frequency shear modulus and a yield stress of at least a few Pa. (See SI Fig. S5 for further information.) Hence, these materials are solid, albeit quite soft. Their closed-cell structure allows the gel to encapsulate a large volume of liquid within a series of robust interior partitions. The potential as a useful delivery vehicle for drugs, dyes, or reagents will be discussed below.
Strong Binding: The stages of destruction
By contrast, when the DOPS content reached a threshold value (approx. 4 mol%), Au-TTMA nanoparticle binding caused complete vesicle disruption in a multi-stage process (Fig. 3). Although each vesicle differed in detail, the stages were common across hundreds of vesicles in dozens of different samples. We begin with a brief synopsis here, then provide details in the following paragraphs, and propose mechanisms in the Discussion section. First, the vesicle diameter steadily decreased over a typical duration of several seconds to minutes as the membrane became loaded with nanoparticles. Vesicles developed nanoparticle-rich spots that diffused on the surface. Vesicles that shrank faster than a rate of 300 μm2/s also developed a single, long-lived pore, through which the interior solution was expelled. Remarkably, these pores maintained stable diameters in the range of 1–10 μm. Finally, these vesicles underwent a complete destruction, wherein the spherical vesicle rapidly shrank until the folding and compression of the surface caused the vesicle to unfurl into a network of lipid tubules coated in nanoparticles. Vesicles that did not form a visible pore earlier in the process continually shrank and then suddenly ruptured and tubulated. There was no discernible dependence of threshold composition or other behavior on the GUV size. (See Fig. S6, which shows GUVs with diameters 11–25 μm at 6 mol%.) The supplemental movie shows this process in dark-field microscopy (SI). In multilamellar vesicles, the outer layers of the vesicle peeled off one by one as they were attacked by the nanoparticles, until only one inner layer remained. A similar sequence of destruction stages was observed in positively-charged vesicles with negatively charged silica nanoparticles (Fig. S7).
Fig. 3.

Microscope images showing the disruption process above threshold binding strength. (A) Bright-field images of 6 mol% DOPS; (B) Confocal fluorescence images of 5 mol% DOPS with <1 mol% Rh-DOPE. (C) Bright-field image of surface spots in GUV with 6 mol% DOPS. (D-F) Images of vesicles with a long-lasting pore. (D) Bright-field image, 6 mol% DOPS. Interior sugar solution can be seen escaping the pore, as indicated by white arrow. (E) Confocal image, 5 mol% DOPS + 1 mol% Rh-DOPE. (F) Dark-field image highlighting nanoparticles, 4 mol% DOPS.
At the start of the disruption process, the diameter of the vesicles steadily decreased. As shown in Fig. 4, vesicles close to one another tended to shrink at similar rates. Fig. 4A shows vesicles that were close to the site of Au-TTMA nanoparticle addition; for most of the shrinkage process, these two vesicles lost apparent surface area at an average rate of approx. 500 μm2/s. Surprisingly, the appearance of a large pore on the surface of each vesicle had no discernible impact on the shrinkage rate. Fig. 4B shows vesicles that were farther from the point of nanoparticle addition, so that the local nanoparticle concentration was reduced by their diffusive spread throughout the sample. The sharp initial decrease in radius was observed in many vesicles and is attributed to excess area in the initial configuration. Following this rapid decrease, the steady area-shrinkage rates were approx. 35 μm2/s, about 14× lower than in Fig. 4A. In separate experiments, we added nanoparticles with a 14× reduced concentration and found that the average shrinkage rate decreased to 0.004 μm2/s, and the rupture process required an hour or more to complete. These data show that the rate of vesicle shrinkage was strongly correlated with nanoparticle concentration. This point will be discussed below.
Fig. 4.

Measured surface areas over time for vesicles attacked by Au-TTMA nanoparticles. (A) 5 mol% DOPS; nanoparticles were added at t = −5 min. The average rate of area reduction was 500 μm2/s. Both vesicles developed a surface pore (visible at t = 21 s), then gradually inverted through the pore as they shrank. (B) 5 mol% DOPS; nanoparticles were added farther away, at t = −50 min. and the local concentration of nanoparticles was lower than in (A). The average rate of area reduction was approx. 35 μm2/s. The vesicles suddenly ruptured at t ≈ 400 s without having first formed a visible pore. (C) A plot of area shrinkage rates of 13 vesicles and various DOPS composition above threshold. All vesicles that shrank faster than 300 μm2/s developed a pore (upper row of symbols) and none of the slower ones did (lower row of symbols).
As the diameters of the vesicles shrank, spots appeared on the surfaces (Fig. 3C). These spots were always similar in size to the microscope’s resolution limit, so that their true size could not be measured accurately. The spots were bright under dark-field imaging (Fig. S8), indicating that they were enriched in Au-TTMA nanoparticles. We never observed nanoparticle aggregates in solution; they were only found on the vesicle surfaces above threshold. These observations indicate an attractive interaction between particles that was mediated by the deformed membrane. Throughout, the spots remained mobile on the vesicles’ surfaces. As the vesicle shrank, these spots visibly increased in concentration but did not increase in apparent size. Every vesicle that we imaged above threshold DOPS had these dark spots in conjunction with surface shrinking. Nanoparticle clusters were not found in solution; they were only found on the vesicle surfaces. Confocal microscope images show lipid-nanoparticle tubules extending toward the vesicle’s interior (Fig. S11). In the Discussion section, we propose that the vesicles turned inside out during the final stage, so that these tubules extended outward.
Formation of an open, micron-sized pore that persisted for at least several seconds is a striking and unique feature of our results. Figures 3D–F show that these pores are truly open. In Fig. 3D, escape of the encapsulated fluid (175 mOsm/L sucrose) can be seen because it has a different index of refraction than the exterior fluid (87.5 mOsm/L sucrose + 90 mOsm/L glucose), leading to a visible fingering effect. Furthermore, the confocal image Fig. 3E shows an open hole without lipid. A time-series of images of this vesicle shows that the bright lipid particle inside the vesicle was pushed out through that pore (Fig. S9). Observing vesicles that contained smaller vesicles inside them, we found expulsion of the interior contents through the pore (Fig. S10). We found a characteristic ‘pearl necklace’ morphology at the outer rim of each pore, consisting of clearly discernible clusters that surrounded the rim of the pore. The dark-field image of Fig. 3F shows that these clusters were enriched in Au-TTMA nanoparticles, visible by their strong scattering.
Figure 4C shows the rate of area shrinkage for 13 vesicles with various DOPS fractions above 4%. In the plot, the shrinkage rate varied from 20 to 2,000 μm2/s. The plot also shows whether or not each vesicle formed a visible pore. There is a striking pattern: only vesicles whose surface area decreased faster than approximately 300 μm2/s formed a visible pore, regardless of the DOPS content of the vesicle (as long as it was above the threshold). Based on our results from the previous paragraph, we conclude that the particle concentration determined the rate of vesicle shrinkage and this rate, in turn, controlled pore formation.
The final stage of the disruption process was the complete destruction of the vesicle structure, resulting in a network of tube-like structures (e.g., Fig. 3A,B). From the optical images, we estimate that tubules had a typical diameter of approximately 1–2 μm. We found no evidence that the initial vesicle size or DOPS content (as long as it was above the threshold) affected the rate of shrinking of the vesicles or the sizes of the remaining tubule structures. The supplemental movie shows this process in dark-field microscopy (SI). In multilamellar vesicles, the outer layers of the vesicle peeled off one by one as they were attacked by the nanoparticles, until only one inner layer remained (Fig. S12).
Computer simulations of nanoparticle binding
We carried out Brownian dynamics computer simulations of spherical nanoparticles binding to adhesive membranes to explore this system in microscopic detail and establish the mechanisms underlying its behavior. Here we set a = 5 nm and the membrane bending modulus,8 κ, equal to 8.2×10−20 J, appropriate for DOPC.37 Like the experiments, simulations were in the regime of τ ≪ κ/a2 and wa2/κ tunable from 0 to more than 1.
Figure 5 shows the steady-state configurations obtained with increasing particle-membrane adhesion for various surface concentrations (area fractions). When wa2/κ < 0.5, simulations showed that particles adhered to the membrane without membrane tubulation or destruction. In the regime where wa2/κ > 0.7, the simulations show a trend from partial buds to tubules to membrane-rupture as the particle area fraction was increased. Tubules began as a linear cluster of two or more particles lying on the plane of the membrane; the cluster was then enveloped by the membrane and reoriented as a tubule. (See Fig. S13 for snapshots of typical trajectories.) In the intermediate regime, 0.5 < wa2/κ < 0.7, the ruptured configuration was pre-empted by linear arrays of particles. It may be that these linear-array states might eventually nucleate tubules, as has been suggested previously.27
Fig. 5.

(A) Diagram showing the steady-state configurations found in simulations as functions of dimensionless adhesion free energy and particle concentration. The symbols correspond to the states illustrated in (B). Particles are rendered in dark blue and membrane headgroups are in violet.
In our experiments, the particle area fraction was not fixed, but increased over time as more particles bound to vesicles. The simulations’ trend of partial buds, tubules and rupture with increasing particle density therefore correspond closely with the observed process of invagination (tubule formation) and pore formation over time in the experiments.
A key result of the simulations is a well-defined value of wa2/κ = 0.5 that defines a crossover from binding to tubule formation. In most cases (especially above 0.6), the simulations showed membrane rupture. This finding is consistent with the threshold behavior seen in the experiments and explains many of the observed stages of destruction.
Discussion: Proposed mechanisms of vesicle adhesion, destruction, and the crossover
Our results show that vesicle adhesion and destruction were triggered by the binding of the nanoparticles, not by osmotic stress. In all of our experiments, there was a large excess of nanoparticles relative to membrane area. The key parameter was the fraction of DOPS or DOTAP in the membrane, which served to tune the adhesion strength w between particles and the membrane by means of an electrostatic double-layer attraction. According to our simulations, the threshold from adhesion to destruction corresponds to a sharp crossover at the particle-scale from weakly-bound to fully-enwrapped particles. The particle-scale wrapping transition is also consistent with our experimental findings and can explain many aspects of the destruction process. Below threshold, the nanoparticles bound to the membrane, were able to diffuse laterally, and spread throughout the outer leaflet of the membrane until the steady-state surface coverage was attained. Above threshold, the membrane continually enveloped particles and left unbound membrane exposed. Such a process of continuous envelopment should continually generate in-plane strain and force an overall remodeling of the membrane shape. A schematic overview is given in Fig. 6.
Fig. 6.

Illustrations of the adhesion (left) and the multi-stage destruction process (right).
In continuum theory, this wrapping transition for a single spherical particle was predicted from the Helfrich model of the membrane, accounting for large-amplitude deformations where linear superposition fails.6,38 When the membrane tension τ = 0 and the interaction is of short range, the transition is discontinuous and occurs when wa2/κ = 2. Membrane tension is predicted to shift the discontinuous transition to higher w,6 while a finite range of interaction softens the transition.8 The threshold can be reduced below 2 if there are many nanoparticles, as suggested in a theoretical study with three or more particles.30 Our computer simulations with many particles showed a crossover to tubulation and destruction at a considerably lower threshold, wa2/κ near 0.5. The linear particle arrays in our simulations (Fig. 5B) and in ref. 24 suggest a crude but simple approximation, in which a linear particle aggregate is treated as a long cylinder lying in the plane of the membrane. The energy of bending around a cylinder of radius a is 4× smaller than for bending around a sphere of radius a because the membrane curves only in one direction. In the continuum limit and with a finite concentration of bound nanoparticles, this implies a wrapping threshold when wa2/κ = ½. Although this approximation does not account accurately for the details of the membrane shape, it is consistent with the simulations. Whatever is the exact numerical value of the threshold, the continuum theory and our simulations all suggest that the dimensionless combination wa2/κ is the key parameter setting the threshold.
For the Au-TTMA/DOPS system, we roughly estimated how much DOPS would be needed to obtain wa2/κ = 0.5, the threshold indicated by the simulations. The estimate is based on the charged double-layer interaction, treating the dopant lipid as a mean-field charge density. Several assumptions are needed, as described in the SI, but the resulting estimates show reasonable agreement with the measured threshold composition of 4 mol%. These estimates also guide predictions of how parameters such as charge density, salinity and membrane modulus in other systems should affect the threshold composition.
For the silica/DOTAP systems, the overall behaviors were similar but the measured thresholds were different from Au-TTMA/DOPS. Even though the silica particles were larger than the Au-TTMA, the thresholds were higher. In part, this is because of the negative potential of the DOPC, which has to be overcome by added DOTAP. It is also likely that the magnitude of the surface charge density of the silica particles differs from that of the Au-TTMA. We did find, however, that the slightly larger particles (by DLS) had the slightly lower threshold. This finding is consistent with our proposal that the threshold corresponds to a constant value of wa2/κ, so that the threshold w should vary as 1/a2.
As particles bind and are wrapped by the membrane, the projected surface area of the membrane shrinks because of the envelopment of each bound nanoparticle. If each nanoparticle-wrapping event reduces the projected membrane surface area by an amount equal to the surface area of the nanoparticle, 4πa2, a steady area-reduction rate of 500 μm2/s on a 15,000-μm2 membrane (as in Fig. 4A) corresponds to a flux of roughly 200 particles/(μm2•s) binding to the membrane. If the particle flux were limited by their diffusion through water, the flux would be 3ϕD/(4πa3R), where D is the nanoparticle diffusion constant, R is the vesicle radius, and ϕ is the volume fraction of nanoparticles. From the known concentration of added nanoparticles, we verified that the diffusion-limited flux is high enough to account for the measured rate of vesicle shrinkage. This model explains why the area reduction rate depends on the local nanoparticle concentration.
We propose that as the effective surface area shrinks, the interior vesicle volume can only decrease at a rate limited by water permeation through the membrane. If the binding is too fast, then τ should increase and eventually reach the lysis tension, at which point the membrane should form a pore. If the area shrinkage is slow, however, the water permeation can keep pace with the area reduction and the tension stays below lysis; in such cases no pore is expected. In our experiments, long-lasting pores were only observed in vesicles whose projected area shrank at a rate faster than 300 μm2/s (Fig. 4C). Using the reported permeability and lysis tension of DOPC membranes,39 we estimated a crossover shrinkage rate of order 0.1 μm2/s, which is far below the measured value. Moreover, our estimate neglects the osmotic stress that would arise if only water and not sugar can permeate the membrane; this would further slow the efflux of water and further increase membrane tension. This difference suggests that the membranes may be far more permeable to water and possibly sugar than expected, perhaps because of particle binding, as proposed previously.23 The possible change of permeability with particle binding remains an important topic for further research.
Without nanoparticles, tension-induced pores generally close very rapidly40 but with nanoparticles in our experiments, the pores were stabilized by the “pearl necklace” arrangement of particle-lipid clusters at the pore’s rim (Fig. 3F). Since we never observed more than one pore on any vesicle, we conclude that pores allowed rapid expulsion of fluid so thatτ remained below the lysis threshold.
The nanoparticle-rich spots (Fig. 3C) indicate clustering of nanoparticles, most likely because of attractive forces induced by the membrane deformation. Previous simulations showed that membrane-mediated attraction between particles occurs when the particles are strongly bound and highly wrapped.28,41 Recent experiments with micron-scale particles confirmed this effect: weakly bound, partially wrapped particles had negligible lateral interactions, while fully-wrapped particles attracted one another over a distance of 3 particle diameters.16 With many particles present, theory and simulations predict that membrane-mediated attraction can lead to linear aggregates24,25 or compact clusters and tubulation.8,26,41 Our experiments clearly show the latter, while adding new information about the threshold behavior and the multiple steps in the tubulation and destruction process.
To form inward-facing, invaginated tubules (seen in our simulations and experiments, Fig. S11), the particles must reside on the interior, concave surface of the tubule. This configuration likely reduces the bending energy needed to enwrap the particles. Previous experimental20–22 and numerical8,25,27,28 studies of spherical particles or viruses also showed a tendency toward tubules with the particles on the inner, concave surface. Alternatively, it is possible that particle binding leads to a contraction of the outer leaflet of the bilayer, which would also favor concave curvature. Previous studies of cationic and anionic particle binding to phosphatidylcholine (PC) lipid membranes, however, indicated that cationic particles should tend to dilate the lipid layer,42,43 which would more plausibly lead particles to favor positive (convex) curvature. Whatever the mechanism, the tubules invaginated such that particles remained on the concave surface while still remaining exterior to the vesicle.
In the final stage of destruction, each vesicle appeared to “erupt” into a network of particle-membrane tubules. This process was too rapid to see clearly with a confocal microscope but from our images we can identify two possible pathways. In the first possible mechanism, the vesicles turn inside out so that the tubules that had extended inward end up facing outward. Why would the vesicles turn inside out? To answer this question, we note that as tubules grew into the vesicle interior, they raised the interior pressure. This pressure is apparent in the time-series showing forcible ejection of an encapsulated lipid-based particle (Fig. S9). This pressure could therefore force the tubules to emerge through the open pore. In the final configuration, the particles still reside on the concave surface of the membrane tubules. In this state, however, the leaflet of the membrane that was initially on the interior (luminal) side ends up on the exterior side: the membrane has inverted its topology.
As a second possibility, it may be instead that outward-extended tubules very rapidly grow from the rim of the pore, rapidly consuming the vesicles’ surface area. This latter mechanism strikes us as the less likely one, at least with the Au-TTMA systems, because in many cases the pores had already existed for extended periods with no discernible tubules growing from them. It seems to us unlikely that several tubules should emerge rapidly and (essentially) simultaneously after a delay. For the silica/DOTAP system, the images are more suggestive of tubules growing from the rim of the pore, but this remains a topic for future investigation.
We anticipate a similar destruction process whenever small spherical particles are added to the exterior of vesicles, provided that the binding energy exceeds the threshold value. On the other hand, if such particles were added to the interior of vesicles or found their way inside through a pore, the same logic would predict outward-growing tubules (consistent with earlier experiments20,21) during the shrinkage stage and possibly a pore. The final state should also consist of a network of nanoparticle-lipid tubules, as was found here.
For a broader view of the full parameter space, it is useful to compare the present results to earlier findings that nanoparticles23 or proteins44 that bind on the exterior leaflet without wrapping can drive tubules extending outward from the vesicle. This finding was explained by a lateral pressure arising from steric interactions among the particles or proteins, leading to a dilation of the outer leaflet that then forms the convex (outer) surface of the tubule. The previous experimental system23 consisted of cationic particles with DOPC lipids (i.e., in the weak-binding regime) and with high enough particle concentration to induce the lateral pressure. In combination with our results, this suggests that systems could be specifically designed to form either outward- or inward-growing tubules, pores, and inverted structures, depending on particle shape, concentration, and wa2/κ.
Conclusions
In our experiments and simulations, we exposed charged lipid bilayer membranes to oppositely-charged nanoparticles to understand how nanoparticle adhesion can be used to reshape the bilayer surface, a mechanism that could potentially be used to design novel responsive materials. We have successfully developed a membrane-particle system with tunable double-layer interactions, leading to the ability to form an adhesive network of vesicles (a bulk gel) or to drive a remarkable, catastrophic destruction of each vesicle leading to a network of tubules. The crossover between the adhesion/gel regime and the destruction regime was driven by the particle-scale crossover from weak binding/deformation to complete wrapping. With cationic Au-TTMA spherical nanoparticles, this crossover threshold was approximately 4% mole fraction DOPS in sugar solution, or 5 mol% in sugar + 20 mM NaCl. For anionic Ludox silica nanoparticles with no added salt, the crossover threshold was 8% and 7% DOTAP for the two different sizes for particles with a = 11.3 and 12.6 nm. These behaviors were consistent despite the differences in particle size, surface functionalization, and lipid composition. According to our simulations, this threshold corresponds to wa2/κ equal to approximately 0.5.
The gel that we found at low w is a macroscopically large aggregate of vesicles that form a cohesive, closed-cell network. The networks can support weight (copper beads) for many hours, indicating that they have a finite shear modulus and yield stress. These gels are more than 99% water. Their closed-cell morphology is reminiscent of cellular tissue but is unusual among synthetic systems. Since the individual vesicles remain intact within the gel, they should be able to encapsulate multiple species in solution inside the gel. We envisage forming two or more different sets of vesicles, each one encapsulating a different reagent; the vesicles could then be dialyzed, mixed, and then made to form a vesicle gel. The two different species of reagent would not react with one another until the gel is ruptured in some way, causing their release.
Above the threshold lipid composition, nanoparticles were fully enveloped by the membrane, causing the vesicle membrane to be loaded with adhered nanoparticles and ultimately causing destruction of the vesicle. The envelope/destruction regime results in complete and irreversible release of the contents of the vesicle. These results may lead to vesicles that are tailor-made to rupture and release only in response to selected particles (that bind strongly) and not to others. Such a system could be very useful for delivery in myriad contexts.
The results obtained with this tunable system show a unified picture that could explain the wide variety of behaviors reported previously with vesicles exposed to nanoparticles, viruses, proteins or polymers. Under conditions of matched osmotic strength (as here) and initially low tension (τa2/κ ≪1), the deformations are caused by particle adhesion energy per area, which competes with membrane bending stiffness. We found that nanoparticle concentration and membrane permeability do not affect the threshold but do play an important role in the dynamics: if the particle flux is high enough, then the vesicles shrink fast enough to form a long-lasting pore.
Materials and Methods
Giant unilamellar vesicles (GUVs) were prepared by electroformation.45 The majority lipid was the zwitterionic mono-unsaturated 1,2-dioleoyl-sn-glycero-3-phosphocholine (18:1 DOPC; Avanti Polar Lipids). Charged lipids with the same fatty-acid tail (to suppress demixing) were added to induce particle adhesion. Anionic 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (18:1 DOPS; Avanti Polar Lipids) was added when using cationic Au-TTMA particles. Cationic 1,2-dioleoyl-3-trimethylammonium-propane (18:1 DOTAP; Avanti Polar Lipids) was added for experiments with anionic silica particles. In some cases, we added a small amount of headgroup-labeled lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Rh-DOPE; Avanti Polar Lipids). All vesicles reported here were formed in 175 mOsm/L sucrose solution and then diluted with an equal volume of 180 mOsm/L glucose solution, then left for a day to make the vesicles slightly floppy (so that τa2/κ ≪ 1). Where explicitly stated, a controlled amount of NaCl was also added to the exterior solution to test for electrostatic effects.
The cationic nanoparticles have a gold core functionalized with surface ligands consisting of a thioalkyl tetra(ethylene glycol)ated trimethylammonium (TTMA) ligand (Fig. 1A).33,46 The tetra(ethylene glycol) spacer was added to keep the particles stable in suspension. Particles were synthesized using the Brust-Schiffrin two-phase synthesis method47 and then functionalized with TTMA ligands via place exchange reactions.48 The core diameter was 2 nm (transmission electron microscopy), the hydrodynamic diameter was 6.7 ± 0.4 nm (dynamic light scattering, DLS), and the zeta potential in suspension was 18.2 ± 0.8 mV (electrophoretic mobility).33 Anionic particles were silica, Ludox AS-30 and Ludox SM (Sigma-Aldrich). The mean particle radii were a = 11.3 nm and 12.6 nm, respectively (DLS, measured in the same solution conditions as our vesicle experiments).
To observe binding dynamics, we injected vesicles into a long, narrow perfusion chamber mounted on an optical microscope (Fig. S1), then added 5 μL of nanoparticle and sugar suspension (178 mOsm/L glucose + sucrose) into one end of a perfusion chamber. See SI for more information.
We performed molecular dynamics simulations to determine how the particle-membrane adhesion strength changed dynamics and the steady-state configuration. We represented the membrane by the coarse-grained solvent-free membrane model,49 which is computationally tractable while capturing the relevant features of biological membranes. The lipids were represented three beads, one bead for the head and two beads for the tails. There are short-ranged attractive interactions between pairs of tail beads that represent hydrophobic effects, and short-range repulsions between pairs of head beads and head-tail pairs (See SI for details.) Nanoparticles and membrane-head beads interacted through a Lennard-Jones potential, with well-depth εatt determining the strength of the nanoparticle-membrane attraction (which was tuned by salt concentration or lipid composition in the experiments). To represent excluded volume, there were also repulsive interactions between nanoparticles and lipid tail beads and nanoparticle-nanoparticle pairs. Membranes were initially planar, approximating the fact that in the experiments the radii of curvature of the initial vesicles was much greater than a. We initialized a 170×170 nm membrane in the center of a box of height 150 nm. Tension was held near zero. We initialized n nanoparticles in the upper half of the box, so that the nanoparticle area fraction (if all nanoparticles adsorbed) was given by ρnp = nπa2/L2, where L is the lateral membrane dimension. Periodic boundary conditions applied in the plane of the membrane, ensuring that nanoparticles remained on one side of the membrane (unless it ruptured).
Supplementary Material
Acknowledgments
We thank Tom Powers, Adrian Parsegian and Joel Cohen for helpful discussions and we thank Rui Cao and Manisha for their technical assistance. We acknowledge the NSF-funded Materials Research Science and Engineering Center (MRSEC) on Bioinspired Soft Materials (DMR-1420382). IKT acknowledges support through the Bio and Soft Matter Research Training (B-SMaRT) REU site at UMass (DMR-1359191). VR acknowledges the NIH (EB022641). MFH and GRL acknowledge the NIH Award Number R01GM108021 from the National Institute of General Medical Sciences. Computational resources were provided by the NSF through XSEDE computing resources (XStream, Maverick, Bridges, Comet) through award number MCB090163 and the Brandeis HPCC, which is partially supported by the Brandeis MRSEC on Bioinspired Soft Materials (DMR-1420382).
Footnotes
Supporting Information Available
The supporting information includes additional information on the sample cells used, additional images that are cited in the main text, additional details of the simulation methods, an estimate of the threshold composition of DOPS from theory, and a movie showing the disruption process in dark-field imaging mode. This material is available free of charge via the Internet.
Cited References
- 1.van Meer G, Voelker DR & Feigenson GW Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zimmerberg J Membrane Biophysics. Current Biol. 2006, 16, R272. [DOI] [PubMed] [Google Scholar]
- 3.Mellor H The Role of Formins in Filopodia Formation. Biochim. Biophys. Acta-Mol. Cell Res. 2010, 1803, 191–200. [DOI] [PubMed] [Google Scholar]
- 4.Richards DM & Endres RG How Cells Engulf: A Review of Theoretical Approaches to Phagocytosis. Rep. Prog. Phys. 2017, 80, 126601. [DOI] [PubMed] [Google Scholar]
- 5.Johannes L, Parton RG, Bassereau P & Mayor S Building Endocytic Pits without Clathrin. Nat. Rev. Mol. Cell Biol. 2015, 16, 311–321. [DOI] [PubMed] [Google Scholar]
- 6.Deserno M Elastic Deformation of a Fluid Membrane Upon Colloid Binding. Phys. Rev. E 2004, 69, 031903. [DOI] [PubMed] [Google Scholar]
- 7.Le Bihan O et al. Cryo-Electron Tomography of Nanoparticle Transmigration into Liposome. J. Struct. Biol. 2009, 168, 419. [DOI] [PubMed] [Google Scholar]
- 8.Bahrami AH et al. Wrapping of Nanoparticles by Membranes. Adv. Colloid Interface Sci. 2014, 208, 214–224. [DOI] [PubMed] [Google Scholar]
- 9.Spangler EJ, Upreti S & Laradji M Partial Wrapping and Spontaneous Endocytosis of Spherical Nanoparticles by Tensionless Lipid Membranes. J. Chem. Phys. 2016, 144, 044901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deserno M & Bickel T Wrapping of a Spherical Colloid by a Fluid Membrane. Europhys. Lett. 2003, 62, 767. [Google Scholar]
- 11.Li NW et al. Curvature-Driven Migration of Colloids on Tense Lipid Bilayers. Langmuir 2017, 33, 600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruiz-Herrero T, Velasco E & Hagan MF Mechanisms of Budding of Nanoscale Particles through Lipid Bilayers. J. Phys. Chem. B 2012, 116, 9595–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen XM, Tian FL, Zhang XR & Wang WC Internalization Pathways of Nanoparticles and Their Interaction with a Vesicle. Soft Matter 2013, 9, 7592–7600. [Google Scholar]
- 14.Chen LP, Xiao SY, Zhu H, Wang L & Liang HJ Shape-Dependent Internalization Kinetics of Nanoparticles by Membranes. Soft Matter 2016, 12, 2632–2641. [DOI] [PubMed] [Google Scholar]
- 15.Santhosh PB et al. Influence of Nanoparticle-Membrane Electrostatic Interactions on Membrane Fluidity and Bending Elasticity. Chem. Phys. Lipids 2014, 178, 52–62. [DOI] [PubMed] [Google Scholar]
- 16.van der Wel C et al. Lipid Membrane-Mediated Attraction between Curvature Inducing Objects. Sci. Rep. 2016, 6, 32825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietrich C, Angelova M & Pouligny B Adhesion of Latex Spheres to Giant Phospholipid Vesicles: Statics and Dynamics. J. Phys. II 1997, 7, 1651–1682. [Google Scholar]
- 18.Koltover I, Radler JO & Safinya CR Membrane Mediated Attraction and Ordered Aggregation of Colloidal Particles Bound to Giant Phospholipid Veislces. Phys. Rev. Lett. 1999, 82, 1991. [Google Scholar]
- 19.Ramos L, Lubensky TC, Dan N, Nelson P & Weitz DA Surfactant-Mediated Two-Dimensional Crystallization of Colloidal Crystals. Science 1999, 286, 2325. [DOI] [PubMed] [Google Scholar]
- 20.Yu Y & Granick S Pearling of Lipid Vesicles Induced by Nanoparticles. J. Am. Chem. Soc. 2009, 131, 14158. [DOI] [PubMed] [Google Scholar]
- 21.Gozen I, Billerit C, Dommersnes P, Jesorka A & Orwar O Calcium-Ion-Controlled Nanoparticle-Induced Tubulation in Supported Flat Phospholipid Vesicles. Soft Matter 2011, 7, 9706–9713. [Google Scholar]
- 22.Ewers H et al. Gm1 Structure Determines Sv40-Induced Membrane Invagination and Infection. Nat. Cell Biol. 2010, 12, 11–U36. [DOI] [PubMed] [Google Scholar]
- 23.Li S & Malmstadt N Deformation and Poration of Lipid Bilayer Membranes by Cationic Nanoparticles. Soft Matter 2013, 9, 4969–4976. [Google Scholar]
- 24.Saric A & Cacciuto A Fluid Membranes Can Drive Linear Aggregation of Adsorbed Spherical Nanoparticles. Phys. Rev. Lett. 2012, 108, 118101. [DOI] [PubMed] [Google Scholar]
- 25.Saric A & Cacciuto A Self-Assembly of Nanoparticles Adsorbed on Fluid and Elastic Membranes. Soft Matter 2013, 9, 6677–6695. [Google Scholar]
- 26.Reynwar BJ et al. Aggregation and Vesiculation of Membrane Proteins by Curvature-Mediated Interactions. Nature 2007, 447, 461. [DOI] [PubMed] [Google Scholar]
- 27.Saric A & Cacciuto A Mechanism of Membrane Tube Formation Induced by Adhesive Nanocomponents. Phys. Rev. Lett. 2012, 109, 188101. [DOI] [PubMed] [Google Scholar]
- 28.Bahrami AH, Lipowsky R & Weikl TR Tubulation and Aggregation of Spherical Nanoparticles Adsorbed on Vesicles. Phys. Rev. Lett. 2012, 109, [DOI] [PubMed] [Google Scholar]
- 29.Yue TT & Zhang XR Cooperative Effect in Receptor-Mediated Endocytosis of Multiple Nanoparticles. ACS Nano 2012, 6, 3196. [DOI] [PubMed] [Google Scholar]
- 30.Raatz M, Lipowsky R & Weikl TR Cooperative Wrapping of Nanoparticles by Membrane Tubes. Soft Matter 2014, 10, 3570–3577. [DOI] [PubMed] [Google Scholar]
- 31.Xiong K et al. Cooperative Wrapping of Nanoparticles of Various Sizes and Shapes by Lipid Membranes. Soft Matter 2017, 13, 4644–4652. [DOI] [PubMed] [Google Scholar]
- 32.Chaudhuri A, Battaglia G & Golestanian R The Effect of Interactions on the Cellular Uptake of Nanoparticles. Phys. Biol. 2011, 8, 046002. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y et al. The Interplay of Size and Surface Functionality on the Cellular Uptake of Sub-10 Nm Gold Nanoparticles. ACS Nano 2015, 9, 9986–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paasonen L et al. Gold Nanoparticles Enable Selective Light-Induced Contents Release from Liposomes. J. Control. Release 2007, 122, 86–93. [DOI] [PubMed] [Google Scholar]
- 35.Needham D & Zhelev D in Perspectives in Supramolecular Chemistry: Giant Vesicles (eds Pier Luigi Luisi & Peter Walde) John Wiley & Sons, 2007. [Google Scholar]
- 36.Wang L & Malmstadt N Interactions between Charged Nanoparticles and Giant Vesicles Fabricated from Inverted-Headgroup Lipids. J. Phys. D-Appl. Phys. 2017, 50, 415402. [Google Scholar]
- 37.Nagle JF, Jablin MS, Tristram-Nagle S & Akabori K What Are the True Values of the Bending Modulus of Simple Lipid Bilayers? Chem. Phys. Lipids 2015, 185, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deserno M & Gelbart WM Adhesion and Wrapping in Colloid-Vesicle Complexes. J. Phys. Chem. B 2002, 106, 5543. [Google Scholar]
- 39.Olbrich K, Rawicz W, Needham D & Evans E Water Permeability and Mechanical Strength of Polyunsaturated Lipid Bilayers. Biophys. J. 2000, 79, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandre O, Moreaux L & Brochard-Wyart F Dynamics of Transient Pores in Stretched Vesicles. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 10591–10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reynwar BJ & Deserno M Membrane-Mediated Interactions between Circular Particles in the Strongly Curved Regime. Soft Matter 2011, 7, 8567. [Google Scholar]
- 42.Li Y & Gu N Thermodynamics of Charged Nanoparticle Adsorption on Charge-Neutral Membranes: A Simulation Study. J. Phys. Chem. B 2010, 114, 2749–2754. [DOI] [PubMed] [Google Scholar]
- 43.Wang B, Zhang LF, Bae SC & Granick S Nanoparticle-Induced Surface Reconstruction of Phospholipid Membranes. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18171–18175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stachowiak JC, Hayden CC & Sasaki DY Steric Confinement of Proteins on Lipid Membranes Can Drive Curvature and Tubulation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herold C, Chwastek G, Schwille P & Petrov EP Efficient Electroformation of Supergiant Unilamellar Vesicles Containing Cationic Lipids on Ito-Coated Electrodes. Langmuir 2012, 28, 5518–5521. [DOI] [PubMed] [Google Scholar]
- 46.Phillips RL et al. Gold Nanoparticle-Ppe Constructs as Biomolecular Material Mimics: Understanding the Electrostatic and Hydrophobic Interactions. Soft Matter 2009, 5, 607–612. [Google Scholar]
- 47.Brust M, Walker M, Bethell D, Schiffrin DJ & Whyman R Synthesis of Thiol-Derivatized Gold Nanoparticles in a Two-Phase Liquid-Liquid System. J. Chem. Soc., Chem. Commun. 1994, 7, 801–802. [Google Scholar]
- 48.Hostetler MJ, Templeton AC & Murray RW Dynamics of Place-Exchange Reactions on Monolayer-Protected Gold Cluster Molecules. Langmuir 1999, 15, 3782–3789. [Google Scholar]
- 49.Cooke IR, Kremer K & Deserno M Tunable Generic Model for Fluid Bilayer Membranes. Phys. Rev. E 2005, 72, 011506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.